

Abstract
Both social defeat stress and environmental enrichment stimulate adrenal glucocorticoid secretion, but they have opposing effects on hippocampal neurogenesis and mood. Hypothalamic-pituitary-adrenal axis dysregulation and decreased neurogenesis are consequences of social defeat. These outcomes are correlated with depressive states, but a causal role in the etiology of depression remains elusive. The antidepressant actions of environmental enrichment are neurogenesis-dependent, but the contribution of enrichment-elevated glucocorticoids is unexplored. Importantly, for both social defeat and environmental enrichment, how glucocorticoids interact with neurogenesis to alter mood is unknown. Here, we investigate causal roles of glucocorticoids and neurogenesis in induction of depressive-like behavior and its amelioration by environmental enrichment in mice. By blocking neurogenesis and surgically clamping adrenal hormone secretions, we showed that neurogenesis, via hypothalamic-pituitary-adrenal axis interactions, is directly involved in precipitating the depressive phenotype after social defeat. Mice adrenalectomized before social defeat showed enhanced behavioral resiliency and increased survival of adult-born hippocampal neurons compared with sham-operated defeated mice. However, mice lacking hippocampal neurogenesis did not show protective effects of adrenalectomy. Moreover, glucocorticoids secreted during environmental enrichment promoted neurogenesis and were required for restoration of normal behavior after social defeat. The data demonstrate that glucocorticoid-dependent declines in neurogenesis drive changes in mood after social defeat and that glucocorticoids secreted during enrichment promote neurogenesis and restore normal behavior after defeat. These data provide new evidence for direct involvement of neurogenesis in the etiology of depression, suggesting that treatments promoting neurogenesis can enhance stress resilience.
Introduction
Chronic uncontrollable psychological stress can precipitate post-traumatic stress disorder, anxiety disorders, and major depression (McEwen and Stellar, 1993; McEwen, 2004). Stress-related disorders are associated with dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis, manifested as abnormal adrenal glucocorticoid (GC) secretion and disrupted hormone feedback loops (Nemeroff et al., 1984; Holsboer et al., 1987). Evidence suggests that normalization of the HPA axis is a requirement for stabile remission of depressive symptomatology (Appelhof et al., 2006; Aihara et al., 2007; Ising et al., 2007).
The hippocampal formation is a powerful upstream modulator of the HPA axis stress response and consequent emotional states (Herman et al., 1989; Brady et al., 1992). Imbalances in circulating levels of GCs can lead to defects in hippocampal functioning, and vice versa (Sapolsky, 2000). Adult neurogenesis in the dentate gyrus (DG) of the hippocampus is sensitive to GC levels and stress context (Cameron and Gould, 1994; Gould and Tanapat, 1999; Mirescu and Gould, 2006; Mirescu et al., 2006). In addition to being influenced by HPA axis activity, hippocampal neurogenesis can regulate the hormonal stress response, and it has been implicated in anxiety and depression (Snyder et al., 2011). Conversely, enhancing neurogenesis can improve mood-related behavior and restore central control over stress response systems (Surget et al., 2011). We hypothesized that GCs secreted during chronic stress reduce neurogenesis and promote depressive-like behaviors and, therefore, that clamping GC secretion would protect against the deleterious effects of chronic stress in wild-type mice with intact neurogenesis but would not afford behavioral protection to transgenic mice with conditional ablation of neurogenesis. We tested this hypothesis by adrenalectomizing mice with and without neurogenesis and subjecting them to social defeat (SD) stress. We found that clamping GC levels in wild-type mice prevented stress-induced decreases in neurogenesis and conferred behavioral resiliency to SD. In contrast, adrenalectomized mice lacking neurogenesis remained behaviorally susceptible.
Elevated GCs do not always reduce neurogenesis or result in psychopathologies. For instance, enriched environments, including voluntary exercise, increase circulating GC levels (Droste et al., 2003, 2007; Stranahan et al., 2008), yet they are also proneurogenic (van Praag et al., 1999a). Despite elevated HPA axis activity, enriched environments mitigate the deleterious effects of adverse stress (distress) on behavior, neurobiology, and endocrine profiles (Greenwood and Fleshner, 2008; Stranahan et al., 2008; van Praag, 2009; Lehmann and Herkenham, 2011). Although counterintuitive, it is possible that these beneficial effects occur as a result of increased GC release. However, the question of whether GCs secreted during enrichment are causally related to improvements in mood and increases in neurogenesis has not been addressed. Thus, we further studied the role of GCs secreted during environmental enrichment (eustress) on behavioral recovery and neurogenesis. We found that mice housed in enriched environments after SD showed behavioral recovery and improved neurogenesis. Remarkably, we found that these beneficial effects were dependent on GC secretion demonstrating that GCs orchestrate divergent effects on mood in a neurogenesis-dependent fashion.
Materials and Methods
Animals.
All experiments were approved by the National Institute of Mental Health Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health guidelines. Experiments were performed using CD-1 and C57BL/6J (C57) mice (aged 8–10 weeks) obtained from National Institutes of Health/National Cancer Institute/ Division of Cancer Treatment (Frederick, MD), and transgenic hGFAP-HSVtk (hGFAPtk) mice backcrossed onto a C57BL/6J background (Schloesser et al., 2010). All animals were housed in 12 h light/dark cycle and tested during the dark phase. Food and water were provided ad libitum.
Conditional ablation of neurogenesis.
Transgenic hGFAPtk mice express herpes-simplex virus thymidine kinase (HSV-tk) under the control of the human GFAP promoter (Schloesser et al., 2010). Valganciclovir (VGCV, Roche) is phosphorylated by HSV-tk to make it toxic to proliferating cells in S-phase of mitosis, rendering the mice neurogenesis-null (NG−). In littermate control mice that do not express the hGFAP-HSV-tk transgene, VGCV administration does not suppress proliferation of GFAP+ cells, leaving the mice neurogenesis-intact (NG+). To control for drug effects, both wild-type control and hemizygous hGFAPtk mice received 15 mg/kg body weight/d VGCV administered in chow for 3 weeks before and throughout the course of experimentation. This dosing regimen is sufficient to eliminate virtually all hippocampal neurogenesis in the hGFAPtk mice without affecting wild-type mice (Schloesser et al., 2010).
An extensive behavioral and pathological examination of hGFAPtk mice was previously conducted by Schloesser et al. (2010). The mice administered VGCV showed no abnormalities in a comprehensive physical examination that included physical characteristics, general behavioral observations, sensorimotor reflexes, motor responses, and olfactory discrimination. This hGFAPtk mouse, unlike one developed previously (Bush et al., 1998), did not develop jejuno-ileitis or any other peripheral pathology, as we documented (Schloesser et al., 2010).
Experimental design, experiment 1: effects of adrenalectomy (ADX) on behavior after SD.
