


Abstract
The imidazopyridine zolpidem (Ambien) is one of the most commonly prescribed sleep aids in the United States (Rush, 1998). Similar to classic benzodiazepines (BZDs), zolpidem binds at the extracellular N-terminal α/γ subunit interface of the GABA-A receptor (GABAR). However, zolpidem differs significantly from classic BZDs in chemical structure and neuropharmacological properties. Thus, classic BZDs and zolpidem are likely to have different requirements for high-affinity binding to GABARs. To date, three residues—γ2Met57, γ2Phe77, and γ2Met130—have been identified as necessary for high-affinity zolpidem binding (Proc Natl Acad Sci USA 94:8824-8829, 1997; Mol Pharmacol 52:874-881, 1997). In this study, we used radioligand binding techniques, γ2/α1 chimeric subunits (χ), site-directed mutagenesis, and molecular modeling to identify additional γ2 subunit residues important for high-affinity zolpidem binding. Whereas α1β2χ receptors containing only the first 161 amino-terminal residues of the γ2 subunit bind the classic BZD flunitrazepam with wild-type affinity, zolpidem affinity is decreased ∼8-fold. By incrementally restoring γ2 subunit sequence, we identified a seven-amino acid stretch in the γ2 subunit loop F region (amino acids 186-192) that is required to confer high-affinity zolpidem binding to GABARs. When mapped to a homology model, these seven amino acids make up part of loop F located at the α/γ interface. Based on in silico zolpidem docking, three residues within loop F, γ2Glu189, γ2Thr193, and γ2Arg194, emerge as being important for stabilizing zolpidem in the BZD binding pocket and probably interact with other loop F residues to maintain the structural integrity of the BZD binding site.
Zolpidem (Ambien), an imidazopyridine, is a popularly prescribed sedative-hypnotic used in the treatment of insomnia. Similar to classic benzodiazepines (BZDs), such as flunitrazepam and diazepam, zolpidem binds to the extracellular N-terminal α/γ subunit interface of the GABA-A receptor (GABAR). Residues that are believed to form the BZD binding site are located in at least six noncontiguous regions of the α and γ subunits, which are by arbitrary convention designated `loops' A to F (Galzi and Changeux, 1995). Although zolpidem binds within the BZD binding site and induces many of the same behavioral effects as other positive modulators of the BZD site, it differs from classic BZDs in both chemical structure (Fig. 1) and neuropharmacological profile (Rush, 1998). Zolpidem binds with high affinity to GABARs containing the α1 subunit isoform and with low affinity for GABARs containing the α2 or α3 subunits, whereas receptors containing the α5 subunit are virtually zolpidem-insensitive (Pritchett and Seeburg, 1990). Furthermore, GABARs containing γ1 or γ3 subunits exhibit little to no zolpidem sensitivity, regardless of the coassembled α subunit isoform (Lüddens et al., 1994, 1998; Sanna et al., 2002). Thus, α1βxγ2 GABARs display the highest affinity for zolpidem. In contrast, classic BZDs show a much broader binding profile, in that all α subunit isoforms (except α4 and α6) and all γ subunit isoforms bind the classic BZDs flunitrazepam and diazepam. Because classic BZDs and zolpidem show differences in subunit selectivity and possess divergent chemical structures, it is likely that they have different structural requirements for high-affinity binding.
Here, we used radioligand binding and γ2/α1 chimeric subunits (χ) to identify γ2 subunit residues important for high-affinity zolpidem binding. Although site-directed mutagenesis and photoaffinity labeling studies have made significant strides in uncovering amino acid residues that contribute to the binding domains of classic BZDs, the residues that preferentially contribute to the binding of unconventional BZD-site ligands, such as zolpidem, remain unresolved. A study exploiting the differences in zolpidem sensitivity between α1 and α5 subunit isoforms identified residues α1Thr162, α1Gly200, and α1Ser204 as contributing to the high zolpidem sensitivity of the α1 subunit isoform (Renard et al., 1999). Furthermore, replacing α1Gly200 with residues of increasing side-chain volume correlates with decreasing zolpidem affinity (Wingrove et al., 2002). Hence, steric hindrance may dictate differences in zolpidem affinity across subunit isoforms because the α1 subunit contains a glycine at position 200, whereas the α2, α3, α4, α5, and α6 subunits possess a glutamate (larger in side-chain volume) at the aligned position.

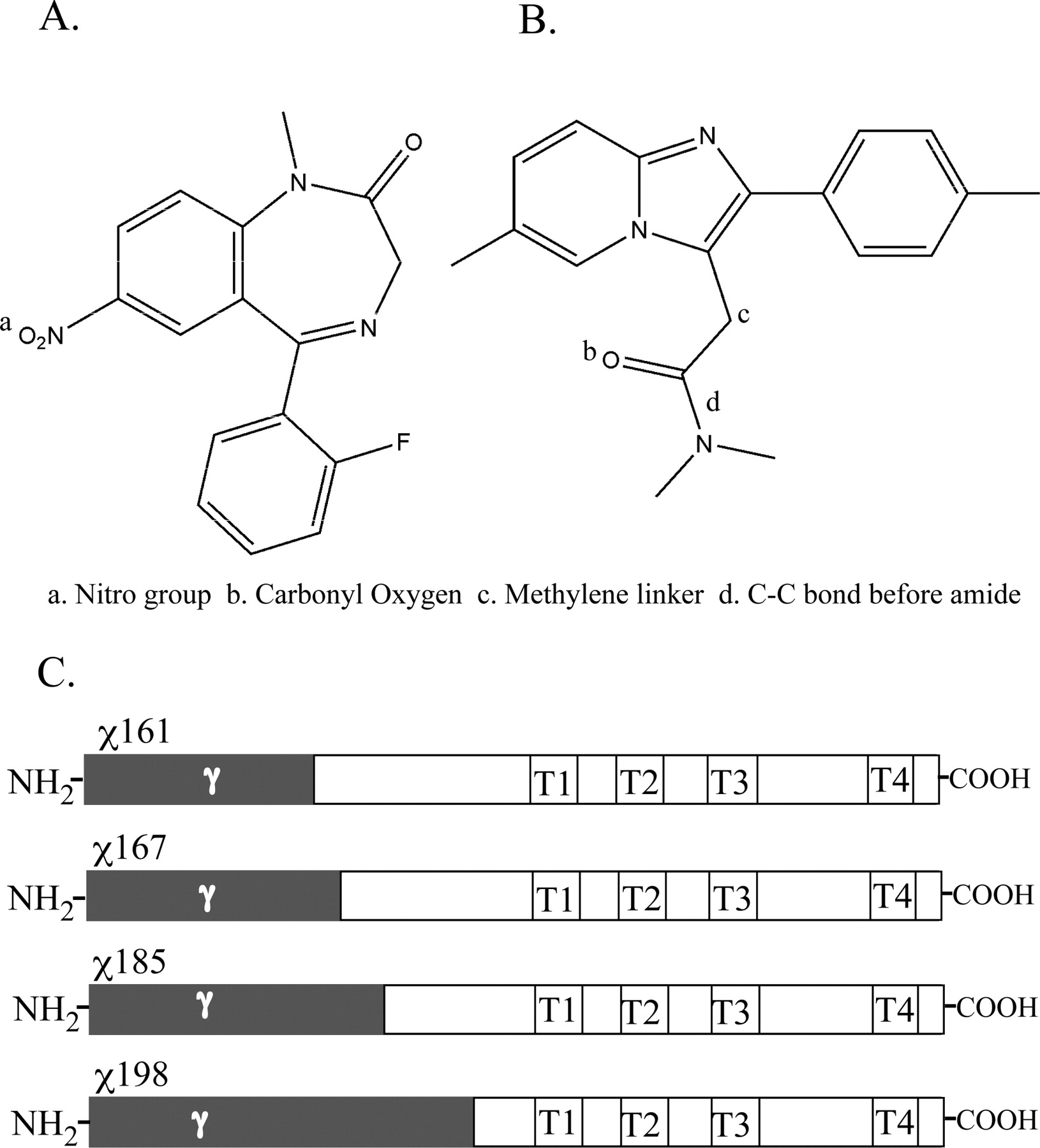
