

Abstract
Spinal hemisection at C2 reveals caudal synaptic pathways that cross the spinal midline (crossed phrenic pathways) and can restore inspiratory activity in ipsilateral phrenic motoneurons. Intermittent hypoxia induces plasticity in the cervical spinal cord, resulting in enhanced inspiratory phrenic motor output. We hypothesized that chronic intermittent hypoxia (CIH) (alternating 11% O2 and air; 5 min periods; 12 hr per night; 7 nights) would strengthen crossed phrenic pathways. Experiments were performed on anesthetized, vagotomized, paralyzed, ventilated, and spinally injured (C2 hemisection) rats that were exposed to either normoxia or CIH before acute injury (preconditioning) or after chronic injury (postconditioning). Spontaneous inspiratory bursts or compound action potentials evoked via stimulation of the ventrolateral funiculus (contralateral to injury) were recorded in both phrenic nerves. Spontaneous or evoked activity in crossed phrenic pathways were minimal or absent in all acutely injured rats regardless of preconditioning. In rats postconditioned with normoxia, crossed phrenic inspiratory bursts were observed occasionally during baseline conditions and always during chemoreceptor stimulation (hypoxia and hypercapnia). However, CIH postconditioned rats had substantially larger crossed phrenic inspiratory bursts during baseline, hypoxia, and hypercapnia (allp < 0.05 vs normoxic group). Short-latency (0.7 msec) evoked crossed phrenic potentials were also enhanced by CIH conditioning in chronically injured rats (p< 0.05). We conclude that CIH induced spinal cord plasticity-enhanced phrenic motor output. This plasticity required preconditions established by chronic spinal injury.
- plasticity
- crossed phrenic phenomenon
- respiratory control
- spinal cord injury
- C2 hemisection
- intermittent hypoxia
Introduction
Impaired respiratory motor function after spinal cord injury (SCI) is debilitating and life-threatening (Mansel and Norman 1990; Jackson and Groomes, 1994). Fortunately, most spinal injuries are incomplete (Young, 1996). Thus, strengthening intact neural pathways in the injured cervical spinal cord may enhance respiratory motor output. Although pharmacological treatments transiently enhance respiratory motor output after cervical SCI (Nantwi and Goshgarian, 2001), an alternative approach is to induce a long-lasting enhancement of existing respiratory neural pathways through endogenous mechanisms (i.e., plasticity).
Intermittent hypoxia enhances respiratory motor output, an effect at least partly attributable to cervical spinal plasticity (Baker-Herman et al., 2001; Fuller et al. 2002a). For example, three 5 min hypoxic episodes evoke a long-lasting (>1 hr) enhancement of phrenic motor output (phrenic long-term facilitation) by a mechanism dependent on spinal serotonin receptor activation and protein synthesis (Fuller et al., 2000; Mitchell et al., 2001; Baker-Herman and Mitchell, 2002). Chronic intermittent hypoxia (CIH) induces additional central neural plasticity (Mitchell et al., 2001). CIH enhances the acute hypoxic phrenic response and phrenic long-term facilitation by serotonin-dependent mechanisms (Ling et al., 2001). CIH augments phrenic motor output during carotid sinus nerve stimulation, indicating an underlying plasticity in central respiratory neurons and networks.
In rats, bulbospinal respiratory neurons project axons bilaterally to phrenic motoneurons (see Fig. 1) (Moreno et al., 1992; Dobbins and Feldman 1994; Lipski et al., 1994). Although most crossed pathways decussate in the brainstem, an apparently ineffective synaptic pathway crosses the spinal midline caudal to C2 (i.e., the “crossed phrenic pathway”) (Goshgarian, 1981; Moreno et al., 1992). This pathway can be revealed after C2 spinal hemisection either by increasing respiratory drive chemically or pharmacologically (Nantwi and Goshgarian, 2001) or by activating spinal serotonin receptors (Ling et al., 1994) of the 5-HT2 receptor subtype (Basura et al., 2001; Zhou et al., 2001).
Because intermittent hypoxia elicits serotonin-dependent spinal plasticity and spinal serotonin receptor activation reveals crossed phrenic pathways, we hypothesized that CIH would enhance crossed phrenic pathways in spinally injured (C2 hemisection) rats. Accordingly, one group of rats was preconditioned with CIH before acute SCI. However, SCI dramatically alters spinal function, creating a “new spinal cord” with (potentially) different physiologic mechanisms regulating motor output (Edgerton et al., 2001). Therefore an additional group of rats was conditioned with CIH after chronic SCI. CIH enhanced crossed phrenic pathways, but only when administered after chronic SCI (i.e., CIH preconditioning had no discernable effect). Electrical stimulation of descending ventrolateral funiculus axons suggested that enhanced spontaneous respiratory responses resulted, at least in part, from spinal mechanisms. Thus, CIH-induced spinal plasticity enhances phrenic inspiratory motor output ipsilateral to SCI, and this effect requires preconditions that are satisfied only after SCI.