As shown in Figure 1B, a 2 × 2 (surgery × stress) factorial design was used to study the effects of ADX on maladaptive behaviors induced by chronic SD. Animals were bilaterally adrenalectomized or sham operated. Adrenalectomized mice were provided 25 μg/ml corticosterone (CORT) in 0.9% NaCl water. After a 3 d recovery period, animals were randomly divided into standard home-cage (HC) housing or SD housing. After 14 d of SD exposure or HC housing, mice were behaviorally phenotyped for mood-related behaviors. In other animals, tail blood was collected either before or 10, 20, 30, 60, or 90 min after SD exposure on the first, seventh, and 14th exposure to SD. Blood was also collected during the diurnal surge period, previously determined to occur 60 min before lights off (Malisch et al., 2008). For each animal, blood was collected once every 7 d according to a schedule that allowed coverage of all the time points across the animals.
We further examined the effects of ADX on newborn neuron survival after SD. The experiment used a 2 × 2 (surgery × stress) factorial design (Fig. 2A). Animals were given five daily injections of BrdU starting 7 d before either ADX or sham surgery. After a 7 d recovery period, mice were exposed to SD or HC housing. Mice were then perfused and brains examined for BrdU+ cells in the hippocampal DG.
Experiment 2: effects of ADX on behavior in mice lacking neurogenesis.
These experiments address the hypothesis that the protective effects of ADX are dependent on adult neurogenesis. First, we examined phenotype differences between mice lacking neurogenesis (NG−) and control (NG+) littermates exposed to either HC or SD. Second, we examined the effects of ADX before SD in mice with conditionally suppressed neurogenesis. For the experimental timeline, see Figure 4A. Using a 2 × 2 (surgery × genotype) factorial design, NG− and NG+ mice were either adrenalectomized or sham operated and then exposed to 14 d of SD.
Experiment 3: effects of environmental enrichment on recovery from SD.
A 2 × 2 (surgery × stress) factorial design was used to study the effects of ADX on the reparative effects of environmental enrichment (EE) in mice previously exposed to SD (see Figure 6A). After either 14 d of SD or HC, animals were either adrenalectomized or sham operated, and all groups were subsequently housed in EE for 21 d and, finally, behaviorally phenotyped.
The effects of SD and housing environment during recovery on circadian CORT activity were examined in a separate cohort of intact mice (2 × 2, stress × environment factorial design). Mice were exposed to either 14 d of SD or HC, and then housed in either HC or EE for 21 d. Tail blood was collected during the diurnal surge (60 min before lights off) every 7 d during SD/HC housing and HC/EE housing to characterize changes in circadian CORT rhythm during SD and subsequent housing.
We further examined the effects of eustress on DG cell survival and modulation of cell survival by ADX using a 2 × 2 × 2 (surgery × stress × housing) factorial design (see Fig. 8A,D). Animals were exposed to either 14 d of SD stress or HC housing. All groups were then transferred to HC housing and given five daily injections of BrdU. One day after the fifth injection, mice were either ADX or sham operated. After a 2 d recovery period, mice were transferred to EE or HC for 3 weeks. Mice were perfused and brains examined for BrdU+ cells.
Housing conditions.
Standard HC dwellings were polycarbonate cages (14.0 cm × 35.5 cm × 13.0 cm, Tecniplast) with woodchip bedding and cotton square nestlet. Cage bedding was replaced once every 7 d. EE housing consisted of a (24.5 cm × 40.5 cm × 18.5 cm) polycarbonate cage (Tecniplast) with wood chip bedding, nesting material, raised platforms, running wheel, and numerous paper and polycarbonate tubes of various sizes and shapes (Bio-Serv) placed in a polycarbonate cage (24.5 cm × 40.5 cm × 18.5 cm, Tecniplast). Polycarbonate tubes and wheels were washed with warm water, dried and replaced in the originating cage once every 7 d to both minimize stress from novel objects and maintain a sanitary environment.
SD.
As described previously (Lehmann and Herkenham, 2011), aggressor CD-1 male mice were single-housed for 1 week. Experimental mice were placed into the resident CD-1 mouse's home cage into which a perforated partition had been placed down the middle to separate the pair. The partition was removed for 5 min/d for 14 d allowing agonistic encounters between the pair. Interaction periods were analyzed for aggressive, submissive, and exploratory behaviors.
ADX and CORT replacement.
Animals were anesthetized using isoflurane. ADX was performed bilaterally through a single dorsal midline incision as previously described (Lehmann et al., 2010). Sham-operated animals underwent the same surgical procedure, but the adrenals were not removed. Adrenalectomized mice were subsequently provided 25 μg/ml CORT (C2505, Sigma) in 0.9% saline as drinking water, which approximates basal CORT levels and mimics the normal circadian pattern of CORT secretion (Jacobson et al., 1988; Gould et al., 1990). After behavioral phenotyping, animals were rapidly decapitated after 30 min of restraint, a duration shown to result in a significant stress response (Pare and Glavin, 1986), and trunk blood assayed for CORT. Successful ADX was defined as mice having levels of plasma CORT ≤100 ng/ml; those with greater CORT values after brief restraint were excluded from the experiment.
Surgical ADX was used in lieu of pharmacological interventions because of the limitations of the available drugs that act on the HPA axis. Metyrapone, a GC-synthesis inhibitor, incompletely blocks GC synthesis (Child et al., 1976), which could mask any experimental effects by permitting the HPA axis to adapt to the drug over the course of a long-term experiment; and RU38486, a GR antagonist, is also a partial GR agonist (Gruol and Altschmied, 1993) and has a high affinity for the progesterone receptor (Gagne et al., 1985).
Behavioral testing.
All behavior was captured and analyzed using automated technology, CaptureStar and TopScan 2.0 (CleverSystems). This software reliably identifies animal shape and body part, and it scores the frequency and duration of multiple different movements (sniffing, ambulation, struggle/swim, and immobility).
To assess anxiety-like behaviors, we used the Light/Dark box (L/D) and the Elevated Zero Maze (EZM) (Schloesser et al., 2010). In brief, the L/D box test uses a 50 × 25 × 30 cm high Plexiglas box divided into a dark (one-third of total area) and a light (40 lux illuminated) compartment separated by an open door. Time spent in the dark compartment and number of crosses between the light and dark compartments were measured as an indicator of anxiety-like behavior. The EZM consisted of alternating open and walled runways raised 65 cm above the ground. Time spent in the open arms was measured.
To assess depressive-like and social avoidance behaviors, we used the Forced Swim Test (FST) and Social interaction (SI) task. For the FST, mice were placed into clear acrylic cylindrical chambers (30 cm in height, 10.2 cm in diameter) two-thirds filled with water heated to 30°C. Swimming and escape behavior versus time spent floating were recorded >6 min and subsequently analyzed. In the SI task, animals were placed into an open-field arena containing two inverted wire mesh cups: one contained the specific dominant CD-1 with which the experimental animal had been housed during the conflict phase, and the empty cup served as a control novel object. Behavior was recorded for 30 min. Behavioral results were reported as a comparison of total time spent by the experimental mouse interacting (i.e., sniffing) with the dominant mouse versus the empty cup. The social interaction ratio (SI ratio) was obtained by dividing the time spent interacting with the dominant mouse by the time spent interacting with the empty cup.
Corticosterone radioimmunoassay.
