

Abstract
Cholinergic neuromodulation controls long-term synaptic plasticity underlying memory, learning, and adaptive sensory processing. However, the mechanistic interaction of cholinergic, neuromodulatory inputs with signaling pathways underlying long-term potentiation (LTP) and long-term depression (LTD) remains poorly understood. Here, we show that physiological activation of muscarinic acetylcholine receptors (mAChRs) controls the size and sign of associative long-term synaptic plasticity via interaction with endocannabinoid signaling. Our findings indicate that synaptic or pharmacological activation of postsynaptic M1/M3 converts postsynaptic Hebbian LTP to presynaptic anti-Hebbian LTD in principal neurons of the dorsal cochlear nucleus (DCN). This conversion is also dependent on NMDA receptor (NMDAR) activation and rises in postsynaptic Ca2+. While NMDAR activation and Ca2+ elevation lead to LTP, when these events are coordinated with simultaneous activation of M1/M3 mAChRs, anti-Hebbian LTD is induced. Anti-Hebbian LTD is mediated by a postsynaptic G-protein-coupled receptor intracellular signaling cascade that activates phospholipase C and that leads to enhanced endocannabinoid signaling. Moreover, the interaction between postsynaptic M1/M3 mAChRs and endocannabinoid signaling is input specific, as it occurs only in the parallel fiber inputs, but not in the auditory nerve inputs innervating the same DCN principal neurons. Based on the extensive distribution of cholinergic and endocannabinoid signaling, we suggest that their interaction may provide a general mechanism for dynamic, context-dependent modulation of associative synaptic plasticity.
Introduction
The behavioral state of the animal and the activation of neuromodulatory systems determine the biological relevance or value of sensory stimuli, thus rendering neuromodulators critical for experience-induced plasticity. Such plasticity leads to information storage, learning, and adaptive behavior (Bear and Singer, 1986; Huerta and Lisman, 1993; Hasselmo, 1999; Reynolds et al., 2001; Weinberger, 2004; Yin and Knowlton, 2006; Froemke et al., 2007), and is mediated, in large part, by long-lasting changes in synaptic strength (Malenka and Bear, 2004; Feldman, 2009). While the cellular mechanisms underlying long-term potentiation (LTP) and depression (LTD) of synaptic strength have been extensively studied, their interaction with specific neuromodulatory systems is less understood.
Cholinergic and endocannabinoid signaling represent two major neuromodulatory pathways in the brain (Bear and Singer, 1986; Blokland, 1995; Rasmusson, 2000; Harkany et al., 2007; Heifets and Castillo, 2009; Regehr et al., 2009) displaying significant overlapping anatomical distribution (Lu et al., 1999; Harkany et al., 2005; Nyíri et al., 2005). Cholinergic inputs from medial septum are required for hippocampal learning and memory formation (Blokland, 1995; Compton et al., 1995), while cholinergic nucleus basalis is critical for activity-dependent cortical receptive field plasticity (Bear and Singer, 1986; Everitt and Robbins, 1997; Xiang et al., 1998; Froemke et al., 2007). In addition, endocannabinoid signaling is one of the major activity-dependent neuromodulatory systems mediating short- and long-term synaptic plasticity (Heifets and Castillo, 2009; Kano et al., 2009; Regehr et al., 2009). Endocannabinoid-mediated LTD, like cholinergic neuromodulation, has been associated with receptive field plasticity in sensory cortex (Liu et al., 2008; Li et al., 2009), with development of GABAergic transmission (Jiang et al., 2010), and with associative learning in hippocampus and amygdala (Marsicano et al., 2002; Varvel et al., 2007). Despite the prevalence of cholinergic and endocannabinoid signaling systems and their remarkable overlap in many brain areas where experience-dependent long-term plasticity is robust, their mechanistic interaction in shaping long-term synaptic plasticity has not been previously addressed.
Here, we test the hypothesis that synaptic activation of cholinergic inputs control long-term synaptic plasticity in the principal neurons of the dorsal cochlear nucleus (DCN). The DCN, an auditory brainstem nucleus thought to mediate adaptive sensory processing (Oertel and Young, 2004; Tzounopoulos and Kraus, 2009), receives robust cholinergic input (Henderson and Sherriff, 1991; Sherriff and Henderson, 1994; Motts et al., 2008), and exhibits endocannabinoid-mediated long-term synaptic plasticity (Tzounopoulos et al., 2007; Zhao et al., 2009). DCN principal cells exhibit Hebbian spike timing-dependent plasticity (STDP); i.e., LTP is typically observed when a postsynaptic spike follows the EPSP by 0–20 ms, while LTD is observed when the order is reversed (Tzounopoulos et al., 2004, 2007). We find that synaptic or pharmacological activation of postsynaptic M1/M3 muscarinic acetylcholine receptors (mAChRs) along with coincident activation of NMDA receptors and rises in postsynaptic Ca2+ convert Hebbian LTP to LTD, termed here anti-Hebbian LTD, because it was induced when a postsynaptic spike followed the EPSP by 0–20 ms. Increased endocannabinoid signaling, via activation of postsynaptic M1/M3 mAChRs, changes the relative strength of LTP and LTD signaling pathways and renders the LTD pathway dominant over the LTP pathway. The prevalence of cholinergic and endocannabinoid signaling in the brain and their mechanistic interaction in shaping and gating long-term synaptic plasticity is expected to unmask cellular mechanisms underlying cholinergic-mediated plasticity important for receptive field plasticity, adaptive sensory processing, and memory formation.
