Article Text

Abstract
Background Mutations in forkhead box protein P1 (FOXP1) cause intellectual disability (ID) and specific language impairment (SLI), with or without autistic features (MIM: 613670). Despite multiple case reports no specific phenotype emerged so far.
Methods We correlate clinical and molecular data of 25 novel and 23 previously reported patients with FOXP1 defects. We evaluated FOXP1 activity by an in vitro luciferase model and assessed protein stability in vitro by western blotting.
Results Patients show ID, SLI, neuromotor delay (NMD) and recurrent facial features including a high broad forehead, bent downslanting palpebral fissures, ptosis and/or blepharophimosis and a bulbous nasal tip. Behavioural problems and autistic features are common. Brain, cardiac and urogenital malformations can be associated. More severe ID and NMD, sensorineural hearing loss and feeding difficulties are more common in patients with interstitial 3p deletions (14 patients) versus patients with monogenic FOXP1 defects (34 patients). Mutations result in impaired transcriptional repression and/or reduced protein stability.
Conclusions FOXP1-related ID syndrome is a recognisable entity with a wide clinical spectrum and frequent systemic involvement. Our data will be helpful to evaluate genotype–phenotype correlations when interpreting next-generation sequencing data obtained in patients with ID and/or SLI and will guide clinical management.
- FOXP1
- intellectual disability
- language impairment
- oromotor dysfunction
- genotype-phenotype correlation
Statistics from Altmetric.com
Introduction
Intellectual disability (ID) and speech and language disorders are common problems in the paediatric population and frequent reasons for encounter with child neurologists and clinical geneticists. They cover a wide range of conditions with heterogeneous phenotypes, making gestalt diagnosis complicated and challenging. In the majority of patients, no molecular defect has been identified thus far, arguing for a genetic heterogeneous and probably multifactorial aetiology.1–4
Meanwhile, over 700 genes have been identified in both isolated ID and ID-associated disorders, and several genes have been associated with speech and language disorders, including FOXP1, FOXP2, SHANK3 and TM4SF20.1 4 5 Additional loci for specific language impairment (SLI), a specific form of speech and language disorders defined by an isolated delayed or absent language acquisition that may however be associated with autism and/or oromotor dyspraxia, have been mapped to chromosomal regions 16q (SLI1; OMIM#606711), 19q (SLI2; OMIM#606712), 13q21 (SLI3; OMIM#607134) and 7q35-q36 (SLI4; OMIM#612514).
The first gene implicated in the pathogenesis of developmental speech and language disorders was FOXP2.6 Later on, similar expression patterns of FOXP2 and FOXP1 in songbird and human brain suggested a functional relationship.7 Since then, several case reports have confirmed FOXP1 defects in language impairment other than verbal dyspraxia,8 9 including both monogenic FOXP1 mutations and more extensive 3p chromosomal deletions encompassing FOXP1. However, until now, no specific phenotype has emerged. The absence of a clear ‘gestalt’ in monogenic causes of SLI, including FOXP1-related SLI, complicates the work-up and delays the diagnosis.
With the introduction of array comparative genome hybridisation (arrayCGH) and next-generation sequencing (NGS) in clinical practice, the concept of reverse genetics allows to identify a recurrent phenotype in patients with similar molecular defects, delineating novel conditions and providing useful information for prognosis, management guidelines and recurrence risk to other family members.
FOXP1 encodes forkhead box protein P1, which belongs to the family of the winged helix/forkhead transcription factors that regulate embryogenesis and maintenance of differentiated tissues through transcriptional repression.10 11 FOXP1 is widely expressed.12–17 Hence, reduced FOXP1 functioning may perturb the development of multiple organ systems.
We report on the molecular and clinical data of 25 novel patients with a FOXP1 defect and compare the data with 23 previously reported patients. This comprehensive description evidences a recognisable ‘gestalt’ for FOXP1-related ID syndrome. Based on the relative frequent occurrence of non-neurological anomalies, we provide recommendations for clinical management and follow-up. We further show that both missense and premature truncation mutations reduce FOXP1 repression activity and/or interfere with protein stability.
