Article Text

Abstract
Background Cytogenetically visible chromosomal translocations are highly informative as they can pinpoint strong effect genes even in complex genetic disorders.
Methods and results Here, we report a mother and daughter, both with borderline intelligence and learning problems within the dyslexia spectrum, and two apparently balanced reciprocal translocations: t(1;8)(p22;q24) and t(5;18)(p15;q11). By low coverage mate-pair whole-genome sequencing, we were able to pinpoint the genomic breakpoints to 2 kb intervals. By direct sequencing, we then located the chromosome 5p breakpoint to intron 9 of CTNND2. An additional case with a 163 kb microdeletion exclusively involving CTNND2 was identified with genome-wide array comparative genomic hybridisation. This microdeletion at 5p15.2 is also present in mosaic state in the patient's mother but absent from the healthy siblings. We then investigated the effect of CTNND2 polymorphisms on normal variability and identified a polymorphism (rs2561622) with significant effect on phonological ability and white matter volume in the left frontal lobe, close to cortical regions previously associated with phonological processing. Finally, given the potential role of CTNND2 in neuron motility, we used morpholino knockdown in zebrafish embryos to assess its effects on neuronal migration in vivo. Analysis of the zebrafish forebrain revealed a subpopulation of neurons misplaced between the diencephalon and telencephalon.
Conclusions Taken together, our human genetic and in vivo data suggest that defective migration of subpopulations of neuronal cells due to haploinsufficiency of CTNND2 contribute to the cognitive dysfunction in our patients.
- Chromosomal
- Copy-number
- Molecular genetics
- Memory Disorders
- Cell biology
Statistics from Altmetric.com
Introduction
In a small subset of patients, apparently balanced structural chromosome rearrangements may associate with neurocognitive problems ranging from mild (attention deficit hyperactivity disorder, dyslexia) to severe (within the autism spectrum disorders and intellectual disability),1 suggesting positions for susceptibility genes resulting from cryptic abnormalities in the vicinity of the breakpoints, gene disruption at the breakpoint or position effects.2 Fine mapping of chromosome rearrangements has often resulted in the identification of novel candidate genes associated with different neurodevelopmental symptoms,3 ,4 and two of the dyslexia candidate genes, DYX1C1 and ROBO1, were identified by this approach.5 ,6
The armadillo repeat family member CTNND2 (delta catenin) is expressed almost exclusively in the brain7 and is a component of the adherens junction complex.8 ,9 The disregulation of cell–cell interactions at these junctions by cadherins and cadherin-associated proteins has been shown to be involved in a wide spectrum of genetic disorders affecting various organ systems from the skin to the central nervous system (CNS). In the CNS, deletion of several cadherin and cadherin-related genes has been linked to various cognitive disorders, such as autism, schizophrenia and bipolar disorder (for a review, see El-Amraoui and Petit10). Most recently, the protocadherins, FAT4 and DCHS1, have been linked to Van Maldergems syndrome,11 an autosomal disorder with intellectual disability and impaired cortical development as one of the characteristics. In humans, CTNND2 is localised on chromosome 5p15.2, in a genome region associated with the mental retardation syndrome, Cri-du-Chat, and recently, Asadollahi et al12 reported one patient and retrieved a further four from the literature and online databases with intragenic heterozygous exonic deletions of CTNND2 and mild intellectual disability. The importance of CTNND2 in neurocognitive functions has previously been demonstrated by multiple in vivo and in vitro studies. Knockout mice show impaired spatial learning behaviour13 despite normal basal levels of synaptic transmission and synaptic ultrastructure. At birth, these mice had normal dendritic complexity, spine density and cortical responsiveness, but they rapidly experienced progressive dendritic retraction, a reduction in spine density and stability, and 5 weeks after birth, a reduced cortical responsiveness.14 ,15 More recently, transient palmitoylation of Ctnnd2 has been shown to be increased following enhanced synaptic plasticity, increasing its interaction with cadherin at the synapse.16 Knockout mice also show an increase in spine and synapse density.17 While other studies have suggested a potential role for CTNND2 in neuronal migration based on both the dynamic expression pattern of Ctnnd2 in the mouse cortex14 and in vitro studies where overexpression led to an increase in motile cell behaviour.9 ,18
Here, we use a combination of low coverage massive parallel whole-genome sequencing and microarray analysis to identify loss-of-function mutations in CTNND2 in three individuals from two unrelated families who show borderline to moderate intellectual dysfunction and specific problems with reading. We also demonstrate that a common CTNND2 polymorphism affects both phonological ability and white matter volume in normally developing children and young adults. Finally, transient knockdown in zebrafish embryos result in ectopic neurons in the zebrafish forebrain.
Materials and methods
Participants
Written informed consent was obtained for all participants, and all protocols were approved by the local ethical boards in Stockholm (Sweden).
Patients 1 and 2
Patient 1, a woman, was the first and only child to non-related parents (figure 2). Her mother (patient 2) had previously had one early miscarriage and experienced an intrauterine death in week 38 of her gestation. No karyotyping had been performed. The maternal grandmother had had two early miscarriages and committed suicide. Some psychiatric problems were also described in a sister of the maternal grandmother, but otherwise, the family history was uneventful. Prenatal karyotyping in patient 1 revealed two apparently balanced translocations, also present in the mother (patient 2). The translocations were not present in the maternal grandfather or the maternal aunt; however, the maternal grandmother was not available for analysis. Phenotypical data are summarised in table 1.
Clinical symptoms of patients with structural abnormalities affecting CTNND2
Patient 3
Patient 3 is a boy of central Asian origin, possibly with distantly related parents originating from a small community of 5000 individuals. He is the fourth child and he has five healthy siblings (four brothers and one sister). There are no other familial cases with a similar phenotype. The family moved to Sweden when the boy was 11 years old. The mother and the siblings were not available for detailed clinical assessment, but they are reported to be healthy and with a normal cognitive development. Phenotypical data are summarised in table 1.