Portions of these data have been published previously in abstract form (Fuller et al., 2001b, 2002b).
Materials and Methods
All procedures were approved by the Animal Care and Use Committee of the University of Wisconsin School of Veterinary Medicine. Experiments were performed on 3- to 5-month-old male Sprague Dawley rats (n = 58; rat colony K-62; Charles River Laboratories, Kingston, NY) that were housed individually with ad libitum access to food and water. Rats were preconditioned with either normoxia or CIH before acute SCI or exposed to the same conditions after chronic SCI (i.e., postconditioning). Thus, four groups of rats were studied: (1) normoxic preconditioning before acute SCI (spontaneous responses,n = 9; evoked responses, n = 7), (2) CIH preconditioning before acute SCI (spontaneous responses,n = 5; evoked responses, n = 8), (3) normoxic postconditioning after chronic SCI (spontaneous responses,n = 8; evoked responses, n = 8), and (4) CIH postconditioning after chronic SCI (spontaneous responses,n = 8; evoked responses, n = 5).
Spinal cord injury. Before chronic SCI, rats were treated with an analgesic (buprenorphine, 0.1 mg/kg), an anti-inflammatory drug (carprofen, 4 mg/kg), and an antibiotic (enrofloxacin, 5 mg/kg). Isoflurane anesthesia was induced in a closed chamber and maintained (2–3%) via nose cone. After C2 laminectomy and durotomy, the spinal cord was hemisected caudal to the C2 dorsal roots with microscissors. A gap (∼1 mm) at the incision site was then created using a blunt-tipped 25 gauge needle connected to a suction pump. Wounds were sutured and rats were monitored post-hemisection and given daily injections (buprenorphine 0.1 mg/kg; carprofen, 4 mg/kg; enrofloxacin, 5 mg/kg) for 2 d. Chronically injured rats were studied 2 weeks after surgery. The same surgical approach was used in urethane-anesthetized rats for acute SCI, and 1–2 hr were allowed before data collection.
Chronic intermittent hypoxia. For 7 consecutive days, rat cages were placed in a Plexiglas chamber that between 6 P.M. and 6 A.M. was flushed at a flow rate of 2 l/min per rat with an air/O2/N2 mixture to achieve quasi-square wave (45 sec equilibration) intermittent poikilocapnic hypoxia [5 min hypoxia (FIO2 = 0.11)/5 min normoxia] (Ling et al., 2001). Between 6 A.M. and 6 P.M. the chamber was flushed with air. Chamber temperature was 22–24°C. CIH exposure after injury began at 1 week and ended at 2 weeks after hemisection. Preconditioned rats were exposed to CIH for the 7 d just before acute spinal hemisection.
Experimental preparation. Isoflurane anesthesia was induced in a closed chamber and maintained (2.5–3.5%) via nose cone while rats were tracheotomized. Rats were mechanically ventilated after tracheal cannulation. After femoral venous catheterization, rats were converted to urethane anesthesia (1.6 gm/kg) and bilaterally vagotomized and paralyzed with pancuronium bromide (2.5 mg/kg, i.v.). Blood pressure was monitored via a femoral arterial catheter and pressure transducer (Gould P23ID, Valley View, OH). End-tidal CO2 was monitored with a rapidly responding analyzer (Novametrix, Wallingford, CT). Arterial partial pressures of O2 (PaO2) and CO2 (PaCO2) as well as pH were determined from 0.2 ml blood samples (ABL-500, Radiometer, Copenhagen, Denmark); unused blood was returned to the animal. Rectal temperature was maintained (37–39°C) with a heated table. Phrenic nerves were isolated with a dorsal approach, cut distally, desheathed, bathed in mineral oil, and placed on bipolar silver electrodes. Nerve activity was amplified (1000–10,000×) and filtered (100–10,000 Hz bandpass; model 1800, A-M Systems, Carlsberg, WA).
Spontaneous phrenic motor output. In all rats, the CO2 apneic threshold for inspiratory activity in the phrenic nerve contralateral to hemisection was determined after waiting a minimum of 1 hr after conversion to urethane anesthesia. In acutely injured rats, between 1 and 2 hr elapsed before the CO2 apneic threshold was set. This delay allowed blood pressure and respiratory motor output to stabilize. The procedure to establish the apneic threshold began by increasing the ventilator frequency until inspiratory activity ceased. Ventilator rate was then decreased slowly until inspiratory activity reappeared. The end-tidal CO2 partial pressure (PETCO2) corresponding to the onset of inspiratory bursting was defined as the CO2 apneic threshold. PETCO2 was maintained 3 mmHg above the apneic threshold by adjusting the ventilator pump rate and inspired CO2 content. After the CO2 apneic threshold and baseline PaCO2 levels were established, 30–45 min were allowed to attain stable baseline conditions. Phrenic responses to isocaspnic hypoxia were tested in both acutely and chronically injured rats with 5 min bouts of hypoxia. The target PaO2 of 40 mmHg was achieved by decreasing inspired O2 concentration. Hypercapnic bouts of 5 min at 60 and 80 mmHg PETCO2 were achieved by raising the inspired CO2 concentration. Hypercapnic responses were tested only in chronically injured rats. Arterial blood samples were drawn in the final minute of each hypoxic and hypercapnic trial (blood gases are reported in Tables 1 and 2).