Chemical structures of flunitrazepam (A) and zolpidem (B) and schematic representations of chimeric (χ) γ2/α1 subunits (C) used in this study. In chimeric subunits (C), N-terminal γ2 sequences are highlighted in gray, and the remaining C-terminal α1 sequences are highlighted in white, with the transmembrane domains labeled (T1, T2, T3, T4). Chimeras are numbered according to the amount of mature γ2 sequence contained in the subunit, where χ161 contains γ2 residues up to and including amino acid 161 of the mature protein sequence and residues C-terminal of 161 are α1 sequence.
Similar site-directed mutagenesis studies have identified residues on the γ2 subunit that confer high zolpidem affinity, including Phe77 (Buhr et al., 1997a; Wingrove et al., 1997), Met57, and Met130 (Buhr and Sigel, 1997; Wingrove et al., 1997). However, γ2Met130 is not solely selective for zolpidem binding, in that mutations of γ2Met130 have also been shown to disrupt high-affinity binding of the classic BZDs flunitrazepam and triazolam (Wingrove et al., 1997). It is noteworthy that introducing both Phe77 and Met130 into the homologous positions in the γ1 subunit was not sufficient to restore its affinity for zolpidem to that of γ2-containing receptors, suggesting that additional γ2 amino acids are necessary for high zolpidem affinity (Wingrove et al., 1997).
The specificity of binding to different subunit isoforms in part dictates the various behavioral endpoints of BZD ligands. For example, GABARs containing the α1 subunit are associated primarily with the sedative properties of classic BZDs, whereas α2/3 containing GABARs are responsible for the anxiolytic effects of classic BZDs (for review, see Sieghart and Ernst, 2005). These pharmacological differences may explain why zolpidem, an α1-selective BZD site ligand, is not a clinically efficacious anxiolytic but rather a sedative-hypnotic. Although classic BZDs such as diazepam are effective in treating anxiety, their use is accompanied by a myriad of side effects, including drowsiness, dependence, and tolerance. These behaviorally ambiguous effects of classic BZDs are thought to result from a relative lack of selectivity in binding affinity and efficacy to GABARs containing different subunit isoforms. As such, identifying the structural determinants involved in the binding of a more selective BZD-site ligand, such as zolpidem, could help in the design and development of more pharmacologically and behaviorally selective BZD site ligands.
Here, we provide evidence that residues γ2Ser186 - Asp192, which make up part of loop F of the BZD binding site, are necessary for conferring high-affinity zolpidem binding to α1β2γ2 GABARs. In addition, by computational docking of zolpidem and flunitrazepam into a structural homology model of α/γ subunit extracellular interface, we describe possible orientations of these ligands in the BZD binding site of the GABAR.
Materials and Methods
Molecular Cloning. γ2/α1 chimeric subunits χ161 and χ167 were created by linearizing and then recircularizing a dual plasmid construct in bacteria by random homologous crossover events as detailed by Boileau et al. (1998). γ2/α1 chimeras χ185, χ192, and χ198 were created using overlapping α/γ complimentary oligonucleotides and standard overlap extension recombinant PCR techniques. Chimeric subunits are numbered according to the amount of mature γ2 N-terminal sequence contained in the subunit, where, for example, χ161 contains γ2 residues up to and including amino acid 161 of the mature protein, and residues C-terminal of 161 are α1 subunit sequence. γ2 subunit point mutations E189R, T193E, and R194D were created using standard recombinant PCR techniques. Wild-type (WT) rat γ2 cDNA in pCEP4 (Invitrogen, Carlsbad, CA) was used as a template for site-directed mutagenesis protocols. Pfu Ultra polymerase (Stratagene, La Jolla, CA) was used for PCR amplification at the recommended specifications. Resulting mutant and chimeric products were subcloned into pCEP4 for transient expression in HEK 293 cells with β2 and α1 cDNAs in pCEP4. All mutant constructs were verified by double-stranded DNA sequencing.
Transient Expression in HEK 293 cells. HEK 293 cells were grown as described previously (Boileau et al., 1998). Cells were cotransfected at ∼70% confluence with WT rat α1, β2, γ2, or mutant subunit cDNAs, and, when necessary, the vector pAdvantage (Promega, Madison, WI), at a ratio of 1:1:1:1.5-2 (3-4 μg of DNA per subunit and 6 μg of pAdvantage per dish) using a standard CaHPO4 precipitation method (Graham and van der Eb, 1973).
