

Abstract
The release of dopamine from terminals in the NAc is regulated by a number of factors, including voltage-gated ion channels, D2-autoreceptors, and nAChRs. Cholinergic interneurons (CINs) drive dopamine release through activation of nAChRs on dopamine terminals. Using cyclic voltammetry in mouse brain slices, nAChR-dependent spontaneous dopamine transients and the mechanisms underlying the origin were examined in the NAc. Spontaneous events were infrequent (0.3 per minute), but the rate and amplitude were increased after blocking Kv channels with 4-aminopyridine. Although the firing frequency of CINs was increased by blocking glutamate reuptake with TBOA and the Sk blocker apamin, only 4-aminopyridine increased the frequency of dopamine transients. In contrast, inhibition of CIN firing with the μ/δ selective opioid [Met5]enkephalin (1 μm) decreased spontaneous dopamine transients. Cocaine increased the rate and amplitude of dopamine transients, suggesting that the activity of the dopamine transporter limits the detection of these events. In the presence of cocaine, the rate of spontaneous dopamine transients was further increased after blocking D2-autoreceptors. Blockade of muscarinic receptors had no effect on evoked dopamine release, suggesting that feedback inhibition of acetylcholine release was not involved. Thus, although spontaneous dopamine transients are reliant on nAChRs, the frequency was not strictly governed by the activity of CINs. The increase in frequency of spontaneous dopamine transients induced by cocaine was not due to an increase in cholinergic tone and is likely a product of an increase in detection resulting from decreased dopamine reuptake.
SIGNIFICANCE STATEMENT The actions of dopamine in the NAc are thought to be responsible for endogenous reward and the reinforcing properties of drugs of abuse, such as psychostimulants. The present work examines the mechanisms underlying nAChR-induced spontaneous dopamine release. This study demonstrates that spontaneous dopamine release is (1) dependent of the activation of nicotinic receptors, (2) independent on the spontaneous activity of cholinergic interneurons, and (3) that cocaine increased the detection of dopamine transients by prolonging the presence and increasing the diffusion of dopamine in the extracellular space. The release of acetylcholine is therefore responsible for spontaneous dopamine transients, and cocaine augments dopamine tone without altering activity of cholinergic interneurons.
Introduction
Dopamine release throughout the striatum, including the NAc, signals the occurrence of salient unpredicted cues and cues with learned associations for reinforcing stimuli, including natural and drug rewards (Liljeholm and O'Doherty, 2012; Berridge and Kringelbach, 2015). Local striatal cholinergic interneurons (CINs) can initiate dopamine release through activation of nAChRs on dopamine terminals (Cachope et al., 2012; Threlfell et al., 2012). CINs fire tonically (Wilson et al., 1990) and may be responsible for previously identified dopamine transients in striatal slices (Zhou et al., 2001) and in vivo (Heien et al., 2005; Cachope et al., 2012). However, because of the infrequent incidence and low amplitude of dopamine transients, few studies have verified their origin or mechanistic underpinnings.
Cocaine increases extracellular dopamine concentrations through inhibition of the dopamine transporter (DAT) (Ross and Renyi, 1967; Roberts et al., 1977; Pettit and Justice, 1989; Yorgason et al., 2011b). The increase in extracellular dopamine activates D2-autoreceptors on dopamine cell bodies within the midbrain (Gantz et al., 2013) and terminals in the NAc to inhibit dopamine cell firing (Einhorn et al., 1988) and terminal release (Adrover et al., 2014). Given this potent mechanism of feedback inhibition, it is surprising that studies measuring dopamine release in vivo consistently report cocaine-induced increases in dopamine transients (Heien et al., 2005; Cheer et al., 2007; Wightman et al., 2007; Aragona et al., 2008; Owesson-White et al., 2009). This suggests that, despite D2-mediated feedback inhibition, dopamine release persists due to continued excitatory activity on dopamine terminals. Previously, the firing rate of CINs was shown to increase upon cocaine application (Witten et al., 2010). Therefore, increased CIN activity may underlie cocaine-induced increases in dopamine transients.
The current study examined mechanisms underlying CIN-dependent dopamine transients in the NAc and the role of CINs in cocaine-induced increases in detection of dopamine transients. Kv channel blockade increased dopamine transient frequency. Transients were blocked by TTX and nAChR antagonism, supporting a role for CIN activity. Manipulations that increased the activity of CINs did not always increase the incidence of dopamine transients, whereas the inhibition of CINs always decreased detection. Cocaine increased the rate and amplitude of spontaneous dopamine transients but did not affect CIN firing or acetylcholine release. The present results indicate that cocaine-induced increases in spontaneous dopamine transients measured with voltammetry are likely mediated by an increase in detection resulting from decreased dopamine reuptake.
Materials and Methods
Animals.
Male and female C57BL/6J (The Jackson Laboratory) mice were given ad libitum access to food and water and maintained on a 12:12 h light/dark cycle. All protocols and animal care procedures were in accordance with the National Institutes of Health Guide for the care and use of laboratory animals and approved by the Oregon Health and Science Institutional Animal Care and Use Committee.
Brain slice preparation and drug application.