Materials and Methods
Coronal brain slices were made from ICR mice (P17–P26). The preparation and use of coronal slices containing DCN has been described in detail (Tzounopoulos et al., 2004). Animals were killed according to methods approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Single cells were visualized with IR interference contrast optics and recorded using patch pipettes in either voltage- or current-clamp mode. Cells in the DCN molecular and fusiform cell layers were identified on the basis of morphological and electrophysiological criteria (for more details, see Tzounopoulos et al., 2004). The external solution contained the following (in mm): 130 NaCl, 3 KCl, 1.2 KH2PO4, 2.4 CaCl2, 1.3 MgSO4, 20 NaHCO3, 3 HEPES, and 10 glucose; saturated with 95% O2/5% CO2. For voltage-clamp experiments pipettes were filled with a Cs+-based solution containing the following (in mm): 130 Cs-gluconate, 10 CsCl, 2 MgCl2, 0.16 CaCl2, 0.5 EGTA, 10 HEPES, 4 Na2ATP, 0.4 NaGTP, and 14 Tris-creatine phosphate. For current-clamp experiments, pipettes were filled with a K+-based internal solution containing the following (in mm): 113 K-gluconate, 4.5 MgCl2, 14 trisphosphocreatine, 9 HEPES, 0.1 EGTA, 4 Na-ATP, 0.3 tris-GTP, and 10 sucrose. All the internal solutions were adjusted to pH 7.3, ∼300 mOsmol. Whole-cell recordings were performed at 31–33°C. For voltage-clamp experiments, series resistance was monitored throughout the experiment from the size and shape of the capacitive transient in response to a 5 mV hyperpolarization. Input resistance was calculated from the sustained response to the same step. Experiments were not included if the series and/or input resistance changed >20% throughout recording. Excitatory postsynaptic responses were evoked by stimulating parallel fiber tracts (see Figs. 2a, 4b1). Auditory nerve responses were evoked by stimulating the deep layer of the DCN (see Fig. 4b1). Cholinergic activation was evoked by placing a bipolar electrode on the termination site of the olivocochlear bundle in the cochlear nucleus (see Fig. 2a, S2). For Figure 3 (coordination of S2 and S1 pairing), activation of cholinergic fibers was revealed by evoking atropine-sensitive long-lasting inward currents as a result of S2 stimulation (see Fig. 2). Only cells that showed atropine-sensitive long-lasting inward currents as a result of S2 stimulation were included in Figure 3 and in other experiments where plasticity was induced with S2 stimulation. The pairing protocol was initiated 10 ms after the termination of S2 stimulation. EPSP/Cs were recorded in the presence of SR95531 (20 μm) and strychnine (0.5 μm). During pharmacological induction of LTD [oxotremorine M (oxo-M) application + pre–post-pairing protocol], the pairing protocol was applied at the peak of mAChR activation for the different experiments (∼10 min after oxo-M application as shown in Fig. 4a1). EPSPs were collected every 5 s (0.2 Hz) before and after pairings. EPSP slope was measured and averaged every minute (12 sweeps), then normalized to baseline. SR95531 and AM-251 were purchased from Ascent Scientific. Atropine, strychnine, and GDP-β-S were purchased from Sigma-Aldrich. oxo-M, U73122, 4-DAMP, WIN-55212–2 (WIN), AP5, γDGG, and APV were purchased from Tocris Cookson and Ascent Scientific. Data were acquired and analyzed using pClamp10.1. All means are reported ±SEM. Statistical comparisons were made using unpaired two-tailed Student's t tests. Statistical significance was based on p values <0.05.
Results
Conversion of Hebbian LTP to anti-Hebbian LTD by endogenous ACh release and activation of mAChRs
Recordings were made from single principal neurons (fusiform cells) in coronal slices of mouse DCN. Fusiform cells were identified by their location in the fusiform cell layer and by their characteristic response to current injection (Tzounopoulos et al., 2004). STDP was induced by pairing EPSPs with postsynaptic action potentials evoked by direct current injection through the recording electrode. Test EPSPs were delivered at 0.2 Hz. A pairing protocol consisted of a shock to parallel fibers followed 5 ms later by a suprathreshold current pulse to the fusiform cell, causing the peak of an action potential to be produced 5 ms after the onset of the EPSP (Fig. 1a1). These pairs were repeated 5 times in 100 ms intervals. This protocol was then repeated 10 times with a frequency of 0.2 Hz (Fig. 1a1, pre–post pairing). Our previous studies have shown that this pairing protocol induces LTP at parallel fiber inputs synapsing onto fusiform cells (Tzounopoulos et al., 2004, 2007). Here we repeated the same protocol and we also observed LTP (Fig. 1a3).

Hebbian LTP in fusiform cells. a1, Plasticity was induced by a protocol comprised of five pairs (subthreshold EPSP produced by S1 stimulation with a current-evoked spike delivered 5 ms later) delivered at 100 ms intervals followed by a 5 s pause, and repeated a total of 10 times. a2, Examples of averaged EPSPs before and 15–20 min after pairing. a3, Time course of induced plasticity (control, LTP, 120 ± 5%, 15–20 min after pairing, n = 6, p < 0.05). All means are reported ±SEM. For statistical significance, p < 0.05. The arrow indicates when the pairing protocol was initiated.
STDP in the DCN depends on opponent processes controlling LTP and LTD that are coactive (Tzounopoulos et al., 2007). LTD is induced by activation of retrograde endocannabinoid signaling leading to reduced presynaptic release, while LTP requires activation of postsynaptic CaMKII and is induced and expressed postsynaptically (Tzounopoulos et al., 2007). Modulators of these two processes by changing the relative strength of LTP and LTD are expected to alter the overall STDP rule. Given that the DCN receives cholinergic input (Henderson and Sherriff, 1991; Sherriff and Henderson, 1994; Motts et al., 2008), we tested the hypothesis that cholinergic neuromodulation by AChRs can change STDP timing rules by shifting the relative strength of LTP and LTD pathways. To test this hypothesis, we stimulated cholinergic inputs during our STDP pairing protocol. To activate cholinergic fibers, we used a second stimulating electrode (S2) positioned on the presumed pathway of the olivocochlear bundle passing by the cochlear nucleus (Fig. 2a, S2). S2 stimulation (100 Hz for 1 s) induced a long-lasting inward (Fig. 2b) current likely caused by the suppression of several potassium conductances mediated by postsynaptic activation of AChRs (Dutar and Nicoll, 1988; Pál et al., 2009). In agreement with this hypothesis, application of atropine (2 μm: a competitive antagonist of mAChRs) blocked the long-lasting component of the inward current (Fig. 2b), indicating that postsynaptic mAChRs were activated during the train delivered by our second stimulating electrode (S2) (Fig. 2a). To test the effect of synaptically activated AChRs on synaptic transmission, we recorded baseline EPSCs by stimulating S1; then, S2 stimulation (100 Hz, 1 s) was applied, and immediately after termination of S2 stimulation, S1 stimulation was applied (Fig. 2c). S2 stimulation had a small but nonsignificant effect on EPSCs evoked by S1 stimulation (Fig. 2c). Therefore, we conclude that S2 stimulation activates postsynaptic mAChRs, but this stimulation has no significant effects on baseline synaptic transmission of parallel fiber EPSCs.