The Nynäshamn cohort
A cohort of 76 (41 males and 35 females) typically developing children and young adults aged 6–25 years from the population registry in Nynäshamn, Sweden, were included in our behavioural imaging longitudinal study. The sample recruitment was done randomly with equal gender distribution in nine age groups (6, 8, 10, 12, 14, 16, 18, 20 and 25 years). The exclusion criteria were: first language other than Swedish, vision or hearing impairment and neurological or neuropsychiatric disorders with exception of dyslexia or ADHD. Based on available reports of parents, in 88.7% of the cases, both the participants and their parents were born in Sweden, 9.3% had at least one parent born outside Sweden but within Europe and the remaining 2% had one or both parents born outside Europe. Informed consent was provided by the participants or by the parents for children younger than 18 years of age. The study was approved by the local ethics committee of the Karolinska University Hospital, Stockholm, Sweden. The longitudinal data collections for both behavioural and imaging assessments were conducted on three time-points, each 2 years apart.
Mate-pair sequencing
To pinpoint the exact positions of the chromosome breaks, we used whole-genome sequencing of the index patient. Libraries were prepared using Illumina's Nextera Mate-Pair Sample Preparation Kit according to the manufacturer's instruction for a gel-free preparation of 2 kb effective insert size library (size distribution mode 2 kb). The libraries were sequenced on an Illumina 2500 sequencer (2×100 bp) resulting in a total of 113 137 889 sequence pairs and 226 275 778 reads, giving an average raw coverage depth of 5×. Raw sequence reads were base-called using CASAVA RTA 1.8. Following Illumina guidelines for mate-pair postprocessing (http://res.illumina.com/documents/products/technotes/technote_nextera_matepair_data_processing.pdf), adapter sequences were removed using Trimmomatic V.0.32. The remaining pairs were aligned to the hg19 human reference genome sequence using bwa 0.7.4-r385 resulting in a 3× mapping coverage. After linker removal, 62% of total reads remain (n=140 133 690/226 275 778) and 58% of total reads were mapped (n=131 856 943/226 275 778). Read mapping was processed using in-house software (https://github.com/vezzi/FindTranslocations) implementing a sliding window analogue of a previously published procedure.19 Samtools20 and custom awk scripts were used to process coverage tracks for visualisation with Circos.21 We previously mapped the four chromosomal breakpoints with FISH to 81–218 kb windows,22 and this information was helpful in optimising the bioinformatic analysis.
Breakpoint junction PCR
Breakpoint PCR was performed by standard methods using phusion taq (Thermo Scientific, Pittsburgh, Pennsylvania, USA) and run out on an agarose gel. Specific products not present in control samples were Sanger sequenced at the KI gene core facility. Primers are given in online supplementary table S1.
Affymetrix SNP array
The CNV detection in patient 3 was performed with the Affymetrix (Santa Clara, California, USA) whole-genome human SNP 6.0 array containing 1.8 million probes with an average spacing of 3.8 kb. All experiments were performed according to the manufacturer's recommendations with minor modifications. The data analysis was performed using the Genotyping Console and the chromosome analysis suite software.
Genotyping of the Nynäshamn cohort
Extraction of genotyped SNPs overlapping with the genomic region of CTNND2 (RefSeq annotation, hg19) from whole-genome human SNP 6.0 array (Affymetrix) data, pruning and filtering was performed using PLINK V.1.07.23 To remove redundant markers, we selected the SNPs overlapping CTNND2 that also fulfilled the criteria of Hardy–Weinberg equilibrium >0.001, minor allele frequency >10% and pair-wise linkage disequilibrium of r2<0.3. The pruning process in PLINK was made using the command–indep–pairwise with a pairwise genotypical correlation of SNPs in a sliding window of 50 SNPs shifting five SNPs in every step. After pruning, 37 out of 267 SNPs remained.
Image acquisition and processing
T1-weighted spin echo scans were collected using a three-dimensional (3D) magnetisation prepared rapid gradient echo sequence with repetition time (TR)=2300 ms, echo time (TE)=2.92 ms, field of view of 256×256 mm2, 256×256 matrix size, 176 sagittal slices and 1 mm3 isotropic voxel size. Generalised autocalibrating partially parallel acquisition with acceleration factor of 2 was used to speed up the acquisition.
Scanning was repeated three times with the same parameters. Voxel-based morphometry was performed on the structural data collected across all three rounds of data using statistical parametric mapping (SPM8), Diffeomorphic Anatomical Registration Through Exponentiated Lie Algebra toolbox. This method segmented the brain into grey matter, white matter and cerebrospinal fluid. The white matter-segmented images were then smoothed with an 8 mm Gaussian kernel to be used in SNP association assessments.
SNP association analyses
As the underlying cause of the difficulties in dyslexia often can be traced to phonological processes,24 ,25 we assessed the effect of genotype on performance in a phonological choice task. This task involves non-word and word reading (and the child should decide which was the word), which requires phonological decoding of the non-words. The test is adjusted for age groups of 6–10 years and ≥12 years, respectively. Basic association analysis was initially performed on the first assessment of performance data using PLINK V.1.0723 with sex and handedness as covariates. Three SNPs, rs2561622 (for group 6–10 years) together with rs16901339 and rs26149 (both ≥12 years) were found in significant association with phonological choice task and were selected for a subsequent association analysis using the total available data with phonological choice task performance assessed three times, 2 years apart.