Isoflurane (Patterson Veterinary) anesthetized mice (>30-d-old) were killed by decapitation, and brains were rapidly removed, sectioned into 220-μm-thick coronal or sagittal striatal slices (Leica VT1000S, Vashaw Scientific), and incubated for 60 min at 34°C in preoxygenated (95% O2/5% CO2) aCSF consisting of (in mm) as follows: 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 21.4 NaHCO3, and 11 d-glucose. Cutting solution also contained either MK801 (0.01 mm; (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine; Abcam) or kynurenic acid (2 mm) for blockade of ionotropic glutamate receptors. At the end of the incubation period, the tissue was transferred to aCSF (34°C) without glutamate receptor blockers. Spontaneous dopamine transients were observed in both coronal and sagittal slices. The following concentrations of drugs (acquired from either Sigma-Aldrich or Tocris Bioscience) were bath applied for slice voltammetry and electrophysiology experiments where specified: 4-aminopyradine (4AP; 30, 100 μm), apamin (100 nm), bestatin (10 μm), cocaine (1–3 μm), dl-threo-β-benzyloxyaspartic acid (TBOA; 100 μm), DNQX (10 μm), hexamethonium (200 μm), JNJ-16259685 (JNJ; 600 nm), [met5] enkephalin (ME; 1 μm), MPEP HCl (MPEP; 300 nm), muscarine (3 μm), reserpine (1 μm), RS-CPP (CPP; 10 μm), scopolamine (1 μm), sulpiride (600 nm), TTX (1 μm), and thiorphan (1 μm).
Voltammetry recordings.
Slices were transferred to the recording chamber and perfused with aCSF (34°C) at a rate of ∼1.8 ml/min. Fast scan cyclic voltammetry recordings were performed and analyzed using Demon Voltammetry and Analysis software (Yorgason et al., 2011a) (RRID:SCR_014468). Carbon fiber electrodes used in voltammetry experiments were made in-house. The carbon fiber (7 μm diameter, Thornel T-650, Cytec) was aspirated into a borosilicate glass capillary tube (TW150, World Precision Instruments). Electrodes were then pulled on a P-87 Horizontal pipette puller (Sutter Instruments) and cut so that 100–150 μm of carbon fiber protruded from the tip of the glass. The electrode potential was linearly scanned as a triangular waveform from −0.4 to 1.2 V and back to −0.4 V (Ag vs AgCl) with a scan rate of 400 V/s (Yorgason et al., 2011a). Before inserting the carbon fiber electrode into the brain slice, the voltage ramp was applied every 16 ms (60 Hz) for a 5–10 min period. After this electrode conditioning period, the scanning interval was changed to 100 ms (10 Hz). If electrodes had low electrical noise (median SD < 0.07 nA averaged across six 1 s bins), then they were used for experiments. Carbon fibers were advanced completely into the tissue at a 20° angle with the tip positioned ∼85 μm below the slice surface. Dopamine release was evoked through electrical stimulation (1 pulse/min) via a glass micropipette (30 μA, monophasic+, 0.5 ms). Paired pulse stimulations were performed at 0.5, 1, 2, 4, 8, 12, and 16 Hz. Spontaneous dopamine transients were measured in between electrical stimulations. Dopamine transients were observed throughout the dorsal and ventral striatum in coronal and sagittal slices. However, the present work focused solely on release in the NAc core, and no direct regional comparisons were made. For experiments examining the effects of TBOA on spontaneous dopamine release, JNJ and MPEP were included in the bath to prevent possible mGluR effects on dopamine release (Zhang and Sulzer, 2003).
Electrophysiology recordings.
Cell-attached recordings were made with glass micropipettes (1.5–2.5 mΩ) filled with aCSF. CINs were identified based on their size, tonic firing, and sensitivity to muscarine. Drugs were applied for 4–6 min. Data were filtered at the amplifier with a 4-pole high-pass Bessel filter at 2–10 kHz (Axopatch 1D; Molecular Devices). Recordings were sampled continuously at 1 kHz with a Powerlab 2/25 digitizer (Chart 5.4.2; AD Instruments) and at 10 kHz using Axograph 1.4.2 (Axograph Scientific).
Data analysis.
Dopamine release was analyzed using Demon Voltammetry software. Dopamine release was measured at peak oxidation currents. Dopamine uptake was measured as the time constant (τ) from a single exponential fit between the peak dopamine current and its return to baseline (Yorgason et al., 2011a). Paired pulse experiments, where the dopamine from the paired stimulation overlapped, used a subtraction method where the dopamine release from a single pulse was subtracted from the paired stimulation to measure the release induced by the second pulse. The paired pulse ratio was calculated as the amplitude of this subtracted pulse over the amplitude of the signal from a single pulse. Custom software was written for detecting and analyzing spontaneous dopamine release events. The software performs a running subtraction on recordings to reduce drift and aliasing noise. Postsubtracted data are then compared across time against known cyclic voltammograms for dopamine, with a low threshold r2 value for initial detection (r2 > 0.3). The location of each putative event (legitimate and spurious) is captured in the software for subsequent manual verification. The program reverts the data back to its nonsubtracted form and performs a new background subtraction (nonrunning) at the time point preceding the putative event. The resultant color plot around that time point, and cyclic voltammogram at the peak current are then examined, and compared against a known dopamine voltammogram to verify similar oxidation potentials. If signals are smaller than the limit of detection (calculated by multiplying the median SD for each file by 3) (Keithley et al., 2011), they are automatically rejected. The events are simultaneously examined for evidence of a false positive caused by drift and aliasing noise coinciding with the oxidation potential. Dopamine concentrations were calculated from calibration values (at 1 μm dopamine). Baseline frequency of dopamine transients was measured during a 10 min period before drug application. This time span was reduced to a 3 min period for evoked dopamine release experiments where stability was determined. Group data from experiments where a drug was applied (evoked and spontaneous dopamine release) were measured across a 3 min period where drug effect stability was determined with evoked release (usually 4–6 min, but up to 60 min for reserpine). For cell-attached electrophysiology recordings, firing rates were analyzed in Axograph 1.4.2. Firing rates for baseline and postdrug conditions were measured across a 3 min period.