Tail blood was rapidly (< 5 min from initiation of handling) collected from awake, unrestrained mice by tail snip. Blood was collected in heparin-coated hematocrit tubes (Safecrit, Iris International), placed on ice, and immediately spun down in a hematocrit microcentrifuge (CritSpin, Iris International) to obtain plasma for subsequent RIA. Additionally, after behavioral testing, ADX mice were stressed and killed, and trunk blood was collected for RIA to confirm complete adrenalectomy. RIA was performed using an I-125 Coat-a-Count Corticosterone In vitro diagnostic Test Kit (Siemens).
BrdU injections and quantification of neurogenesis.
BrdU ((+)-5′ bromo-2′-deoxyuridine; 97%; Sigma-Aldrich) (200 mg/kg, i.p.) was administered once daily for five consecutive days (Schloesser et al., 2010). Four weeks after BrdU injection, mice were perfused, tissue collected, and the entire DG (−0.9 mm to −4.2 mm from bregma) sectioned at 50 μm thickness.
Fluorescence immunolabeling was used for cell quantification, density analysis, and cell phenotyping. For BrdU and NeuN double labeling, free-floating sections were denatured in 50% formamide in 2× SSC for 2 h at 65°C, rinsed in 2× SSC, incubated in 2 N HCl for 30 min at 37°C, rinsed in 0.1 m Na borate, blocked, and incubated overnight at 4°C in monoclonal rat anti-BrdU antibody (1:500; Accurate) and mouse anti-NeuN (1:1000; Millipore). Sections were rinsed and incubated for 60 min in the appropriate secondary antisera (Alexa-488 for BrdU and Alexa-555 for NeuN; Invitrogen). Hoechst 33342 was used to counterstain. Sections were mounted and coded until completion of data analysis.
Quantification of BrdU-labeled cells was performed on every 10th section covering the entire DG. Cells were counted under fluorescent illumination at 400× using a Nikon Eclipse E800 microscope. Cells located in the outermost plane of focus were excluded. The granule cell layer in each section was traced under the 4× objective using StereoInvestigator Version 9.01 Software (MBF Biosciences). The granule cell reference volume was determined by summing the traced granule cell areas for each section multiplied by the distance between sections sampled. The number of BrdU-labeled cells was then related to granule cell layer sectional volume and multiplied by the reference volume to estimate total number of BrdU+ cells. We separately analyzed the rostral (−0.9 to −2.4 mm from bregma) and caudal (−2.4 to −4.2 mm from bregma) subdivisions of the DG. Fifty BrdU+ cells per animal were analyzed for coexpression of BrdU and NeuN using a Zeiss Meta confocal microscope (Model LSM 510).
Peroxidase immunolabeling for BrdU was used for white-light visualization purposes as previously described (Tang et al., 2007). Briefly, sections were incubated in monoclonal rat anti-BrdU antibody (1:500, Accurate) followed by biotinylated anti-rat IgG (1:250; Vector), and avidin-biotin-horseradish peroxidase (AB; 1:100; Vector). Tissue was developed in a nickel-enhanced DAB (Ni-DAB) peroxidase substrate (Vector Laboratories), slide-mounted, and counterstained with eosin Y.
For double-cortin (DCX) fluorescence immunolabeling, free-floating sections were washed, blocked in 0.3% Tween 20 PBS with 3% donkey serum for 90 min and subsequently incubated for 5 d at 4°C with goat anti-DCX antibody (1:400; Santa Cruz Biotechnology). Sections were rinsed and incubated for 120 min in AlexaFluor-488 anti-goat secondary (Invitrogen) and counterstained with Hoechst 33342.
Statistical analysis.
Data for all experiments were analyzed using parametric statistics with ANOVA, or multifactors ANOVA, as appropriate using SPSS software. ANOVA analysis was followed by post hoc tests or planned comparisons as projected from the design of each experiment.
Results
Experiment 1: maladaptive behavioral responses to SD are alleviated by prior adrenalectomy
We hypothesized that clamping the HPA axis output during SD would blunt depressive behaviors induced by SD. We initially characterized CORT responses to repeated SD in intact mice (Fig. 1A). A robust CORT surge was detected during each of the sampling days, and this surge was significantly elevated up to 90 min after SD compared with baseline measurements (Pre). Although a two-way repeated-measures ANOVA found a significant effect of sampling day (F(2,105) = 8.95, p < 0.03) and time after SD (F(5,105) = 8.16, p < 0.001), Bonferroni comparisons for each sampling time point found no significant differences in CORT across each sampling day (p > 0.05). In the ADX group, SD had no effect on clamped plasma CORT levels. Both intact and ADX mice had comparable CORT levels before SD across all testing days. After SD, CORT levels in ADX mice were significantly attenuated compared with intact mice across all testing days and most sampling time points (surgery effect: day 1, F(1,12) = 135.18; day 7, F(1,12) = 27.52; day 14, F(1,12) = 11.21). Post hoc significance was found for all time points, except 10 min after SD on day 7 and day 14.

Maladaptive behavioral responses to SD stress are prevented by ADX. A, An acute and prolonged elevation in plasma CORT was observed after SD exposure during each sampling day (D1, D7, and D14). *p < 0.05 versus baseline (Pre) (post hoc test). ADX prevented SD-induced increases in CORT (n = 8 for intact, 6 for ADX). B, Diagram depicting experimental groups and study design. C, Lit compartment exploration was significantly reduced in defeated mice during the L/D (stress effect, F(1,50) = 34.3, p < 0.0001). D, The significant reduction in transitions between light and dark compartments observed in defeated mice was abrogated by prior ADX (interaction, F(1,50) = 16.78, p < 0.001; stress effect, F(1,50) = 10.52, p < 0.005; surgery effect, F(1,50) = 5.99, p < 0.01). E, ADX prevented the defeat-induced avoidance of open arms exhibited by defeated shams in the EZM (interaction, F(1,50) = 10.33, p < 0.01). F, ADX before SD prevented the stress-induced increase in floating time in the FST (interaction, F(1,50) = 4.69, p < 0.05; stress effect, F(1,50) = 35.85, p < 0.0001). G, Strong social aversion displayed by defeated sham mice was significantly reduced in mice ADX before defeat stress (interaction quotient; duration on mouse (s)/duration on object; interaction, F(1,50) = 7.74, p < 0.01; stress effect, F(1,50) = 29.47, p < 0.0001). H, Heat maps representing time spent exploring CD-1 mouse chamber and empty chamber. Error bars indicate mean ± SE (n = 12–14). *p < 0.05 versus control group (post hoc test). **p < 0.01 versus control group (post hoc test). ***p < 0.001 versus control group (post hoc test).