Whole-brain exploratory analysis was then performed on white matter-segmented images to find the white matter-specific regions associated with the variations in the CTNND2 SNP associated with the phonological processing. The SNP was entered as a main factor in a flexible factorial design second-level SPM analysis (http://www.fil.ion.ucl.ac.uk/spm/software/spm8) that modelled whether the main effects were from the same or different participants by including subject and testing round as factors. The model included the participants with and without repeated measures. Age, sex, handedness and total white matter volume were entered as covariates. SNP interactions with age and sex were also added to the model.
Fish maintenance and lines used
In order to analyse a subpopulation of forebrain neurons, an available isl:GFP line was used. For in situ hybridisations, wildtype TL lines were used. All fish were maintained on a 14 h day/night cycle at the Karolinska Institute zebrafish core facility. Embryos were produced via light-induced spawning and raised at 28°C.
Morpholino knockdown of CTNND2 orthologues
Splice-blocking morpholinos (MO) were used to knockdown ctnnd2a and ctnnd2b. For each of the duplicate copies, two non-overlapping splice blockers were used to control for specificity of the observed phenotype. For ctnnd2a, MOs were used to target exon4/intron4 (ctnnd2a MO1; see online supplementary table S3) or exon2/intron2 (ctnnd2a MO2; online supplementary table S3). For ctnnd2b, MOs were designed against exon3/intron3 (MO1; online supplementary table S3) or intron2/exon3 (ctnnd2b MO2; online supplementary table S3). A standard control MO (cont MO) that has been shown previously to have no toxic effects in zebrafish was used as a control for MO dose (Genetools, Std cont MO; online supplementary table S2). MOs were diluted to desired concentrations and injected into newly fertilised embryos at the one-cell stage following standard methods26 and raised to the desired stage.27 All injections were repeated at least twice. The efficacy of the MOs was confirmed by sequencing PCR products amplified from cDNA samples of MO injected and uninjected wild-type Tupfel longfin zebrafish embryos (online supplementary figure S2).
Confirmation of splice blockers
In order to confirm mis-splicing of ctnnd2a and ctnnd2b, RNA was extracted from fish at 2 dpf using Trizol as per manufacturer’s (Invitrogen) instructions. Random hexamer-primed cDNA was synthesised using Superscript III First strand synthesis kit (Invitrogen) and used in PCR reactions. Primers are given in online supplementary table S1.
Immunofluorescence
Embryos were fixed in 4% (w/v) paraformaldehyde (PFA) overnight at 4°C or for 2 h at RT, and brains were dissected using fine forceps. The brains were subsequently processed for immunofluorescence as described previously28 using an anti-HuC antibody (cat #A21271 life technologies, Carlsbad, California, USA) or antiacetylated tubulin (cat# T6793; Sigma) and a goat antimouse Alexa 594 secondary antibody (cat # A11032 Life Technologies, Carlsbad, California, USA), mounted in Gelvatol and imaged.
In situ hybridisation
Eight hundred and sixty base pairs of CTNND2b were amplified from zebrafish cDNA using the specific primers (see online supplementary table S1), and products were subsequently gel-purified and cloned into pGEMT-easy (Promega). PCR templates were amplified from pGEMT-easy clones using T7 and Sp6 primers to facilitate incorporation of RNA polymerase binding sites. The resulting PCR products were then used as templates in a Dioxigenin-labelling reaction (F Hoffman-La Roche, Basel, Switzerland) to synthesise RNA antisense and sense control probes using a T7 or SP6 RNA polymerase. Whole-mount in situ hybridisation was carried out as previously described.29
Confocal imaging
Embryos fixed in 4% PFA were mounted rostrally in gelvatol mounting medium and imaged using an inverted Olympus FV1000 CSLM. Acquired Z-stacks were compiled using Image J and analysed for the presence of ectopic isl:GFP neurons. The significance in difference of phenotypical penetrance was analysed using the χ2 test both in the separate experimental groups and in the pooled samples. Ectopic isl:GFP cells between the telencephalon and diencephalon at the region of the optic recess were subsequently counted and compared using a standard t test; 3D reconstruction of images was carried out using IMARIS software.
Results
Identification of two balanced translocations in a mother and daughter, both with borderline intellectual disabilities
This study was initiated when a prenatally identified carrier of two balanced reciprocal chromosomal translocations, t(1;8)(p22;q24) and t(5;18)(p15;q11) (patient 1; figures 1A, B and 2A), turned out to have learning problems within the dyslexia spectrum, attention deficit, speech delay, myopia, normal growth and recurrent upper airway infections. This prompted us to ask whether one of the chromosomal breakpoints had disrupted a locus important for cognitive development. Both translocations were inherited from the mother (patient 2) who presented with a similar phenotype, including learning difficulties, normal growth, no obvious dysmorphic features and refraction error (figure 2A).

Illumina Nextera mate-pair whole-genome sequencing of a patient with two reciprocal chromosomal translocations. (A) and (B) Two chromosomal breakpoints were characterised that result in two balanced translocations, t(1;8)(p22;q24), t(5;18)(p15;q11). The karyotype of the first translocation is shown in (A), and the second in (B) with the aberrant chromosomes to the right. (C) The circle diagram illustrates the multiple reads supporting the location of the genomic breakpoints. The locations of CTNND2 and ZSCAN30 are shown. (D) Direct sequencing of the t(1;8) junction revealed multiple sequence homologies (bold text). Additionally, 7 bp were deleted from chromosome 1 directly adjacent to the breakpoint. The 1/8 junction sequence is marked in blue and 8/1 in green. Correlations with chromosome 1p22 (top) and chromosome 8q24 (bottom) are indicated by vertical lines. Sanger sequencing traces are shown in (E) (1/8) and (F) (8/1). (G) The t(5;18) chromosomal junction as in (A). The sequence may be interpreted in two different ways: (1) a 1 bp microhomology is present at the junction (pink) and at chr18:32849313 a novel single nucleotide variants (SNV) is found in the patient (purple) or (2) a 2 bp duplication has occurred on chromosome 5 (chr5:11291110-11291111) and a deletion of 2 bp on chromosome 18 (chr18:32849313-32849314) framed in black. The top track is chromosome 5p15, the middle the junction sequence and the bottom track chromosome 18q11 as above. Sanger sequencing traces are shown in H (5/18) and I (18/5), the novel SNV is annotated with a red star.