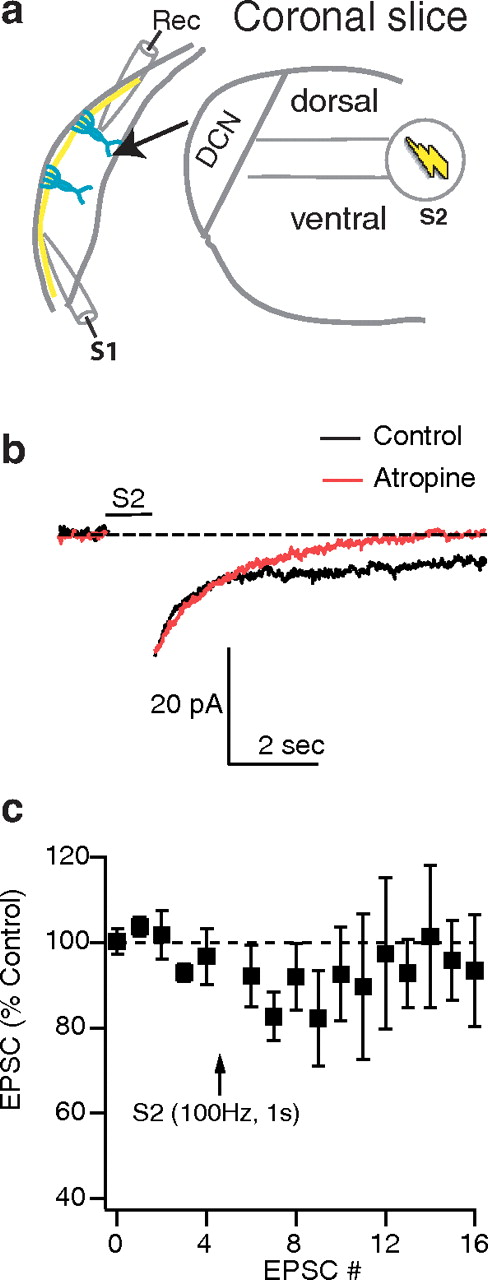
Synaptic activation of cholinergic inputs. a, Schematic of the slice preparation illustrates recording (rec.) and stimulation (S1: parallel fiber; S2: cholinergic fibers) sites. The arrow indicates a “blow-up” of the DCN. b, Postsynaptic activation of atropine-sensitive long-lasting inward currents by S2 stimulation (100 Hz, 1 s). Time course of evoked long-lasting inward currents in the absence (control, black trace) or in the presence of atropine (atropine, red trace). All responses were obtained in the presence of blockers of excitatory and inhibitory synaptic transmission (SR95531, 20 μm; strychnine, 0.5 μm; NBQX, 20 μm; APV, 50 μm; and γDGG, 30 μm). c, Effect of S2 (100 Hz, 1 s) stimulation on baseline synaptic transmission. S1 stimulation was given every 5 s. EPSCs plotted here are the result of S1 (parallel fiber) stimulation. Application of S2 stimulation (cholinergic pathway) did not cause any significant effect on the EPSC (97 ± 9% last three EPSCs, not significant; 87 ± 6% the first three EPSCs, n = 6, not significant). All means are reported ±SEM. For statistical significance, p < 0.05. The arrow indicates when the pairing protocol or S2 train was initiated.
To examine the effect of synaptically activated mAChRs on STDP timing rules, S2 stimulation (100 Hz, 1 s) was applied before every set of EPSPs and action potentials of the pre–post pairing protocol (Fig. 3a1). Surprisingly, S2 stimulation converted LTP to LTD (Fig. 3a2). The conversion of LTP to LTD was blocked by atropine (Fig. 3a2), suggesting that mAChR activation by physiological levels of ACh changed the polarity of plasticity. This is a critical finding, as this is the first study that provides evidence that physiological activation of cholinergic system switches the polarity of induced synaptic plasticity. Together, these findings indicate that synaptic activation of mAChRs during pre–post pairing converts Hebbian LTP to anti-Hebbian LTD.

Conversion of Hebbian LTP to anti-Hebbian LTD by endogenous ACh release and activation of mAChRs. a1, S2 stimulation (100 Hz for 1 s) preceded every cycle of pairing protocol that was described in Figure 1a1. The pairing protocol was initiated 10 ms after the termination of S2 stimulation. a2, LTD was induced when S2 was stimulated, which was converted into LTP in the presence of mAChR antagonist (atropine, 2 μm). Time course of induced plasticity (control, anti-Hebbian LTD, 70 ± 7%, 15–20 min after pairing, n = 8, p < 0.05; atropine, Hebbian LTP, 129 ± 7%, 15–20 min after pairing, n = 7, p < 0.05; induced plasticity without S2 stimulation as in Fig. 1a3 is in red line for comparison). b, Associativity and temporal dependence of STDP when S2 is stimulated. Summary graph showing synaptic plasticity induced by a pairing protocol as shown in: (pre–post pairing, control, Δt = 5 ms, 20 ± 5%; S2 + control, Δt = 5 ms, −30 ± 7%; S2 + control + atropine, Δt = 5 ms, 29 ± 7%; S2 + control, Δt = 20 ms, 5 ± 5.1%; S2 + control, Δt = −5 ms, −3 ± 4.2%), where Δt is the time interval between the EPSP onset and spike peak; for positive Δt values, EPSP precedes the spike, while for negative Δt values, the spike precedes the EPSP, n = 6–9 per point. All means are reported ±SEM. Asterisks indicate statistical significance, p < 0.05. The arrow indicates when the pairing protocol was initiated.
To determine whether the sign and size of induced plasticity are dependent on the relative order of presynaptic and postsynaptic firing when cholinergic inputs are activated, we coordinated stimulation of cholinergic fibers with a pairing protocol in which postsynaptic action potentials preceded the EPSPs by 5 ms. This protocol (Δt = −5 ms) resulted in no significant changes in synaptic strength (Fig. 3b). To determine the timing window over which coordinated presynaptic and postsynaptic activity can induce long-lasting changes in synaptic strength, we used a pairing protocol where the spike followed the EPSP by 20 ms. This manipulation (Δt = 20 ms) also resulted in no changes in synaptic strength (Fig. 3b). Together, these results indicate that the observed STDP rule during cholinergic activation is associative (coordinated presynaptic and postsynaptic activity is required), depends on the relative order of presynaptic and postsynaptic firing in an anti-Hebbian manner (pre–post pairing leads to LTD), and operates on a timing window of <20 ms difference between EPSP and spike.