Radioligand Binding. Approximately 48 h after transfection, membrane homogenates were prepared, and homologous competition and saturation binding experiments were performed as described by Boileau et al. (1998). In brief, for competition binding, membrane homogenates (100 μg) were incubated at room temperature for 1 h with sub-Kd concentrations of radioligand ([3H]flunitrazepam; PerkinElmer Life and Analytical Sciences, Boston, MA) in the absence or presence of seven different concentrations of unlabeled ligand in a final volume of 250 μl. Data were fit by nonlinear regression analysis to a single site competition defined by the equation y = Bmax/[1 + (x/IC50)], where y is bound 3H-labeled ligand (in disintegrations per minute), Bmax is maximal binding, x is the concentration of displacing ligand, and IC50 is the concentration of unlabeled ligand that inhibits 50% of 3H ligand binding (Prism; GraphPad Software, San Diego, CA). KI values were calculated using the Cheng-Prusoff/Chou equation (Cheng and Prusoff, 1973; Chou, 1974): KI = IC50/[1 + (L/KD)], where KI refers to the equilibrium dissociation constant of the unlabeled ligand, KD refers to the equilibrium dissociation constant of the radioactive ligand and L refers to the concentration of radioactive ligand (Prism software).
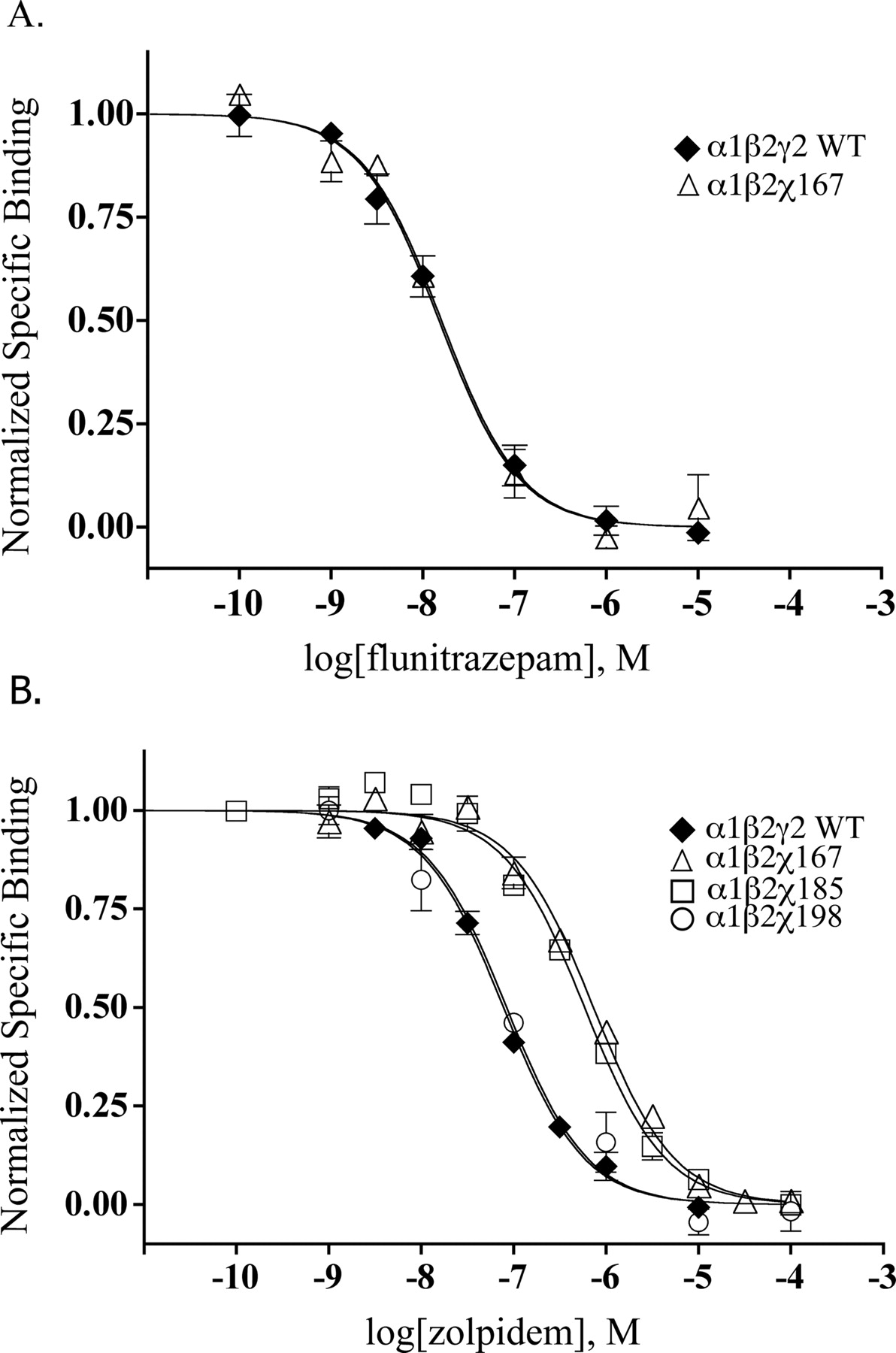
γ2/α1 chimeric subunit constructs differentially affect zolpidem binding in α1β2χ receptors. Representative radioligand binding curves depict the displacement of [3H]flunitrazepam binding by flunitrazepam (A) and zolpidem (B) in α1β2γ2WT(♦), α1β2χ167 (▴), α1β2γ2χ185 (□), and α1β2γ2χ198 (○) receptors, where each point is the mean ± S.E.M. of triplicate measurements. Data were fit by nonlinear regression analysis as described under Materials and Methods. KI values are reported in Table 1.
For saturation binding, membrane homogenates were incubated at room temperature with seven to nine concentrations of [3H]flunitrazepam. Data were fit by nonlinear regression analysis to a single site using the equation y = Bmax × x/(KD + x), where y is specifically bound 3H ligand (in disintegrations per minute), Bmax is maximal binding, and x is concentration of 3H ligand. In both cases, specific binding was defined as 3H-drug bound in the absence of displacing ligand minus the amount bound in the presence of displacing ligand. Unlabeled flunitrazepam was obtained as a gift from Dr. Sepinwall (Hoffman-La Roche, Nutley, NJ). Unlabeled zolpidem was obtained from RBI/Sigma (Natick, MA).