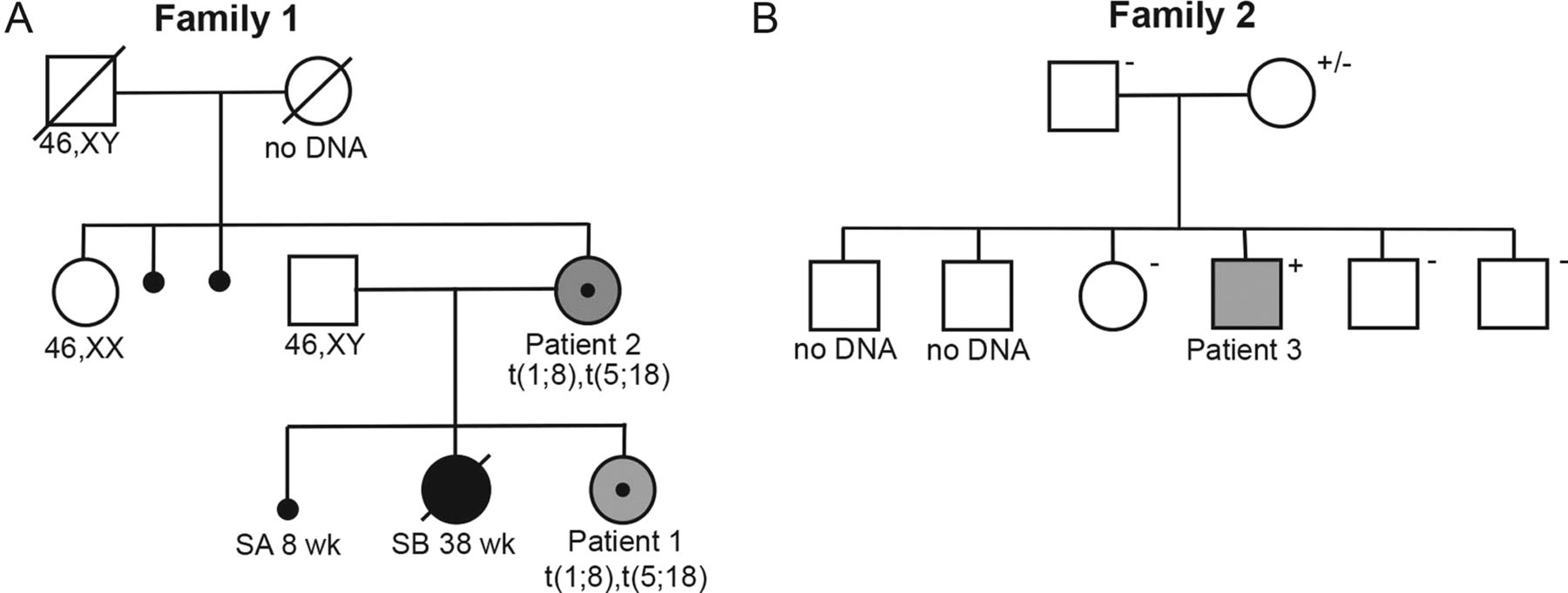
Pedigrees of the families with CTNND2 mutations. (A) and (B) Pedigrees of Family 1 and Family 2. Squares denote males and circles denote females, small black circles indicate a spontaneous miscarriage. The large blackened symbols represent a severely affected stillborn child, and the small blackened circles are first-trimester spontaneous abortions. Grey symbols indicate learning difficulties. Individual identification numbers are given below the symbols. Karyotypes are given for those individuals in Family 1 where a chromosome analysis has been performed and deletion carrier status is annotated in Family 2 as +, − or ± (mosaic carrier).
Whole-genome mate-pair sequencing and breakpoint PCR
To pinpoint the exact positions of the chromosomal breaks, we used low-coverage mate-pair whole-genome sequencing of patient 1. The average coverage was that the two chromosomal translocations were supported by multiple hybrid reads in the whole-genome sequencing data (n=40 for t(1;8) and n=63 for t(5;18); figure 1C). The breakpoint regions were narrowed down to 205 bp (chr 1), 70 bp (chr 18), 150 bp (chr 5) and 315 bp (chr 8). We then designed primers and performed PCR spanning the 1/8 and 5/18 translocation junctions (online supplementary table S1). The exact genomic coordinates for the breakpoints were chr1:78931219–78931227 (+ strand) and chr8:137813394–137813393 (− strand) for t(1;8) and chr5:11291109–11291112 (+ strand) and chr18:32849314–32849311 (− strand) for t(5;18) (figure 1D, I, all coordinates in hg19).
The detailed analysis of the junction fragment signatures reported here revealed that in the t(1;8) (p22;q24) junction, two stretches of four nucleotide tandem repeats were present (TCCT and AAGG), and seven nucleotides were deleted on chromosome 1 directly adjacent to the break. In t(5;18)(p15;q11) the junction sequence may be interpreted in two ways: (1) there was a one-nucleotide microhomology (G) at the breakpoint and a potentially novel point mutation (G to T) was present on chromosome 18; one-nucleotide from the junction, or (2) a 2 bp duplication has occurred on chromosome 5 (chr5:11291110–11291111) masking a deletion of 2 bp on chromosome 18 (chr18:32849313–32849314) (figure 1D, I).