Methods
Patients
All patients were evaluated by experienced clinical geneticists and recruited through direct referral or through the Decipher website. Patient 19 (Decipher 252324) was already published by Thevenon et al 18 although a more detailed phenotypic description is presented here. Previously published patients with FOXP1 mutations were identified through a National Center for Biotechnology Information (NCBI) PubMed literature search (www.ncbi.nlm.nih.gov/pubmed).9 18–29 We excluded the four patients reported by Chang et al 12 and Srivastava et al 30 because of the unavailability of phenotypic data. We also excluded patients with interstitial 3p deletions if the precise chromosomal breakpoints were not provided and therefore the involvement of FOXP1 unknown.
The study was approved by the Ethics Committee of the Ghent University Hospital. For the publication of clinical and molecular data, we obtained informed consent from all individuals or their legal representatives. For publication of clinical pictures, we obtained a specific consent.
Phenotypic analysis
A clinical summary, photographs and a checklist were provided by the referring clinicians. To reduce interobserver variability, all data were re-evaluated by IM and BCa. Missing data were excluded from statistical analyses. Subgroup analysis, comparing patients with 3p deletions and monogenic FOXP1 defects, and patients with FOXP1 deletions or truncating mutations (nonsense, frameshift and splice site mutations) and FOXP1 missense mutations, was performed using Fisher’s exact tests (SPSS Statistics V.22).
Molecular analysis
Molecular analysis was performed at each referring centre with varying methodologies for arrayCGH analysis, direct Sanger sequencing and NGS of ID gene panels. All NGS detected FOXP1 mutations were confirmed by Sanger sequencing. Sequences were compared with the wild-type sequence as submitted to NM_032682.5 (Ensembl Accession number ENST00000318789.4). Nucleotides were numbered starting from the first base of the initiation codon (ATG) of the cDNA reference sequence. Amino acid residues are numbered from the first methionine residue of the reference sequence. CNVs were mapped against the human genome build hg19/GRCh37.
Functional analysis
Luciferase assay
To evaluate transcriptional repression activity, we used a standard luciferase assay.9 23 26 Mutations were introduced into the full length coding sequence of FOXP1 longest isoform (FOXP1a; obtained from Kazusa DNA Research Institute, Kisarazu, Japan) and subcloned into the mammalian expression vector pcDNA4His (Invitrogen). HEK293 cells were then cotransfected using Fugene 6 (Roche), in 24-well plates, with 400 ng of pcDNA4His without an insert or containing the wild-type FOXP1 (wt-FOXP1) or the mutant FOXP1 cDNA, along with 50 ng of pGL3-promoter construct (Promega) (in which the SV40 promoter drives a firefly luciferase reporter). To normalise for transfection efficiency and variation in cell number, cells were also cotransfected with 50 ng of a Renilla luciferase construct (pRL-TK; Promega) driven by the HSV-thymidine kinase promoter which is not affected by FOXP1. Cells were lysed 48 hours after transfection. Firefly and Renilla luciferase activities were quantified using the Synergy H4 Hybrid Multiplate Reader (BioTek). Protein expression was verified by western blotting (see below). Student’s t-test was performed to evaluate statistical significance.
Western blotting
Expression of the different exogenously expressed proteins was evaluated using standard western blotting. To enable monitoring and detection of FOXP1, we fused it to an n-terminal tag (Xpress) and used Xpress Monoclonal Antibody (Thermo Fisher Scientific) for detection, thereby avoiding simultaneous detection of endogenous FOXP1. Ten micrograms of total protein were loaded per lane of a 12% acrylamide gel (Bio-Rad). Transfer onto a low fluorescence polyvinylidene difluoride (PVDF) membrane was done using turbo blot.
Splicing efficiency
Splice site mutations were tested in the pSPL3 splicing vector.31 32 The FOXP1 exons flanked by 200 bp of intronic sequence containing the putative splice mutation were cloned in between two b-globin exons. HEK293 cells were transfected with these constructs and RNA was collected 48 hours post-transfection. Splicing was analysed by sequencing the RT-PCR fragment generated by the fused b-globin mini-gene.