We next investigated the effects of ADX on changes in mood-related behaviors elicited by SD (Fig. 1B). After 14 d of SD stress, adrenalectomized mice showed significant resilience to the effects of SD and displayed a behavioral profile comparable with nondefeated HC mice. In L/D text (Fig. 1C,D), defeated mice spent significantly less time in the light compartment. Although no effect of surgery was found for time in light compartment, when we measured the number of transitions between the light and dark compartment (Fig. 1D), which is considered a more salient measure of anxiety-like behavior in the L/D test (Crawley, 1985), adrenalectomized defeated mice showed levels of transitions comparable with nondefeated control groups. The anxiolytic effect of ADX was further confirmed in the EZM (Fig. 1E); ADX prevented the SD-induced decline in open-arm exploration time. In tests for depressive-like behavior, SD significantly increased immobility during the FST, and this effect was abrogated in adrenalectomized mice (Fig. 1F).
We previously showed that avoidance of an aggressor mouse is a maladaptive response coincident with anxiety and depressive-like behaviors (Schloesser et al., 2010). Here, we tested the effects of ADX on social avoidance behavior after SD stress. We found that sham-operated defeated mice displayed a pronounced avoidance of the dominant mouse, whereas adrenalectomized defeated mice showed reduced social avoidance (Fig. 1G,H), suggesting that ADX exerts a protective action against social stress. ADX did not alter several baseline measures of locomotor activity in the FST or anxiety-like behavior in nondefeated control groups. Together, the behavioral resiliency observed in ADX defeated mice suggests that chronic adrenal activation during SD reduces positive adaptive responses and leads to the development of a prodepressive phenotype.
The effects of SD and ADX on body mass changes were also examined. Defeated mice with intact adrenals had a significantly higher body mass (25.01 ± 0.24 g) compared with HC controls (22.79 ± 0.29 g) and both ADX groups (ADX→HC, 23.46 ± 0.43; ADX→SD, 22.8 ± 0.38) (interaction, F(1,50) = 6.78, p < 0.05). Body mass was not significantly different between adrenalectomized HC and SD groups.
Finally, we examined the interaction between SD and ADX on newborn neuron survival along the rostral-caudal axis of the DG (Fig. 2A). The rostral, septal pole zone is primarily involved with cognitive and spatial functions, whereas the caudal zone incorporates the ventral hippocampus and parahippocampal cortical circuitry that has been shown to be related to emotional and behavioral affect (for review, see Bannerman et al., 2004; Fanselow and Dong, 2010). BrdU+ cell counts of the DG showed that the effects of chronic social stress and ADX on cell survival were limited to the caudal divisions of the DG; no treatment effects were observed in the rostral DG (Fig. 2B,C). In sham groups, chronic social stress resulted in a substantial decrease in BrdU+ cells. This decrease was attributed to CORT secreted during SD because mice given ADX before SD showed substantial increases in the survival of BrdU+ cells compared with sham-SD mice and ADX-control mice (Fig. 2B). A combination of ADX and defeat exposure significantly increased BrdU+ cells compared with ADX alone (p < 0.001).

Clamping adrenal hormone secretions during SD stress prevents a reduction in neurogenesis in the caudal DG. A, Experimental design testing the interaction between ADX and SD exposure on newborn cell survival in the DG. Mice administered BrdU were adrenalectomized or given sham surgery and then housed in HC or SD for 2 weeks. B, Quantification of BrdU+ cells in the rostral and caudal subdivisions of the DG. Quantification was made in fluorescently labeled tissue. SD significantly reduced survival of BrdU+ cells in the caudal DG. Mice adrenalectomized before SD showed a robust increase in BrdU+ cells compared with sham defeated mice and nondefeated ADX mice (interaction, F(1,20) = 27.05 p < 0.0001; surgery effect, F(1,20) = 9.89, p < 0.005). C, Representative photomicrographs of Ni-DAB-stained BrdU+ cells in the rostral and caudal DG of sham and ADX mice exposed to SD (counterstained with eosin Y) are shown for clarity. D, Confocal photographs of caudal DG BrdU and NeuN double immunostaining in defeated mice with intact adrenals (Sham-SD) and mice ADX before SD (ADX-SD); arrows indicate BrdU+ cells. E, Example of NeuN+ neuron colabeled with BrdU. Error bars indicate mean ± SE (n = 6). **p < 0.01 (significant post hoc comparisons to respective control groups). ***p < 0.001 (significant post hoc comparisons to respective control groups). Scale bars: C, 100 μm; D, 40 μm; E, 10 μm. GCL, Granule cell layer; HIL, hilus.
Analysis of cell phenotype using confocal microscopy showed a large majority of BrdU+ cells colocalized with NeuN (Fig. 2D,E). The average percentage of BrdU+ cells staining for NeuN was comparable across all treatment groups (sham→HC, 81.7 ± 3.1%; sham→SD, 79.0 ± 3.8%; ADX→HC, 82.3 ± 4.5%; ADX→SD, 80.0 ± 3.5%), suggesting that SD or ADX did not alter the phenotypic differentiation of new cells.
Experiment 2: adrenalectomy does not confer stress resiliency to mice with conditionally suppressed neurogenesis
Experiment 1 showed a strong correlation between survival of adult-born hippocampal neurons, GCs, and mood-related behavior. We next examined whether the protective effects of ADX during SD would persist in mice lacking neurogenesis. Neurogenesis was conditionally suppressed in hGFAPtk mice after 3 weeks of VGCV administration (NG− mice), indicated by the absence of doublecortin (DCX)-positive newborn neurons (Fig. 3A). We initially examined the effect of SD or HC housing on mood-related behaviors in NG− mice and mice with intact neurogenesis (NG+ mice) (Fig. 3B). Ablation of neurogenesis produced no significant behavioral changes (Fig. 3C–H) in nondefeated HC control mice. Exposure to SD significantly increased maladaptive behaviors in a similar manner for both NG− and NG+ mice. For measures of anxiety-like behavior, defeated NG− and NG+ mice showed similar reductions in light compartment exploration (Fig. 3C), number of transitions between compartments in the L/D test (Fig. 3D), and reductions in open-arm exploration in the EZM (Fig. 3E). Similar effects of SD were observed in the FST (Fig. 3F); defeated NG− and NG+ mice spent significantly more time immobile compared with nondefeated groups, and no effect of genotype was observed. We further assessed social interactions with the dominant male mouse versus a neutral object. Defeated mice had a significantly decreased interaction ratio compared with nondefeated groups (Fig. 3G,H), and the expression of avoidance behaviors was similar between NG+ and NG− mice. Both defeated NG+ and NG− mice showed similar increases in body weight (NG+, 25.68 ± 0.38 g; NG−, 25.41 ± 0.47 g) compared with nondefeated groups (NG+, 23.65 ± 0.65 g; NG−, 23.21 ± 0.52 g) (stress effect, F(1,26) = 12.42, p < 0.001). The data confirm our earlier finding that repeated SD confers a depressive phenotype in mice both with and without NG (Schloesser et al., 2010).