Automated Ligand Docking. Our GABAR model was constructed using SYBYL 7.1 (Tripos Inc., St. Louis, MO) as described previously (Mercado and Czajkowski, 2006). In brief, the extracellular N-terminal domain of the GABAR was built by residue replacement on the crystal structure of the acetylcholine binding protein (Brejc et al., 2001). The N-terminal domain was then merged with a model of the transmembrane domains created analogously from the refined 4-Å resolution cryo-electron microscopy of the nicotinic acetylcholine receptor (Unwin, 2005). Atom potentials were assigned from the Tripos force field, and the entire structure was minimized by the Powell conjugate gradient method. The resulting structure was then refined by manual adjustment to correct gross distortions of side chains and local electrostatic incompatibilities uncovered by evaluation with the program PROCHECK (Laskowski et al., 1996).
BZD ligands were constructed in SYBYL 7.1 and docked at the α1/γ2 interface using Flo+ version of QXP (Quick eXPlore) (McMartin and Bohacek, 1997). The initial placement of the ligand was determined ad hoc, based on the extensive literature amassed on the requirements for BZD binding at the α and γ subunit interface, implicating loops A, B, and C on the α subunit and loops D, and E on the γ subunit. The automated docking strategy used a Monte Carlo search followed by simulated annealing technique, assuming a vacuum (no solvent) environment for the ligand-protein interaction. First, the ligands were docked by 1000 cycles of the default Monte Carlo search, which included both ligand and residue side chains within 4 Å of the initial ligand coordinates. Poses were scored by default, according to energy evaluations using the AMBER force field. The Monte Carlo search yielded 25 low energy receptor-ligand complexes that deviated from one another by at least 0.4 Å root-mean-square-deviation (RMSD). Output poses were binned into spatial clusters (RMSD less than 2 Å), where all members from a given cluster represented a single common binding mode. Average cluster energies were then taken from all members residing in the cluster.
To further optimize receptor-ligand interactions, the lowest energy representative pose from each cluster was treated with simulated annealing molecular dynamics to further optimize interactions in the receptor-ligand complex. The simulated annealing procedure consisted of 100 cycles of variable temperature molecular dynamics. In each cycle, the temperature was scaled from 600 to 50 K over an interval of 9 ps followed by quenching by the default Polak-Ribiere conjugate gradients minimization to 0.1 kcal·mol-1Å-1. The lowest energy pose among the 100 generated was selected as the final representative pose. Ligands converged upon the same set of modes regardless of starting coordinates for docking, suggesting a robust search procedure that was not strongly influenced by the initial ligand placement. All images were produced using PyMOL (DeLano Scientific, LLC, San Carlos, CA).
Results
Flunitrazepam and Zolpidem Binding to αβχ Receptors. Four chimeric γ/α constructs, χ161, χ167, χ185, and χ198, containing varying amounts of γ2 subunit N-terminal sequence (Fig. 1) were expressed with WT α and β subunits in HEK 293 cells. Binding affinities (KI) for flunitrazepam and zolpidem were measured by displacement of [3H]flunitrazepam binding (Fig. 2). Wild-type α1β2γ2 receptors bound flunitrazepam and zolpidem with KI values of 8.4 and 56.3 nM, respectively (Table 1). All chimeric receptors retained WT binding affinity for flunitrazepam (Table 1, Fig. 2A), indicating that unique γ2 subunit residues C-terminal to 161 are not crucial for high-affinity flunitrazepam binding. In contrast, only α1β2χ198 retained WT apparent binding affinity for zolpidem (KI = 54.3 nM), while α1β2χ161, α1β2χ167, and α1β2χ185 displayed 8- to 9-fold decreases in zolpidem affinity (p < 0.01; Table 1, Fig. 2B). Thus, γ2 subunit residues from the N terminus to residue 185 are not sufficient to confer high-affinity zolpidem binding to α1β2χ receptors. Because the addition of γ2 residues Ser186 to Leu198 restored zolpidem affinity in α1β2χ198 receptors to WT values, we hypothesized that a subset of residues within this region is required for high-affinity zolpidem binding. To further delineate residues within this region that selectively contribute to high-affinity zolpidem binding, we constructed another γ/α chimera, χ192. α1β2χ192 receptors bound zolpidem with high affinity (KI = 16.5 nM, Table 1), indicating that residues Ser186 to Asp192, in loop F of the γ2 subunit, are necessary for conferring high-affinity zolpidem binding.
Binding affinities of flunitrazepam and zolpidem for α1β2γ2s WT and α1β2ϰ receptors
Values shown are the mean ± S.E.M. for n number of independent experiments. KI is the equilibrium dissociation constant (apparent affinity) of the unlabeled ligand. KI values for flunitrazepam were determined by [3H]flunitrazepam saturation and competition binding experiments. KI values for zolpidem were determined by the displacement of [3H]flunitrazepam. Statistical differences between WT and chimera logKI values were determined using one-way analysis of variance with Dunnett's post test.
Molecular Docking of Flunitrazepam and Zolpidem. As depicted in a model of the N-terminal extracellular BZD binding domain of the GABAR, the γ2Ser186-Asp192 region is part of a lengthy and flexible region of ambiguous secondary structure known as Loop F/9 (Fig. 3, A and B). The model provides insight into the structure of the BZD binding pocket, where loop F is nestled among various residues that have previously been implicated in BZD binding (Fig. 3B), highlighting the potential importance of loop F in forming part of the binding pocket. We performed simulated molecular docking of flunitrazepam and zolpidem within the α/γ BZD binding interface to help illuminate possible residues within loop F that participate in zolpidem binding and to examine the compatibility between the predictions made by computational modeling and those conclusions drawn from experimental observations.
