

Abstract
Two determinants of dopamine release from terminals in striatal and limbic structures are the pattern and rate of dopamine neuron firing in the ventral midbrain. This activity is regulated in part by somatodendritic release of dopamine and subsequent feedback inhibition through activation of D2 receptors on dopamine neuron cell bodies and dendrites. This study describes stimulus-dependent long-term depression (LTD) of IPSCs mediated by dopamine. This LTD was blocked by chelation of postsynaptic intracellular calcium, was dependent on the activation of D2 receptors and was independent of glutamate-mediated transmission. Application of a high concentration of dopamine mimicked depression of the IPSC and prevented additional attempts to induce LTD, suggesting that the mechanism of the depression is agonist-dependent receptor activation. Using extracellular recording, there is an inhibition of firing that follows electrical stimulation, and after the induction of LTD the duration of that inhibition was decreased. Reduced inhibition could increase burst firing and action potential-dependent release of dopamine in terminal regions in vivo.
Introduction
Dopamine neurons in the ventral midbrain are important for initiation of movement, learning, and memory and contribute to the etiology of Parkinson's disease, schizophrenia, and drug abuse. Dopamine neuron firing communicates a signal in response to the presentation of an unexpected reward or associated conditioned stimulus (Schultz, 2002). This reinforces the behavioral processes that produced the reward, implicating dopamine in salience and incentive sensitization (Berridge and Robinson, 1998). Glutamate input from regions such as the prefrontal cortex stimulates midbrain dopamine neurons, promoting burst rather than single spike firing (Grace and Onn, 1989; Taber et al., 1995; Grillner and Mercuri, 2002; Floresco et al., 2003). Burst firing facilitates the supra-additive release of dopamine in terminal projection fields such as the olfactory tubercule and the nucleus accumbens. The resulting increase in extracellular dopamine overwhelms uptake transporters and produces the phasic response ultimately responsible for many of the behavioral consequences of dopamine release (Chergui et al., 1994; Floresco et al., 2003; Phillips et al., 2003).
Burst firing also facilitates dopamine release in somatodendritic regions of the midbrain (Bjorklund and Lindvall, 1975; Geffen et al., 1976; Kalivas and Duffy, 1991; Rice et al., 1997; Jaffe et al., 1998; Adell and Artigas, 2004), where dopamine neurons make dendrodendritic synapses onto other dopamine neurons (Wilson et al., 1977; Groves and Linder, 1983; Nirenberg et al., 1996). By using a combination of electrophysiological and genetic tools we recently identified an IPSC mediated by vesicular dopamine release activating D2 dopamine autoreceptors in the ventral tegmental area (VTA) and substantia nigra (Beckstead et al., 2004, Ford et al., 2006). D2 receptor activation increases an inhibitory G-protein coupled inwardly rectifying potassium conductance (GIRK) that hyperpolarizes cells on a millisecond time scale and may contribute to the pause typically observed after a burst of action potentials (Lacey et al., 1987; Davila et al., 2003; Beckstead et al., 2004; Koyrakh et al., 2005). D2 dopamine receptor activation in the midbrain strongly inhibits dopamine cell firing (Pucak and Grace, 1994). Therefore, use-dependent plasticity of dopaminergic synapses could have important consequences for cell excitability, burst firing, terminal dopamine release, and dopamine-mediated behaviors.
The factors that affect synaptic strength have not been described for dopamine-mediated synaptic transmission. This study reports a long-term depression (LTD) of a G-protein coupled receptor-mediated IPSC resulting from dendritic release of dopamine in the ventral midbrain. The mechanism of this depression is through a postsynaptic desensitization of D2 autoreceptors.
Materials and Methods
All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at Oregon Health and Science University. Dopamine, MK-801, DNQX, calmidazolium, staurosporine, prazosin, picrotoxin, cyclopiazonic acid, and l-glutamic acid were obtained from Sigma (St. Louis, MO). Baclofen, sulpiride, and hexamethonium were from Research Biochemicals International (Natick, MA). CGP 56999a was a gracious gift from Novartis Pharmaceuticals (Basel, Switzerland). LY 341495 and AM 251 were obtained from Tocris Bioscience (Ellisville, MO).