Conditional genetic ablation of adult neurogenesis has no effect on baseline behavior and has no effect on the induction of maladaptive behaviors after SD. A, The DG of control mice (NG+) contains many DCX+ neuroblasts (green), whereas these cells are virtually abolished in hGFAPtk mice treated with VGCV for 3 weeks (NG−). B, Experimental design testing the interaction between adult neurogenesis and SD exposure on behavior. Defeated mice with intact (NG+) and ablated (NG−) neurogenesis showed similar and significant increases in anxiety- and depressive-like behavior. Anxiety-like behaviors were measured in the L/D and EZM. SD significantly reduced (C) light compartment exploration (stress effect, F(1,26) = 13.56), (D) transitions between light and dark compartments (stress effect, F(1,26) = 30.49), and (E) open-arm exploration in the EZM (stress effect, F(1,26) = 113.1). Defeated mice (F) showed increased immobility in the FST (stress effect, F(1,26) = 23.66) and (G) spent significantly less time with the aggressor mice in the SI task (stress effect, F(1,26) = 22.41). H, Heat maps representing time spent exploring CD-1 mouse chamber and empty chamber. Scale bar: A, 50 μm. Error bars indicate mean ± SE (n = 7 or 8). **p < 0.01, ***p < 0.001.
The first experiment showed that clamping adrenal hormones protected mice from SD-induced depressive-like states and decreases in new cell survival, suggesting that adrenal hormones may drive stress-induced changes in mood and neurogenesis. Next, we investigated the interaction between neurogenesis and ADX in defeated mice to determine whether neurogenesis is required for the protective effects of ADX. NG− and NG+ mice were sham-operated or adrenalectomized and exposed to 14 d of SD. After SD, all groups were tested for mood-related behaviors (Fig. 4A).

Adrenalectomy does not confer stress resiliency to mice lacking neurogenesis. A, Experimental design testing the interaction between ADX and neurogenesis on behavior in mice exposed to chronic SD. ADX reduced anxiety- and depressive-like behaviors in defeated NG+ mice but had no effect in defeated NG− mice. B, C, In the L/D test, ADX NG+ mice spent more time in the light compartment compared with sham NG+ mice (B, surgery effect, F(1,22) = 6.58, p < 0.01). C, ADX NG+ mice showed an elevated number of transitions between light and dark compartments compared with all other groups (interaction effect, F(1,22) = 9.84, p < 0.005; surgery effect, F(1,22) = 12.03, p < 0.002). D, ADX increased open-arm exploration in the EZM in NG+ mice only (interaction, F(1,22) = 6.75, p < 0.02; surgery effect, F(1,22) = 6.21, p < 0.03). E, ADX reduced immobility time in NG+ mice during the FST (surgery effect, F(1,22) = 4.96, p < 0.05). F, ADX significantly increased social interaction in NG+ mice, whereas no effect of ADX was detected in NG− mice (interaction, F(1,22) = 5.02, p < 0.05; surgery effect, F(1,22) = 4.33, p < 0.05). G, Heat maps representing time spent exploring CD-1 mouse chamber and empty chamber. Error bars indicate mean ± SE (n = 6–8). *p < 0.05, **p < 0.01, ***p < 0.001.
The protective effects of ADX persisted only in mice with intact neurogenesis. In the L/D and EZM test, ADX had robust anxiolytic effects in NG+ mice only; no behavioral effect of ADX was observed in NG− mice (Fig. 4B–D). In measures of depressive-like behavior, ADX significantly reduced FST immobility (Fig. 4E) in NG+ mice only. We next examined social interaction (Fig. 4F). Compared with all other groups, adrenalectomized NG+ mice displayed a significantly higher interaction quotient (Fig. 4G). Although divergent behavioral effects of ADX were observed between defeated NG+ and NG− mice, both groups had similar but significantly lower body weights compared with sham NG+ and NG− mice (sham NG+, 25.2 ± 0.45 g; sham NG−, 24.78 ± 0.47 g; ADX NG+, 22.61 ± 0.71 g; ADX NG−, 22.1 ± 0.5 g) (surgery effect, F(1,22) = 27.49, p < 0.001). These behavioral tests for SD-induced changes in affect confirm the results from Experiment 1 and further show that intact adult neurogenesis is required for the behavioral protection of ADX.
Experiment 3: adrenal GCs are required for the antidepressant effects of environmental enrichment in defeated mice
Enriched housing environments that provide opportunities for voluntary physical exercise are eustressors that stimulate the HPA axis and increase GC levels both acutely during exercise and during circadian surge periods (Droste et al., 2003; Stranahan et al., 2006). These elevations in GC levels may contribute to the cognitive benefits and increased emotional stability attributed to EE (Stranahan et al., 2008; Hajisoltani et al., 2011). Furthermore, we demonstrated that EE facilitates recovery from maladaptive behaviors generated during SD exposure (Schloesser et al., 2010). Therefore, we hypothesized that increased adrenal activity during EE exposure is responsible behavioral recovery in defeated mice.
We initially characterized the effects of SD and EE on GC levels during the diurnal surge, 60 min before lights OFF. We chose this period to comparably measure CORT levels in these two conditions because mice are inactive, and the acute effects of unpredictable running patterns on HPA axis activity are minimized. Mice housed in SD showed a marked increase in diurnal surge compared with HC mice (Fig. 5A), indicating that SD alters HPA axis activity beyond the immediate context of defeat exposure and disrupts normal circadian variation in CORT levels. Defeated or nondefeated HC mice were then exposed to EE or HC during recovery. EE housing significantly increased day 7 surge levels in nondefeated mice compared with all other groups (Fig. 5B). However, after 2 weeks of EE, both EE groups (HC→EE and SD→EE) were elevated compared with both HC groups (HC→HC and SD→HC). Thus, the prior defeat experience transiently (at 7 d) blunted the EE-induced CORT surge; and after 14 d of EE housing, previously defeated mice showed CORT levels comparable with nondefeated EE controls.

SD and EE increase the magnitude of diurnal CORT surges. Mice exposed to HC or SD for 2 weeks were transferred to either EE or HC housing for 3 weeks. Tail blood was assayed for CORT every 7 d during the diurnal surge, 60 min before lights off. A, SD exposure markedly increased CORT during the diurnal surge (main stress effect, F(1,90) = 24.12, p < 0.0001). B, Prior stress exposure and the type of recovery housing (HC vs EE) affected diurnal CORT surges during the recovery period (2-way mixed-model ANOVA, stress × recovery housing interaction, F(6,56) = 4.272, p < 0.001; effect of prior SD, F(3,28) = 13.13, p < 0.001). During the recovery period, surge levels were significantly increased in nondefeated mice housed in EE (HC→EE) on day 7 compared with all other treatment conditions (interaction, F(1,28) = 17.61, p < 0.001; housing effect, F(1,28) = 4.28, p < 0.05). On days 14 and 21, CORT during the diurnal surge was significantly higher in both EE groups; this effect occurred independent of prior stress exposure (day 14 housing effect, F(1,28) = 18.05, p < 0.001; day 21 housing effect, F(1,28) = 24.95, p < 0.0001). Prolonged EE housing increased diurnal CORT surges in previously defeated mice (SD→EE) (repeated-measures ANOVA across days, F(2,23) = 11.55, p < 0.001) (n = 16 per group during induction, n = 8 per group during recovery). *p < 0.05 (significant post hoc comparison).