Results
Molecular characteristics
The molecular data of 25 novel and 23 previously reported patients are shown in figure 1 and see online supplementary table 1. In our cohort, all defects originated de novo (22/22) (in three patients the analysis of parents was not possible). Six patients have an interstitial 3p deletion (6/25) affecting more proximally and/or more distally located genes with sizes of the deleted region ranging from ~3 Mb to ~6 Mb. Nineteen patients have a monogenic FOXP1 defect (19/25), including five partial FOXP1 deletions (5/19) (~120 kb to ~630 kb) and 14 point mutations (14/19), comprising nonsense (6/14), missense (2/14), frameshift (3/14) or splice site (3/14) alterations. Three patients harbour the c.1573C>T, p.(Arg525*) mutation, previously reported by Hamdan et al.9
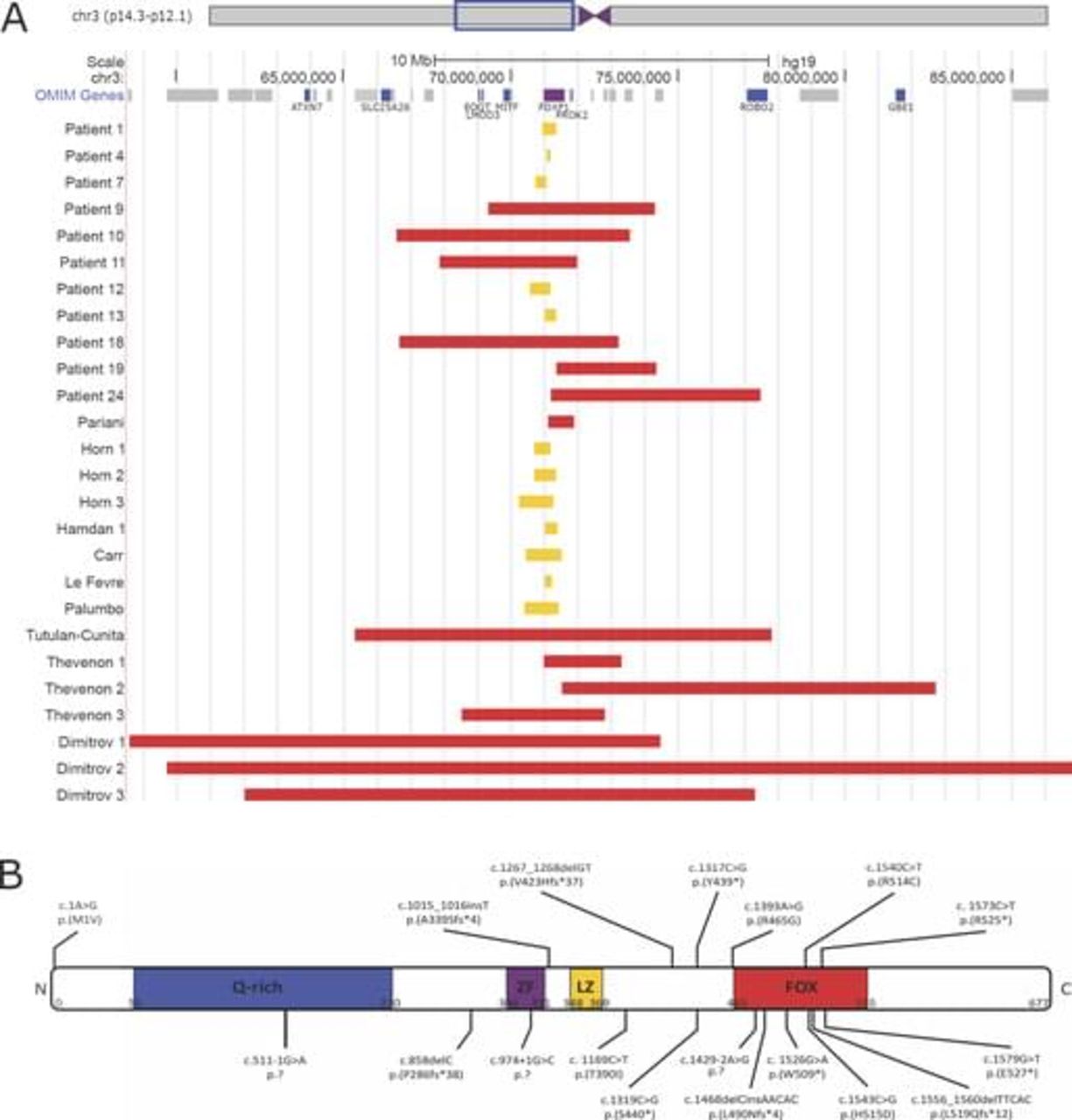
Molecular data of 25 novel and 23 previously reported patients with FOXP1 defects. (A) CNVs affecting FOXP1. Interstitial 3p deletions encompassing FOXP1 are depicted in red, CNVs restricted to FOXP1 are shown in yellow. (B) Small indels and point mutations identified in FOXP1. Previously reported mutations are shown above the schematic representation of the FOXP1 protein, while novel mutations identified in this study are shown below. Four patients harbour the c.1573C>T, p.(Arg525*) mutation. (Q-rich, glycine-rich domain; ZF, zinc finger domain; LZ, leucine zipper domain; and FOX, DNA-binding forkhead box domain).
Comparable, all previously reported cases have de novo (19/19) defects (data were missing for four patients), including extended 3p deletions (8/23) (~800 kb to ~27.2 Mb), whole FOXP1 deletions (1/15) (~200 kb to ~1 Mb), partial FOXP1 deletions (6/15) and seven point mutations (7/15) (2 nonsense, 3 missense and 2 frameshift). One patient has a balanced chromosomal rearrangement with breakpoints in 3p13 (affecting FOXP1) and 10q21.2 (1/15).
Most pathogenic FOXP1 variants result in premature truncation (10/18). Missense mutations (5/18) and splice site mutations (3/18) are present in a minority of the patients. Figure 1B illustrates that nonsense mutations, frameshift mutations and splice site mutations appear throughout the whole protein, while all but one missense mutations are located in or close to the DNA-binding FOX domain.