The reciprocal translocations disrupt candidate loci
Next, we turned our attention to potential candidate genes affected by the chromosomal breakpoints. No genes were affected on chromosome 1 or chromosome 8. The 5p breakpoint was located in intron 9 of the CTNND2 gene (MIM 604275; NM_001332; figure 3), and the 18q break affected an uncharacterised gene, ZSCAN30 (syn.ZNF917, ZNF397OS). As the genes were fused in opposite orientation, both were considered to be loss-of-function mutations. A thorough literature search revealed little information on ZSCAN30; there was no evidence of brain expression.30 However, in situ hybridisation in mouse showed transcript expression in postnatal (P56) mice (Allen mouse brain atlas; http://mouse.brain-map.org/experiment/show?id=695309). By contrast, the armadillo repeat family member CTNND2 (delta catenin) is expressed almost exclusively in the brain7 and is a component of the adherens junction complex.8 ,9 Given these findings, we focused our efforts on CTNND2.

A schematic illustration of CTNND2. The exon–intron structure of CTNND2 gene (A). Exons are denoted by black boxes and introns by lines. Black stars indicate the genomic position of the TaqMan copy number assays (hs06067454_cn and hs06128465_cn). The breakpoint in intron 9 is indicated as a dashed line vertical red line, and the microdeletion of exons 12–18 in patient 3 is shown as a solid red horizontal bar in (A) and (B). The predicted protein structure is shown in (B) compared with its orthologues in several commonly used model organisms.
Identification of a third patient with cognitive difficulties and CTNND2 deletion
We then browsed the Database of Chromosome Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER; http://decipher.sanger.ac.uk) and identified an additional individual with similar phenotypical presentation and a maternally inherited microdeletion exclusively involving CTNND2 (Patient 3; table 1; figures 2B and 3). This boy had more pronounced learning disabilities and, specifically, difficulties in reading suggestive of dyslexia. Some dysmorphic features were present as well as joint laxity in the fingers and a marfanoid habitus.
The heterozygous deletion was identified by chromosomal microarray analysis. A total of 162 probes were deleted on chromosome 5p15.2, located 11055920 to 11218488 bp from the p telomere, affecting exons 12–18 of the CTNND2 gene (online supplementary figure S1). The deletion was confirmed by qPCR using two TaqMan copy number (CN) assays performed according to the manufacturer's recommendations (hs06067454_cn and hs06128465_cn). Segregation analysis in the family using the same qPCR assays showed that the mother was mosaic for the 5p15.2 microdeletion (CN Calculated=1.63; minCN=1.47 maxCN=1.83; online supplementary figure S1), and the microdeletion was absent in the patient's healthy siblings (online supplementary figure 2B, data not shown).
Taken together, the segregation of the disruptive chromosome 5/8 translocation in Family 1 and the intragenic deletion in Family 2 strengthen the hypothesis that loss-of-function mutations of CTNND2 are involved in the development of learning difficulties and reading disabilities in these patients.
Association of a CTNND2 SNP with changes in white matter volume and reading performance
Since all three patients reported here presented with clear reading difficulties, we decided to further evaluate the effect of CTNND2 on reading performance and variability of white matter volume. We used longitudinal data from a cohort31 of 76 normally developing children and young adults aged 6–25 years and studied associations to polymorphisms in CTNND2. Interestingly, using sex and handedness as covariates and considering three repeated measures, we found rs2561622 to be significantly associated with performance in non-word reading in children of age 6–10 years (p=1.54×10−4; n=28; AA: 11, AG: 13, GG: 4). The association remained significant after adjustment for multiple testing (Bonferroni correction for 37 SNPs; p<0.0013), with the G allele of rs2561622 associated with a better test performance. By contrast, the rs2561622 association with phonological choice tasks for individuals aged 12–14 years (p=0.816; n=18; AA: 8, AG: 6, GG: 4) and above 16 years (p=0.018; n=28; AA: 10, AG: 16, GG: 2) did not survive the multiple comparison correction. The allele frequencies for all 37 SNPs are shown in online supplementary table S2.
Finally, we used the SPM8 software to test genetic associations to white matter structure by a flexible factorial model considering three different time-points of our longitudinal MRI data. Image acquisition and analyses were performed as described previously.31 Briefly, the SNPs were entered separately as main factor, and the model was corrected for age, sex and handedness. The gene interactions by age and sex were also entered as covariates. The SNP rs2561622 showed significant effect on white matter volume in the left frontal lobe (peak coordinate: −47, 25, 22, p=1.28×10−5; corrected at the cluster level Pfamilywise error, FWE<0.01) with higher white matter volume for GG allele carriers (figure 4A, B). The significant cluster partly overlapped with the arcuate fasciculus, superior longitudinal fasciculus and the inferior fronto-occipital fasciculus in the left white matter area. Moreover, the boundary of this significant cluster extended to grey matter mainly in the left inferior frontal gyrus and partially to the middle frontal cortex (figure 4C).

White matter structure is influenced by CTNND2 polymorphisms. (A) Main effect of CTNND2 polymorphisms (rs2561622) on white matter structure shown in sagittal section. The significant cluster partly overlaps with the left arcuate fasciculus and the inferior fronto-occipital fasciculus. (B) Distribution of residuals of white matter volume in the cluster associated with rs2561622 across different genotypes after correction for age, sex and handedness (error bars: ±1 SEM). (C) The extension of the significant cluster to its adjacent cortical regions: left inferior frontal gyrus and the middle frontal cortex.