Pharmacological activation of mAChRs mediates input-specific conversion of Hebbian LTP to anti-Hebbian LTD
S2 stimulation indicated that physiological levels of ACh are involved in the modulation of synaptic plasticity. While inhibitory inputs were pharmacologically blocked during coordinated S2 stimulation and pairing protocol, S2 stimulation activated cholinergic as well as glutamatergic inputs. To study the effect of specific activation of mAChRs, and to determine whether specific mAChR activation is necessary in converting Hebbian LTP to anti-Hebbian LTD, we used a specific agonist of mAChRs. Application of oxo-M (5 μm, specific agonist of mAChRs) for 3 min resulted in a transient decrease in synaptic strength (Fig. 4a1). In agreement with synaptic stimulation of mAChRs leading to a transient inward current (Fig. 2b), oxo-M resulted in a transient increase in the input resistance (Fig. 4a2), further suggesting that postsynaptic activation of mAChRs causes the suppression of potassium conductances. However, when the same pharmacological activation of mAChRs was coordinated with the pre–post-pairing protocol (the pairing protocol was applied at the peak of mAChR activation), synaptic response strength was reduced and no recovery of synaptic strength was observed, indicating that LTD is induced (Fig. 4b3, black squares). This result shows that specific activation of mAChRs, when paired with a pre–post-pairing protocol, is necessary and sufficient to converting Hebbian LTP to anti-Hebbian LTD.

Pharmacological activation of mAChRs controls associative synaptic plasticity in an input-specific manner. a1, Bath application of mAChR agonist (oxo-M, 5 μm) for 3 min reversibly decreases EPSP amplitude. Time course of the effect of oxo-M on parallel fiber EPSPs (95 ± 5%, 40–45 min after oxo-M application, n = 6, not significant). a2, Bath application of oxo-M for 3 min reversibly increases input resistance. Time course of the effect oxo-M on input resistance (95 ± 6%, 40–45 min after oxo-M application, n = 6, not significant). b1, Schematic of the slice preparation illustrates recording electrode (rec.) and stimulation electrodes: S1 for stimulating parallel fibers and S1 (AN) for stimulating auditory nerve fibers (experiments from auditory nerve fiber stimulation are shown in d). b2, Examples of averaged EPSPs before and 40–45 min after oxo-M application and pre–post pairing as shown in Figure 1a1. b3, Bath application of oxo-M allows induction of anti-Hebbian LTD after pre–post pairing as in Figure 1a1 (STDP protocol). Time course of induced plasticity (oxo-M + pre–post-pairing protocol: 58.5 ± 5.4%, 40–45 min after oxo-M application, n = 7, p < 0.05; application of oxo-M without pre–post pairing is in red line for comparison taken from a1). c, Associativity and temporal dependence of STDP when mAChRs are pharmacologically activated. Summary graph showing synaptic plasticity induced by a pairing protocol as shown in: (oxo-M + pre–post pairing protocol, Δt = 5 ms, −41.5 ± 5.4%; oxo-M + pre–post-pairing protocol, Δt = −5 ms, −8% ± 5.1%; S2 + control + atropine, oxo-M + pre–post-pairing protocol, Δt = 20 ms, −8% ± 7.8%), where Δt is the time interval between the EPSP onset and spike peak; for positive Δt values, EPSP precedes the spike, while for negative Δt values, the spike precedes the EPSP, n = 5–9 per point. d, Pharmacological activation of mAChRs did not induce anti-Hebbian LTD in auditory nerve inputs. Pre–post-pairing protocol (as shown in Fig. 1a1) did not elicit long-term plasticity in the auditory nerve inputs, in control or in the presence of oxo-M. Time course of induced plasticity (pre–post pairing: 103.1 ± 7.8%, n = 6, not significant; oxo-M + pre–post pairing: 108 ± 13%, 30–35 min after oxo-M application, n = 6, not significant). All means are reported ±SEM. Asterisks indicate statistical significance, p < 0.05. The arrows indicate when the pairing protocol was initiated.
To determine whether the sign and size of induced plasticity is dependent on the relative order of presynaptic and postsynaptic firing when mAChRs are pharmacologically activated, we coordinated application of oxo-M with a pairing protocol in which action potentials preceded the EPSPs by 5 ms (Δt = −5 ms). This protocol resulted in no significant changes in synaptic strength (Fig. 4c). Test of the timing limits between EPSP and spike required for LTD induction revealed that no synaptic plasticity was observed when the interval between EPSP and spike was 20 ms (Fig. 4c) (Δt = 20 ms). Together, these results indicate that the observed STDP rule during pharmacological activation of mAChR activation is associative and operates on a timing window of <20 ms difference between EPSP and spike.
Fusiform cells integrate plastic parallel fiber inputs synapsing onto apical dendrites with auditory nerve inputs synapsing onto the basal dendrites (Oertel and Young, 2004). To determine whether activation of mAChRs leads to input-specific anti-Hebbian LTD, we tested whether mAChR activation modulates synaptic plasticity in the auditory nerve inputs. To activate auditory nerve EPSPs, we positioned our stimulation electrode in the deep layer of the DCN (Fig. 4b1). We tested whether pairing of auditory nerve EPSPs with postsynaptic spikes (same pre–post pairing as in Fig. 1a1) revealed STDP. Contrary to parallel fiber inputs (Fig. 1a3), auditory nerve input did not show STDP (Fig. 4d, black squares). In addition, application of oxo-M did not reveal any synaptic plasticity in the synapses between auditory nerve inputs and fusiform cell (Fig. 4d, white circles), thus indicating that activation of mAChRs leads to input specific anti-Hebbian LTD.
Activation of M1/M3 mAChRs enhances endocannabinoid signaling and converts Hebbian LTD to anti-Hebbian LTD via a presynaptic mechanism
To investigate expression mechanisms of the anti-Hebbian LTD, we used coefficient of variation (CV) analysis, an assay that is sensitive to changes in neurotransmitter release (Faber and Korn, 1991; Tsien and Malinow, 1991; Larkman et al., 1992). Consistent with our previous studies indicating that postsynaptic induction and expression mechanisms underlie Hebbian LTP (Tzounopoulos et al., 2007), 1/CV2 did not change after induction of LTP (Fig. 5a), However, 1/CV2 was reduced after LTD induction (Fig. 5a), suggesting that anti-Hebbian LTD is expressed presynaptically, via a decrease in probability of release (Pr). Given the critical role of endocannabinoid signaling in mediating presynaptically expressed LTD in DCN interneurons (Tzounopoulos et al., 2007), we tested whether the anti-Hebbian LTD in fusiform cells is mediated by activation of endocannabinoid signaling. Application of AM-251 (1 μm, a selective CB1 receptor antagonist) blocked anti-Hebbian LTD when we used the same induction protocol as in Figure 4b2 (oxo-M + pre–post pairing) (Fig. 5a). In addition, application of AM-251 unmasked LTP (Fig. 5b), thus indicating that it is the enhancement of endocannabinoid signaling via activation of mAChRs that converts Hebbian LTP into anti-Hebbian LTD in fusiform cells. To determine whether physiological activation of mAChRs converts Hebbian LTP to anti-Hebbian LTD via the same mechanism, we tested the effect of AM-251 on the anti-Hebbian LTD that was induced by coordination of S2 stimulation and conditioning (pre–post pairing) protocol. In agreement with pharmacological activation of mAChRs, synaptic activation of mAChRs converted anti-Hebbian LTD to Hebbian LTP (Fig. 5c). These data confirm that synaptic and pharmacological anti-Hebbian LTD share the same induction and expression mechanisms. These experiments also indicate that physiological or pharmacological activation of mAChRs promotes endocannabinoid signaling during coincident presynaptic and postsynaptic activity and this promotion leads to the induction of endocannabinoid-mediated anti-Hebbian LTD that is expressed by a decrease in Pr. Finally, these results show that activation of mAChRs does not affect the induction of postsynaptic Hebbian LTP, as the amounts of LTP obtained in control (pre–post pairing) or after pharmacological or synaptic activation of mAChRs during blockade of presynaptic CB1Rs by AM-251 were similar in amplitude (Fig. 5d). If activation of mAChRs were affecting LTP induction, we would expect to unmask different amounts of LTP in the presence of AM-251, when compared to control LTP. Together, these results indicate that activation of mAChRs switches the sign of induced synaptic plasticity by promoting endocannabinoid signaling and LTD induction, but without affecting LTP induction.