As shown in figure 2, further functional evaluation of p.Arg525*, p.Trp509*, p.His515Asp and p.Thr390Ile mutant proteins shows that FOXP1-Arg525* and FOXP1-Trp509* (both stably expressed from plasmid cDNA vectors) fail to reduce luciferase levels as compared with wt-FOXP1 (p<0.001), indicating that both truncating mutations impair FOXP1s ability to repress the SV40 promoter. The missense mutation FOXP1-His515Asp (also stably expressed in vitro) maintains some residual repressor activity (p<0.005), while FOXP1-Thr390Ile (which has limited protein expression in vitro) has rather normal repressor activity.

Functional analysis in selected group of FOXP1 mutations. (A) Results of western blot evaluating the effects of FOXP1 variants on protein expression from cDNA plasmid vectors. FOXP1-p.Arg525*, p.Trp509* and p.His515Asp are stably expressed, while p.Thr390Ile results in decreased protein expression. The detection of endogenous produced beta actin insured equivalent loading of protein in all western blot tracks. (B) Luciferase reporter assay assessing the impact of the p.Arg525*, p.Trp509*, p.His515Asp and p.Thr390Ile mutations on FOXP1 transactivation activity in transfected HEK293 cells. Wild-type FOXP1 and FOXP1-Thr390Ile significantly inhibited pGL3-promoter (SV40) transcriptional activity (p<0.001) as compared with empty vector (pcDNA4)-transfected cells only (comparisons for each pair using Student’s t-test). In contrast, FOXP1-Arg525* and FOXP1-Trp509* failed to repress the pGL3-promoter activity (**p<0.001). FOXP1-His515Asp had a reduced repression activity compared with FOXP1 (*p<0.005). Results (mean of four independent experiments, each performed in triplicate) are shown as relative firefly luciferase activity, which is driven by SV40 promoter, normalised with Renilla luciferase and driven by herpes simplex virus thymidine kinase (HSV-TK) promoter activity. The control signal value was obtained with cells transfected with the empty expression vector (pcDNA4/HisMax) and both pGL3-promoter (firefly luciferase) and pRL-TK (Renilla luciferase) constructs.