Statistics were performed using Prism 5 (GraphPad) and NCSS 8 (NCSS). Statistical significance was determined for groups of 2 variables using a two-tailed Student's t test. Experiments with >2 groups, but only one factor, were tested for significance using a one-way ANOVA; or when data were from multiple time points from the same experiment, a repeated-measures ANOVA was performed. For experiments that examined multiple factors, and possible interactions, two-way ANOVA or repeated-measures ANOVAs were used. Tukey's HSD and Bonferroni correction methods were used for post-ANOVA analysis.
Results
Dopamine release: electrically evoked and spontaneous
Fast scan cyclic voltammetry was used to detect extracellular dopamine in coronal slices of the NAc (Fig. 1A,B). Electrical stimulation (1 pulse, 0.5 ms) resulted in a reliable dopamine release. In addition, unstimulated (spontaneous) dopamine release was also measured, although signals were much smaller in amplitude (Fig. 1C; evoked: 645 ± 35 nm; spontaneous: 21 ± 3 nm; two-tailed unpaired t test: t(37) = 17.93, p < 0.0001). Thus, spontaneous release likely represents activity from fewer release sites than evoked release. Example cyclic voltammograms from electrically evoked and spontaneous dopamine release events are shown in Fig 1D–F.

Spontaneous and electrically evoked dopamine release in NAc brain slices. A, Voltammetric recording of spontaneous and electrically evoked dopamine release events. Insets, Cyclic voltammograms at indicated peaks, showing similar peak oxidation potentials for spontaneous and evoked dopamine release. B, Background subtracted color plot for traces shown in A. C, Evoked release amplitude is greater than spontaneous dopamine release (unpaired two-tailed t test). D–F, Example cyclic voltammogram traces of evoked (black) and spontaneous (red and magenta) dopamine release from three different experiments. Respective y-axis scales are shown in black (evoked) and red (spontaneous). Bar plots represent mean. Error bars indicate SEM. ***p < 0.001.
In control conditions, spontaneous transients in coronal slices occurred at low frequencies (0.3 ± 0.08 events/min). To investigate whether voltage-gated potassium (Kv) channels regulate dopamine release, the nonselective Kv channel blocker 4AP was applied. The frequency of spontaneous dopamine transients was increased by 4AP from 0.28 ± 0.11 to 6.69 ± 0.78 events/min at 30 μm and 0.35 ± 0.12 to 10.08 ± 1.59 events/min at 100 μm 4AP [30–100 μm, Figure 2A,B; two-way repeated-measures ANOVA (4AP × concentration); 4AP: F(1,10) = 112.6, p < 0.0001; concentration: F(1,10) = 4.729, p = 0.0547; interaction: F(1,10) = 4.767, p = 0.0539]. In addition, 4AP increased the mean amplitude by 100%–340% [30–100 μm; Figure 2C; two-way ANOVA (4AP × concentration); 4AP: F(1,295) = 6.08, p = 0.014; concentration: F(1,295) = 1.87, p = 0.173; interaction: F(1,295) = 2.06, p = 0.152]. Electrically evoked release was examined to determine whether 4AP had a direct action on dopamine terminals as opposed to an increase in ACh release from CINs (Fig. 2D–F). The nAChR antagonist hexamethonium (200 μm) reduced evoked dopamine release to ∼15% of predrug values (Fig. 2E; one-way repeated-measures ANOVA: F(2,17) = 81.41, p < 0.0001). In the presence of hexamethonium, 4AP (30 μm) increased evoked release amplitude to ∼48% of baseline (Fig. 2F). Thus, Kv channel blockade directly increased dopamine terminal release, facilitating detection of spontaneous dopamine transients.

Spontaneous dopamine release is increased by 4AP. A, NAc spontaneous dopamine release (¤) increases in the presence of 4AP (30–100 μm). B, C, Group data (mean ± SEM) showing increases in spontaneous release frequency (B), and release amplitude (C) after 4AP application (Tukey's post test). D, Evoked dopamine release is reduced by hexamethonium (Hex, 200 μm) and increased by 4AP (30 μm). E, Normalized group data (mean ± SEM) from experiments described by D, showing time course of drug effects on evoked dopamine release. F, Group average and individual peak release data showing the excitatory effects of 4AP on evoked dopamine release (Tukey's post test; baseline removed for clarification). *p < 0.05. ***p < 0.001.
Spontaneous and evoked dopamine release is vesicular and action-potential dependent
The vesicular monoamine transporter inhibitor reserpine (1 μm; in 30–100 μm 4AP) was applied to deplete dopamine vesicular stores (Shore, 1966). Electrically evoked and spontaneous dopamine transients were both reduced by reserpine (Fig. 3A,B; evoked: one-way repeated-measures ANOVA: F(59,236) = 57.49, p < 0.0001; spontaneous: one-way repeated-measures ANOVA: F(4,12) = 18.76, p < 0.0001). The voltage-gated sodium channel blocker TTX (1 μm; in 30–100 μm 4AP) blocked evoked dopamine release (two-tailed paired t test: t(4) = 43.26, p < 0.0001) and reduced the frequency of dopamine transients (Fig. 3C,D; two-tailed unpaired t test: t(5) = 4.724, p = 0.0052). Thus, both evoked and spontaneous dopamine transients are dependent on vesicular packaging and action potential-dependent mechanisms.

