Article Text

Abstract
A diagnosis of demyelination carries important therapeutic and prognostic implications. In most cases the diagnosis is made clinically, and involvement of the histopathologist is largely confined to postmortem confirmation and clinicopathological correlation. However, every now and then, accurate diagnosis of the presence or cause of demyelination before death hinges on the histopathological assessment. Recognition of demyelination depends on an awareness of this as a diagnostic possibility, and on the use of appropriate tinctorial and immunohistochemical stains to identify myelin, axons and inflammatory cells. In biopsy specimens, the critical distinction is usually from ischaemic or neoplastic disease, and the types of demyelinating disease most likely to be encountered are multiple sclerosis, acute-disseminated encephalomyelitis, progressive multifocal leucoencephalopathy and extrapontine myelinolysis. Interpretation of the pathology has to be made in the context of the clinical, radiological and biochemical findings. Freezing of a small amount of fresh tissue allows for later virological studies, and electron microscopy is occasionally helpful for demonstration of viral particles.
- ADEM, acute-disseminated encephalomyelitis
- AHL, acute haemorrhagic leucoencephalitis
- CNS, central nervous system
- CPM, central pontine myelinolysis
- CSF, cerebrospinal fluid
- EPM, extrapontine myelinolysis
- MRI, magnetic resonance imaging
- PCR, polymerase chain reaction
- PML, progressive multifocal leucoencephalopathy
Statistics from Altmetric.com
- ADEM, acute-disseminated encephalomyelitis
- AHL, acute haemorrhagic leucoencephalitis
- CNS, central nervous system
- CPM, central pontine myelinolysis
- CSF, cerebrospinal fluid
- EPM, extrapontine myelinolysis
- MRI, magnetic resonance imaging
- PCR, polymerase chain reaction
- PML, progressive multifocal leucoencephalopathy
The term demyelination describes a loss of myelin with relative preservation of axons. This results from diseases that damage myelin sheaths or the cells that form them. These diseases should be distinguished from those in which there is a failure to form myelin normally (sometimes described as dysmyelination). Although axons that have been demyelinated tend to atrophy and may eventually degenerate, demyelinating diseases exclude those in which axonal degeneration occurs first and degradation of myelin is secondary.
What follows is an approach to the pathological diagnosis of demyelinating diseases before and after death. The emphasis is on distinguishing between various causes of demyelinating disease, differentiating demyelination from other disease processes with which it may be confused, and making best use of limited amounts of tissue to establish the diagnosis when dealing with small biopsy samples. This article covers demyelinating diseases of the central nervous system (CNS) only.
CLASSIFICATION
Demyelinating diseases of the CNS can be classified according to their pathogenesis into several categories: demyelination due to inflammatory processes, viral demyelination, demyelination caused by acquired metabolic derangements, hypoxic–ischaemic forms of demyelination and demyelination caused by focal compression. Some of these distinctions are rather simplistic in that there is overlap in pathogenesis between the entities in the different categories, but the classification provides a conceptual framework that may be useful in accurate diagnosis.
INFLAMMATORY DEMYELINATION
Three diseases fall into this category: multiple sclerosis, acute-disseminated encephalomyelitis (ADEM) and acute haemorrhagic leucoencephalitis (AHL). The commonest of these, multiple sclerosis, is pathologically and pathogenetically heterogeneous, and has been divided according to clinical and pathological features into four main subtypes (classical, acute, neuromyelitis optica and concentric sclerosis) with further subdivision of plaque types on the basis of a combination of morphological and immunohistochemical findings.
Multiple sclerosis
Aetiology, pathogenesis and epidemiology
This is the commonest of the demyelinating diseases. It is thought to be caused by the interaction of multiple genetic and environmental factors. The risk of developing multiple sclerosis is increased 100-fold to 190-fold if an identical twin has the disease, 20-fold to 40-fold in a full sibling, 7-fold to 13-fold in a half-sibling and 5.5-fold in an affected parent (for a review, see Kenealy et al1). The concordance rate is 25–30% among monozygotic twins compared with 2–5% between dizygotic twins with multiple sclerosis.1,2 The strongest evidence for the involvement of environmental factors comes from prevalence and migration studies. The prevalence of multiple sclerosis varies markedly geographically, from below 5/100 000 in many areas of Africa, South America and Asia, to over 100/100 000 in Scotland and parts of Scandinavia and Canada.3,4 Migration before 15 years of age from a high-prevalence to a lower-prevalence area reduces the likelihood of developing multiple sclerosis. Migration to a higher-prevalence area between 11 and 45 years of age increases the risk of developing multiple sclerosis, even above that of the natives.5,6 Genetic studies have shown linkage of multiple sclerosis to the region of the major histocompatibility complex on chromosome 6p21; among northern European populations, this seems to be largely attributable to association with the human leucocyte antigen-DR2 allele.7,8
Pathological similarities between multiple sclerosis and experimental allergic encephalomyelitis, and the detection of T cells reactive against components of myelin, have been interpreted as evidence that multiple sclerosis is an autoimmune disease.9 As evidence to the contrary, other researchers point to a failure to induce demyelination by transfer of patient serum or T cells and a lack of specific immune markers for most forms of the disease.10 Several epidemiological, serological and virological studies have suggested a role for viruses in multiple sclerosis, those most consistently implicated being Epstein–Barr virus and human herpesvirus 6.11,12 Possible mechanisms include direct viral injury to the CNS that results in immunogenic exposure of myelin antigens and subsequent autoimmune damage to myelin, and molecular mimicry between viral antigens and myelin, whereby the immune response that is directed at the virus also results in damage to structurally homologous constituents of the myelin sheath.