Furthermore, we evaluated the effects of FOXP1 c.974+1G>C, c.1429-2A>G and c.551–1G>A mutations on splicing. Failed sequencing of the RT-PCR fragment of the c.974+1G>C donor splice site mutation and the c.1429-2A>G acceptor splice site mutation could result from nonsense-mediated mRNA decay or exon skipping. The c.974+1G>C mutation resulted in normal splicing, although with a lower splicing efficiency. For the c. 1429-2A>G mutation, the exon is skipped in 100%, compared with 50% in wt-FOXP1 in this system. The c.551–1G>A acceptor splice site mutation results in alternative splicing skipping 6 bp of the exon (resulting in deletion of two glutamine residues) (data not shown).
Phenotypic characteristics
The phenotypic characteristics of all novel and previously reported patients are summarised in table 1. For extended clinical data see online supplementary table 2.
Overview of clinical features in FOXP1 patients
Demographic characteristics
Patients originate from Belgium (9/25), France (1/25), Germany (1/25), the UK (5/25), Spain (3/25), Italy (1/25), the Czech Republic (1/25), the USA (3/25) and New Zealand (1/25). The mean age at last follow-up is 6.48 years, and the median age is 7.25 (range 0.42–23). Two patients died (2/25). Patient 3 died at the age of 16 years due to an aspiration pneumonia. Patient 9 died at the age of 5 months due to complications of a diaphragmatic hernia. The male:female ratio is 20:5.
Neurological phenotype
Common neurological features are developmental language delay (23/23; 100%), neuromotor delay (NMD) (22/24; 92%) and ID (23/24; 96%). ID is mostly mild to moderate although one-third have severe ID. The severity of the NMD correlates well with ID in this cohort. Of note, two patients (17 and 22) harbouring a frameshift and a splice site mutation, respectively, have a normal neuromotor development, and intellect is normal in patient 22. Behavioural problems (including hyperactivity, aggression, mood lability, obsessions and compulsions) are common (63%). Autistic features occur in 14 out of 20 patients (70%).
Expressive language is more affected than receptive language. One-third of the patients (8/21) were able to speak a single word by the age of 2 years, and about 60% acquires the ability to speak a few words. Seven patients use short sentences (32%). Poor grammar (5/7) and articulation problems (9/11) are frequent, and more than half of the patients experience oromotor dysfunction (10/17) and/or feeding difficulties (16/24).
Neurological examination reveals hypotonia in half of the patients (10/21), either generalised (6) or axial (4). Hypertonia is present in six patients (6/21; 29%), being generalised in four. Two patients had peripheral hypertonia in combination with axial hypotonia. Spasticity and contractures (10/19; 53%) can be present from birth on and can be progressive or transient. Seizures are documented in three patients (3/22; 14%). Half of the patients show structural brain abnormalities (11/23), including cerebral and/or cerebellar atrophy, cortical, subcortical and deep white matter abnormalities, and/or cerebellar abnormalities.
Staturoponderal evolution
Failure to thrive is reported in eight patients and 30% (6/20) has a body mass index (BMI) ≤10th centile at last follow-up. None of them are obese (BMI ≥90th centile). Nine patients (43%) have a head circumference above the 90th centile, and eleven patients have a relative macrocephaly (head circumference more than 25 centiles above the height).