Arterial partial pressure of CO2(PaCO2, mmHg) during spontaneous phrenic bursting (A) and evoked phrenic responses (B)
Arterial partial pressure of O2(PaO2, mmHg) during spontaneous phrenic bursting (A) and evoked phrenic responses (B)
Evoked phrenic potentials. Rats were hyperventilated (PaCO2 <30 mmHg) to prevent spontaneous inspiratory efforts. A monopolar tungsten electrode (5 MΩ) (A-M Systems) was inserted contralateral to the spinal hemisection and ∼1.0 mm rostral to the C2 dorsal roots. The electrode tip was placed in or in close proximity to the ventrolateral funiculus (1.8–2.3 mm below the dorsal root entry zone). Electrode position was selected by maximizing the amplitude of a short-latency (<1.0 msec) evoked potential in the phrenic nerve contralateral to SCI (Fuller et al., 2002). Stimulus–response relationships were obtained by applying current pulses (20–1000 μA, 0.2 msec duration) with a stimulator (model S88, Grass Instruments, Quincy, MA) and stimulus isolation unit (model PSIU6E, Grass Instruments). Phrenic potentials were digitized and analyzed with P-CLAMP software (Axon Instruments, Foster City, CA).
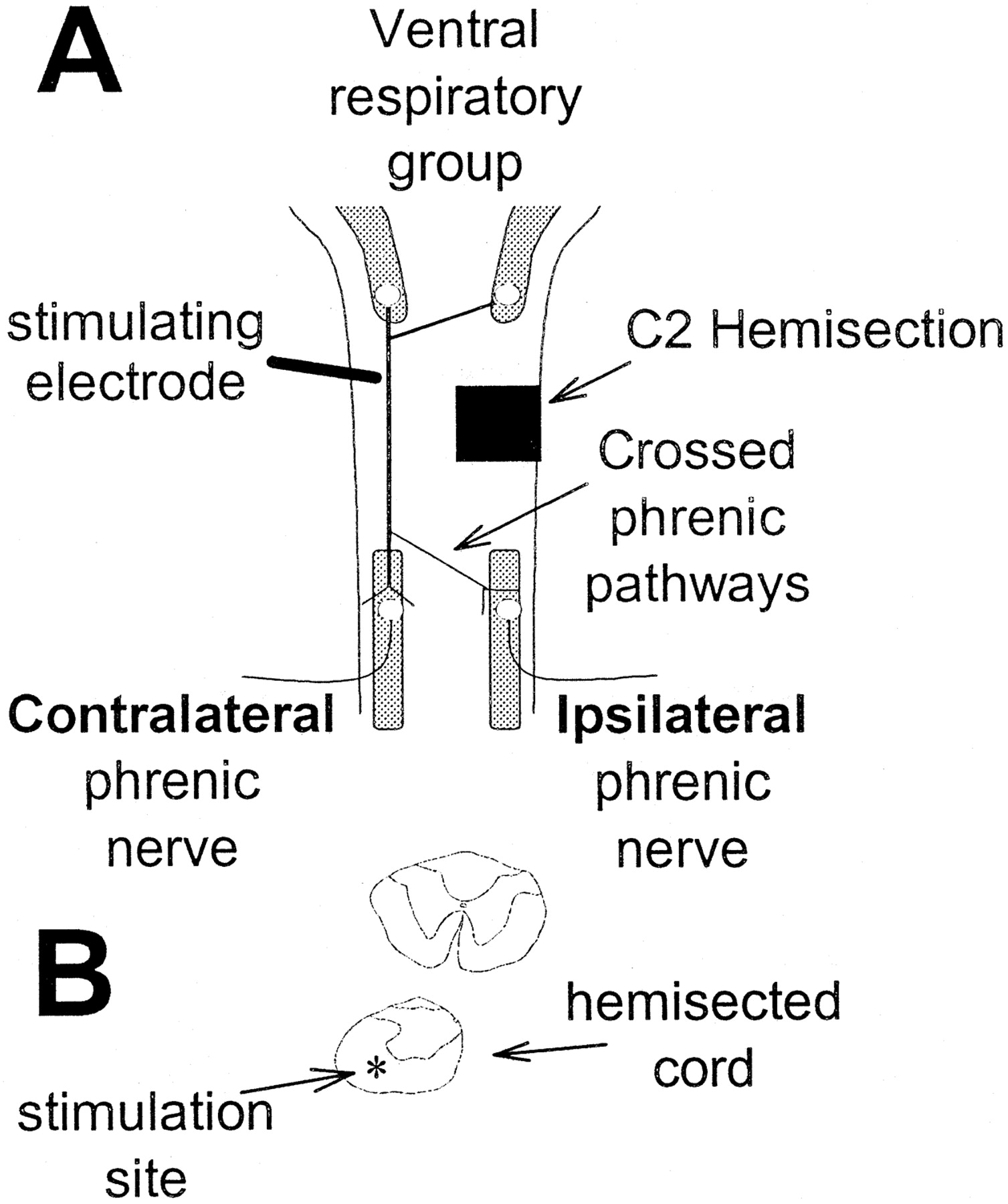
Hemisection confirmation. The hemisection was created by aspirating a 1.0 mm section of the cervical spinal cord and confirmed visually at the time of surgery. In acutely injured rats, the hemisection completeness was verified by the lack of ipsilateral inspiratory phrenic activity in the presence of contralateral inspiratory phrenic activity. In all chronically hemisected rats, the cervical spinal cord was examined histologically after the experiment. Rats were killed by systemic perfusion with 4% paraformaldehyde, and the cervical spinal cord was removed, cryoprotected, and sectioned (90 μm) (Fig.1B). Tissue sections were slide mounted, Nissl stained, and examined with light microscopy.