Brain slices were prepared from young adult [postnatal day 32 (P32)–P95] male or female C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) as described previously (Williams et al., 1984). Homozygous (−/−) β-arrestin 2 (arrestin 3) knock-out mice were used for one experiment (Kohout et al., 2001). Briefly, mice were placed in a chamber, anesthetized with halothane, and killed by decapitation. Brains were quickly removed and placed in a 0°C physiological solution (modified Krebs' buffer) containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.4 NaH2PO4, 25 NaHCO3, and 11 d-glucose. Horizontal slices of the ventral midbrain (220 μm) were obtained and incubated for at least 25 min at 35°C in Krebs' containing MK-801 (10 μm). Slices were placed in a recording chamber attached to an upright microscope (Carl Zeiss, Oberkochen, Germany) and maintained at 35°C with Krebs' solution perfused at a rate of 1.5 ml/min. Infrared illumination was used to visualize dopamine cells in the substantia nigra pars compacta (∼80%) and anterior lateral VTA (∼20%), which were identified by visual inspection of their location in relation to the midline, the third cranial nerve, and the medial terminal nucleus of the accessory optic tract. Physiological identification was based on the presence of spontaneous pacemaker firing of wide (∼2 ms) action potentials at 1–5 Hz, a large hyperpolarization-induced IH current, and sensitivity to an iontophoretic application of dopamine. Whole-cell patch-clamp recordings were obtained with large glass electrodes (1.5–2.0 MΩ; World Precision Instruments, Sarasota, FL) and an internal pipette solution containing the following (in mm): 115 K-methylsulfate, 20 NaCl, 1.5 MgCl2, 2 ATP, 0.2 GTP, 10 phosphocreatine, and either 10 BAPTA, 1 EGTA, 0.1–0.4 EGTA, or 0.4 Fluo-4, pH 7.30–7.35, 270–280 mOsm. Cells were voltage clamped at −60 mV with an Axopatch 1D amplifier (Molecular Devices, Foster City, CA) to prevent spontaneous cell firing. Series resistance was monitored to insure sufficient and stable electrical access (<7 MΩ) to the inside of the cell was maintained. Drugs were applied by bath perfusion, and dopamine was also applied iontophoretically. Iontophoretic pipettes were pulled from thin-wall glass microelectrodes (resistance 100–150 MΩ) filled with dopamine (1 m) and placed with the tip within 10 μm of the soma. Leakage was prevented by application of a negative backing current (1.0–5.0 nA) and dopamine was ejected as a cation with application of + 10–190 nA for 1–5 s with an Axoclamp 2A amplifier (Molecular Devices). This brief, high dopamine concentration produced a maximal D2 receptor-mediated current that recovered rapidly (<3 min).
The D2 receptor-mediated IPSC was obtained as described previously (Beckstead et al., 2004) in the presence of receptor blockers picrotoxin (100 μm), prazosin (100 nm), MK-801 (10 μm), CGP 56999a (100 nm), and in some experiments DNQX (10 μm), hexamethonium (50 μm), or LY 341495 (100 μm). A bipolar platinum stimulating electrode was placed into the slice 50–200 μm caudal to the cell and IPSCs were evoked by electrically applying a train of five action potentials at 40 Hz once every 50–100 s. The CB1 cannabinoid receptor antagonist AM 251 (1 μm) was added for the GABAB synaptic experiment to inhibit retrograde endocannabinoid inhibition of GABA terminal release, as well as sulpiride (50–200 nm) to block D2 dopamine receptor signaling.
For the cell-firing experiment, dopamine neurons were loosely patched with slightly higher resistance electrodes (2–3 MΩ) using a modified Krebs' internal solution. Externally, the bath solution contained picrotoxin (100 μm), prazosin (100 nm), MK-801 (10 μm), CGP 56999a (100 nm), DNQX (10 μm), and hexamethonium (50 μm). Neurons that exhibited spontaneous firing in a pacemaker pattern were maintained in the cell-attached patch configuration for the duration of the experiment. Often (∼50% of the time) the loose patch began to break into the neuron (whole cell) before the end of the experiment, evidenced by visual identification of an afterhyperpolarization potential and an increase in spontaneous firing rate. In these cases, the experiment was immediately terminated.