We next examined whether ADX would disrupt behavioral recovery in defeated mice housed in EE. Mice exposed to SD or HC housing were sham-operated or adrenalectomized, transferred to EE housing for 3 weeks, and subsequently examined for changes in mood-related behaviors (experimental design shown in Fig. 6A).

Environmental enrichment facilitates behavioral recovery in socially defeated mice through adrenal hormone secretions. A, Experimental design testing the role of adrenal activity during EE housing on recovery from SD. Clamping adrenal secretions during EE severely disrupted recovery in defeated mice (SD→ADX→EE), whereas defeated mice with intact adrenals (SD→sham→EE) showed behavioral responses comparable with nondefeated controls. Surgery had no effect on behavior in nondefeated groups. SD→ADX→EE mice showed (B) a significant decrease in light compartment activity (interaction, F(1,44) = 18.15, p < 0.0001; surgery effect, F(1,44) = 54.44, p < 0.0001) and (C) a significant decline in number of transitions between light and dark compartments (interaction, F(1,44) = 10.96, p < 0.001; surgery effect, F(1,44) = 5.39 p < 0.02). D, Likewise, SD→ADX→EE mice showed significant decreases in open-arm exploration of the EZM compared with SD→sham→EE mice and nondefeated adrenalectomized mice (interaction, F(1,44) = 8.30, p < 0.005; stress effect, F(1,44) = 20.58, p < 0.0001). E, In the FST, SD→ADX→EE mice showed a substantial increase in immobility time relative to all other groups (interaction, F(1,44) = 4.79, p < 0.05; stress effect, F(1,44) = 6.63, p < 0.02; surgery effect, F(1,44) = 9.31, p < 0.005). F, G, In the SI task, SD→sham→EE mice showed an interaction preference with a social target comparable with nondefeated controls, whereas SD→ADX→EE mice showed a significant increase in social avoidance compared with all tested groups, indicative of a strong prodepressive phenotype (interaction, F(1,44) = 10.43, p < 0.005; stress effect, F(1,44) = 21.95, p < 0.0001; surgery effect, F(1,44) = 8.55, p = 0.005). Error bars indicate mean ± SE (n = 12). *p < 0.05, **p < 0.01, ***p < 0.001.
Defeated mice showed marked improvement in mood if the adrenals remained intact during EE housing (SD→sham→EE). In contrast, the SD-induced mood disturbances persisted in mice adrenalectomized before EE (SD→ADX→EE). For example, in the L/D and EZM (Fig. 6B–D), anxiety-like behaviors induced by SD were significantly remediated after EE only if the adrenals remained intact. ADX before EE significantly reduced light compartment exploration (Fig. 6B) and transitions between compartments (Fig. 6C) in previously defeated mice. Similarly, ADX before EE significantly attenuated exploration of the open arms in the EZM (Fig. 6D) in defeated mice. In contrast, defeated mice with intact adrenals showed levels of exploration comparable with nondefeated HC mice. In the FST for depressive-like behaviors (Fig. 6E), EE ameliorated the effects of SD in sham mice but not in adrenalectomized mice. In the SI task (Fig. 6F,G), SD→ADX→EE mice showed a significant increase in social avoidance compared with all tested groups, indicative of a strong depressive phenotype that still endured 3 weeks after the last defeat experience. The persistence of these mood-related behaviors in the defeated ADX group suggests that adrenal activity during EE is required for restoration of normal phenotype. It is unlikely that differences in behavior between defeated sham and ADX mice were the result of sham mice engaging in higher levels of activity during EE because similar levels of running activity were observed between treatment groups (Fig. 7A,B). However, sham groups had significantly higher body weights compared with ADX groups, although no significant effect of prior stress exposure was observed. (HC→sham→EE, 23.94 ± 0.35 g; SD→sham→EE, 24.3 ± 0.23 g; HC→ADX→EE, 22.78 ± 0.25 g; SD→ADX→EE, 22.63 ± 0.34 g) (surgery effect, F(1,44) = 22.56, p < 0.001).

Effects of SD and ADX on wheel running activity. A, Diagram depicting experimental groups and study design. Mice were exposed to 14 d of HC or SD. Animals then received either ADX or sham surgery (Sham) and were then housed in enriched housing with running wheels for 21 d. B, Characteristics of running activity during a 24 h period were examined every 3 d, including distance run, time spent running, and average velocity of running speed. No noticeable effects of surgery or prior stress exposure were detected.
Last, we examined the interaction between EE and ADX on the survival of new cells in the DG after either HC or SD housing (Fig. 8A). ADX significantly reduced numbers of BrdU+ cells in the caudal DG of mice housed in EE, suggesting that CORT secreted during EE facilitated cell survival during EE (Fig. 8B,C). This effect occurred regardless of prior stress exposure. Prior SD stress significantly reduced cell survival in the rostral DG, but the effects were limited to mice with intact adrenals (stress effect, F(1,20) = 4.43, p < 0.05, Fig. 8C). These effects of stress and surgery extend only to mice housed in EE during the recovery period; cell survival was not affected by stress or surgery in animals housed in HC during the recovery period (Fig. 8D–F).