Spontaneous dopamine release is vesicular and action potential dependent. A, Example traces of spontaneous dopamine release (¤) from voltammetry experiments (in 30–100 μm 4AP) before and after reserpine (1 μm) application. B, Group data (mean ± SEM) showing the time course of effects of reserpine-mediated depletion on evoked dopamine release amplitude and spontaneous (Spon) release frequency. C, Traces depicting spontaneous dopamine release (in 30–100 μm 4AP) before and after TTX (1 μm). D, Time course for overlaid group data (n = 6) from TTX experiments.
Dopamine release is dependent on nicotinic receptor activity
CINs are tonically active and are thought to underlie the spontaneous dopamine release events observed in slices of dorsal striatum (Zhou et al., 2001). In the NAc, hexamethonium (200 μm; in 30–100 μm 4AP) reduced the frequency of dopamine transients that fully reversed upon wash (Fig. 4A,C; one-way repeated-measures ANOVA: F(2,18) = 33.14, p < 0.0001). Electrically stimulated dopamine release was also inhibited by nAChR blockade (Fig. 2E). However, in the presence of 4AP, hexamethonium had mixed effects: typically attenuating (Fig. 4B), but sometimes enhancing evoked dopamine release (Fig. 4D,E). The variability in the action of hexamethonium was associated with the frequency of spontaneous dopamine events measured before the application of hexamethonium (Fig. 4D). In experiments where the frequency of dopamine transients was high, hexamethonium facilitated evoked release, whereas when the frequency of dopamine transients was low, hexamethonium caused a further decrease (r = 0.8111, n = 7, p = 0.0268). The interpretation of this observation was that spontaneously released dopamine driven by ACh tone activated D2-autoreceptors. Blocking the ACh tone removed the D2-auoreceptor activation, resulting in greater electrically stimulated release. Although this statistical association could also result from other mechanisms, the results show that activation of nAChR underlying spontaneous dopamine transients can also regulate electrically stimulated dopamine release.

Nicotinic receptor activation mediates spontaneous dopamine release. A, Example of spontaneous dopamine release (¤) before and after application of the nicotinic receptor antagonist hexamethonium (Hex, 200 μm). B, Raw evoked dopamine trace (in 4AP) showing hex-induced reduction in dopamine release. C, Group data (mean ± SEM) of spontaneous dopamine release frequency. Hex application reduced spontaneous release frequency (Tukey's post test). D, Group data (mean ± SEM) showing effects of hex on evoked dopamine release (in 30–100 μm 4AP). Evoked release is typically reduced by hex but occasionally enhanced. E, Pearson correlation and linear regression of hex data in D (y-axis) compared with prehex spontaneous release frequency (x-axis). In experiments where baseline spontaneous release frequency was high, hex increased evoked release. ***p < 0.001.
Modulation of cholinergic activity and spontaneous dopamine transients
Cell-attached recordings were used to study the association between CIN firing and spontaneous dopamine transients (Fig. 5). The firing of CINs was increased or decreased pharmacologically while measuring dopamine transients. Application of 4AP (30–100 μm) resulted in an increase in the firing rate of CINs (Fig. 5A,B; 4–6 min, mean firing normalized to baseline, 30 μm: 210 ± 45%, 100 μm: 316 ± 93%, one-way repeated-measures ANOVA: F(2,14) = 11.11, p = 0.0013, Tukey's post hoc p < 0.05 for baseline vs 100 μm) and an increase in spontaneous dopamine transients (Fig. 5C; one-way repeated-measures ANOVA: F(19,57) = 5.057, p < 0.0001). Blockade of excitatory amino acid transporters with TBOA (100 μm) increased the firing of CINs (222 ± 35%) within 5–6 min, which was reversed with DNQX+CPP (10 μm, Fig. 5D,E; 127 ± 19%, one-way repeated-measures ANOVA: F(2,10) = 8.822, p = 0.0062, Tukey's post hoc, p < 0.05 for baseline vs TBOA and TBOA vs DNQX+CPP). However, TBOA did not change the frequency of dopamine transients (Fig. 5F; one-way repeated-measures ANOVA: F(29,203) = 0.8543, p = 0.6835). CINs express calcium-activated small conductance potassium channels (Sk) that maintain a pacemaker pattern at a low frequency (Goldberg and Wilson, 2005). Blocking Sk channels with apamin (100 nm) promoted a shift in cholinergic firing, from low-frequency tonic firing to burst firing (Fig. 5G,H; 1.4 ± 0.2 Hz, with 13.3 ± 5.3 APs in a burst, one-tailed paired t test, t(7) = 2.305, p = 0.0273). Despite the pronounced effect on CIN firing within 1–2 min, apamin had no apparent effect on dopamine transient frequency (Fig. 5I; one-way repeated-measures ANOVA: F(20,120) = 0.9976, p = 0.4703). Next, the frequency of dopamine transients was examined following inhibition of CIN firing. The μ/δ-opioid peptide agonist ME (1 μm) inhibited CIN firing within the first 2 min (Fig. 5J,K; one-way repeated-measures ANOVA: F(2,10) = 9.755, p = 0.0045, Tukey's post hoc, p < 0.05 for baseline and wash vs ME). Inhibition of CIN firing was variable (baseline coefficient of variation = 0.635 vs ME coefficient of variation = 1.178) in that the inhibition was transient in some neurons. On average, the firing rate was decreased to 10 ± 7.5% (after 2–3 min) predrug values and returned after 3–4 min wash (117 ± 31%). The frequency of dopamine transients was also reduced by ME (Fig. 5L; one-way repeated-measures ANOVA: F(2,18) = 6.269, p = 0.0086). Complete inhibition of dopamine transients was temporary, although there was some inhibition throughout the ME application. The prolonged inhibition of ME on dopamine transients was most likely due to additional effects of opioid receptors on CIN terminals. Importantly, ME-mediated inhibition of dopamine release is likely through direct effects on CINs because μ/δ-opioid receptor agonists are abundantly expressed in CINs, and nAChR-dependent effects on evoked dopamine release are precluded with saturating μ/δ-opioid receptor selective agonists (Britt and McGehee, 2008). Therefore, inhibition of CIN firing decreased spontaneous dopamine transients, whereas excitation of CINs did not always increase dopamine transients.