Knockdown of ctnnd2b in zebrafish results in misplaced isl:GFP cells
The zebrafish genome contains two orthologues of human CTNND2, named ctnnd2a and ctnnd2b, (figure 3B). To knock down these two orthologues, we used non-overlapping MOs targeting two different splice junctions in either of the two genes (Genetools, Philomoth, USA; online supplementary table S2). Initial independent injections of the two morpholinos against ctnnd2a in isl:GFP embryos up to a dose of 10 ng did not result in any gross morphological defects at 48–54 hpf (ctnnd2a MO1; n=104, ctnnd2a MO2 n=36), or in the misplacement of isl:GFP cells in the forebrain or hindbrain (figure 5H and data not shown). By contrast, injection of ctnnd2b MO1 or ctnnd2b MO2 resulted in a small percent of embryos with gross abnormalities (10 ng ctnnd2b MO1: 11% n=13/118, 5 ng ctnnd2b MO2: 14% n=6/43; online supplementary figure S3D) compared with control MO-injected embryos (5% n=3/60). The rest of the embryos were indistinguishable from uninjected or control MO-injected embryos (online supplementary figure S3C). Additionally, similarly to ctnnd2a MO-injected embryos, analysis of isl:GFP expressing hindbrain motoneurons showed no significant defects in migration (data not shown).

Knockdown of zebrafish ctnnd2b results in misplaced isl:GFP (isl-1) neurons. Confocal z-stack images of whole-mount zebrafish embryos are shown in (A)–(D) rostral facing with dorsal to the top. Three-dimensional projections of (C) and (D) are shown in (F), (F′) and (G), (G′), respectively, with (F), (G) being rostral projections and (F′), (G′) lateral. Embryos were injected with 10 ng of standard control MO (cont MO), 10 ng ctnnd2a MO1, 10 ng ctnnd2a MO2, 10 ng ctnnd2b MO1 or 5 ng ctnnd2b MO2. Morpholino penetrance is shown in H. Injection of either morpholino targeting ctnnd2b resulted in the presence of ectopic isl:GFP cells between the telencephalon and diencephalon at both 48 hpf (B) and 54 hpf (red rectangle D, yellow cells in G and G′) compared with injection of a control MO (red rectangle A, yellow cells in F and F′) or ctnnd2a MO (not shown). The number of embryos presenting this phenotype was significant for both ctnnd2b MO1 (**p<0.01) and ctnnd2b MO2 (*p<0.05) injected embryos when compared with cont. MO injected embryos. The number of ectopic neurons demarcated by the red square in C-D were counted (E) and shown to be significantly increased compared with controls (***p<0.001). The χ2 test was used to compare phenotype penetrance. For comparison of the number of ectopic isl:GFP cells, the t test was used. Error bars denote SE of the mean.
We then analysed a subpopulation of neurons in the rostral forebrain in morphologically normal embryos at 48 and 54 hpf and noted the presence of ectopic isl:GFP cells between the telencephalon and diencephalon at both 48 and 54 hpf when compared with control MO-injected embryos (figure 5). Expression of ctnnd2b in this region at 48 hpf was confirmed via FISH on whole-mount brains, as previously described32 (see online supplementary figure S3A,B). Misplaced neurons were observed directly adjacent to the optic recess, which defines the frontal horn of the forebrain ventricle. At 54 hpf, a total of 42% (n=13/31) and 48% (12/25) of embryos injected with either ctnnd2b MO1 or ctnnd2b MO2 showed this misplacement of cells. The number of ectopic isl:GFP cells around the optic recess proved to be significantly increased in affected ctnnd2b knockdown embryos compared with control MO-injected embryos (10 ng ctnnd2b MO1; mean: 9.1, SE of mean (SEM): 0.82 and 5 ng ctnnd2b MO2; mean: 8.6, SEM: 0.76) compared with controls (control MO; mean: 2.4, SEM 0.40; figure 5).
As the transcription factor islet-1 (isl) has been previously shown to be expressed specifically in a subpopulation of early neuron progenitors in the zebrafish telencephalon and diencephalon,33 we wanted to investigate whether a more general misplacement of neuronal cells was observed. In order to analyse all neurons at 48 hpf–54 hpf, embryos injected with ctnnd2b MO1 or MO2 were stained with the postmitotic neuronal marker, HUC.34 No significant difference in the positioning of the gross neuronal population was observed (figure 6A–C). Additionally, analysis at an earlier stage in 30 hpf embryos injected with either of the two ctnnd2b MOs showed normal specification of isl:GFP cells at that stage and normal formation of the forebrain commissures, indicating normal patterning of the forebrain (see online supplementary figure S3E,F). Specific analysis of single slice confocal sections 0.50 μm of the identified mispositioned isl:GFP cells in proximity to the forebrain ventricle (figure 6D–D″) revealed that the majority of these cells did not overlap with Huc C/D expression or showed only weak onset of expression in contrast with isl:GFP cells that had integrated into the diencephalic cluster (figure 6D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ectopic isl:GFP (isl1) cells fail to express the postmitotic neuronal marker HUC. Single optical slices (0.50 µm) at the level of the optic recess are shown in (A)–(D″) with rostral facing and dorsal to the top. isl:GFP cells are shown in green and cells labelled with the pan-neuronal marker anti-HUC in red. A magnified view of the orange rectangle in B is shown in (D)–(D″); where D’ shows Huc-stained cells, D″ isl:GFP cells and D the overlay. Knockdown of ctnnd2b does not affect the general neuron population (red labelled cells in B and C) compared with controls (A), migrating isl:GFP cells that have not yet integrated into the diencephalon can be observed near the ventricles (yellow rectangle in B) many of which have not yet started expressing HUC (filled arrow heads D) or show weak expression (unfilled arrowhead D) in contrast with cells that have integrated into the diencephalon (blue rectangle in D).