Schematic depicting the experimental preparation and bulbospinal pathways to phrenic motoneurons. A, Bulbospinal respiratory neurons have cell bodies in the medulla (Ventral respiratory group) and project bilaterally to phrenic motoneurons. Bulbospinal projections that cross the spinal midline in the cervical spinal cord are known as crossed phrenic pathways. Spontaneous inspiratory activity or compound action potentials (evoked via ventrolateral funiculus electrical stimulation) were recorded in the phrenic nerves of spinally injured (C2 Hemisection) rats. The terms ipsilateral and contralateral are used relative to the hemisection. B, Camera lucida drawings of cervical spinal tissue sections (90 μm) taken rostral to (top drawing) and at the hemisection (bottom drawing). The drawings show a representative C2 hemisection and the approximate site of ventrolateral funiculus electrical stimulation (asterisk).
Data analyses. The peak amplitudes of the integrated inspiratory phrenic bursts and evoked phrenic potentials were quantified as (1) absolute voltage, (2) relative to baseline, and (3) relative to activity in the contralateral nerve. Normalization was required because the absolute voltage can be influenced by factors beyond experimental control (e.g., nerve–electrode contact, etc.).
Spontaneous inspiratory phrenic nerve activity was averaged over 30 sec periods immediately before the first hypoxic episode (baseline), at the end of each hypoxic episode, and at the end of each hypercapnic bout. The following variables were determined: peak integrated phrenic amplitude (∫Phr), phrenic burst frequency (bursts per minute), and minute phrenic activity (∫Phr × frequency). Statistical comparisons of neurogram amplitude and blood pressure were made using a two-way ANOVA with a repeated measures design, followed by the Student–Newman–Keuls post hoc test (Sigma Stat, Jandel Scientific, St. Louis, MO). Statistical differences between CO2 apneic thresholds were tested using one-way ANVOA. Comparisons between ipsilateral and contralateral phrenic neurograms were made with an unpaired t test. Significance was designated as p ≤ 0.05.
Results
Blood pressure
Mean arterial pressure at baseline was not different between normoxic and CIH preconditioned rats (Table3). However, hypoxia-induced hypotension was significantly attenuated by CIH preconditioning (p < 0.05) (Table 3) as reported previously for spinally intact rats (Fuller et al., 2001a). Mean arterial pressure was not different between normoxic and CIH postconditioned rats at baseline or during hypoxia. Thus, in contrast to preconditioning, CIH postconditioning did not alter hypoxic blood pressure regulation. Mean arterial pressure was significantly lower at baseline in all chronically versus acutely injured rats (p < 0.05) (Table 3). Rats with C2 hemisection regain normal mean arterial pressures within 1–2 months after injury (Golder et al. 2001a,b). The relative hypotension seen in our rats 2 weeks after hemisection may reflect inadequate time (i.e., 2 vs 4–8 weeks) for endogenous compensatory mechanisms to overcome sympathetic neuron atrophy and reorganization (Krassioukov and Weaver, 1996).
Mean arterial pressure (MAP, mmHg) during spontaneous phrenic bursting
CO2 apneic threshold
Preconditioning
After acute SCI, eight of nine normoxic and four of five CIH preconditioned rats showed no phrenic bursts ipsilateral to SCI under any condition. Accordingly the apneic threshold in this nerve could not be determined. However, the apneic threshold for the phrenic nerve contralateral to SCI was greater in normoxic (37 ± 1 mmHg) than CIH preconditioned rats (32 ± 2 mmHg; p = 0.04), a finding consistent with spinally intact rats (Ling et al., 2001).