Craniofacial phenotype
Clinical photographs of some of the patients are shown in figure 3. The most common craniofacial features in patients with FOXP1 mutations include a high, broad and prominent forehead (96%), a frontal hair upsweep (42%), hypertelorism (46%), downslanting palpebral fissures (58%), ptosis (67%), blepharophimosis (17%), bent palpebral fissures (31%) and a short nose (71%) with a broad tip (79%). In most patients, the mouth is wide with full reddish lips (58%), a pronounced vermilion border (73%) and downturned corners (58%). Patients often have an open mouth appearance (60%) with pronounced nasolabial folds (42%). About half of the patients have a prominent chin, mostly with a horizontal crease. Occasionally, unilateral or bilateral hypoplastic upper ear helices (21%), prominent digital pads (9%), a single palmar crease (38%) and clinodactyly (29%) are reported.

{kind=link}
{kind=link}
{kind=link}
Clinical features of patients with monogenic FOXP1 defects (patient 1 at age 2 years (A), patient 2 at age 2 years (B), patient 6 at age 11 years (C), patient 13 at age 7 years (D), patient 20 at age 3 years (E), patient 22 at age 9 years (F), patient 23 at age 10 years (G), patient 25 at age 8 years (H)) or interstitial 3p deletions (patient 10 at age 4 years (I), patient 18 at age 5 years (J)). Note the high and broad forehead, downslanting and/or bent palpebral fissures, ptosis, bulbous nasal tip and pronounced nasolabial folds. Full red lips are most prominent in patients with monogenic defects (A–H).
Other organ involvements
Other commonly affected organ systems are the eye (81%), heart (47%) and genitourinary tract (52%). Ophthalmological abnormalities include strabismus and/or refractive errors, mainly hypermetropia. Optic nerve hypoplasia is reported in one patient. Cardiac defects are minor and often restricted to atrial septal defects. More than half of the boys have unilateral or bilateral cryptorchidism. Four patients had an inguinal hernia. Miscellaneous findings that remain to be confirmed in other patients with FOXP1 mutations are renal malformations (1), diaphragmatic hernia (1), hepatic and bile duct abnormalities (1), transient splenomegaly (1), oesophageal dysmotility (1) and constipation (1), subclinical hypothyroidism (1), hearing impairment (2), developmental dysplasia of the hip (2), spina bifida occulta (1), sacral dimple (2) and anal malformations (1).
Literature review and subgroup analysis
The previously published cases confirm the higher male:female ratio (14:9) and the major neurological features, including variable NMD, ID, SLI (with more severely affected expressive language), behavioural traits and autistic features. Seventy per cent developed speech, but with poor articulation. Oromotor dysfunction (40%) and feeding difficulties (73%) are commonly reported. Hypotonia (50%) and hypertonia (43%) are within similar ranges. Spasticity or contractures are reported in 8 out of 10, and seizures in 4 out of 11 patients (36%). Anatomical brain defects are present in 10 out of 14 (71 %) patients. In 15 patients with reported staturoponderal measurements, failure to thrive is reported in 47%, and two patients are obese. Absolute macrocephaly or relative macrocephaly is reported in three patients (3/15; 20%). Associated organ systems in the previously reported cases include the eye (refractive disorders (4), strabismus (3), iris coloboma (2) and aniridia (1)), heart (secundum atrial septal defect (5)) and genitourinary tract (cryptorchidism (3)). Three patients have hearing impairment (3/9; 33%).
Using data from the total cohort, we compared the occurrence of the studied phenotypic features between the group with more extended 3p deletions and the group with monogenic FOXP1 defects. Severe NMD, severe ID, feeding difficulties and failure to thrive, spasticity and contractures, and hearing impairment were more frequently encountered in the 3p interstitial deletion group (p<0.05, table 2). Further comparison of phenotypic characteristics between FOXP1 deletions, truncating mutations and splice site mutations and FOXP1 missense mutations showed no difference in severity of NMD and/or ID between both groups. Neither was there any difference in the other evaluated phenotypic features between both groups, except for the presence of prominent finger pads (p<0.05, table 3).