M1/M3 mAChR activation enhances endocannabinoid signaling and converts Hebbian LTP to anti-Hebbian LTD. a, Presynaptic expression mechanisms underlie anti-Hebbian LTD. 1/CV2 analysis before and after LTD induction suggests that LTD is expressed presynaptically (LTD: 1/CV2 = 0.68 ± 0.06 of control, n = 6; LTP: 1/CV2 = 1.1 ± 0.1 of control, n = 6). b, Pharmacologically induced anti-Hebbian LTD is converted to LTP when endocannabinoid signaling is blocked by application of CB1R antagonist (AM-251, 1 μm). Time course of induced plasticity (oxo-M + pre–post-pairing protocol + AM-251: 127 ± 8%, 40–45 min after oxo-M application n = 8, p < 0.05; oxo-M and pre–post-pairing is in red line for comparison, taken from Fig. 4b3). c, Synaptically induced anti-Hebbian LTD is converted to LTP when endocannabinoid signaling is blocked by application of CB1R antagonist (AM-251, 1 μm). Time course of induced plasticity (S2 + pre–post-pairing protocol + AM-251; 140 ± 18%, 20–25 min after S2 stimulation, n = 7, p < 0.05; S2 stimulation and pre–post pairing is in red line for comparison, taken from Fig. 3a2). d, LTP induction is not affected by mAChR activation when endocannabinoid signaling is blocked. Summary graph showing comparison of LTP in control (20 ± 5%; n = 6, same as in Fig. 1a3), or after pharmacological or synaptic activation of mGluRs when CB1Rs are blocked (oxo-M + pre–post pairing + AM-251; 27 ± 8%; n = 8; S2 + pre–post pairing + AM-251: 40 ± 18%; n = 7, not significant between the three different conditions). e, Application of M1 and M3 mAChR antagonists (pirenzepine, 10 μm and 4-DAMP, 1 μm) block mAChR-dependent conversion of Hebbian LTP to anti-Hebbian LTD and unmasks Hebbian LTP. Time course of induced plasticity induced by pre–post pairing (Δt = 5 ms), and coordinated synaptic activation of mAChRs (S2 stimulation) (135 ± 13%, 20–25 min after S2 stimulation, n = 7, p < 0.05; S2 stimulation and pre–post pairing is in red line for comparison, taken from Fig. 3a2). f, LTP magnitude is not affected by potential tonic mAChR activation. Summary graph showing comparison of LTP in control (20 ± 5%; n = 6, same as in 1a3), or after synaptic activation of mGluRs when M1/M3 mAChRs are blocked (S2 + pre–post pairing + atropine; 29 ± 7%, n = 7, same as in Fig. 3a2; S2 + pre–post pairing + M1/M3 antagonists: 35 ± 13%; n = 7, not significant between the three different conditions). All means are reported ±SEM. Asterisks indicate statistical significance, p < 0.05. The arrows indicate when the pairing protocol was initiated.
Next, we determined the specific mAChR subtype mediating the modulation of associative synaptic plasticity. Pharmacological activation of both M1 and M3 mAChRs has been shown to mediate enhancement of endocannabinoid-mediated short-term plasticity (Kim et al., 2002; Ohno-Shosaku et al., 2003; Narushima et al., 2007), yet their action on endocannabinoid-mediated long-term plasticity remains unknown. To determine whether M1/M3 are mediating the enhancing effect of mAChRs on endocannabinoid signaling and whether this enhancement can also promote long-term plasticity, we used pirenzepine (10 μm, specific M1 antagonist) and 4-DAMP (1 μm, specific M3 antagonist) to block M1 and M3 receptors, respectively. Application of pirenzepine and 4-DAMP blocked physiologically (S2 + pre–post pairing) induced anti-Hebbian LTD and unmasked LTP (Fig. 5e), suggesting that activation of M1/M3 mAChRs mediates the conversion of Hebbian LTP to anti-Hebbian LTD. The amounts of LTP obtained in control (pre–post pairing), or during physiological activation of cholinergic fibers (S2 + pre–post-pairing) in the presence of atropine or pirenzepine and 4-DAMP were similar in amplitude (Fig. 5f). These results suggest that there is neither unexpected cholinergic stimulation nor tonic activation of mAChRs leading to LTD (or affecting LTP in any other way) during pre–post pairing (control). If unexpected cholinergic activation or tonic activity of mAChRs (M1/M3) were inducing LTD during pre–post pairing, we would expect to unmask bigger LTP in the presence of atropine or pirenzepine and 4-DAMP. Together, these results show that physiological activation of M1/M3 mAChRs converts Hebbian LTP to anti-Hebbian LTD, by promoting endocannabinoid release and inducing LTD without affecting LTP induction. Previous studies have activated mAChRs by applying agonists, with no studies of how, or indeed whether, synaptically released ACh can influence synaptic plasticity via the activation of endocannabinoid signaling. Thus, another critical and novel finding of our study is that physiological activation of M1/M3 mAChRs converts Hebbian LTP to anti-Hebbian LTD by promoting endocannabinoid signaling.