Clinical features
Multiple sclerosis affects twice as many women as men. Most patients are between about 15 and 55 years of age at the time of initial presentation. Both younger and older ages of onset are well documented but rare. Acute inflammatory demyelination in a patient <10 years of age is more likely to be due to ADEM.13
The clinical manifestations of multiple sclerosis are wide ranging. Common presenting features include weakness, paraesthesia or focal sensory loss, optic neuritis, diplopia, ataxia and vertigo. Autonomic motor abnormalities of bladder, bowel and sexual function are common. Other manifestations can include painful muscle spasms, trigeminal neuralgia, fatigue and depression, subtle cognitive difficulties, psychiatric disturbances and seizures. Lesions are usually seen on magnetic resonance imaging (MRI), and oligoclonal bands of immunoglobulins on electrophoresis of the cerebrospinal fluid (CSF). Neurophysiological investigation often shows delayed visual or other sensory-evoked responses.
Typically, multiple sclerosis pursues a relapsing and remitting course but it can be progressive from the outset or can become progressive after initial remissions. The interval between relapses is variable. The latent phase between the first manifestation of multiple sclerosis and the first relapse can be many years. Several sets of criteria have been proposed for the clinical diagnosis of multiple sclerosis; at present the criteria most widely used are those of McDonald et al.14
Variants and their pathological features
Classical multiple sclerosis
Plaques of demyelination of varied size and shape can involve cerebral cortex and subcortical white and grey matter, cerebellar white matter, brain stem and spinal cord. Periventricular white matter, optic pathways and spinal cord are often extensively affected. Several schemes have been proposed for subdividing plaques according to disease activity, stage and presumed pathogenesis.15,16 Some of these schemes are quite complex and, although invaluable in shedding light on the mechanisms of demyelination and disease heterogeneity, are primarily of use in research. A simple practical approach relies on a combination of a myelin stain (such as luxol fast blue/cresyl violet or solochrome cyanin; fig 1), a macrophage marker (eg, antibody to CD68) and a stain for axons (eg, Palmgen silver impregnation or immunohistochemistry for neurofilament proteins) to subdivide plaques into the following:

Plaques of demyelination in multiple sclerosis. (A) Active plaque containing sheets of lipid-laden macrophages. Cellularity is particularly marked along the junction with surrounding white matter. (B) Higher magnification through the edge of an active plaque, showing sheets of macrophages and perivascular cuffs of lymphocytes. As is often the case around active plaques, the surrounding white matter is oedematous and slightly hypercellular, and the edge of the plaque quite poorly delineated. (C) Edge of a chronic active plaque. A zone of debris-laden macrophages separates hypocellular plaque (asterisk) from myelinated white matter (top left of figure). (D) Small perivenous plaque; in isolation this appearance could cause confusion with acute-disseminated encephalomyelitis. (E) Shadow plaque, in which myelin staining is reduced but not absent. Arrowheads indicate the edge of the plaque. (F) Chronic inactive plaque in the medulla. Note the preservation of neurones in the inferior olivary nucleus. All sections stained with luxol fast blue and cresyl violet.
-
Active plaques, containing perivascular infiltrates of lymphocytes and macrophages and large numbers of lipid-laden macrophages distributed throughout the plaque parenchyma; the macrophages contain myelin debris, which can be observed with appropriate stains.
-
Inactive plaques, which are hypocellular and densely gliotic, and in which silver impregnation or immunohistochemistry for neurofilament proteins usually shows a reduced density of axons.
-
Chronic active plaques, which are centrally hypocellular and densely gliotic but include a peripheral zone that is densely infiltrated by lipid-laden macrophages, within some of which myelin debris can usually be seen.