Subgroup analysis of monogenic FOXP1 defects versus interstitial deletions
Subgroup analysis of FOXP1 deletions or truncating mutations versus FOXP1 missense mutations
Discussion
Mutations in FOXP1 are a generally accepted monogenic cause of ID and SLI. In contrast to FOXP2 mutations they occur de novo.6 Our results indicate a loss of function for all tested FOXP1 mutations. Most of the mutations have the potential to result in premature truncation and nonsense-mediated mRNA decay.33 Although western blotting did show the presence of truncated protein if overexpressed from a cDNA plasmid vector, this does not rule out nonsense-mediated mRNA decay in the in vivo situation as this complex process may require the genomic context to be properly activated. Nonetheless, our reporter assay, normalised for transfection efficiency and cell count using an additional Renilla reporter, demonstrates that in case the truncated proteins are produced, their transcriptional repression activity is impaired. A significantly reduced transcriptional repression was also observed for the p.His515Asp missense mutation. This reduced transcriptional repression might be due to impaired DNA-binding of the mutant protein, due to its impact on dimerisation by three-dimensional domain swapping.34–36 In contrast, the p.Thr390Ile missense mutation shows normal repression activity compared with wt-FOXP1, although the resulting phenotype is typical for FOXP1-related ID and the mutation occurred de novo. The p.Thr390Ile locates outside the DNA-binding FOX domain, in a region where currently only truncating mutations have been identified. We could confirm reduced protein levels for this mutation from Western blot (figure 2A), since the level of protein resulting from the different transfections was normalised according to the Renilla level.
Besides the previously reported link of FOXP1 mutations with ID, SLI and autistic features, our study shows a broader neurological phenotype with more general neurodevelopmental delay (including NMD and ID), spasticity and/or contractures, structural brain abnormalities and epilepsy, as well as involvement of other organ systems including the ocular, cardiovascular and genitourinary systems. Moreover, most patients show a recognisable craniofacial phenotype, that so far has not been routinely recognised. Besides the earlier reported features of relative macrocephaly and a broad and high forehead,20 23 37 other distinctive features were downslanting and/or bent palpebral fissures, ptosis and/or blepharophimosis, marked nasolabial folds, and a wide mouth with downturned corners and full red lips. The chin becomes more prominent with advancing age. Hand and foot anomalies, such as single palmar crease, clinodactyly and syndactyly are noted, although finger pads, previously considered part of the syndrome,20 are only observed in two patients of our cohort.
Previous reports were contradictory on the occurrence of autism.9 20 In our cohort 70% of patients present with autistic features, confirming this association. It should however be noted that autism remains a difficult diagnosis in patients with ID and SLI, which might be overdiagnosed in these patients. In about two-thirds other behavioural problems are reported, including hyperactivity, aggression, mood lability, obsessions and compulsions.9 37 Oromotor dysfunction and articulation problems may aggravate the apparent SLI20 37 and correlate well with the feeding difficulties (Fisher’s exact test p=0.006). Feeding difficulties were also associated with failure to thrive (Fisher’s exact test p<0.001). The observation of obesity in two previously reported patients25 remains thus far unconfirmed. Remarkably, we observe a male predominance (34:14). Sex predisposition in autosomal disorders remains largely unexplained, although it has been observed in other entities including Wolf-Hirschhorn syndrome, Brugada syndrome, and long QT syndrome.38–40 Fisher’s exact test comparing the presence of each studied phenotypic characteristic between male and female revealed no statistically significant difference in the severity of the phenotype between both sexes. Therefore, the observed sex difference cannot be explained by either an increased lethality (less likely) or otherwise a higher occurrence of mild unrecognised cases in women.
Of note, all reported frequencies of the phenotypic characteristics are solely based on available data and did not take into account the amount of missing data. Therefore, the presence of a reporting bias cannot be excluded. Initially, most patients were reported as a 3p- syndrome, involving FOXP1.41–45 However, NGS panel analysis for ID genes and exome analysis clearly shows that monogenic FOXP1 defects are at least twice as frequent as interstitial 3p- deletions. Five FOXP1 mutations were found in a cohort of 2000 patients referred for panel analysis because of epilepsy and ID or autism (DL, personal communication). Therefore, mutations in FOXP1 in this cohort were as frequent as ARID1B and DYRK1A mutations, establishing it as a relatively frequent, but often unrecognised cause of monogenic ID.