Activation of mAChRs converts Hebbian LTP to anti-Hebbian LTD via postsynaptic G-protein-coupled pathway, leading to PLC activation
Next, we investigated the mechanism via which activation of M1/M3 mAChRs promote endocannabinoid signaling. Endocannabinoid signaling has a postsynaptic (endocannabinoid synthesis and release) and a presynaptic (activation of cannabinoid receptors, CB1Rs) component. To determine whether M1/M3 mAChR-dependent conversion of Hebbian LTP to anti-Hebbian LTD is mediated through a postsynaptic activation of mAChRs and initiation of a G-protein-coupled receptor intracellular signaling cascade known to enhance endocannabinoid short-term synaptic plasticity (Maejima et al., 2001; Varma et al., 2001; Kim et al., 2002), we tested the effect of GDP-β-S (2 mm, a nonhydrolyzable analog of GDP). Based on our findings that pharmacological and physiological activation of mAChRs convert Hebbian LTP to anti-Hebbian LTD via identical induction and expression mechanisms (Figs. 2⇑⇑–5), we only used pharmacological activation of mAChRs for the rest of the experiments. Loading of fusiform cells with GDP-β-S blocked anti-Hebbian LTD (Fig. 6a), suggesting that activation of mAChRs is mediating the induction of anti-Hebbian LTD through a postsynaptic G-protein-coupled receptor pathway. However, GDP-β-S antagonizes all G-protein molecular pathways in the postsynaptic cell and could potentially affect synaptic and intrinsic properties of fusiform neurons. To determine whether GDP-β-S causes changes in baseline synaptic strength, we investigated its effects on baseline EPSCs in the absence of any pairing protocol and/or oxo-M application. Application of GDP-β-S did not cause any significant change in synaptic strength (Fig. 6b). Application of GDP-β-S caused a decrease in the input resistance of the fusiform cell (Fig. 6c). However, this change in input resistance cannot explain the effect of GDP-β-S in blocking anti-Hebbian LTD, as a decrease in input resistance could potentially cause a small decrease in EPSPs and thus could reveal an even “larger” depression of EPSPs that could be considered as more robust LTD. However, GDP-β-S, by blocking LTD, causes an overall enhancement of the EPSP compared to control, further suggesting a role of a postsynaptic G-protein-coupled pathway involved in the induction of anti-Hebbian LTD. However, the small change in input resistance may explain why LTP is not induced after application of GDP-β-S, as it was seen after application of AM-251 or M1/M3 antagonists under the same conditions (Fig. 5b–f).

M1/M3 mAChR activation enhances endocannabinoid signaling and converts Hebbian LTP to anti-Hebbian LTD via a postsynaptic G-protein-coupled pathway that involves PLC activation. a, Application of intracellular nonhydrolyzable analog of GDP (GDP-β-S, 2 mm applied in the recording pipette) blocks the effect of oxo-M in converting LTP to LTD. Time course of induced plasticity (oxo-M + pre–post pairing + GDP-β-S: 102.6 ± 7%, 40–45 min after oxo-M application, n = 7, not significant; oxo-M and pre–post pairing is in red line for comparison, taken from 4b3). b, Application of intracellular nonhydrolyzable analog of GDP-β-S does not affect baseline EPSCs. Time course of EPSCs (87 ± 11%; n = 5, 40–45 min after oxo-M application, not significant). c, Application of intracellular GDP-β-S decreases input resistance. Time course of input resistance (78 ± 10%; n = 5, 40–45 after oxo-M application, p < 0.05). d, Incubation of slices and subsequent bath application of PLC inhibitor (U73122, 5 μm) blocks the effect of oxo-M in converting LTP to LTD. Time course of induced plasticity (oxo-M + pre–post pairing + U73122: 112.3 ± 6.4%, 40–45 min after oxo-M application, n = 6, not significant). All means are reported ±SEM. For statistical significance, p < 0.05. The arrows indicate when the pairing protocol was initiated.
Thus, our electrophysiological results indicate that the conversion of Hebbian LTP to anti-Hebbian LTD is mediated by M1/M3 mAChRs that are postsynaptically localized. This is consistent with other electrophysiological studies showing postsynaptic actions of M1/M3 mAChRs in DCN principal neurons (Pál et al., 2009). Together, our results indicate that GDP-β-S blocks a postsynaptic G-protein-coupled receptor pathway that is activated by postsynaptic M1/M3 mAChR activation and that causes anti-Hebbian LTD via activation of endocannabinoid signaling.
Next we determined the biochemical pathway via which M1/M3 mAChRs enhance endocannabinoid signaling after activating a postsynaptic G-protein-coupled receptor pathway. Our previous studies have shown that 2-arachidonoylglycerol (2-AG) is the endocannabinoid mediating synaptic plasticity in the DCN (Zhao et al., 2009). Given that G-protein-coupled receptor pathways have been shown to promote synthesis of 2-AG through activation of phospholipase C (PLC) (Hashimotodani et al., 2005), we tested whether PLC activation is necessary for mAChR-mediated enhancement of endocannabinoid signaling. Incubation of slices and subsequent bath application of U73122 (5 μm, PLC blocker) blocked anti-Hebbian LTD (Fig. 6d), suggesting that activation of PLC through postsynaptic mAChRs is necessary for the induction of anti-Hebbian LTD.
However, activation of other G-protein-coupled receptors is also signaling via PLC, such as mGluRs. To determine whether PLC activation was solely dependent on mAChRs, we tested for the requirement of mGluRs on the induction of anti-Hebbian LTD. Given the involvement of group I metabotropic glutamate receptors (mGluRs) in activating PLC and in promoting endocannabinoid-mediated LTD in other systems (Chevaleyre and Castillo, 2003), we tested the role of mGluR1 and mGluR5 on the induction of LTD. Bath application of 100 μm LY367385 and 4 μm MPEP, selective antagonists of mGluR subtypes 1 and 5, respectively, had no effect on fusiform cell LTD (Fig. 7a), indicating that mGluR1/5 activation is not necessary for LTD induction. This result further supports our hypothesis that it is the specific activation of postsynaptic M1/M3 that leads to the enhancement of endocannabinoid signaling via PLC activation.