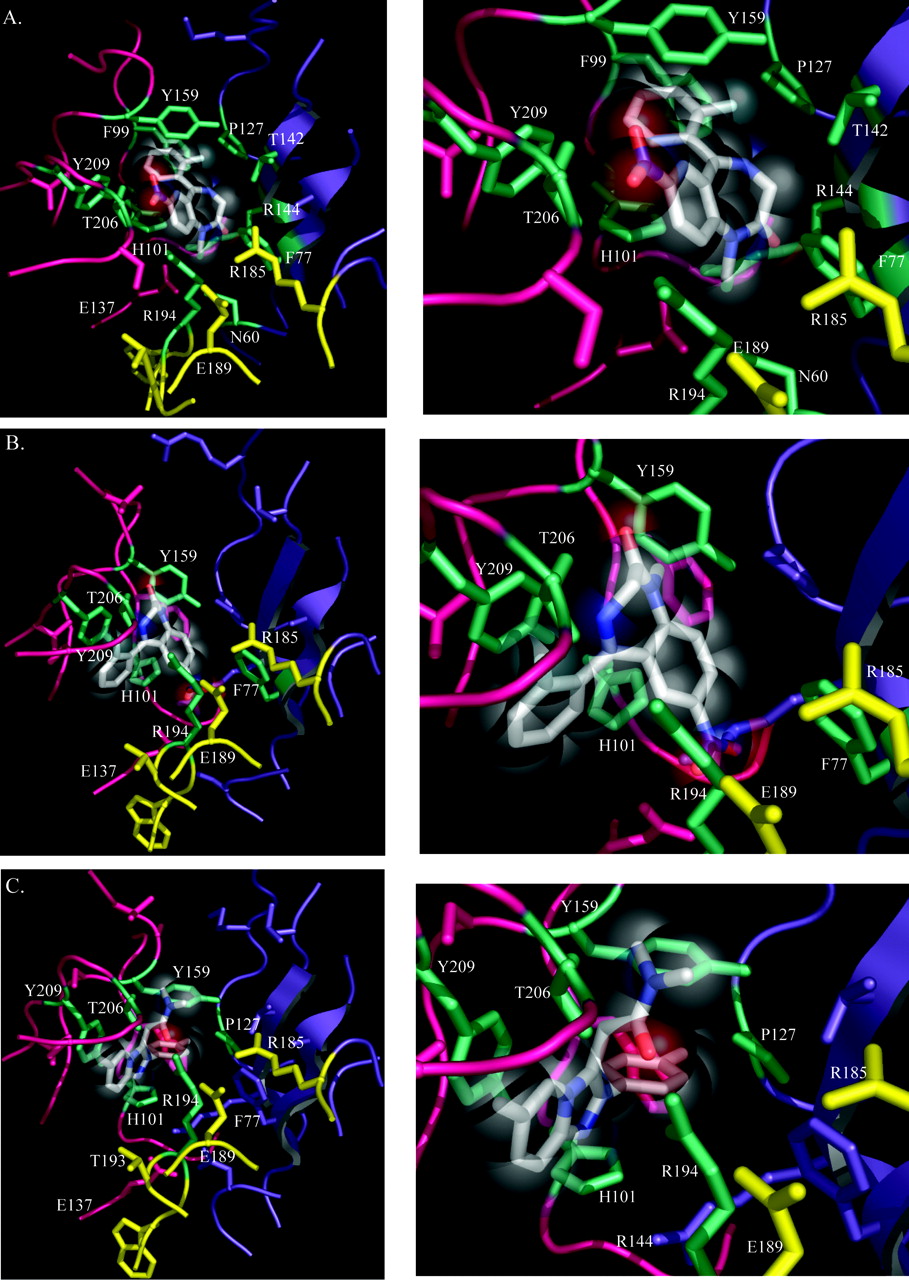
Flunitrazepam docking yielded three different orientations of the ligand within the binding pocket (Fig. 4, A and B, Table 2). In contrast, zolpidem docking and subsequent energy minimization converged to a single pose of low energy (-43.5 kJ/mol; Fig. 4C). Of the three flunitrazepam poses, the one with the lowest energy (-34 kJ/mol, 32% populated; Table 2) shown in Fig. 4A most closely predicts the α1 and γ2 residues involved in high-affinity BZD binding, as previously determined experimentally. The most obvious difference between the lowest energy (Fig. 4A) and most populated (Fig. 4B) flunitrazepam poses is in the π-π stacking interactions of the pendant phenyl group of flunitrazepam, which in the lowest energy pose is sandwiched between the aromatic rings of α1Tyr159 and α1His101, whereas in the most populated pose it stacks exclusively with α1Tyr209. The variability in flunitrazepam orientation may result from a greater conformational freedom of flunitrazepam relative to zolpidem within the BZD binding site.
Zolpidem and flunitrazepam docking energies
Docking simulations were carried out using the Flo + version of QXP (Quick eXPlore), as detailed under Materials and Methods. Docking yielded one orientation of zolpidem and three orientations of flunitrazepam (1-3). The lowest and population average energies after the MC and after SA procedures are shown (see Materials and Methods). Values are the mean ± S.E.M., where error is reported.
Given the technical and methodological constraints of current molecular simulation techniques, it is difficult to fully recapitulate the true biological environment within which protein-protein and protein-ligand interactions occur. Nevertheless, to gain insight into the potential interactions between zolpidem or flunitrazepam and the GABAR in our docked receptor-ligand complexes, we measured the distances between residue atoms of the protein and atoms of each ligand (Fig. 5). We assigned proximity ratings to each interaction; the higher the proximity rating number, the closer the ligand is to a given subunit residue, ranging from 6 (2.5 Å) to 1 (5 Å). Functional groups within less than 4 Å of one another possess potential for forming salt bridges and hydrogen bonds (Kumar and Nussinov, 1999), whereas functional groups greater than 7 Å apart from one another probably do not participate in meaningful electrostatic interactions (Schreiber and Fersht, 1995). Hydrophobic interactions, on the other hand, can occur at much longer distances (Israelachvili and Pashley, 1982).
Residues α1His101, α1Tyr159, α1Thr206, α1His101, γ2Phe77, and γ2Thr142, all previously implicated in both flunitrazepam and zolpidem binding and/or efficacy (Mihic et al., 1994; Wingrove et al., 1997; Amin et al., 1997; Buhr et al., 1997a,b; Sigel et al., 1998), are located within 4 Å of both flunitrazepam and zolpidem (Fig. 5, A and B). Thus, these residues could potentially participate in a myriad of noncovalent intermolecular interactions with the ligand, including van der Waals, salt bridges, and hydrogen bonds. In addition, our docks predict that α1Thr162, α1Ser204, and α1Glu208 come within 5 Å of zolpidem, but not flunitrazepam (Fig. 5A), and potentially participate in longer range (4-7 Å) molecular interactions with zolpidem. It is noteworthy that α1Thr162 and α1Ser204 have previously been shown to confer binding selectivity for zolpidem to the α1 subunit (Renard et al., 1999). That our simulated molecular docking results are consistent with past experimental findings lends credence to the accuracy of our homology model and the prediction that flunitrazepam and zolpidem are situated differently within the BZD binding pocket.