Postconditioning
In chronically injured normoxic rats, the PETCO2 at which inspiratory bursts began was significantly greater in the ipsilateral (43 ± 2 mmHg) versus contralateral (36 ± 1 mmHg; p = 0.02) phrenic nerve. CIH postconditioned rats had similar apneic thresholds for the ipsilateral (38 ± 2 mmHg) and contralateral (37 ± 2 mmHg) phrenic nerves (p = 0.6).
Acute versus chronic injury
The apneic threshold in the contralateral phrenic nerve was not different between acutely (37 ± 1 mmHg) and chronically (36 ± 1 mmHg) injured normoxic rats (p > 0.05). However, the contralateral phrenic apneic threshold in CIH preconditioned rats (32 ± 1 mmHg) was significantly lower than the corresponding value in CIH postconditioned rats (37 ± 2;p < 0.05).
Spontaneous inspiratory phrenic activity
Preconditioning
The phrenic nerve ipsilateral to SCI did not display inspiratory bursts at baseline in acutely injured rats, regardless of CIH preconditioning (Figs. 2,3). Small inspiratory bursts in the ipsilateral nerve were induced by hypoxia in one of eight control and one of five CIH rats. Contralateral to SCI, inspiratory burst amplitude in the phrenic nerve was similar between treatment groups at baseline and hypoxia (Figs. 2, 3). Inspiratory burst frequency was not different between control and CIH preconditioned rats (Fig. 3).

Representative phrenic neurograms from rats conditioned with normoxia or CIH before acute SCI (i.e., preconditioning) (top panel) and after chronic SCI (i.e., postconditioning) (bottom panel). The ipsilateral phrenic nerve is silent at baseline and during hypoxia in acutely injured rats, regardless of CIH preconditioning (A). The ipsilateral nerve is also silent at baseline in the rat postconditioned with normoxia (B). However, the CIH postconditioned rat shows robust inspiratory bursts at baseline (B). Moreover, the CIH postconditioned rat displays considerably greater inspiratory phrenic burst amplitude during hypoxia versus the normoxia-treated rat (B). Contralateral phrenic inspiratory motor output was not consistently different at baseline or hypoxia in acutely (C) or chronically injured (D) rats, regardless of CIH conditioning.

Inspiratory phrenic nerve activity in rats treated with normoxia or CIH before acute SCI (i.e., preconditioning) (top panel) and after chronic SCI (i.e., postconditioning) (bottom panel). Top panel, Inspiratory bursts were never present at baseline in acutely injured rats, regardless of CIH preconditioning (A, B). Furthermore, in the majority of acutely injured rats the ipsilateral phrenic nerve remained silent during hypoxia (no differences between normoxia or CIH preconditioned rats) (A, B). There were no differences in contralateral phrenic inspiratory motor output at baseline or hypoxia in acutely injured rats, regardless of CIH preconditioning (C). Phrenic burst frequency was not different between groups at any time point (D).Bottom panel, After chronic SCI, ipsilateral phrenic inspiratory burst amplitude was significantly greater at baseline and hypoxia in CIH versus normoxia postconditioned rats (E, expressed as an absolute voltage, p= 0.01; G, relative to the contralateral phrenic amplitude, p = 0.007). Contralateral phrenic nerve burst amplitude was not significantly different at any time point (F). Phrenic burst frequency was not different between groups at any time point (H). *CIH postconditioned is significantly greater than normoxia postconditioned.
Postconditioning
Inspiratory burst amplitude in the phrenic nerve ipsilateral to SCI was enhanced in chronically injured rats exposed to CIH (Figs.2-4). At baseline, inspiratory bursts in the phrenic nerve ipsilateral to SCI were present more frequently in CIH rats (normoxia = four of eight rats; CIH = seven of seven rats). Moreover, ipsilateral phrenic burst amplitude was greater during baseline, hypoxia, and hypercapnia in CIH versus normoxic rats (Figs.2-4). Inspiratory burst amplitude in the contralateral phrenic nerve was not affected by CIH during baseline, hypoxia, or hypercapnia (Figs.2, 3). Inspiratory burst frequency was not different between control and CIH postconditioned rats.

Hypercapnic phrenic responses in rats treated with normoxia or CIH after chronic SCI (i.e., postconditioning). A small but distinct ipsilateral phrenic inspiratory burst is present at baseline in the normoxia postconditioned rat shown in A. Hypercapnic stimulation increases ipsilateral burst amplitude in this rat (A). However, the CIH postconditioned rat shows considerably larger ipsilateral phrenic inspiratory bursts at baseline and hypercapnia (A). On average, ipsilateral phrenic burst amplitude was significantly greater in CIH versus normoxia postconditioned rats during hypercapnia (B, C). D shows representative contralateral phrenic neurograms in chronically injured normoxia and CIH postconditioned rats. Contralateral phrenic amplitude was not different at baseline or hypercapnia between normoxia or CIH post-conditioned rats (E). Phrenic inspiratory burst frequency was not different between groups at any time point (F). *CIH postconditioned is significantly greater than normoxia postconditioned.