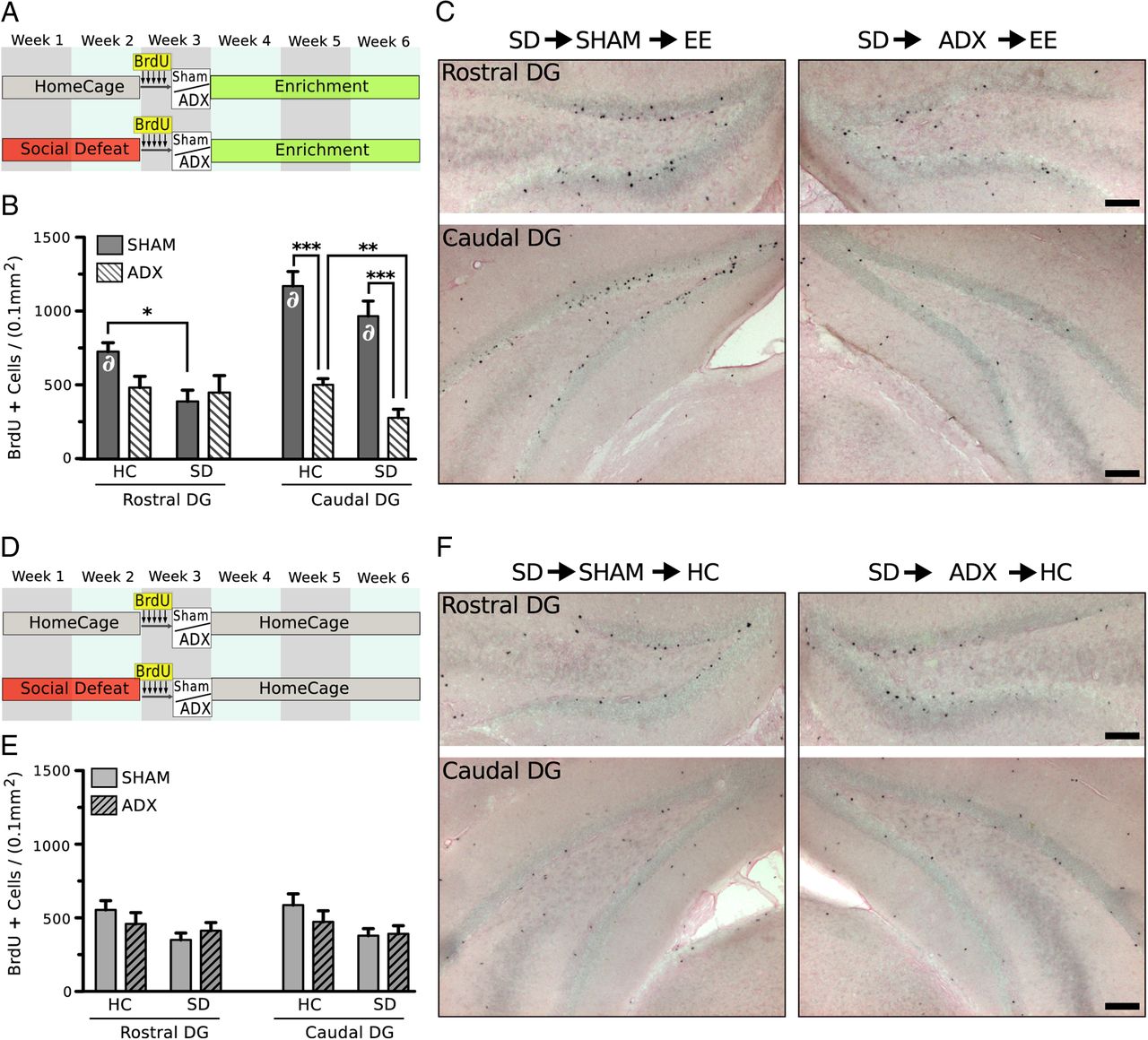
EE enhances neurogenesis through adrenal hormones. A, Experimental design examining the interaction between ADX and prior SD on newborn cell survival in the DG during EE. B, Quantification of BrdU+ cells in the rostral and caudal subdivision of the DG. CORT secreted during EE facilitated cell survival during EE, but only in the caudal DG. ADX significantly diminished numbers of BrdU+ cells in the caudal DG of mice housed in EE during recovery (surgery effect, F(1,20) = 76.33, p < 0.001), an effect that occurred regardless of prior stress exposure (HC→EE, p < 0.001; SD→EE, p < 0.001). SD stress also significantly reduced cell survival in the rostral DG, but the effects were limited to mice with intact adrenals (stress effect, F(1,20) = 4.43, p < 0.05). C, Representative photomicrographs of BrdU+ cells in the rostral and caudal DG of defeated mice sham and ADX before housing in EE (counterstained with eosin Y). D, Experimental design examining the interaction between ADX and prior SD on newborn cell survival during HC housing recovery from SD. Mice are housed in standard control housing during recovery. E, Quantification of BrdU+ cells in the rostral and caudal subdivision of the DG. Two-way ANOVA (surgery × stress) showed no significant effect of surgery and no effect of CORT secreted during standard housing on cell survival. A significant effect of prior stress was detected in both the rostral (stress effect, F(1,20) = 4.11, p < 0.05) and caudal (stress effect, F(1,20) = 4.92, p < 0.05) subdivisions of the DG, but no significant post hoc comparisons were observed. F, Representative photomicrographs of BrdU+ cells in the rostral and caudal DG of defeated mice sham and ADX before housing in HC (counterstained with eosin Y). Quantification of BrdU+ cells was made in fluorescently labeled tissue. Ni-DAB labeled tissue is shown for clarity. Error bars indicate mean ± SE (n = 6). ∂, significantly elevated from comparable group housed in HC during recovery: *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars, 100 μm.
We further compared the effects of differential housing environments during recovery (EE vs HC) on cell survival. We found that, in sham groups, EE significantly increased cell survival in the caudal DG compared with HC-housed mice (F(1,20) = 52.29, p < 0.001), and this effect occurred regardless of prior stress exposure. ADX abolished any potentiating effect of EE in the caudal DG; levels of cell survival in both defeated and HC housed mice adrenalectomized before EE were comparable with those housed in HC after ADX. An effect of housing environment was also detected in the rostral DG; EE increased cell survival, but this effect was limited to nondefeated mice with intact adrenals (HC→sham→EE vs HC→sham→HC) (F(1,20) = 4.62, p = 0.045).
Differentiation of surviving BrdU+ cells was examined by immunofluorescent double labeling for BrdU and neuronal marker NeuN. BrdU+ cells were predominately colocalized with NeuN across all groups. We found a comparable percentage of BrdU+ cells staining for NeuN in each experimental group housed in EE during recovery (HC→sham, 87.3 ± 2.4%; SD→sham, 82.3 ± 4.0%; HC→ADX, 81.3 ± 2.1%; SD→ADX, 81.7 ± 4.1%). Similar percentages of BrdU+ cells colabeled with NeuN were observed for groups housed in HC during recovery (HC→sham, 84.0 ± 3.9%; SD→sham, 82.3 ± 3.6%; HC→ADX, 83.0 ± 2.2%; SD→ADX, 83.6 ± 2.3%). No effect of stress or surgery on the phenotypic differentiation of newborn cells was detected.
Discussion
Figure 9 summaries the three main experiments and the major findings and conclusions drawn from them. We showed that GCs secreted during SD exposure were required for the induction of maladaptive behaviors and for the reduction of newborn hippocampal neurons in defeated mice (Experiment 1). Mice showed behavioral resilience to SD and enhanced survival of newborn hippocampal neurons when adrenal activity was surgically clamped before SD exposure (ADX→SD). The protective effects of ADX against SD-induced changes in mood were not extended to mice lacking neurogenesis (NG−), suggesting that intact adult neurogenesis is required for the behavioral protection afforded by ADX (Experiment 2). The data showed that ablation of hippocampal neurogenesis, even in the absence of high levels of CORT, is sufficient to eliminate stress resilience in mice. The data imply that increased adrenal hormone secretion during SD decreases neurogenesis, and this decline is permissive for the induction of maladaptive behaviors.

Summary of experimental conditions, results, and conclusions made in the study.
We further demonstrated that increased GC output, usually causing dysfunctional affect and reduced neurogenesis (Sapolsky, 2000; McEwen, 2004; Mirescu and Gould, 2006), is necessary for the antidepressant and proneuronal survival effects of EE (Experiment 3). EE and running-wheel exercise elevated HPA axis activity (Droste et al., 2003; 2007; Stranahan et al., 2008), and EE after chronic SD exposure (SD→sham→EE) normalized behavior and increased newborn cell survival in the caudal DG. Strikingly, when we adrenalectomized defeated mice before EE housing (SD→ADX→EE), the restorative effects of EE on new cell survival and mood-related behavior were ineffective. The combined results suggest that, during SD, HPA axis stimulation promotes maladaptive behavior and reduced neurogenesis, whereas during EE, it promotes behavioral recovery and increased neurogenesis.