Within loop F of the γ2 subunit, only Arg194 was found to come within at least 3 Å of flunitrazepam (Fig. 5B). In the most energetically favorable (lowest energy) pose, Arg194 comes within 2.5 Å of flunitrazepam (Figs. 4A and 5B), where the guanidine group of the arginine is pointing toward the nitro group of flunitrazepam. However, the nitro group of flunitrazepam (Fig. 1A) may also participate in hydrogen bonding with the side-chain functional group of α1Thr206, which also comes within 2.5 Å of the substrate (Fig. 4A). In the most highly populated binding mode for flunitrazepam (Fig. 4B), Arg194 is pointing toward the pendant phenyl of flunitrazepam, coming within 2.5 Å of the substrate. None of the residues in the γ2Ser186-Asp192 region are near flunitrazepam, falling greater than 5 Å away from the substrate (Figs. 4, A and B, and 5B), which may explain why mutations in this region do not disrupt high-affinity flunitrazepam binding.
Our model of the zolpidem pharmacophore predicts that γ2 loop F residue Arg194 comes within 2.5 Å of zolpidem (Fig. 5B). What distinguishes the interaction observed between Arg194 and zolpidem from that of Arg194 and flunitrazepam is the potential for strong hydrogen bonding exclusively between the guanidine group of the arginine and the carbonyl oxygen (Fig. 1B) on zolpidem (Fig. 4C). In addition, γ2Thr193 could potentially participate in longer-range molecular interactions with zolpidem, even possibly hydrogen bonding, positioned within 5 Å of the substrate (Figs. 4C and 5B). It is surprising that the loop F residues γ2Ser186 to Asp192, which we have shown in experiments to be important in zolpidem binding, do not seem to participate, given our docking results, in any direct interactions with zolpidem. However, there are some notable intrasubunit interactions within loop F, including potential electrostatic interactions between R185, Glu189, and Arg194 (Fig. 4C). It is important to note that a residue can influence ligand binding in multiple ways. Some residues may directly contact the ligand, some may be important for maintaining the structural integrity of the binding site, and others may mediate local conformational movements near the binding site. Therefore, although residues γ2Ser186 to Asp192 may not directly contact zolpidem, they are likely to contribute to zolpidem binding by maintaining the structural integrity of this loop region and strategically orienting Arg194 within the binding pocket. To test the importance of residues γ2Glu189, γ2Thr193, and γ2Arg194 in zolpidem binding, we mutated each to their aligned α1 subunit residue. Given our sequence alignment, these residues show the most dramatic divergence in the chemical properties of their side chains (Fig. 6A).

Homology model of the extracellular α1/γ2 subunit interface of the GABAR. The α1 subunit is pink, the γ2 subunit is purple, and loop F is highlighted in yellow (A and B). A close-up view of the BZD binding pocket is illustrated in B, where several residues previously implicated in benzodiazepine binding are highlighted in teal. Data from this study suggest that residues within loop F of the γ2 subunit, γ2Ser186 to Asp192, shown in yellow, are involved in conferring high-affinity zolpidem binding to α1β2γ2 GABARs.
The Effects of Point Mutations in Loop F of the γ2 Subunit on Zolpidem and Flunitrazepam Binding. Of the three mutations tested, γ2E189R, γ2T193E, and γ2R194D, none disrupted high-affinity zolpidem binding (Fig. 6B). Both α1β2γ2T193E and α1β2γ2E189R receptors had affinities for zolpidem comparable with WT (KI = 40 and 85.6 nM, respectively), whereas α1β2γ2R194D receptors displayed a 3.5-fold increase in affinity for zolpidem (KI = 16.6 nM, p < 0.01; Fig. 6B; Table 3). Therefore, the results suggest that residues γ2Glu189, γ2Thr193, and γ2Arg194 are not individually responsible for conferring high-affinity zolpidem binding. It is noteworthy that γ2E189R resulted in a 5.5-fold increase in flunitrazepam affinity (Table 3). Introducing an arginine at position γ2Glu189 would disrupt the predicted salt-bridge triad with γ2Arg185 and γ2Arg194, suggesting that these electrostatic interactions are not necessary for high-affinity flunitrazepam or zolpidem binding. Given the purported structural flexibility of loop F and the presence of other charged residues within the region, it is possible that alternate electrostatic interactions compensate for the introduction of a positive charge at γ2Glu189.
Binding affinities of flunitrazepam and zolpidem for α1β2γ2s WT and α1β2γ2s mutant receptors
Values shown are the mean ± S.E.M. for n number of independent experiments. KI is the equilibrium dissociation constant (apparent affinity) of the unlabeled ligand. KI values were determined by the displacement of [3H]flunitrazepam. Statistical differences between WT and chimera logKI values were determined using one-way ANOVA with Dunnett's post test.
To compliment our experimental findings, molecular docking of zolpidem was carried out on γ2E189R and γ2R194D mutant receptors. Although the binding energy and the position of zolpidem in the binding pocket of the γ2E189R mutant were comparable with wild-type, the amino acid side chains adopted slightly different orientations within the site, which may explain why the γ2E189R mutation had little effect on zolpidem binding affinity (Table 2). In the γ2R194D mutant background, the binding energy for zolpidem was comparable with that of wild-type receptors and the amino acid side chains maintained positions similar to those in the WT dock. It is noteworthy that zolpidem adopted a flipped orientation within the site, with the amide group on zolpidem coming within 4.5 Å of the carboxyl group of the aspartic acid at position 194. The reorientation of zolpidem within the binding pocket may account for the inability of the γ2R194D mutation to reduce the apparent affinity of zolpidem (Table 2). When considering our mutagenesis findings in the context of our docking data, it becomes clear that residues Ser186 to Asp192 are probably not contact residues, because the docking results suggest that these residues are not located near zolpidem, and our mutagenesis results indicate that individual point mutations fail to disrupt high-affinity zolpidem binding. Taken collectively, these findings indicate that residues within loop F of the γ2 subunit probably work in a synergistic fashion to maintain the proper configuration of the BZD binding site.
Discussion
Multiple methods have been used to identify residues within loops A to E that are involved in the efficacy and/or binding of BZD site ligands. In this study, we demonstrate that γ2Ser186 to Asp192 in loop F is necessary for high-affinity binding of the imidazopyridine zolpidem. In contrast, unique γ2 subunit loop F residues are not required for high-affinity flunitrazepam binding, because affinity for flunitrazepam is preserved in α1β2χ167 receptors (Table 1). To our knowledge, this study is the first to identify residues in loop F of the γ subunit that are important for the action of a BZD-site ligand.