Relationship between cholinergic firing and spontaneous dopamine release. A, Cell-attached recording of CIN firing before and after 4AP (100 μm), depicting typical increased firing after of Kv channel blockade. B, Firing rates (mean ± SEM) of CINs under baseline (BL) conditions and in the presence of 4AP (30, 100 μm). C, Group data (mean ± SEM) from voltammetry experiments measuring the excitatory effects of 4AP (100 μm) on dopamine transient frequency. D, Cell-attached recordings demonstrating excitatory effects of excitatory amino acid transporter blockade via TBOA (100 μm) on cholinergic firing. TBOA-induced increases in firing are reversed by the ionotropic glutamate receptor antagonists DNQX+CPP (10 μm). E, Firing rates (mean ± SEM) of CINs under BL conditions, in TBOA (100 μm) and in DNQX+CPP (10 μm) mixture. F, Dopamine transient frequency (mean ± SEM) from voltammetry experiments was unchanged by TBOA. G, Cell-attached recordings of cholinergic interneuron firing showing the effects of apamin (100 nm) application. Apamin increases burst firing in cholinergic interneurons. H, Firing rates (mean ± SEM) of CINs under BL conditions and in apamin (100 nm). I, Time course of dopamine transient frequency (mean ± SEM) before and after apamin application. J, Cholinergic firing is transitorily inhibited by the opioid ME (1 μm). K, Firing rates (mean ± SEM) of CINs under BL, ME (1 μm), and post-ME conditions. L, Dopamine transient frequency (mean ± SEM) is reduced by ME application. *p < 0.05. **p < 0.01. ***p < 0.001.
DAT blockade increased spontaneous dopamine transients
Dopamine release is spatially restricted by the DAT (Torres et al., 2003; Ford et al., 2010). Therefore, blocking dopamine uptake may increase detection of ongoing release events that are normally confined to the synapse and undetected by the voltammetry probe. The monoamine transport blocker cocaine (1–3 μm) enhanced electrically evoked (1 pulse) dopamine release (1 μm: 151 ± 5%, one-way repeated-measures ANOVA: F(2,10) = 92.59, p < 0.0001; 3 μm: 161 ± 4%, one-way repeated-measures ANOVA: F(2,10) = 92.97, p < 0.0001). The evoked dopamine signal was further potentiated by the D2 antagonist sulpiride (600 nm; 1 μm Coc+Sulp: 169 ± 4%; 3 μm Coc+Sulp: 189 ± 7%), indicating that cocaine increased dopamine tone, resulting in the activation of terminal autoreceptors. The action of cocaine (1 μm) and sulpiride (600 nm) on the frequency and amplitude of spontaneous dopamine transients was examined. Cocaine increased the frequency of spontaneous dopamine events, an action that was further enhanced with the addition of sulpiride (Fig. 6A–C; one-way repeated-measures ANOVA: F(2,16) = 28.5, p < 0.0001). The amplitude of the spontaneous release events was increased by cocaine (Fig. 6D; one-way ANOVA: F(2,538) = 3.648, p = 0.0267) and sulpiride did not cause a further increase in amplitude. Cocaine increased the clearance time (τ) of evoked and transient dopamine release (Fig. 6E,F), with no significant difference between evoked and transient release clearance [two-way ANOVA (cocaine × release type): cocaine: F(1,54) = 22.03, p < 0.0001; release type: F(1,54) = 0.009159, p = 0.9241; interaction: F(1,54) = 0.944, p = 0.3356]. Thus, by decreasing dopamine uptake, the frequency of dopamine transients was facilitated. The further increase in the frequency of dopamine transients induced by sulpiride suggests that increased dopamine levels from cocaine block of dopamine reuptake recruits D2-autoreceptors that reduced spontaneous release.

Cocaine increases spontaneous dopamine release. A, Raw voltammetric traces showing the effects of cocaine (1 μm) and sulpiride (600 nm) on spontaneous dopamine release (¤). B, Time course from an experiment where cocaine and sulpiride increased frequency and amplitude of spontaneous dopamine release events. C, Group data (mean ± SEM) of spontaneous dopamine release frequency under baseline (BL) conditions and in the presence of cocaine (Coc) and sulpiride (Sulp; Tukey's post test). D, Spontaneous release amplitude was increased by cocaine and sulpiride application (Tukey's post test). E, Averaged normalized traces from evoked (top) and spontaneous (bottom) dopamine release events (n = 20 for each). F, Clearance rates (τ) for evoked and spontaneous (Spon) release events before and after cocaine (Bonferroni post test). *p < 0.05. **p < 0.01. ***p < 0.001.