The hippocampus provides powerful inhibitory control over the HPA axis. Defects in hippocampus structure and function may be manifested by HPA axis dysregulation (Schloesser et al., 2009; Snyder et al., 2011; Surget et al., 2011), which is associated with many forms of affective disorders (Nemeroff and Vale, 2005). SD has been shown to produce HPA axis dysfunction and adrenal hyperactivity in rodents (Buwalda et al., 1999; Bhatnagar and Vining, 2003; Wood et al., 2010; Lehmann et al., 2012), findings that concur with our observations that SD markedly elevated CORT secretions both acutely and during the diurnal surge period. We further showed that elevated CORT during SD is required for the development of subsequent alterations in mood-related behavior because adrenalectomized animals failed to show behavioral dysfunction. This finding is consistent with previous studies that use the GC receptor antagonist mifepristone (Wu et al., 2007) or adrenalectomy (Goshen et al., 2008) to block behavioral and neural alterations induced by chronic stressors. The mechanisms by which distress-induced pathological secretions of CORT disrupt mood may be related to the induced decline in new cell survival in the DG. The effects of chronically elevated GCs on hippocampal cytoarchitecture and new cell survival are well documented (Dranovsky and Hen, 2006; Mirescu and Gould, 2006). Here, we showed, for the first time, that the reduction in newborn neurons is a consequence of GC stimulation during SD. Thus, the antidepressant-like effect conferred by ADX in SD very likely results from enhanced or preserved neurogenesis. Indeed, newborn neurons appear to be an important target for antidepressant drug efficacy (Santarelli et al., 2003; David et al., 2009), and treatments that enhance neurogenesis mitigate activation of stress-integrative corticolimbic circuits during chronic stress exposure (Surget et al., 2011).
Not all stressful events have adverse consequences. For example, exercise and EE potently elevate GC levels (Droste et al., 2003), but they enhance cognitive function, increase resistance to further stress, and aid in stress recovery (Duman et al., 2008; Lehmann and Herkenham, 2011). EE and voluntary running also enhance learning and memory and increase synaptic complexity, neurogenesis, and brain-derived neurotrophic factor levels (van Praag et al., 1999a,b). Voluntary exercise is therapeutic in depressed patients and improves mood in healthy subjects (Salmon, 2001; Cotman et al., 2007). We previously showed that blockade of neurogenesis abolished the EE-mediated recovery from SD-induced behavioral dysfunction (Schloesser et al., 2010). Here, we found that clamping adrenal activity during EE reduced neurogenesis and also prevented behavioral recovery in defeated mice. Together, these results support the hypothesis that GCs facilitate neurogenesis during EE and suggest a critical role for neurogenesis in behavioral recovery in previously defeated mice.
Collectively, our results show that both SD and EE stimulate the HPA axis but have opposing effects on cell survival and mood-related behavior. These paradoxical influences of the HPA axis may follow an inverted U-shaped relationship that varies with the severity and controllability of the stressor. For instance, in the hippocampus, moderate elevation of plasma CORT levels favors hippocampal-dependent memory consolidation (Hui et al., 2004) and spatial memory formation (Lehmann et al., 2010; Salehi et al., 2011), whereas either a dearth or an excess of CORT impairs these processes (de Quervain et al., 1998). Similarly, either excessive or substantially reduced GCs impair hippocampal cell survival (Wong and Herbert, 2004). However, it is also probable that there are other factors that interact with GCs to direct their influence. For example, the degree of behavioral control an animal has over an aversive stressor can alter behavioral and neurochemical consequences of the event (Maier and Watkins, 1998). In our paradigm, EE allows an animal to have control over exercise stress vis-à-vis voluntary wheel running. In contrast, during SD, an animal has little control over stressor onset, duration, or intensity, and thus it is likely that the behavioral control model applies for EE and SD as well. Such behavioral control may be influenced by serotonin activity via 5HT1A receptors and raphe-to-hippocampal serotonergic neurotransmission (Lanfumey et al., 2008). Serotonin, which enhances neurogenesis through 5HT1A receptors (Banasr et al., 2004), may be dose-dependently driven by GCs in opposite directions (Judge et al., 2004). For example, chronic social stress (Flugge et al., 1998) and prolonged CORT elevation (Chalmers et al., 1993) decrease hippocampal 5HT1A receptor expression, whereas EE (Rasmuson et al., 1998) increases it.
Altered levels of testosterone may also drive the differential effects of GCs on 5HT1A receptor expression and neurogenesis. Testosterone has been reported to enhance neurogenesis (Spritzer and Galea, 2007), and levels of circulating androgens are severely reduced after chronic SD (Flugge et al., 1998), whereas EE has the opposite effect (Mitra and Sapolsky, 2012). Treatment of defeated animals with testosterone can reverse the downregulation of hippocampal 5HT1A receptors (Flugge et al., 1998). These reports implicate a role for androgens in the current findings. Future experiments will be necessary to understand the complicated interactions between circulating hormones and serotonin on the regulation of neurogenesis.
We demonstrated that the survival of newborn neurons in the caudal/ventral hippocampus is key to distress and eustress effects of GCs. The hippocampus is divided into two functionally distinct regions (dorsal, involved in spatial memory; and ventral, involved in affective and emotional behaviors) (Bannerman et al., 2004; Fanselow and Dong, 2010). Several recent studies have assessed the regional changes in neurogenesis along the septo-temporal hippocampal axis after chronic stress or EE, showing reductions in the ventral hippocampus after chronic stress (Jayatissa et al., 2006; Tanti et al., 2012) and increases in both dorsal and ventral subregions after EE (Tashiro et al., 2007), observations that are in accordance with our data. Given the reciprocal connections between the ventral hippocampus and regions involved with stress regulation, it is likely that the ventral hippocampus is more sensitive to the effects of stress and GCs. This sensitivity may be mediated in part by the differential functions and expression of GC and mineralocorticoid receptors across the septo-temporal axis (Robertson et al., 2005; Maggio and Segal, 2009).
We proposed that the protective effects of ADX during SD are mediated by hippocampal neurogenesis. Our finding that mice lacking neurogenesis are not protected from the deleterious effects of SD by ADX identifies a causal link between decreased neurogenesis, distress-induced chronic adrenal stimulation, and mood-related behavior. It is noteworthy that SD is able to induce depressive-like behavior in nonadrenalectomized NG− mice (Schloesser et al., 2010) and that NG− mice do not exhibit depressive-like behavior at baseline. Thus, it is clear that the etiology of depression is not entirely the result of decreased neurogenesis. However, our results demonstrate that neurogenesis is required for stress resilience. Therefore, it is likely that the CORT-induced decline in neurogenesis during SD is permissive to a secondary set of stress-induced neurobiological changes that result in depressive behavior. Mice in which neurogenesis is protected by ADX are also protected against the deleterious effects of chronic stress. Based on the findings shown here, we hypothesize that other treatments that prevent stress-induced reductions in neurogenesis would also buffer the negative effects of SD on behavior.
Footnotes
This work was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health. We thank Jason Snyder for helpful comments on the manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Michael Lehmann at the above address. Michael.lehmann{at}nih.gov





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}