Acute versus chronic SCI
Phrenic burst amplitude ipsilateral to SCI was greater at baseline and during hypoxia in all chronic versus acutely injured rats (p < 0.05) (Fig. 3). Indeed, the ipsilateral phrenic nerve was usually silent after acute SCI (Fig. 2). Neither inspiratory burst amplitude contralateral to SCI nor burst frequency was different between acutely and chronically injured rats (p > 0.05) (Fig. 3).
Evoked phrenic potentials
Preconditioning
Potentials in the phrenic nerve ipsilateral to injury were evoked in three of eight normoxic and four of eight CIH preconditioned acutely injured rats. Neither the threshold current (normoxic = 900 ± 50 μA; CIH = 800 ± 50 μA) nor the amplitude (Figs. 5,6) of the ipsilateral phrenic potential was different between control and CIH preconditioned rats. One rat had an ipsilateral evoked potential amplitude >10-fold greater than any of the others and was excluded from the analysis as an outlier (p < 0.01; Dixon's test for outliers) (Sokal and Rohlf, 1995); exclusion of this rat did alter our conclusions. The threshold stimulus current for the phrenic nerve contralateral to SCI appeared to be greater in normoxic (210 ± 40 μA) versus CIH rats (96 ± 25 μA), but this difference did not attain statistical significance (p = 0.07). Evoked potential amplitude in the contralateral phrenic nerve was not different between groups at any stimulus current (Figs. 5, 6). Stimulus latencies (time to peak) were not different between groups in either nerve and ranged from 0.5 to 0.7 msec.

Representative evoked phrenic potentials from rats treated with normoxia or CIH before acute SCI (i.e., preconditioning) (top panel) and after chronic SCI (i.e., post-conditioning) (bottom panel). Evoked potentials were absent in the ipsilateral phrenic nerve of acutely injured rats, regardless of CIH preconditioning (A) and also were absent in the chronically injured normoxia-conditioned animal (B). In contrast, the rat conditioned with CIH after chronic hemisection displays a clear evoked potential in the ipsilateral phrenic nerve at stimulus currents of 800 and 1000 μA (B). Thus, crossed phrenic pathways are enhanced by a spinal mechanism after CIH. Evoked responses in the contralateral phrenic nerve were not altered by CIH preconditioning (C) or postconditioning (D).

Evoked compound action potential amplitude in the phrenic nerve ipsilateral (left panels) and contralateral (right panels) to hemisection in rats treated with normoxia or CIH before acute SCI (i.e., preconditioning) or after chronic SCI (i.e., postconditioning). Evoked potentials in the ipsilateral phrenic nerve were consistently present only in chronically injured CIH postconditioned rats and only when the stimulus current was >700 μA (compare A–D). Evoked ipsilateral phrenic potential amplitude was significantly greater in these rats than in normoxia postconditioned rats or normoxia and CIH preconditioned rats. Contralateral phrenic evoked potentials were not affected by CIH preconditioning or postconditioning (compareE–F). *CIH postconditioned is significantly greater than normoxia postconditioned.
Postconditioning
The amplitude of the evoked phrenic potential ipsilateral to SCI was significantly greater in CIH versus normoxic rats at stimulus intensities >700 μA (Figs. 5, 6). However, the threshold current for evoked ipsilateral phrenic potentials was not statistically different between groups (normoxic = 860 ± 96 μA; CIH = 630 ± 70 μA; p = 0.11). Similarly, the threshold current for evoked phrenic potentials contralateral to SCI was not different between normoxic (60 ± 20 μA) and CIH-treated rats (100 ± 28 μA; p = 0.15). Evoked potential amplitude contralateral to SCI was not different between groups (Figs.5, 6). Stimulus latencies were not different between groups in either phrenic nerve (0.5–0.7 msec).
Discussion
Although CIH preconditioning had no discernable effect on phrenic motor output, CIH after chronic SCI (i.e., postconditioning) enhanced spontaneous inspiratory phrenic burst amplitude ipsilateral to spinal hemisection at C2. Electrical stimulation of the spinal cord indicated that the strength of synaptic pathways crossing the spinal midline caudal to C2 (crossed phrenic pathways) was enhanced when CIH was applied after SCI. Collectively, these results indicate that chronic SCI established necessary preconditions that allowed CIH-induced plasticity in crossed-spinal pathways to phrenic motoneurons.