Discussion
In the present study, we show loss-of-function mutations in CTNND2 in three individuals from two unrelated families, all presenting with mild cognitive dysfunction and reading disabilities.
Mechanisms underlying the two balanced translocations in Family 1
The fundamental mechanisms underlying the formation of chromosomal translocations are still incompletely understood. Even though approximately 1 out of 500 individuals are carriers of a balanced structural chromosome rearrangement, the occurrence and cotransmission of two reciprocal translocations in a family are very rare. From our breakpoint data, it is clear that the two translocations are separate rearrangements. Segregation did not reveal any indication of mosaicism, as both mother and daughter carried both translocations in all analysed cells. From the family history, we suspected that the maternal grandmother was a carrier of at least one translocation (two miscarriages and psychiatric illness resulting in suicide), but no DNA was available to test this hypothesis and no other family members are carriers (figure 2A).
By characterising the translocation breakpoints at the DNA level, we had an opportunity to study potential mechanisms underlying the translocational events. Most previous studies of breakpoint features of genomic structural variants have focused on either genomic CNVs (deletions and duplications)35 or acquired somatic rearrangements associated with human malignancies.36 The dominating mechanistic models hypothesised to explain such alterations, particularly non-recurrent events, are non-homologous end-joining (NHEJ) and replicative mechanisms (such as fork stalling and template switching/microhomology-mediated break-induced replication, FoSTeS/MMBIR).35 ,37 Not many balanced constitutional reciprocal translocations have been studied at the breakpoint level yet, but in a large study using massive parallel sequencing, Chiang et al38 suggested NHEJ as the predominant mechanism. The microarchitecture at the DNA breakpoints reported here suggests that the two translocations have been formed separately, but precludes us from decisively determining the exact nature of the underlying mechanism of formation. For instance, the observed junction signatures were similar to CNV breakpoints generated by replication-based repair mechanisms. Detailed studies of breakpoints of duplications have shown that the combination of microhomology, a few base-pair deletions, insertions and novel point mutations flanking the junctions may indicate FoSTeS/MMBIR.39–41 However, since FoSTeS/MMBIR generates non-reciprocal rather than reciprocal events, further studies are needed to determine whether replicative mechanisms have a role in reciprocal translocations. Therefore, our results support NHEJ as the underlying mechanism of formation of the translocation events that resulted in the disruption of CTNND2.
CTNND2 haploinsufficiency and learning disability
Haploinsufficiency of CTNND2 was recently linked to mild intellectual disability with or without autistic traits.12 Here, we report two additional cases with heterozygous structural variants resulting in CTNND2 loss-of-function mutations with low normal intelligence and learning disabilities borderlining on intellectual disability (table 1) and a third patient with mild intellectual disability. Some similarities with the patients reported by Asadollahi et al can be observed possibly outlying the CTNND2 haploinsufficiency phenotype. Wechsler intelligence scale for children tests (WISC) performed on Patient 1 (WISC-III) and patient 3 (WISC-IV) gave uneven profiles similar to the WISC result of Patient 1 in the report by Asadollahi et al, with a better verbal than non-verbal result for both. Both, Patients 1 and 2 in our report, had significant problems throughout middle school (grades 4–9); were moved to special class and repeated a grade, respectively, but then caught up and were able to finish normal high school. Hence, compared with Asadollahi et al, these two patients present with a milder phenotype with IQ in the low normal range, and thus, fit a type 4 classification as set out recently by Beaudet.42 However, the cognitive profile and neurological manifestations of patient 3 reported here, and patient 2 in Asadollahi’s report,12 both present with low IQ, distinct facial features and hyperextensibility of the finger joints. These cases may represent the more severe end of the CTNND2 haploinsufficiency phenotypical spectrum.
Hemizygous loss of CTNND2 in Cri-du-chat patients correlates with an increase in the severity of the syndrome.43 However, the region deleted in Cri-du-chat contains many other genes, including genes coding for axon guidance molecules, such as semaphorin, suggesting that the clinical pathology is a result of various perturbed developmental processes acting either synergistically in the same pathway or parallel in an additive manner. Further supporting an involvement of CTNND2 in the Cri-du-chat-cognitive phenotypes is provided by a recent report describing a Cri-du-chat patient with a complex CTNND2 rearrangement and a milder cognitive phenotype including an improvement in reading.44
To date, nine dyslexia susceptibility loci (DYX1-DYX9) have been proposed, and four strong-effect candidate genes have been identified: DYX1C1 (DYX1),45 DCDC2 (DYX2),46 KIAA0319 (DYX2)47 and ROBO1 (DYX5).6 We have previously demonstrated that SNPs from the DYX1C1, DCDC2 and KIAA0319 genes influence white matter volume.31 Here, we show a similar result for a common SNP in CTNND2 and rs2561622, which is significantly associated with both performance in non-word reading and white matter volume. The significant white matter cluster is located in the left frontal lobe and extends over to grey matter mainly in the left inferior frontal gyrus and partially to the middle frontal cortex (figure 4C). These findings support the argument that CTNND2 is involved in reading, as the inferior frontal gyrus includes Broca's area, a key region in phonological processing.48 ,49 Further, dyslexic readers have previously been reported to have lower activity in the left inferior frontal gyrus during reading and phonological processing.50 ,51
The polymorphism reported here, rs2561622, is a non-conserved common SNP of yet unknown functional significance and located in a Long interspersed nuclear element (LINE) in an intron of the gene CTNND2 on chromosome 5p15.2. To the best of our knowledge, this SNP does not overlap with previous replicated linkage regions for developmental dyslexia (for an overview, see Kere.52). Furthermore, it is not found significantly associated with reading and writing in recently published genome-wide association analysis using quantitative measures of reading or writing skills.53–55 The community sample used here for assessing the role of rs2561622 in phonological processing is of limited size, and further, targeted studies in larger independent sample sets are needed to verify the role of rs2561622 alleles on susceptibility to developmental dyslexia.