Postsynaptic activation of NMDARs and rises in postsynaptic Ca2+ are required for the conversion of Hebbian LTP to anti-Hebbian LTD, while mGluRs are not necessary. a, Anti-Hebbian LTD was induced in the presence of 100 μm LY367385 and 4 μm MPEP, selective antagonists of mGluR subtypes 1 and 5, respectively (68 ± 7%, 40–45 after oxo-M application, n = 6, oxo-M and pre–post pairing is in red line for comparison, taken from Fig. 4b3). The magnitude of LTD was not significantly different from control LTD (p > 0.05). b, Failure to induce anti-Hebbian LTD after synaptic cholinergic stimulation (100 Hz for 1 s, repeated 10 times at a 0.2 Hz frequency) in the absence of pre–post-pairing protocol. Otherwise, the protocol was the same as Figure 3a (S2 stimulation: 93 ± 13%, n = 5, not significant; S2 stimulation and pre–post pairing is in red line for comparison, taken from Fig. 3a2). c, Failure to induce anti-Hebbian LTD in the presence of 50 μm APV (96 ± 12%, 40–45 after oxo-M application, n = 6, not significant; oxo-M and pre–post pairing is in red line for comparison, taken from Fig. 4b3). d, Failure to induce anti-Hebbian LTD in the presence of intracellular MK-801 (20 μm; 103 ± 10%, 40–45 min after oxo-M application, n = 6, not significant; oxo-M and pre–post pairing is in red line for comparison, taken from Fig. 4b3). e, Failure to induce anti-Hebbian LTD in the presence of intracellular BAPTA (10 mm; 94 ± 12%, 40–45 min after oxo-M application, n = 6, not significant; oxo-M and pre–post pairing is in red line for comparison, taken from 4b3). f, Summary graph showing comparison of anti-Hebbian after pharmacological activation of mAChRs in control (41.5 ± 5.4%, n = 6, p < 0.05, same as in Fig. 4b3), when mGluRs are blocked (32 ± 7%, n = 6, p < 0.05), when NMDARs are blocked and when rises in postsynaptic Ca2+ are blocked by intracellular application of BAPTA (oxo-M + pre–post-pairing + APV: 4 ± 12%, n = 6; oxo-M + pre–post-pairing + BAPTA: 6 ± 12%, n = 6; oxo-M + pre–post-pairing + iMK-801: −3 ± 12%, n = 6, negative sign indicates increase in synaptic strength). All means are reported ±SEM. Asterisks indicate statistical significance, p < 0.05. The arrows indicate when the pairing protocol was initiated.
Our findings indicate that mAChR activation is necessary and sufficient in inducing anti-Hebbian LTD only when it is coordinated with a pre–post-pairing protocol. Specifically, application of oxo-M led only to a transient decrease of synaptic strength (Fig. 4a1), while synaptic activation of mAChRs by application of one round of cholinergic stimulation (100 Hz, 1s) did not lead to any changes in synaptic strength (Fig. 2c). To confirm that stronger synaptic stimulation of cholinergic inputs do not lead to LTD, S2 stimulation was given 10 times (exact same amount of cholinergic stimulation as in the protocol that induced anti-Hebbian LTD in Fig. 3a) but now in the absence of the pairing protocol. No LTD was induced (Fig. 7b), further supporting our conclusion that the pairing protocol is necessary for the induction of LTD under our experimental conditions. These results suggest that synaptic activation of NMDARs and postsynaptic rises in Ca2+ during pre–post-pairing protocol may be necessary for the induction of anti-Hebbian LTD. The NMDAR antagonist dl-2-amino-5-phosphonovalerate (APV, 50 μm applied to the bath) (Fig. 7c) or MK-801 (20 μm, applied intracellularly) blocked anti-Hebbian LTD (Fig. 7d), as did intracellular application of 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA, 10 mm) (Fig. 7e). Therefore, the induction of anti-Hebbian LTD requires a postsynaptic rise in Ca2+, probably mediated by postsynaptic NMDARs. Together, these results indicate that while activation of NMDARs and postsynaptic Ca2+ lead to LTP (Tzounopoulos et al., 2007), when M1/M3 receptors are simultaneously activated, Ca2+ elevation and Gq/11-coupled receptor activation convert Hebbian LTP to anti-Hebbian LTD. This is an important finding suggesting that coactivation of different signaling pathways can lead to cooperative effects capable of reversing the effects of activation of either pathway alone, thus unmasking a nonclassical mechanism of determining synaptic strength.
Discussion
Our results indicate that cholinergic activation controls synaptic plasticity in the DCN via a postsynaptic interaction with endocannabinoid signaling. During pairing of presynaptic and postsynaptic activity, postsynaptically induced and expressed, LTP is observed under control conditions. However, the same pairing of presynaptic and postsynaptic activity leads to presynaptic LTD, when postsynaptic M1/M3 mAChRs are activated by synaptic stimulation of cholinergic fibers. Coincident synaptic activation of postsynaptic M1/M3 mAChR and NMDARs promotes endocannabinoid signaling via a Ca2+-assisted, Gq-coupled pathway leading to activation of PLC. Increased PLC activity enhances endocannabinoid synthesis and release and thus activates presynaptic CB1Rs and converts postsynaptic LTP to presynaptic LTD (Fig. 8).

Interactions between M1/M3 mAChRs, NMDARs, and endocannabinoid signaling determine synaptic plasticity. Hebbian LTP arises from activation of NMDARs, rises in postsynaptic Ca2+ levels, and activation of CaMKII (postsynaptically induced and expressed) (Tzounopoulos et al., 2007). However, synaptic activation of postsynaptic M1/M3 mAChR in coordination with activation of postsynaptic NMDARs and rises in postsynaptic Ca2+ now enhance endocannabinoid release and lead to anti-Hebbian LTD (postsynaptic induction and presynaptic expression).
Plasticity mechanisms and neuromodulatory inputs
Recent studies have established the existence of parallel, opposing, signaling pathways for LTP and LTD. In these cases, the overall plasticity rule results from the balance or competition between separately activated induction pathways for LTP and LTD (Liu et al., 2004; O'Connor et al., 2005; Wang et al., 2005; Bender et al., 2006; Nevian and Sakmann, 2006; Wittenberg and Wang, 2006; Seol et al., 2007; Tzounopoulos et al., 2007; Shen et al., 2008). The distinction of LTP and LTD signaling pathways underlying STDP has been recently unmasked (Bender et al., 2006; Nevian and Sakmann, 2006; Seol et al., 2007; Tzounopoulos et al., 2007; Shen et al., 2008). For LTP, postsynaptic NMDARs are the coincident detectors and the Ca2+ source, while LTD involves retrograde endocannabinoid signaling leading to a decrease in probability of release and thus suggesting a common signaling motif found in many presynaptic forms of LTD (Sjöström et al., 2003; Bender et al., 2006; Nevian and Sakmann, 2006; Tzounopoulos et al., 2007). LTP and LTD pathways interact to produce the overall STDP rule, and this interaction suggests that modulation of either pathway can change dramatically the overall plasticity rule. Our results indicate that the relative dominance of signaling pathways is modulated by cholinergic activation. Cholinergic activation, by promoting endocannabinoid signaling, allows for domination of LTD pathway over the LTP pathway during pre–post pairs, thus converting a Hebbian synaptic plasticity rule to an anti-Hebbian one.