For the calcium-imaging experiment, the recording electrode was filled with Fluo-4 (40 μm) and no other calcium chelators. Changes in intracellular calcium concentrations were determined by monitoring real-time fluorescence with a confocal microscope imaging system (Solamere Technology, Salt Lake City, UT).
Data was collected on a Macintosh (Apple, Sunnyvale, CA) G4 computer using Axograph 4.5.1 (Molecular Devices) and Chart 4.0.1 (Molecular Devices), and stored for offline analysis at a later time. One- and two-way ANOVA was used to assess effects of independent variables, followed when appropriate by Tukey's (all comparisons) or Dunnett's (versus a control) post hoc test. When only two groups were involved, comparisons were made with paired (within cells) and unpaired t tests. Statistical significance was defined a priori with α = 0.05. Data are presented as mean ± SE with n designating the number of cells obtained in each experimental group.
Results
Long-term depression of D2- but not GABAB receptor-mediated IPSCs
The recent identification of a synaptic potential mediated by dopamine (Beckstead et al., 2004) made possible an examination of stimulation-induced synaptic changes that could be important for adaptations observed in vivo. Whole-cell recordings were obtained from dopamine neurons in the lateral VTA (20%) or substantia nigra pars compacta (80%) in horizontal slices of the mouse ventral midbrain. A bipolar platinum stimulating electrode was placed 50–200 μm caudal to the dopamine neuron and used to evoke dendrodendritic dopamine IPSCs. Using a control intracellular solution that contained EGTA (0.4 mm), low-frequency electrical stimulation (0.5 ms, 2 Hz for 300 s) induced a robust LTD of the dopamine IPSC (Fig. 1a,c) in 12 of 13 neurons tested. No recovery from LTD was ever observed (up to 60 min after the low-frequency stimulation).

Low-frequency stimulation induces LTD of dopamine but not GABAB IPSCs. a, Low-frequency stimulation (LFS, 0.5 ms, 2 Hz for 300 s) induced depression of dopamine IPSCs (1, pre-LFS; 2, post-LFS; n = 13). b, In contrast, low-frequency stimulation produced temporary depression but failed to induce LTD of GABAB receptor-mediated IPSCs (1, pre-LFS; 2, post-LFS; n = 5; p = 0.0005). c, Depression of the dopamine IPSC persisted for the length of the experiment (c), and administration of the antagonist CGP 56999a (100 nm) or sulpiride (50–200 nm) confirmed that the IPSCs were mediated by GABAB or D2 receptors, respectively.
GABAB receptors activate the same GIRK conductance in dopamine neurons as D2 receptors (Lacey et al., 1988; Davila et al., 2003; Beckstead et al., 2004; Koyrakh et al., 2005). Thus, the receptor specificity of the LTD was next tested by examination of GABAB receptor-mediated IPSCs. Low frequency stimulation (0.5 ms, 2 Hz for 300 s) decreased the GABAB IPSC, an effect that recovered completely within 15–20 min (Fig. 1b). It is unclear whether this short-term synaptic depression was presynaptic or postsynaptic, but the duration of the inhibition did not resemble LTD of D2 receptor IPSCs (Fig. 1c). This indicates a distinct difference between plasticity of D2 and GABAB receptor synapses and suggests that all GIRK conductances are not susceptible to long-term depression subsequent to this stimulation protocol.
Depression of the dopamine IPSC is blocked with strong calcium chelation
The induction of LTD at glutamate synapses is dependent on a rise in intracellular calcium that can often result from the activation of NMDA or metabotropic glutamate receptors (mGluRs) (Malenka and Bear, 2004). This was not the case in the present study as NMDA receptors were blocked during the LTD experiments (with MK-801, 10 μm). Similarly the AMPA receptor blocker DNQX (10 μm) and the nicotinic acetylcholine receptor antagonist hexamethonium (50 μm) did not prevent depression of the dopamine IPSC (n = 13, LTD −41.8 ± 4.0% change) (data not shown). Thus, calcium entry through these ionotropic receptor channels is not necessary to induce LTD.