Respiratory function after cervical SCI in rats
After C2 spinal hemisection, rats maintain blood gases (Goshgarian 1981) and breathe with increased frequency and decreased tidal volume (Golder et al. 2001a). Alterations in breathing pattern are (at least partly) mediated vagally but may also reflect reorganization of the medullary respiratory control network (Golder et al. 2001b). During quiet breathing (i.e., eupnea), the phrenic nerve or hemidiaphragm ipsilateral to C2 hemisection is silent (i.e., no inspiratory bursting) in the days to weeks after the injury (Goshgarian, 1981; Nantwi et al., 1999; Golder et al., 2001a,b). However, chemical or pharmacological stimulation of respiratory drive produces rhythmic inspiratory phrenic nerve activity below the hemisection via the activation of existing but ineffective crossed-spinal synaptic pathways to phrenic motoneurons (Goshgarian, 1981; Nantwi and Goshgarian, 1998; Nantwi et al., 2001;Zhou et al., 2001). This effect has been termed the crossed phrenic phenomenon (Goshgarian, 1981). Crossed phrenic inspiratory bursts occur at the same frequency as inspiratory bursts in the contralateral phrenic nerve, but amplitude is considerably less (Goshgarian, 1981;Golder et al., 2001b).
One to 2 months after C2 hemisection, spontaneous inspiratory bursts during eupnea (i.e., quiet breathing) are observed in the phrenic nerve or hemidiaphragm ipsilateral to injury (Nantwi et al., 1999; Golder et al. 2001b). However, this spontaneous functional recovery reportedly is not present at 2 weeks after hemisection (Nantwi et al., 1999). Conversely, 50% of our normoxia postconditioned rats demonstrated weak inspiratory bursts in the ipsilateral phrenic nerve at baseline conditions 2 weeks after hemisection. This quantitative discrepancy may reflect differences in the baseline PaCO2values [not reported by Nantwi et al. (1999)] or could result from rat substrain (Fuller et al., 2000) or gender differences (Hauben et al., 2001).
Phrenic motoneuron morphology is also altered by C2 hemisection (Sperry and Goshgarian, 1993; Mantilla et al., 2002). An increase in the number of dendrodendritic appositions and synaptically active zones is observed in the ipsilateral phrenic motor nucleus a few hours after C2 hemisection (Sperry and Goshgarian, 1993). These rapid morphological effects are blunted when animals are given a serotonin synthesis inhibitor (p-chlorophenylalanine) before hemisection (Hadley et al., 1998). The surface area of ipsilateral phrenic motoneuron somata is significantly decreased 2 weeks after C2 hemisection, suggesting that motoneuron excitability actually increases (Mantilla et al., 2002).
CIH preconditioning did not affect phrenic motor output
Although CIH enhances respiratory motor output in spinally intact rats (Ling et al., 2001; Peng et al., 2001), we found no obvious effect of CIH preconditioning on phrenic motor output in acutely injured rats. However, observed cardiorespiratory plasticity confirmed that CIH preconditioning was physiologically active. First, the CO2 apneic threshold in the phrenic nerve contralateral to SCI was lower in CIH versus control rats, similar to reports on spinally intact animals (Fuller et al., 2001; Ling et al., 2001). Second, the threshold current for electrically evoked potentials in the phrenic nerve contralateral to SCI tended to be lower after CIH preconditioning (however, p = 0.07 versus control). Finally, CIH preconditioning prevented or minimized the hypotension associated with hypoxia (Table 3) (Fuller et al., 2001).
Because CIH was physiologically active, the possibility must be considered that CIH-enhanced phrenic motor output was masked by inhibitory influences associated with acute SCI (e.g., hemorrhage, swelling, etc.) (Ramer et al., 2000). Such inhibitory influences could prevent crossed phrenic pathway activation leading to erroneous conclusions about the efficacy of CIH preconditioning. On the other hand, robust crossed phrenic responses have been reported in acutely injured animals after a preconditioning lesion (cervical dorsal rhizotomy) (Fuller et al. 2002c) that enhances serotonergic terminal density and neurotrophin concentration in the cervical spinal cord (Kinkead et al., 1998; Johnson et al., 2000). Moreover, the serotonin precursor 5-hydroxytryptophan reveals evoked (Ling et al., 1994) and spontaneous crossed phrenic activity in acutely injured animals (Fig. 7). Thus, putative inhibitory effects from acute hemisection, if present, do not always mask crossed phrenic activity. Although CIH preconditioning may strengthen synaptic pathways contralateral to SCI, it does not appear to strengthen crossed phrenic pathways.

Crossed phrenic pathways can be activated in rats after acute spinal hemisection. Systemic delivery of the serotonin precursor 5-hydroxytryptophan (5-HTP) (5 mg/kg) reveals robust spontaneous inspiratory crossed phrenic activity <2 hr after C2 hemisection. These results demonstrate that acute responses to SCI (e.g., inflammation, etc.) do not prevent spontaneous inspiratory crossed phrenic activity.
CIH after chronic hemisection strengthens crossed phrenic pathways
In rats with chronic SCI, CIH post-treatment significantly increased inspiratory burst amplitude in the phrenic nerve ipsilateral to hemisection (i.e., crossed phrenic pathways were enhanced). Crossed phrenic inspiratory activity occurs rapidly (seconds to minutes) after chemical or pharmacological stimulation in normoxic rats (Goshgarian, 1981; Ling et al., 1994; Golder et al., 2001a,b; Nantwi and Goshgarian, 2001). Therefore, the synaptic pathways underlying crossed phrenic activity are present but not physiologically effective under most conditions. We hypothesize that the CIH increased the efficacy of existing spinal synaptic pathways to phrenic motoneurons (vs formation of new synaptic connections).