-
Shadow plaques, which are sharply circumscribed regions of reduced but not absent myelin staining, due to the presence of thinly remyelinated axons.
Astrocytic gliosis in plaques is usually evident in sections stained with haematoxylin and eosin, but can be observed more obviously by immunolabelling the astrocytes—for example, with antibody to glial fibrillary acidic protein.
A combination of different plaque types is diagnostic of multiple sclerosis. Other features strongly suggestive of multiple sclerosis include plaques of variable size and shape; markedly asymmetrical cerebral, cerebellar or brain stem involvement; and plaques in the cerebral cortex (especially in the subpial region), deep cerebral grey matter or spinal cord. When demyelination is not due to multiple sclerosis, the strongest indication is usually the clinical history. Although a precise diagnosis can usually also be made on the basis of the neuropathological findings at autopsy, this is not the case for small biopsy specimens.
The following are some of the features to bear in mind in evaluating loss of myelin in white matter. Distinction of demyelination from destructive processes such as infarction depends largely on the demonstration of axons within regions of white matter that are devoid of myelin. This distinction is more obvious when grey matter is affected: in multiple sclerosis neurones are preserved (fig 1F), but this is not the case for infarction or other destructive processes that cause loss of myelin. Small, active multiple sclerosis plaques are often centred on venules (fig 1D), and this may suggest ADEM. However, in ADEM the demyelination is generally confined to a much narrower perivenous or perivenular zone that extends in a sleeve-like fashion along the blood vessels; also all the lesions show similar disease activity. A uniform pattern of disease activity is likewise a feature of central pontine myelinolysis and extrapontine myelinolysis, but these have a characteristic clinical history and stereotyped distribution of demyelination (see later) that are unlike those in multiple sclerosis. The leucodystrophies are progressive, usually inherited, disorders of myelin metabolism, and tend to be of onset in childhood and to produce symmetrical, diffuse loss of myelin and degeneration of white matter, sometimes associated with accumulation of metachromatic material or multinucleated cells. The leucodystrophies most likely to be confused with multiple sclerosis are adrenoleucodystrophy and adrenomyeloneuropathy, in both of which there is inflammation and demyelination that can, particularly in the brain stem, appear plaque-like. However, the clinical features and diffuse nature of the rest of the disease should prevent misdiagnosis of multiple sclerosis. Patients with adrenoleucodystrophy and adrenomyeloneuropathy also have high levels of saturated, very long-chain fatty acids in their brain and plasma. Inflammatory demyelination has rarely been reported in patients given 5-fluorouracil or its derivatives.
Acute (Marburg-type) multiple sclerosis
This term has been given to a rare, fulminant variant of multiple sclerosis. It is believed, based on anecdotal evidence, to affect children and young adults usually. Some patients diagnosed with this disease may have had aggressive forms of ADEM.17 This designation may also overlap acute inflammatory demyelination presenting as a space-occupying lesion (see later).18,19
Neuromyelitis optica (Devic’s disease)
This variant is characterised by the development of optic neuritis and acute transverse myelitis within days, weeks or occasionally months of each other. Most patients present with visual loss and subsequently develop paraplegia and sensory loss, but the order may be reversed.
Neuromyelitis optica is pathogenetically distinct from most other types of multiple sclerosis in that the demyelination is antibody dependent and complement mediated.20,21 During the acute disease, the CSF contains large numbers of polymorphonuclear leucocytes. Oligoclonal bands of immunoglobulins are much less often seen in neuromyelitis optica than in other types of multiple sclerosis,22 and in many patients the serum contains autoantibodies with high specificity for the disorder.23
The disease usually causes extensive demyelination of optic nerve and of the affected segments of the spinal cord. In the acute phase, the cord is swollen and includes an infiltrate of foamy macrophages, perivascular neutrophils and eosinophils (fig 2A,B), but relatively few T cells. Immunoglobulins, particularly IgM (fig 2C), and C9neo antigen are often immunohistochemically demonstrable in the vicinity of small blood vessels.20 The vessel walls become thickened and fibrotic. In patients who survive the acute stage, the spinal cord often shows considerable axonal degeneration, possibly because of additional ischaemic damage due to the combination of oedema and microvascular changes. Affected segments of cord are often severely gliotic and atrophic. Multiple sclerosis plaques are usually present elsewhere in the CNS, but may be small and sparsely distributed.

Acute neuromyelitis optica (Devic’s disease). (A) Perivascular accumulation of foamy macrophages in affected cord. Note the hyaline thickening of small blood vessel walls (arrows). (B) Oedematous demyelinated tissue containing scattered eosinophils (arrows). (C) Immunohistochemical demonstration of perivascular immunoglobulin M in demyelinated spinal white matter.