To determine the role of intracellular calcium in the depression of the dopamine IPSC, low-frequency stimulation was applied in the presence of different levels of intracellular calcium chelation. The induction of LTD was blocked when intracellular calcium was chelated to very low concentrations using an internal solution with BAPTA (10 mm and no added Ca2+) (Fig. 2a,b). A small amount of LTD was observed after low frequency stimulation using an internal containing EGTA (1 mm and no added Ca2+) (Fig. 2b).

LTD of the dopamine IPSC is blocked by chelating intracellular calcium. a, b, Strongly chelating intracellular calcium with BAPTA (10 mm) prevented induction of LTD (a; 1, pre-LFS; 2, post-LFS), and mildly chelating intracellular calcium with EGTA (1 mm) reduced the amplitude of the synaptic depression (b). Sulpiride (50–200 nm) was used to confirm that the IPSCs were mediated by D2 receptors. Dunnett's test indicated a nonsignificant difference from control conditions for EGTA (1 mm; p = 0.068) and a significant difference from control conditions for BAPTA (10 mm; p = 0.00002). n = 8–13 in each group. c, LTD induction was maintained in the presence of the mGluR antagonist LY 341495 (100 μm, n = 6) or after depletion of intracellular calcium stores with cyclopiazonic acid (CPA) (10 μm; n = 6).
Activation of mGluRs can increase intracellular calcium, resulting in the induction of plasticity at glutamate synapses onto dopamine neurons (Bellone and Luscher, 2005). To determine whether mGluR activation was necessary for depression of dopaminergic synapses, experiments were conducted in the presence of the mGluR antagonist LY 341495 (100 μm). LTD was not inhibited by mGluR antagonism, nor when intracellular calcium stores were depleted with cyclopiazonic acid (10 μm) (Fig. 2c). This suggests that LTD of the dopamine IPSC was not caused by a rise in internal calcium resulting from release from intracellular stores or from calcium entry secondary to mGluR activation. LTD was also not induced by perfusion with a high concentration of glutamate (1 mm; change from baseline, −1.7 ± 4.6%; n = 5) (data not shown) in the absence of AMPA and mGluR blockers. This result confirms that AMPA and mGlu receptor activation are not involved in depression of the dopamine IPSC. Finally, LTD was not induced by a series of postsynaptic depolarizations (to 0 mV for 10 ms, 2 Hz for 300 s; change from baseline, +7.0 ± 8.6%; n = 7) (data not shown). This suggests that unlike LTD at glutamate synapses on dopamine neurons (Jones et al., 2000; Thomas et al., 2000), LTD of the dopamine IPSC cannot be reproduced by a depolarization-induced influx of calcium through voltage-gated channels. Together, the results suggest that depression of the dopamine IPSC is dependent on a physiological level of calcium and not dependent on a transient rise in intracellular calcium.