Enhancement of spontaneous inspiratory activity below SCI could occur via several mechanisms. Increased peripheral chemosensitivity after CIH could augment inputs to medullary respiratory neurons and enhance descending inputs to phrenic motoneurons during hypoxia (Peng et al., 2001). However, this same influence should occur after CIH preconditioning, but does not. Because CIH enhanced synaptic inputs to phrenic motoneurons during baseline, hypoxia, and hypercapnia, a common mechanism is suggested that is not specifically associated with increased chemoreceptor sensitivity. Because short-latency (time to peak = 0.5–0.7 msec) evoked phrenic potentials were also enhanced in the phrenic nerve ipsilateral to SCI, increased efficacy of a monosynaptic pathway is suggested. Thus, we hypothesize that augmented spontaneous crossed phrenic inspiratory burst amplitude after CIH arises from increased synaptic strength of the crossed phrenic pathway.
Transformation of silent synapses to functionally effective synaptic connections has considerable precedent (Atwood and Wojtowicz, 1999). For example, silent glutamatergic synapses in the rat spinal dorsal horn are transformed into functional synapses by serotonin (Li and Zhuo, 1998). Serotonin may be of particular importance because CIH enhances phrenic motor output via serotonin-dependent mechanisms in spinally intact rats (Ling et al., 2001). Serotonin is also linked to functional recovery of locomotion after SCI (Hashimoto and Fukuda, 1991; Saruhashi et al., 1996), and crossed phrenic pathways are less robust after serotonin depletion (Hadley et al., 1998). The hypothesis that CIH enhanced crossed phrenic pathways after SCI by a serotonin-dependent mechanism requires further experimentation.
Neurotrophins such as brain-derived neurotrophic factor (BDNF) may be involved in spinal cord plasticity after CIH. BDNF is critical for several forms of neuroplasticity (e.g., hippocampal long-term potentiation) (Schinder and Poo, 2000) and is upregulated in the cervical spinal cord after episodic hypoxia (Baker-Herman et al., 2001). BDNF may strengthen crossed phrenic pathways in rats because a chronic preconditioning lesion (cervical dorsal rhizotomy) increases BDNF protein levels in the ventral spinal cord (Johnson et al., 2000) and enhances crossed phrenic pathways (Fuller et al., 2002). BDNF and receptor tyrosine kinase B mRNA expression transiently increase after C2 hemisection but return to control levels within 2 weeks; the elevation in BDNF protein levels is more prolonged (Mantilla et al., 2002). The influence of CIH on spinal BDNF levels in spinally injured rats is currently unknown.
Significance
After partial cervical SCI in humans, adequate minute ventilation is generally maintained at rest and during chemoreceptor stimulation, although the breathing pattern is altered (increased frequency, decreased tidal volume) (Pokorski et al., 1989; Loveridge and Dubo, 1990). Reduced tidal volume reflects diminished respiratory motoneuron recruitment and respiratory muscle weakening (Roth et al., 1997). Although therapeutic training protocols designed to enhance respiratory muscle function are often successful in SCI patients (Rutchik et al., 1998; Liaw et al., 2000), the neural (vs muscular) effects of respiratory muscle training in SCI patients have not been addressed. Our data show that CIH-induced neuroplasticity can enhance respiratory motor output after SCI.
Repeated intermittent activation of the respiratory neural control network with CIH may have therapeutic potential in restoring respiratory function after SCI. However, there may be shortcomings that limit or constrain the potential of CIH as a therapeutic tool. For example, certain CIH protocols elicit pathophysiology such as systemic hypertension (Fletcher et al., 1992), altered sympathetic chemoreflexes (Greenberg et al., 1999), and hippocampal apoptosis (Gozal et al., 2001). These pathophysiological effects probably depend on the duration and severity of hypoxia. Other CIH protocols (altered severity and duration of hypoxia) may elicit beneficial effects without the attendant pathophysiology. As our understanding of CIH and its detailed cellular mechanism(s) advances, alternative therapeutic strategies may be developed that do not involve repetitive hypoxia, bypassing the mechanisms leading to pathophysiology.
Footnotes
This work was funded by the National Institutes of Health (HL/NS 69064 and HL 65383). D.D.F. was supported by a Parker B. Francis fellowship in pulmonary research. We thank Dr. F. J. Golder for his careful critique of this manuscript.
Correspondence should be addressed to Dr. David D. Fuller, Department of Comparative Biosciences, School of Veterinary Medicine, University of Wisconsin, 2015 Linden Drive, Madison, WI 53706. E-mail: fullerd{at}svm.vetmed.wisc.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}