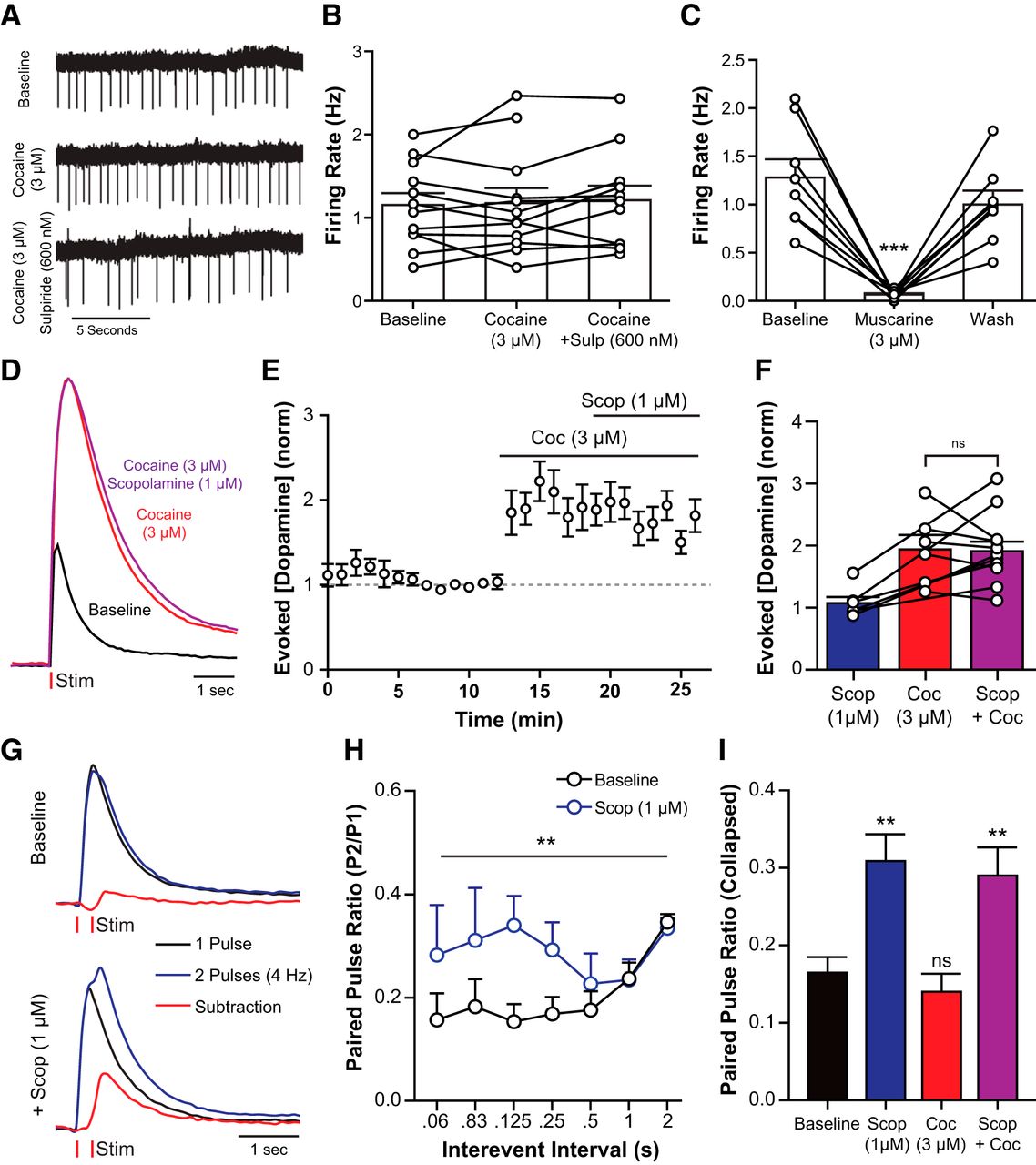
Cocaine did not change the activity of cholinergic neurons
Cocaine may increase spontaneous dopamine release by an action directly on CINs (Witten et al., 2010). The spontaneous activity of CINs was measured using cell-attached recording in the absence and presence of cocaine (3 μm) and cocaine plus sulpiride (600 nm). The rate and pattern of CIN firing were unaffected by cocaine (Fig. 7A,B) or sulpiride (one-way ANOVA: F(2,32) = 0.0296, p = 0.9709). Although CIN firing was stable in the presence of cocaine, it is possible that cocaine increased acetylcholine release onto dopamine terminals independent of CIN firing. Activation of the muscarinic acetylcholine receptor (mAChR) with muscarine (3 μm) inhibited cholinergic firing (Fig. 7C; one-way repeated-measures ANOVA: F(2,14) = 26.69, p < 0.0001). Furthermore, mAChR activation decreased evoked dopamine release (Shin et al., 2015). If there were a cocaine-induced increase in acetylcholine release, one expectation was that there would also be an increase in the activation of mACh autoreceptors that would oppose any further release. This possibility was examined by measuring the action of the mAChR antagonist scopolamine (1 μm) on electrically evoked dopamine release. If there were an increase in acetylcholine tone, the prediction was that scopolamine would increase dopamine release. Scopolamine had no effect in the absence or presence of cocaine (absence, two-tailed paired t test: t(5) = 0.9236, p = 0.3981; presence, Fig. 7D–F; one-way repeated-measures ANOVA: F(2,21) = 6.427, p = 0.0066). Paired pulse stimulation (0.06–2 s interspike interval; in 600 nm sulpiride) was used to increase mAChR activity on dopamine release. Scopolamine resulted in an increase in dopamine release on the second pulse (Fig. 7G,H). The increase in the paired pulse ratio was detected with an interpulse interval of 60–250 ms, with a peak at 125 ms [Figure 7H; two-way ANOVA (scopolamine × interval): scopolamine: F(1,114) = 7.59, p = 0.007; interval: F(6,114) = 0.89, p = 0.50; interaction: F(6,114) = 0.83, p = 0.55]. Unlike the change in paired pulse ratio seen with scopolamine, the paired pulse release ratio was not changed by cocaine (Fig. 7I; one-way ANOVA: F(3,173) = 6.803, p = 0.0002). Thus, it is unlikely that cocaine increased acetylcholine release. Coapplication of scopolamine and cocaine produced an increase of the paired pulse ratio that was similar to scopolamine alone. Therefore, cocaine did not increase CIN firing or activate mAChR in these brain slices.