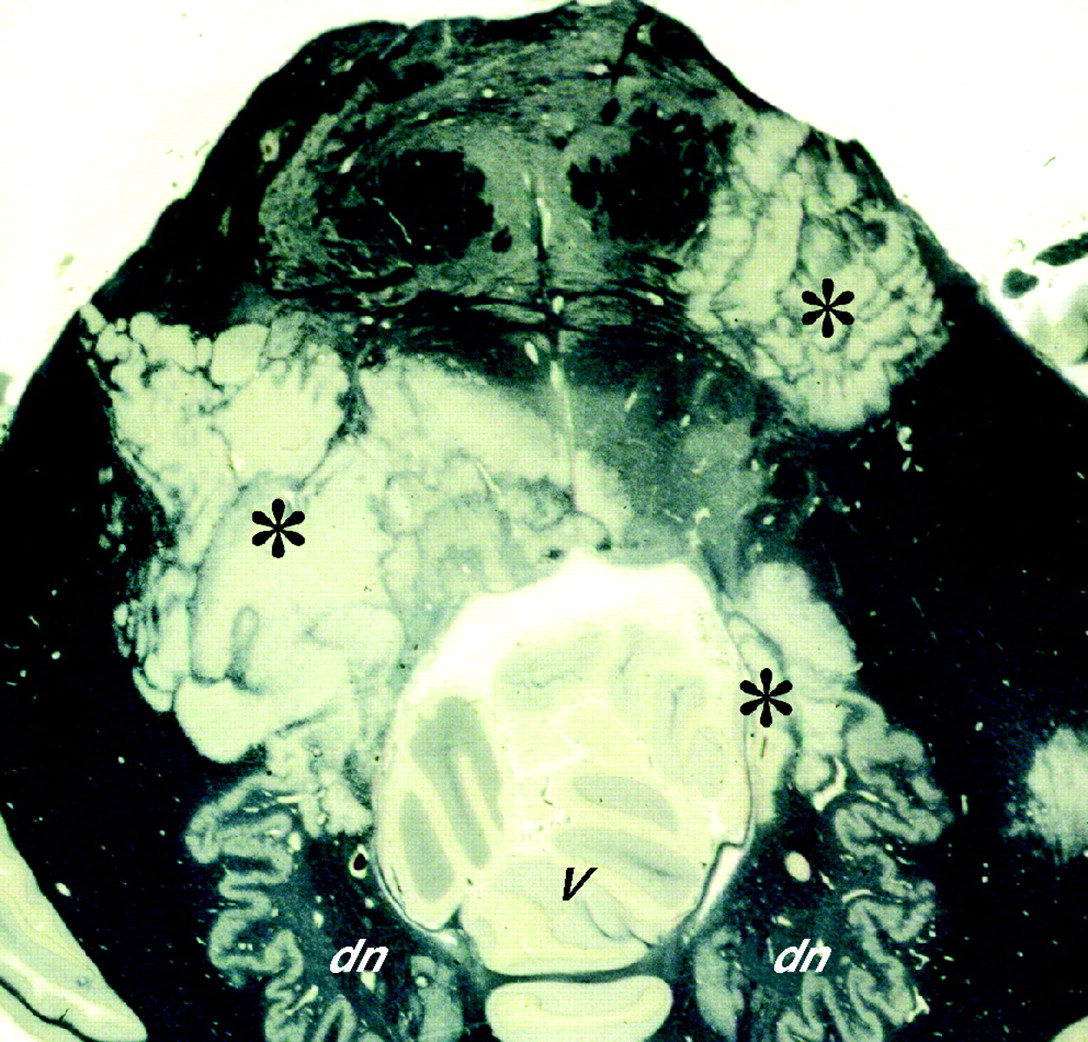
Concentric sclerosis (Baló’s sclerosis)
This rare and unmistakable variant of multiple sclerosis is characterised by lesions composed of alternate bands of demyelinated and myelinated white matter, forming concentric rings or irregular stripes (fig 3). Concentric sclerosis is often rapidly progressive. However, the distinctive lesions may be admixed with other, typical plaques and can occur in chronic multiple sclerosis. Stadelmann et al24 have provided some evidence that the release of mediators of oxidative stress, such as nitrous oxide and superoxide, by macrophages and microglia in the bands of demyelination, causes protective preconditioning of oligodendrocytes in the immediately adjacent white matter, which is therefore preserved from demyelination as the inflammatory process spreads outwards until it reaches the next region with oligodendrocytes that are still susceptible.