While it is well established that STDP varies among different brain regions (Caporale and Dan, 2008) and even among different cell-types within the same nucleus (Tzounopoulos et al., 2004), few studies have addressed mechanisms of STDP modulation in one neuron (Seol et al., 2007; Shen et al., 2008). In the visual cortex, cholinergic and β-adrenergic modulation control the polarity of STDP in single cells by phosphorylating AMPARs at sites that serve as specific tags for LTP and LTD (Seol et al., 2007). According to this mechanism, the phosphorylation status of postsynaptic AMPARs limits the magnitude of LTP and LTD, and the relative activation of mAChRs determines the polarity of STDP through phosphorylation of postsynaptic AMPARs. Contrary to a pure postsynaptic mechanism of modulation, our findings indicate that mAChR activation enhances endocannabinoid signaling and thus allows for domination of presynaptic LTD over postsynaptic LTP. In addition, while physiological activation of mAChRs enhances LTP in mouse hippocampal slices (Shinoe et al., 2005) and induces LTD in NMDAR EPSC via a postsynaptic mechanism modulating NMDAR endocytosis (Jo et al., 2010), our study reveals, for the first time, conversion of Hebbian LTP to anti-Hebbian LTD by synaptically released ACh.
Conceptually similar results have been reported in the striatum, where dopamine has been shown to modulate the polarity of STDP (Kreitzer and Malenka, 2008; Surmeier et al., 2009). In principal striatal medium spiny neurons (MSNs), D1 and D2 dopamine receptors interact with adenosine receptors, NMDARs, mGluRs, and Cav1.3 channels in establishing bidirectional Hebbian STDP. Following depletion of dopamine, pairing of presynaptic and postsynaptic activity—regardless of the order—induces LTP in D2-expressing MSNs and LTD in D1-expressing MSNs. These results suggest that dopamine shapes Hebbian synaptic plasticity that can be converted in anti-Hebbian when DA is depleted (Shen et al., 2008). The conceptual similarity between cholinergic and dopaminergic modulation suggests that similar principles organize the effects of these two major neuromodulatory systems on LTP and LTD. According to these principles, in many areas where LTP and LTD are governed by “opponent processes” that interact at presynaptic and postsynaptic sites and determine the size of the synaptic response. Acetylcholine and dopamine may determine the final outcome of induced plasticity by enhancing or inhibiting these opposing processes.
Interaction between endocannabinoid signaling and modulatory systems
Neuromodulatory systems usually exert their effect by controlling the release of endocannabinoids. Activation of some types of Gq-coupled receptors promotes endocannabinoid release from the dendrites of cells throughout the brain (Kim et al., 2002; Hirasawa et al., 2004; Haj-Dahmane and Shen, 2005; Narushima et al., 2007; Oliet et al., 2007; Best and Regehr, 2008; Kola et al., 2008). While several studies have established that pharmacological activation of mAChRs modulates endocannabinoid-mediated short-term plasticity (Kim et al., 2002; Ohno-Shosaku et al., 2003; Narushima et al., 2007), the effect of mAChRs in modulating endocannabinoid-mediated long-term plasticity remains unknown. Our study establishes that activation of M1/M3 mAChRs and, presumably, NMDAR-mediated rises in postsynaptic Ca2+ enhance endocannabinoid release and mediate anti-Hebbian LTD induction. Given the dependence of mAChR-mediated endocannabinoid release on postsynaptic NMDAR activation and elevations of postsynaptic Ca2+, we suggest that the lack of effect of synaptic activation of mAChRs on synaptic responses is probably due to limited Ca2+ rise in the postsynaptic cell (Fig. 2c). Although Ca2+-assisted, receptor-driven endocannabinoid release (RER) has been implicated in short-term plasticity (Varma et al., 2001; Kim et al., 2002), here we report a novel mechanism that links NMDARs, postsynaptic Ca2+, mAChRs, and endocannabinoid release and shapes associative long-term, synaptic plasticity.
Functional role of Hebbian LTP and anti-Hebbian LTD
Recent studies in mice lacking M1, M3, or M5 mAChRs displayed normal auditory brainstem responses (Maison et al., 2010). However, these studies assess baseline auditory brainstem response, and based on our results, M1 and M3 mAChRs are expected to shape auditory response during learning or during adaptive processing (Tzounopoulos and Kraus, 2009). Our results indicate that activation of cholinergic inputs converts Hebbian LTP to anti-Hebbian LTD in the DCN, and thus this transition may be involved in associative mediated learning or adaptation. Hebbian STDP has been linked with associative forms of learning such as the creation a memory traces that sensitizes the circuit to particular profiles of subsequent sensory stimuli (Yao and Dan, 2001) and with training and deprivation-induced receptive field plasticity in sensory cortex (Feldman and Brecht, 2005; Dan and Poo, 2006). Anti-Hebbian STDP has been observed in different brain areas (Bell et al., 1997; Fino et al., 2005). Anti-Hebbian STDP has been linked with the prediction and the correction of motor errors and motor learning in the cerebellum (Ohyama et al., 2003) and with the cancelation of predictable sensory consequences of the organism's own motor actions or the cancelation of redundant, spatially diffused sensory inputs in the cerebellar-like structures of the electric fish (Bell et al., 2008; Harvey-Girard et al., 2010). Therefore, Hebbian synaptic learning rules may create a memory trace (Yao and Dan, 2001) that sensitizes the DCN to particular profiles of subsequent auditory and nonauditory stimuli, while anti-Hebbian may be responsible for constructing negative images of any ongoing auditory activity that is correlated with parallel fiber activity. Information about the position of the head and neck relayed by parallel fibers may provide the “raw material” that would be needed to cancel predictable consequences of movements on auditory input. Also, anti-Hebbian plasticity may aid in responding to novel sounds by suppressing the response to self-generated or expected sounds. Interaction of cholinergic inputs with endocannabinoid signaling is expected to provide the cellular mechanism underlying activity-dependent transition from computational tasks that create a memory trace (positive image) to computational tasks that lead to the cancellation of ongoing predictable sensory activity (negative image).
Footnotes
-
This work was supported by National Institutes of Health–National Institute on Deafness and Other Communication Disorders Grant R01 DC-007905 to T.T. We thank Dr. E. Aizenman for helpful discussions and for critical reading of the manuscript.
- Correspondence should be addressed to Thanos Tzounopoulos, Department of Otolaryngology, University of Pittsburgh School of Medicine, Biomedical Science Tower 3, 3501 Fifth Avenue, Room 10021, Pittsburgh, PA 15261. thanos{at}pitt.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}