Many dyslexia candidate genes, such as DYX1C1, DCDC2, KIAA0319, KIAA0319 L, S100B and ROBO1 have been linked to defects in neuronal migration both in vivo and in vitro.56–63 In particular, disruption of Dyx1c1 in adult rodent brains via RNAi was shown to cause neuronal migration defects,59 and in vitro perturbation of Dyx1c1 altered the expression of genes involved in cell migration and CNS development in vitro.64 Even more conclusive evidence for abnormal neuronal migration, as one potential neurodevelopmental mechanism of dyslexia, stems from the postmortem examination and brain imaging studies of human patients. Neuroanatomical examinations of four male and three female patients revealed various abnormalities, including cerebral asymmetries, focal architectonic dysplasias and neuronal ectopias, the latter of which is indicative of neuronal migration defects.65 ,66 More recent results come from studies using structural MRI in conjunction with neuropsychological testing in patients with periventricular nodular heterotopia (PNH), anatomically defined as the presence of misplaced grey matter nodules along the ventricular wall, due to a disruption of radial neuronal migration from the ventricular zone to the cortical plate. Most patients with PNH have average intelligence despite the rather severe anatomical presentation, but suffer from epilepsy and frequently have a form of dyslexia specifically affecting reading fluency.67
Here, we show that knockdown of one of the two Ctnnd2 orthologues resulted in the presence of ectopic isl1 neurons in the zebrafish forebrain suggestive of potential defects in neuronal migration. Recently, an orthologous subpopulation has been shown to differentiate into cholinergic neurons in mouse.68 Additionally, a somatic loss of CTNND2 in human glioblastomas has been shown to lead to a mesenchymal transformation associated with an invasive progression and a more aggressive prognosis69 suggesting an in vivo role of CTNND2 in cell motility. Interestingly, the misplaced neurons reported here show no or only weak expression of the postmitotic neuronal marker, HUC, suggesting that the ectopic cells are not yet fully differentiated (figure 6). Further work is required to assess whether this lack of differentiation is the cause of the misplaced neurons, as shown previously in cases of periventricular neuronal heterotopias, due to loss of cadherin receptor ligand pairs, DCHS1 and FAT4,11 or an after effect, due to inappropriate integration into the neuroepithelium.
In summary, we show loss-of-function mutations in CTNND2 and learning difficulties within the dyslexia spectrum in three individuals from two unrelated families. The gene was identified by low-coverage massive parallel whole-genome sequencing of a patient with learning difficulties within the dyslexia spectrum and two balanced reciprocal translocations. This illustrates the capability of whole-genome end sequencing to detect balanced structural variation,70 which is re-emerging as part of a comprehensive whole-genome test in leading clinical testing laboratories.71 It serves as a complement to balanced CNV analysis.72 CTNND2 function was further evaluated first in a community cohort of normal developing children where a CTNND2 polymorphism was significantly associated with phonological processing and white matter volume in the left inferior frontal lobe, close to Broca's area, a region previously linked to phonological processing and developmental dyslexia. Second, we performed in vivo studies in zebrafish embryos and revealed the presence of ectopic neurons in the zebrafish forebrain after MO knockdown of ctnnd2. The combined data suggest that loss of CTNND2 is involved in learning difficulties possibly through defects in neuronal migration.
Acknowledgments
We are grateful to the patients and their families for their cooperation and enthusiasm during this study. We also gratefully acknowledge the use of computer infrastructure resources at UPPMAX, Project b2011157, and the Knut and Alice Wallenberg foundation for funding the CLICK facility at the Karolinska Institutet. We would also like to thank the zebrafish core facility for maintenance of our fish, Satish Kitambi for use of his islet:GFP fish stock, Miriam Armenio for her technical assistance and Dr Myriam Peyrard-Janvid for genotyping of BrainChild samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online table S1
- Data supplement 3 - Online table S2
- Data supplement 4 - Online table S3
Footnotes
-
Contributors WH: conceived and designed the experiments, performed the experiments, analysed the data, contributed to the writing of the manuscript. DN: conceived and designed the experiments, analysed the data, contributed to the writing of the manuscript. AT and B-MA: analysed the data, contributed to the writing of the manuscript, contributed reagents/materials/analysis tools. FD and HM: performed the experiments, analysed the data, contributed to the writing of the manuscript. ITP: conceived and designed the experiments, contributed to the writing of the manuscript. TK and VW: conceived and designed the experiments, contributed reagents/materials/analysis tools, contributed to the writing of the manuscript. LS: performed the experiments, analysed the data. FV: performed the experiments, analysed the data, contributed to the writing of the manuscript. JK: conceived and designed the experiments, contributed reagents/materials/analysis tools, contributed to the writing of the manuscript. MN: conceived and designed the experiments, contributed reagents/materials/analysis tools, contributed to the writing of the manuscript. ESL: conceived and designed the experiments, contributed to the writing of the manuscript. AL: conceived and designed the experiments, analysed the data, contributed reagents/materials/analysis tools, contributed to the writing of the manuscript.
-
Funding This work was funded by the Swedish Brain Foundation, the Harald och Greta Jeanssons Foundation, the Nilsson-Ehle Foundation, Erik Rönnbergs Foundation and the Swedish Research Council grant 2012-1526 to AL, 2010-3286 to ESL, 2012-2279 to JK and 2011-4592 to MN.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The local ethical boards in Stockholm (Sweden).
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement All the data is reported and no unpublished data is available in the current study.