Concentric sclerosis (Baló’s sclerosis). A horizontal section through the pons and adjacent part of the cerebellum includes several Baló-type plaques (asterisks), composed of zones of demyelination intersected by irregular stripes of myelinated tissue that has been stained dark grey or black (by Holmes’s method). dn, dentate nucleus; v, vermis.
Acute-disseminated encephalomyelitis
Aetiology, pathogenesis and epidemiology
This inflammatory demyelinating disease mainly affects children. It typically occurs within 3 weeks of infection, vaccination or giving drugs, and is thought to be due to a T cell hypersensitivity reaction.25–27 The infections most often responsible are viral (usually measles, mumps, varicella, rubella or infectious mononucleosis), but Mycoplasma pneumoniae, Campylobacter jejuni, group A streptococci or other bacteria are sometimes the causative agents. Vaccination is a less common antecedent, and drug-induced ADEM is rare. In some patients, no preceding illness or other initiating event is identified. The incidence of recorded ADEM is about 0.4/100 000/year, but this is probably an underestimate as in children with encephalopathy the diagnosis is readily missed unless MRI is carried out.27
Clinical features
The most prominent clinical features are usually ataxia, headache and weakness.25,27 Other manifestations can include vomiting, slurring or impairment of speech, extraocular or other cranial nerve nerve palsies, agitation, seizures, lethargy, delirium and stupor. Approximately 80% of patients make a full recovery. Although ADEM is classically a monophasic disease, relapses have been reported in 5–10% of cases (multiphasic disseminated encephalomyelitis); if relapse occurs on more than one occasion, a diagnosis of multiple sclerosis rather than multiphasic disseminated encephalomyelitis is probably appropriate. A recurrent form of demyelinating perivenous encephalomyelitis can occur in patients with familial erythrophagocytic lymphohistiocytosis.28
Pathological findings
Lesions are usually present bilaterally, although not exactly symmetrically, within the cerebral white matter and brain stem, and sometimes the cerebellum and spinal cord. Small veins and venules in the affected white matter are surrounded by lymphocytes, macrophages and occasional plasma cells (fig 4A). A sleeve of adjacent white matter is oedematous and demyelinated (fig 4B,C). Occasional foci of demyelination may be more extensive. Arteries and arterioles are relatively free of inflammation. There may be small perivascular haemorrhages and some axonal fragmentation. Inflammatory cells may be present in the leptomeninges, and subpial demyelination in the brain stem and spinal cord.

Acute-disseminated encephalomyelitis. (A) Perivenular (arrowheads) accumulation of lymphocytes and foamy macrophages in the corpus callosum. Note the lack of inflammation around an adjacent arteriole (arrow). (B) Staining of an adjacent section with luxol fast blue and cresyl violet shows a sleeve of demyelination and inflammation around a venule (arrowheads). The arrow indicates a small arteriole. (C) In this section axons have been immunostained for neurofilament protein (brown), and a solochrome cyanin counterstain has been used to show myelin (blue). Nerve fibres in the immediate vicinity of the blood vessel show loss of myelin.
Acute haemorrhagic leucoencephalitis
This rare, usually fatal, disease is thought to be a hyperacute variant of ADEM. Like ADEM, AHL may be preceded by viral or M pneumoniae infection. Other rare associations include ulcerative colitis, Crohn’s disease, septicaemia and some drugs. In many patients there is no obvious precipitant.
Clinical features
A typical presentation is pyrexia, headache, vomiting, multifocal neurological deficits and seizures, progressing within 2 or 3 days through drowsiness and coma to death. The outcome is usually death or severe disability, but good recovery has been documented after aggressive medical and surgical reduction of raised intracranial pressure and after giving intravenous immunoglobulin.29
Pathological findings
The abnormalities are often more asymmetrical than those in ADEM and may be confined to a single lobe or hemisphere. Affected parts of the brain are oedematous and contain parenchymal blood vessels that have undergone fibrinoid necrosis and are surrounded by zones of demyelinated, acutely haemorrhagic or necrotic tissue, containing neutrophils, mononuclear inflammatory cells and nuclear debris (fig 5). Ring-shaped and ball-shaped haemorrhages often predominate, and should be distinguished from the scattered petechial haemorrhages that may result from microemboli (especially fat emboli) and coagulopathies.

Acute haemorrhagic leucoencephalitis. Biopsy from a patient with a suspected glioblastoma shows perivascular demyelination, centred on a small blood vessel with a narrow surrounding zone of necrosis. Towards the lower edge of the figure is a collection of mononuclear inflammatory cells and nuclear debris. The white matter includes a small ball-shaped haemorrhage.