Molecular docking of flunitrazepam (A and B) and zolpidem (C). A, the lowest energy orientation of flunitrazepam within the binding site, after Monte Carlo (MC) and simulated annealing (SA) analysis. B, an alternative, most highly populated (based on the MC) binding mode for flunitrazepam after MC and SA (see Materials and Methods). C, the orientation of zolpidem after MC and SA. On the right are zoom-in views of the docked ligands in A, B, and C. The α1 subunit is pink, and the γ2 subunit is purple. The substrate is colored such that carbon is white, nitrogen is blue, oxygen is red, and fluorine is light blue. Loop F residues γ2Arg185 to Arg197 are highlighted in yellow. Residues within 3 Å of the docked ligand are highlighted in teal. Results from the molecular docking procedure are summarized in Table 2.
Molecular docking reveals that zolpidem and flunitrazepam probably inhabit overlapping, yet partially distinct, locations within the BZD binding pocket. The dockings accurately identify residues in loops A to C of the α1 subunit, and loops D and E of the γ2 subunit, that previous experiments have shown to contribute to the binding of both classic BZDs and zolpidem. On the α1 subunit, several aromatic residues stabilize zolpidem and flunitrazepam in the binding site. For example, the aromatic ring α1Tyr159 (loop B) stacks with the pendant phenyl group of both flunitrazepam (lowest energy pose, Fig. 4A) and zolpidem (Fig. 4C). Removing the aromatic ring at α1Tyr159 completely abolishes diazepam-mediated potentiation of GABA current in α1β2γ2 receptors (Amin et al., 1997) and we predict, based on our modeling, that this residue may similarly abolish the actions of zolpidem. In addition, the aromatic ring of α1His101 (loop C) stacks with the fused phenyl ring on flunitrazepam (Fig. 4A) and zolpidem (Fig. 4C), potentially forming π-π interactions. In the most populated flunitrazepam dock (Fig. 4B), the pendant phenyl of flunitrazepam also participates in π-π stacking with α1His101, underscoring the importance of an aromatic residue at this position. Studies have shown that an aromatic ring at position 209 is crucial for maximal diazepam mediated modulation of GABA current as well as high-affinity zolpidem, flunitrazepam, and diazepam binding in α1β2γ2 GABARs (Amin et al., 1997; Buhr et al., 1997b). In our dock, a potential hydrogen bond between α1Thr206 (loop C), and the nitro group (Fig. 1A) of flunitrazepam (lowest energy pose, Fig. 4A), or the fluorine of the pendant phenyl (most populated pose, Fig. 4B) is also observed. Previous findings have shown that the mutation α1T206A significantly disrupts high-affinity binding of classic BZDs, as well as zolpidem (Buhr et al., 1997b; Sigel et al., 1998). It is noteworthy that our model predicts that α1T206C and the carbonyl oxygen of zolpidem interact via hydrogen bonding, suggesting that α1T206C may also influence zolpidem affinity (Fig. 4C).
Although there is a significant amount of overlap in residues forming the recognition site for flunitrazepam and zolpidem, our findings suggest that there are differences in their binding requirements. On the α subunit, α1His101 (loop A), α1Thr162 (loop B), and α1Ser204 (loop C) lie within the core of the zolpidem binding pocket. Our docking results reveal that the pendant phenyls of both zolpidem and flunitrazepam participate in π-π stacking with the aromatic ring of α1His101 (loop A) (Fig. 4, A and C). α1His101 has long been noted as a crucial residue in high-affinity classic BZD binding. Zolpidem is also sensitive to mutations at this position. For example, mutating α1His101 to arginine abolishes flunitrazepam and zolpidem binding, as well as the in vivo behavioral effects of zolpidem (Wieland et al., 1992; Crestani et al., 2000). However, the converse mutation at the aligned position in the α6 subunit (α6R100H), which restores sensitivity of α6β2γ2 receptors to classic BZDs, does not restore high-affinity zolpidem binding (Wieland et al., 1992). In addition, GABARs photoincorporated with flunitrazepam have drastically reduced affinities for classic BZDs, whereas the binding affinity of zolpidem remains intact (McKernan et al., 1998). These data suggest that zolpidem may have more tolerance for substitutions at α1His101 and requires additional residues in the α1 for high-affinity binding. We predict that two residues in particular, α1Thr162 and α1Ser204, are selectively involved in zolpidem binding because they reside within at least 4.5 Å of zolpidem but not flunitrazepam (Fig. 5). α1Ser204 in particular comes within 4 Å of zolpidem, making this residue a candidate for hydrogen bonding with the substrate. Replacing the corresponding residues in the zolpidem-insensitive α5 subunit with the aligned residues from the α1 subunit (i.e., α5P166T and α5T208S) significantly increased zolpidem affinity in α5β2γ2 mutant receptors (Renard et al., 1999), further supporting our predictions.
On the γ2 subunit, Phe77 (loop D) is positioned within 4 Å of zolpidem as well as flunitrazepam and is probably involved in hydrophobic and/or electrostatic interactions with both ligands (Fig. 4&5). Mutagenesis, radioligand binding, and studies on γ2F77I transgenic knockin mice indicate that γ2Phe77 is critical for the high-affinity binding of classic BZDs and zolpidem, as well as the in vivo behavioral effects of zolpidem (Wingrove et al., 1997; Sigel et al., 1998; Cope et al., 2004; Ogris et al., 2004). In addition, γ2R144 (loop E) is positioned within 4 Å of both flunitrazepam and zolpidem and potentially H-bonds with the carbonyl oxygen of flunitrazepam (Fig. 4A). It is noteworthy that γ2R144 seems to be involved in a potential intersubunit salt-bridge with α1E137 and may be important for stabilizing the structure of the BZD binding site (Fig. 4, A-C). A study by Harrison and Lummis (2006) suggests that the aligned residue in the GABA-C receptor ρ1 subunit (ρ1R170; i.e., GABAR γ2R144) is important for proper receptor function and is probably involved in intersubunit salt-bridge interactions. Finally, γ2Thr142 (loop E) is positioned within 4.5 Å of both flunitrazepam and zolpidem (Fig. 5) and has previously been shown to influence the efficacy of classic BZDs as well as zolpidem (Mihic et al., 1994).