Cocaine does not affect CIN activity or acetylcholine release in the NAc core. A, Representative traces from cell-attached recordings of a cholinergic interneuron testing the effects of cocaine (3 μm) and sulpiride (600 nm) on firing activity. B, Cholinergic firing rate was not significantly changed by cocaine or sulpiride (Tukey's post test). C, Muscarine application inhibits cholinergic firing (Tukey's post test). D–F, Voltammetry experiments examining muscarinic activity on evoked dopamine release in the presence of cocaine. D, Representative traces showing the effects of cocaine and scopolamine on evoked dopamine release. E, F, Group data (mean ± SEM) from experiments depicted in D. Cocaine increases evoked release, but addition of scopolamine has no apparent effect (Tukey's post test). G, H, Paired pulse stimulations in the NAc can be used to increase muscarinic effects on dopamine release. G, Representative traces for electrically evoked dopamine release with 1 pulse (black), or 2 pulses (blue) separated by 250 ms at baseline (top) and during scopolamine (1 μm) application. Signals from single pulse experiments are subtracted from paired pulse data to unmask the release elicited by the second pulse (red). H, Group data (mean ± SEM) from paired pulse voltammetry experiments showing that scopolamine increases the amount of dopamine elicited from a second pulse (Tukey's post test). I, Group data from experiments depicted in G and H (mean ± SEM) collapsed across interpulse intervals. Scopolamine (1 μm) significantly increased the paired pulse ration. Cocaine (3 μm) had no effect. **p < 0.01. ***p < 0.001.
Discussion
This study examined the mechanisms underlying spontaneous dopamine transients. The results indicate that dopamine transients are dependent on CIN activity. Manipulations that increased dopamine transmission (increased release or decreased uptake) also increased the frequency of spontaneous events. The increase in dopamine transients induced by cocaine likely results from an increase in the spread of dopamine in the extracellular space and not a direct action on CINs.
Optogenetic activation of CINs drives dopamine release through nAChR-dependent depolarization of dopamine terminals (Cachope et al., 2012; Threlfell et al., 2012; Kress et al., 2014; Wang et al., 2014). The inhibition of dopamine release resulting from block of nAChRs following electrical stimulation was dependent on the frequency of stimulation. The inhibition was smaller with low-frequency stimulation and larger at higher frequencies (Rice and Cragg, 2004). The present study found that nAChR blockade increased evoked dopamine release when the frequency of nAChR-dependent dopamine transients was high. The biphasic effect of nAChR antagonists on evoked release is generalizable to other receptors that inhibit dopamine release, including D2 and GABAB receptors (Pitman et al., 2014). Therefore, the attenuation in evoked release observed during periods of high spontaneous dopamine activity likely represents feedback inhibition, which could include D2 autoreceptors and nAChR desensitization.
Kv channel blockade increases dopamine terminal excitability, increasing spontaneous transients
Kv channels play an important role in restoring membrane potentials during an action potential, and modulating electrical excitability (Jan and Jan, 2012). In dopamine neurons, Kv blockade prolonged action potentials, reduced afterhyperpolarizations, and increased neuronal excitability (Kimm et al., 2015). The results from the present study confirm that blocking Kv channels increased dopamine release (Fulton et al., 2011; Martel et al., 2011) independent of the activity of CINs. Kv blockers are thought to increase transmitter release by prolonging the action potential, recruiting additional voltage-gated calcium channels (Martel et al., 2011), thus increasing the probability of vesicular release. Therefore, 4AP plays two direct roles in increasing dopamine release. First, it increases depolarization propagation efficiency. Second, it increases calcium entry to facilitate vesicle release. Block of Kv with 4AP likely increased acetylcholine release onto dopamine terminals and may facilitate the depolarizing effects of nAChR activity on dopamine terminals.
Spontaneous cholinergic activity is greater than spontaneous dopamine transients
The frequency of dopamine transients in 4AP (6–10 /min) was less than the firing rate of CINs (1–2 Hz). Acetylcholine is released spontaneously onto NAc medium spiny neuron mAChRs at rates similar to CIN firing (Mamaligas and Ford, 2016). Therefore, it is possible that dopamine terminals detect acetylcholine release at higher frequencies, but that ongoing nAChR activity is insufficient to evoke dopamine release. Several lines of evidence support the idea that nAChR-dependent dopamine release is temporally and spatially restricted. For instance, dopamine release measured by voltammetry can be evoked with whole-field optogenetic stimulation of CINs, but not activation of a single CIN (Cachope et al., 2012; Threlfell et al., 2012). This suggests that acetylcholine release from a single neuron does not typically provide enough nAChR activity to elicit dopamine release. Furthermore, high-frequency optogenetic stimulation of CINs produced dopamine release that was similar to that observed with a single pulse (Cachope et al., 2012; Threlfell et al., 2012). Thus, the release of dopamine induced by nAChRs appears to be temporally limited, perhaps by desensitization of nAChRs.