VIRAL DEMYELINATION
Progressive multifocal leucoencephalopathy
The principal viral demyelinating disease in humans is progressive multifocal leucoencephalopathy (PML) caused by the papovavirus, JC virus. Approximately 50% of adolescents and 75% of adults have serological evidence of JC virus infection, but it is usually asymptomatic. The virus establishes latent infection in B cells, kidney and possibly CNS.30,31 Reactivation occurs under conditions of impaired cell-mediated immunity—for example, after organ transplantation, in patients with leukaemia or lymphoma or in those with AIDS.32
Clinical features
Patients usually present with insidious onset of neurological deficits that often affect motor function, speech, vision, personality and cognition. Conventional examination of the CSF is usually normal, but JC viral nucleic acids can be shown by polymerase chain reaction (PCR).33,34 MRI typically shows multiple small lesions in the white matter, but these may increase rapidly in size, and occasionally cause mass effect.35 Until a few years ago, PML was usually a rapidly progressive fatal disease. However, successful reversal of impairment of cell-mediated immunity—for example, by administration of antiretroviral drugs to patients with AIDS, can lead to remission of PML.
Pathological findings
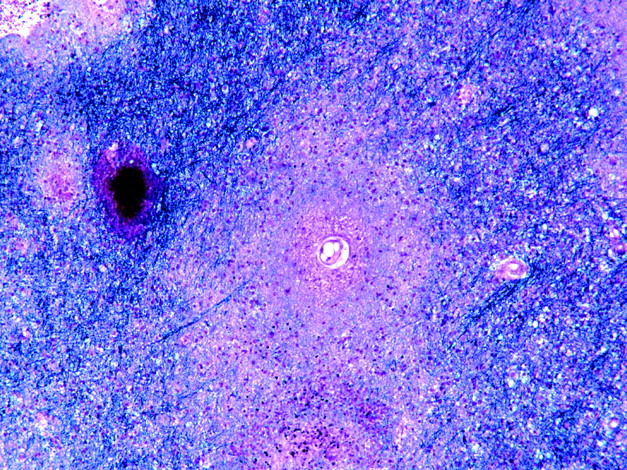
Affected white matter contains multiple foci of demyelination (fig 6A,B). Small round foci of demyelination with good preservation of axons are often present along the junction between cerebral cortex and white matter. Foamy macrophages are usually abundant, but lymphocytes very sparse. In more advanced disease the white matter contains large, confluent foci, some with central cavitation. Towards the periphery of foci of active demyelination are oligodendrocytes with enlarged nuclei that contain homogeneous amphophilic inclusions (fig 6C). The demyelinating lesions may also contain large astrocytes with bizarre, pleomorphic, hyperchromatic nuclei. The oligodendroglial inclusions and bizarre astrocytes are immunopositive for papovavirus (fig 6D). In chronic disease, especially after successful treatment of the underlying immunosuppression (eg, in AIDS), the lesions may lack viral inclusions or antigen. Administration of highly active antiretroviral treatment to patients with AIDS and PML may cause a lymphocytic inflammatory response to the virus and exacerbation of clinical disease.36,37

Progressive multifocal leucoencephalopathy. (A,B) Adjacent sections through the biopsy of a patient with lymphoma having a lesion in the cerebral white matter. Although there is good preservation of axons, as shown by Palmgren silver impregnation (A), a solochrome cyanin myelin stain reveals focal demyelination (arrow in B). (C) At high magnification, some of the oligodendrocytes have enlarged nuclei that show homogeneously amphophilic staining (arrow). (D) Antibody to papovavirus labels the oligodendroglial viral inclusions (arrows) as well as several enlarged astrocytes (arrowheads).
Other viral demyelinating diseases
HIV infection can cause a range of white matter abnormalities, including microglial nodule or multinucleated giant cell encephalitis, diffuse leucoencephalopathy and vacuolar myelopathy. In addition, patients with AIDS receiving highly active antiretroviral treatment may occasionally develop severe inflammatory demyelination that is not due to PML.38–40 There is an intense perivascular inflammatory infiltrate of lymphocytes and macrophages. At least occasional macrophages (many, in some reported cases) are immunopositive for HIV. Viral RNA is usually abundant.
Subacute sclerosing panencephalitis, due to measles virus, causes perivascular inflammation and gliosis of white matter (as well as grey matter). This may be associated with patchy or extensive loss of myelinated fibres. Axonal loss is usually commensurate with that of myelin but, particularly towards the edges of severely affected white matter, axons are sometimes relatively preserved. Measles virus intranuclear inclusion bodies may be seen, but tend to be sparse. In most cases the clinical and serological findings are diagnostic, and viral RNA can be detected by PCR.