No specific genotype–phenotype correlations emerged considering monogenic FOXP1 defects only. Indeed, wide phenotypic variation exists between the four patients who harbour exactly the same c.1573C>T, p.(Arg525*) mutation. Moreover, mild phenotypes were observed in association with both frameshift and splice site mutations. Furthermore, no relevant clinical findings differed significantly between patient groups harbouring missense versus premature truncating mutations. In contrast, patients with extended 3p deletions are at risk of a more severe delay (Fisher’s exact test p<0.05), anterior eye segment abnormalities and hypogenitalism compared with patients with monogenic FOXP1 defects, despite the absence of known disease-modifying candidate genes in the overlapping regions of the deletions. In addition, hearing impairment only occurs in patients with extended 3p deletions (Fisher’s exact p<0.001) for which MITF, associated with Waardenburg type IIA syndrome (MIM: 193510), emerges as a plausible candidate gene. One patient with hearing impairment did not show a deletion of MITF, but a positional effect cannot be excluded.
The differential diagnosis of SLI is wide (see online supplementary table 3), but the craniofacial ‘gestalt’ may guide towards an underlying FOXP1 defect and may be helpful in correlating NGS data with clinical presentation.
In case of unclear pathogenicity and absence of sufficient patient material, in vitro functional assessment of variants is straightforward and feasible, showing loss of function either by alternative splicing, reduced repression activity or reduced protein expression.
Patients with FOXP1 defects should be screened for associated organ system involvement including a detailed clinical examination, eye and hearing evaluation (especially in those patients with extended 3p interstitial deletions), echocardiography and ultrasound of the genitourinary tract. Indeed, single nucleotide variants (SNVs) in FOXP1 have recently been associated with congenital anomalies of the kidneys and urinary tract.46 An electroencephalogram should be performed if there is any doubt of seizures, and brain MRI should be considered in the presence of epilepsy.
Follow-up should include repeated assessment of the neuromotor development, oromotor development (especially in case of failure to thrive), language development and behavioural problems in order to enable early initiation of therapy. Physiotherapy might be beneficial to prevent progressive contractures in case of spasticity. The management and follow-up of associated organ system involvements should be adjusted to the specific underlying problems and the patient’s needs.
In conclusion, we have delineated the FOXP1-related ID syndrome as a recognisable entity that seems to be more frequent than expected based on the rather rare previous case reports. Moreover, the ‘gestalt’ may help clinicians in interpreting genotype–phenotype correlations.
References
Footnotes
Contributors We hereby declare that all authors have contributed to this work: IM and BCa contributed to the conception and design of the article, data acquisition, analysis and interpretation of the clinical, molecular and functional data, and writing of the manuscript. DL contributed to the conception and design of the article and to the acquisition of clinical and molecular patient data. DR, GAR, FFH, JLM and PAD contributed to the acquisition of the functional data, and analysis and interpretation of these data. NRe, JP, CC, JM, JR, NRa, SGM, PL, MPB, MÁM, StM, VB, SM, NB, AO, OV, MV, TJLdR, DM, JS, KKV, NDD, AKHK, LH, BD, AN, LT, JA, MJP, KN, BCe, A-SS, DP, MaH, MiH, MB, AD and BM contributed to the acquisition of clinical and molecular patient data. All authors have approved the manuscript and none have reported a conflict of interest.
Funding BCa is a Senior Clinical Investigator, funded by Fund for Scientific Research Flanders (FWO). This project was cofunded by FWO project G044615N and Czech grants 00064203 and NF-CZ11-PDP-3-003-2014.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Commissie voor Medische Ethiek Universitair Ziekenhuis Gent.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement This study makes use of data generated by the DECIPHER Consortium. A full list of centres who contributed to the generation of the data is available from and via email from decipher@sanger.ac.uk. Funding for this project was provided by the Wellcome Trust.