In the present work, both TBOA and apamin increased CIN firing but did not increase the frequency of dopamine transients, further suggesting that timing of dopamine release evoked by CINs is somehow restricted. A number of mechanisms may regulate the ability of nAChRs to evoke dopamine release. First, the membrane properties of dopamine terminals may restrict excitatory activity from nAChRs. Decreasing Kv conductance with 4AP robustly increased spontaneous dopamine transients, possibly through an increase in terminal excitability. Second, the degradation enzyme acetylcholinesterase rapidly terminates acetylcholine signaling and is abundantly expressed in the striatum (Mesulam et al., 2002; Wevers, 2011). Indeed, low concentrations of acetylcholinesterase inhibitors potentiate spontaneous acetylcholine and dopamine release (Zhou et al., 2001; Zhang et al., 2004; Mamaligas and Ford, 2016). Third, dopamine terminals express α4α6β2-containing nAChRs (Jones et al., 2001; Exley et al., 2011), which desensitize quickly and recover slowly from desensitization (Luetje and Patrick, 1991; Fenster et al., 1997). The potent nAChR agonist nicotine and high concentrations of acetylcholinesterase inhibitors reduced evoked and spontaneous dopamine transients (Zhou et al., 2001; Zhang et al., 2004). Fourth, mAChRs on CINs act as autoreceptors to decrease ACh release and thereby decrease nAChR-dependent dopamine release (Shin et al., 2015). Dopamine release from paired pulse stimulation was reduced via mAChRs, suggesting that these receptors may be differentially activated during burst firing and tonic firing that is the hallmark of CIN activity. Because pauses and rebound bursts in CIN firing are involved in encoding associative learning (Brown et al., 2012), it is tempting to speculate that pauses in CIN activity are optimized to prime dopamine terminals for sensing a “burst” in acetylcholine activity. Together, the frequency of dopamine transients driven by CINs is most likely lower than the frequency CINs due to a requirement for coincident activation of dopamine release.
Cocaine increased detection of spontaneous dopamine transients
Dopamine diffusion in the striatum is tightly regulated by the DAT (Torres et al., 2003; Ford et al., 2010). Cocaine-induced increases in the frequency and amplitude of dopamine transients were not the result of increases in CIN activity. Therefore, the increased rate of transients after cocaine likely reflects ongoing release that is undetectable in control conditions. Dopamine release from a single electrical pulse is insensitive to D2 autoreceptor antagonism, suggesting minimal “tone” during baseline conditions (Kennedy et al., 1992; Phillips et al., 2002). However, cocaine (1–10 μm) reduced dopamine uptake sufficiently to produce D2-dependent feedback inhibition (Adrover et al., 2014). Importantly, cocaine is also thought to increase mobilization of synapsin-tethered vesicles (Venton et al., 2006; Kile et al., 2010), which could further increase the release, the spread of dopamine, and the activation of more D2 receptors that would decrease release.
Cocaine and cholinergic interneuron interactions
Dopamine activity on striatal CINs produces diverse effects that are regionally dependent. In the dorsal striatum, and to a smaller extent in the NAc core, CINs are hyperpolarized through D2 receptor activation (Chuhma et al., 2014; Straub et al., 2014). On the other hand, CINs in the NAc shell are excited during dopamine release through a dopamine-independent mechanism (Chuhma et al., 2014). Because cocaine did not significantly affect CIN firing in the NAc core, perhaps a higher concentration of cocaine is needed to affect the activity of NAc CINs. Interestingly, cocaine also blocks nAChRs with an IC50 on release of ∼4 μm (Acevedo-Rodriguez et al., 2014). Therefore, higher cocaine concentrations would likely reduce dopamine transients. Considering the regional differences in CIN dopamine sensitivity, it would be informative to examine the action of cocaine on dopamine transients across regions of the striatum. Interestingly, nAChR blockers delivered locally to the NAc attenuate cocaine-induced dopamine increases (Zanetti et al., 2007) and cocaine self-administration (Mark et al., 1999). Therefore, the increases in spontaneous dopamine transients observed after cocaine in vivo likely involve CIN-initiated dopamine release and appear to contribute to the reinforcing effects of cocaine.
In conclusion, increasing dopamine terminal excitability results in higher rates of dopamine transients. Dopamine transients are dependent on nAChRs, and inhibition of CINs decreased the frequency of transients. However, increasing CIN activity did not always increase the frequency of dopamine transients. By limiting its reuptake, cocaine decreases the spread of dopamine, thus increasing detection of ongoing dopamine transients without affecting CIN activity. The activity of CINs is implicated in neuropsychiatric disorders ranging from movement disorders to psychostimulant addiction, so elucidating the mechanisms of CIN-dependent dopamine release will be important for both understanding their etiology as well as developing better pharmacological treatments.
Footnotes
This work was supported by National Institutes of Health Grants T32 DA DA6297007 and F32 DA040409 to J.T.Y. and Grant R01 DA004523 to J.T.W.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. John T. Williams, Vollum Institute, Oregon Health & Science University, 3181 SW Sam Jackson Park Road, Portland, OR 97239. williamj{at}ohsu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}