Rarely, in patients with AIDS, varicella-zoster virus causes multifocal lesions in the cerebral white matter that are partly demyelinating.41 Viral inclusions and antigen can be seen in oligodendrocytes.
ACQUIRED METABOLIC DEMYELINATION
The most common diseases in this category are central pontine myelinolysis (CPM) and extrapontine myelinolysis (EPM). Very rarely, demyelination occurs in association with chronic alcoholism and malnourishment (Marchiafava–Bignami disease).
Central pontine and extrapontine myelinolysis
CPM is a monophasic demyelinating disease of the pons and lower midbrain. It most often occurs in association with alcoholic liver disease or correction of hyponatraemia (especially if the hyponatraemia is marked and the correction rapid).42 Another clinical context in which CPM is increasingly common is in patients with liver transplant, in whom high ciclosporin levels may have a role.43 CPM rarely occurs in patients who are normonatraemic or hypernatraemic. About 25–50% of patients with CPM also have EPM; this usually affects the cerebellum but may also affect parts of the cerebrum. In up to 25% of patients the demyelination is exclusively extrapontine.
The presentation is usually rapid onset of confusion, limb weakness (often progressing to spastic tetraparesis) and mutism. Other frequent manifestations include ataxia, dysphagia and hypotension. Movement disorders (dystonia, choreoathetosis and parkinsonism) occur in some patients and are probably related to EPM. With good supporting care, most patients with CPM and EPM now survive the acute disease, but many have residual neurological deficits.
Pathological findings
CPM affects the base of the pons and is usually central and symmetrical (fig 7A), but also can be neither (fig 7B). EPM usually affects the cerebellar folia but can affect the lateral geniculate body, capsula externa or extrema, superficial subcortical cerebral white matter, basal ganglia, thalamus, or internal capsule; again, the lesions are usually approximately symmetrical. The lesions are sharply demarcated and, during active disease, contain sheets of lipid-laden macrophages and large numbers of reactive astrocytes. Infiltration by lymphocytes is sparse or absent. Axons and neurones are mostly well preserved. In patients with multiple foci of pontine or extrapontine demyelination, all lesions are of the same age and demyelinating activity (unlike those in multiple sclerosis). The presence of symmetrically spared “islands” of myelinated white matter is common in CPM, but is not a feature of multiple sclerosis. Occasionally, suboptimal fixation can impair staining of myelin in the central part of the pons, so that there appears to be demyelination. However, the lack of foamy macrophages and poorly defined outline of the “demyelination” should prevent confusion with CPM.

Central pontine myelinolysis (CPM). (A) Typical central demyelination in the base of the pons. (B) In this case of CPM, the demyelination (arrows) is neither central nor exactly symmetrical. Both sections stained with luxol fast blue and cresyl violet.
HYPOXIC–ISCHAEMIC DEMYELINATION
Brain tissue generally undergoes necrosis rather than demyelination in response to hypoxia or ischaemia. However, in some circumstances the myelinating oligodendrocyte bears the brunt of the hypoxic or ischaemic damage.
-
In patients with severe small vessel cerebrovascular disease, usually in association with hypertension, the subcortical white matter often shows ischaemic damage. Quite often, these patients develop dementia with superimposed focal neurological deficits that are related to lacunar infarcts. Pathology shows marked arteriosclerosis and arteriolosclerosis, particularly within the deep cerebral white matter, which is rarefied and gliotic and usually includes both cavitating and poorly circumscribed demyelinating lesions. These tend to be hypocellular, but may contain small to moderate numbers of foamy macrophages. Lymphocytic inflammation is not a feature.
-
Rarely, global brain hypoxia due to cardiac arrest, asphyxia or depression of cardiorespiratory function by drug overdose causes a leucoencephalopathy with relative sparing of grey-matter structures.44 The damage is predominantly necrotic, but there may also be some demyelination.
-
Exposure to carbon monoxide, which causes hypoxaemia by binding to haemoglobin with 200-fold greater affinity than oxygen, can cause damage to white matter as well as grey matter structures such as the globus pallidus. White matter damage becomes evident only in patients who survive the acute intoxication. The likelihood of sustaining this damage depends on the level and duration of exposure. The subcortical U fibres tend to be spared. The deep cortical white matter is diffusely rarefied and gliotic, due to a mixture of axonal degeneration and demyelination (fig 8). As in small vessel cerebrovascular disease, the demyelinated white matter may contain foamy macrophages but shows depletion of oligodendrocytes.