Our docking and experimental findings indicate that the binding requirements for zolpidem and flunitrazepam also differ in the loop F region of the γ2 subunit. γ2Thr193 and γ2Arg194 are positioned within 5 Å of zolpidem (Fig. 5B). Although γ2Arg194 is also located near flunitrazepam, we speculate that it plays a more critical role in orienting zolpidem, in that the guanidine group of the arginine probably participates in a hydrogen bond with the carbonyl oxygen (Fig. 1B) of zolpidem (Fig. 4C). Furthermore, α1β2γ2R194D receptors bound zolpidem with even higher apparent affinity than WT, whereas flunitrazepam apparent affinity was unaltered, supporting the conclusion that this residue may play a more critical role in dictating zolpidem affinity. It is possible that a negatively charged aspartate at position 194 preferentially or more favorably interacts with the partial positive charge of the amide moiety on zolpidem (Fig. 1B), instead of the carbonyl oxygen, because the smaller side-chain volume of the aspartate would be able to better accommodate the bulkier amide group. Binding studies using a variety of phenylimidazopyridine derivatives have shown that shortening the methylene linker or lengthening the carbon chain before the amide group (Fig. 1B) of the ligand results in a significant decrease in apparent affinity (Trapani et al., 1997). The spacer length between the imidazopyridine nucleus and the amide group is probably important for correctly positioning the hydrogen bond accepting site, the carbonyl oxygen of zolpidem, within the BZD binding pocket. Consistent with this hypothesis, when we performed molecular docking of zolpidem in the α1β2γ2R194D receptor background, zolpidem assumed a different orientation within the binding pocket such that the amide group of zolpidem moved within 4.5 Å of the carboxyl side chain of the aspartate at position 194.

Proximity of BZD binding site residues to flunitrazepam and zolpidem. The distances between various amino acid side-chains and flunitrazepam (lowest energy pose in white, and most populated mode in gray) or zolpidem (black) on the α1 subunit (A) and the γ2 subunit (B) were measured. The proximity ratings of these residues to the docked ligands are plotted, where 1 = 5Å, 2 = 4.5Å, 3 = 4Å, 4 = 3.5Å, 5 = 3Å, and 6 = 2.5Å. Distances were measured from the atom on the residue that is closest to the docked ligand. Note that within loop F of the γ2 subunit, two residues come within 5 Å of zolpidem (γ2Thr193 and γ2Arg194).

Point mutations in loop F of the γ2 subunit do not disrupt high-affinity zolpidem binding. In A, sequence alignments of the loop F region of WT α1 and γ2 subunits and the chimeric χ192 subunit are shown. γ2 sequence is depicted in black, and α1 sequence is depicted in gray; each star represents a residue in α1 that does not have a corresponding residue in γ2. Residues in loop F of the γ2 subunit implicated in zolpidem binding by molecular docking (circled) were mutated to their corresponding α1 residues. Of these residues, one (γ2Glu189) is contained within the region defined by our study (γ2186-192) as being selectively involved in zolpidem binding. B, representative radioligand binding experiments depict displacement of [3H]flunitrazepam binding by zolpidem in α1β2γ2 WT (♦) and α1β2γ2E189R (□), α1β2γ2T193E (○), and α1β2γ2R194D (▴) mutant receptors, where each point is the mean ± S.E.M. of triplicate measurements. Data were fit by nonlinear regression analysis as described under Materials and Methods. KI values for zolpidem and flunitrazepam are summarized in Table 3.
We demonstrate experimentally that γ2Ser186 to Asp192 is important for high-affinity zolpidem binding. Based on molecular docking, these residues do not come within 5 Å of zolpidem (Fig. 5), suggesting that residues Ser186 to Asp192 contribute to zolpidem binding not by forming direct contacts with the ligand but by maintaining the secondary structure of the binding site. It is important to note, however, that the positioning of loop F is open to interpretation; the crystal structure of acetylcholine binding protein, upon which our model is based, is poorly resolved within loop F (Brejc et al., 2001). Furthermore, loop F protein sequence is poorly conserved among GABAR subunit isoforms and other ligand-gated ion channels, making sequence alignment difficult. It is possible that γ2Ser186 to Asp192 plays a role in zolpidem binding by a complex series of interactions, perhaps serving to properly position γ2Arg194 in the binding pocket. Loop F is likely to be a highly mobile and adaptable structure, thus garnering the ability to compensate for conservative alterations in sequence identity. Our finding that point mutations in the γ2 loop F region do not disrupt high-affinity zolpidem binding supports our hypothesis, suggesting that multiple loop F residues act together to preserve the structure of the BZD site and stabilize zolpidem within the binding pocket.
Our finding that Loop F of the γ2 subunit is involved in BZD binding compliments previous reports that residues in the homologous regions of the GABA binding site interface, as well as the related nicotinic acetylcholine and 5-HT3 receptors, participate in orthosteric agonist binding (Lyford et al., 2003; Newell and Czajkowksi, 2003; Thompson et al., 2006). Studies using molecular dynamics simulation, hydrophobic photolabeling, and the substituted cysteine accessibility method have shown that loop F of the acetylcholine binding protein, as well as the nicotinic acetylcholine GABA-A and 5-HT3 receptors, undergoes conformational changes during channel activation and/or agonist binding (Leite et al., 2003; Lyford et al., 2003; Newell and Czajkowski, 2003; Gao et al., 2005; Thompson et al., 2006). Given these findings, it is reasonable to hypothesize that loop F of the γ subunit undergoes structural rearrangements upon binding of BZDs. It is possible that loop F is important for regulating not only BZD binding but also BZD efficacy. These are intriguing possibilities that need to be explored in future experimental endeavors.
Acknowledgments
We thank Dr. Ken Stayshur for assistance with the construction of the structural homology model.
Footnotes
-
This work was supported by National Institutes of Health grant NS34727 (to C.C.) and National Research Service Award GM07507 (to F.S.).
-
ABBREVIATIONS: BZD, benzodiazepine; GABAR, GABA-A receptor; PCR, polymerase chain reaction; WT, wild type; HEK, human embryonic kidney; RMSD, root-mean-square-deviation; 5-HT, 5-hydroxytryptamine; χ, chimera; MC, Monte Carlo; SA, simulated annealing.
- Received August 4, 2006.
- Accepted September 28, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}