-
Mitochondrial toxins, such as cyanide45,46 and hydrogen sulphide,47,48 are very rare causes of “histotoxic” hypoxia. Demyelination resembling that in multiple sclerosis can also occur in those subtypes of the mitochondrial disease Leber’s hereditary optic neuropathy that are caused by the G11778A49,50 or T14484C mutation51 in mitochondrial DNA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
White matter damage in carbon monoxide poisoning. The deep white matter appears rarefied, but the subcortical U fibres are relatively spared.
COMPRESSION-INDUCED DEMYELINATION
This is rarely encountered in routine diagnostic neuropathology. The best documented example of compression-induced demyelination is that responsible for many cases of trigeminal neuralgia and due to compression of trigeminal nerve root fibres by an overlying artery or vein, close to the zone of entry of the nerve root into the pons. This produces a focal region of non-inflammatory demyelination, up to about 2 mm across, in the proximal (CNS) part of the nerve root where the compression has occurred.52 In this there are often groups of demyelinated fibres in close juxtaposition, which has been suggested to allow non-synaptic, direct (ephaptic) transmission of nerve impulses from one to another.
APPROACH TO THE DIAGNOSIS OF DEMYELINATION IN BIOPSY SPECIMENS
Make sure you are aware of the clinical, radiological and laboratory features
Accurate interpretation of the pathological findings in demyelination depends to a large extent on knowing the results of clinical, radiological and other laboratory investigations. This is particularly important when it comes to interpreting and making best diagnostic use of the biopsy material. In addition to the age of the patient, signs and symptoms, the course of disease, presence or absence of a preceding illness, number and distribution of lesions within the CNS, electrolyte concentrations, and results of CSF examination may all be relevant in making the diagnosis.
Acute demyelination may present as a rapidly expanding space-occupying lesion
Multiple sclerosis and ADEM can simulate an intrinsic brain neoplasm, both clinically and radiologically.18,19,53 The pathological findings can also cause confusion, as astrocytes in active demyelinating lesions often have strikingly enlarged, pleomorphic nuclei and can even contain mitotic figures. To avoid misdiagnosis (a) always bear in mind the possibility of demyelination; (b) review the clinical, radiological and laboratory findings with the relevant clinicians; (c) stain for myelin (the solochrome cyanin stain is quick and simple; luxol fast blue/cresyl violet is a bit more time-consuming but provides better cytological detail) and axons (either by immunohistochemistry for neurofilament proteins or by silver impregnation such as the Palmgren method); and (d) stain for macrophages (eg, with antibody to CD68)—foamy macrophages, in particular, are a constant feature of active demyelination but are rarely numerous and widely distributed in tumours.
Demyelination occasionally occurs at the edge of neoplastic lesions
Occasionally, malignant glial tumours, or rarely other primary or secondary neoplasms, are surrounded by a zone of demyelination and reactive changes that is the only tissue included in a biopsy specimen. This is most likely to be a problem in dealing with small biopsy materials, in which case one should be particularly cautious in making a diagnosis of demyelinating disease. Repeat neuroimaging after the procedure may be useful in confirming the site of biopsy. Rarely, glial tumours have arisen within lesions of multiple sclerosis.54–57
Make best use of the tissue
If warned about the possibility of demyelination, be sure to freeze some of the fresh tissue and, ideally, also fix some for possible electron microscopy. Once it has been established that the biopsy shows demyelination rather than tumour or some other disease, the frozen tissue can be used to look for viral DNA or RNA. Electron microscopy remains useful for showing virus particles—particularly in suspected PML. Formalin-fixed paraffin wax-embedded tissue is suitable for most of the other histological and immunohistochemical techniques that are applicable to the diagnosis of demyelinating disease. The main tinctorial and immunohistochemical methods have been described earlier. Several other antibodies are used to identify oligodendrocytes and to characterise the precise type of demyelination in multiple sclerosis. These are rarely needed for routine diagnostic work; however, antibodies to myelin proteins are invaluable for demonstrating demyelination within the grey matter. The combination of neurofilament immunohistochemistry combined with a solochrome cyanin counterstain, on 10–12-μm sections, is sometimes helpful, especially for biopsies.
If in doubt, seek further advice
Befriend a neuropathologist.
Take-home messages
-
Make sure you are aware of the clinical, radiological, and laboratory features.
-
Think of demyelination.
-
In the case of biopsies, do not rely entirely on paraffin histology: freeze some of the tissue, and fix some for possible electron microscopy.
-
If demyelination is a possibility, use specific stains for axons and myelin, not only haematoxylin and eosin.
-
If in doubt, seek advice (before cutting through the block).
REFERENCES
Footnotes
-
Competing interests: None.