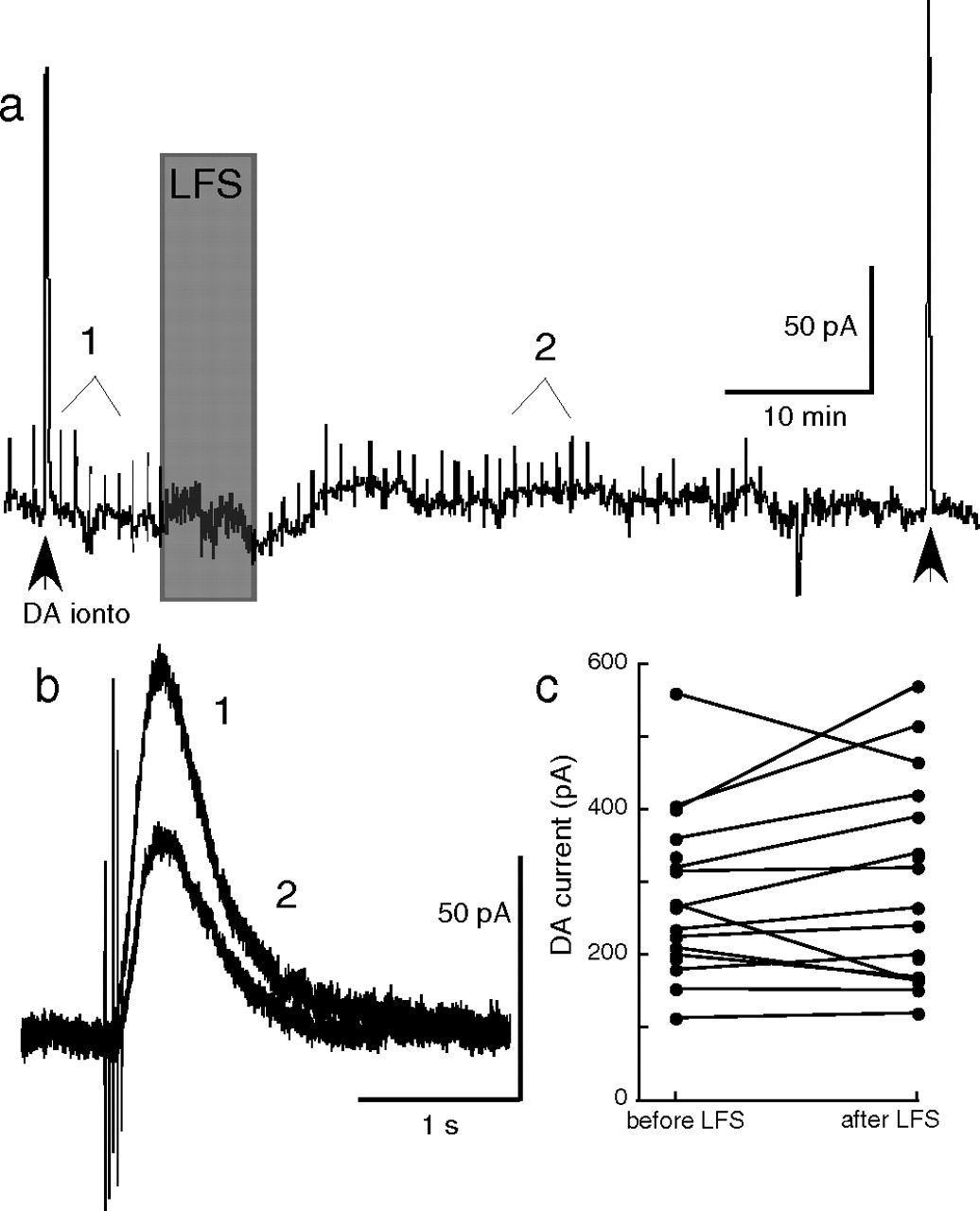
There was no change in the amplitude of the current induced by iontophoretic application of dopamine measured before and after LTD (Fig. 3). Although the location of dopamine release sites is not known, a dendritic site is more likely than somatic areas. Iontophoretic application of dopamine was focused at the cell body, so the current evoked by synaptic release and iontophoretic application of dopamine may result from the activation of different populations of D2-receptors. The observation that synaptic current was depressed after the LTD protocol whereas the current induced by iontophoretic application of dopamine was unaffected also suggests a segregation of receptors that were activated by synaptic and iontophoretic application of dopamine.

Low-frequency stimulation does not affect currents elicited by exogenous dopamine. a, One of the tests used to confirm that neurons being recorded were dopaminergic (see Materials and Methods) was a brief application of exogenous dopamine delivered via an iontophoretic pipette at the beginning of each recording (DA ionto arrowhead). In some neurons, a second iontophoretic pulse was applied at the end of the experiment (second arrowhead) to assess changes in maximum dopamine current. b, This sample trace is from a neuron that expressed LTD (1, before LFS; 2, after LFS). c, There was no consistent change in the iontophoretic current throughout the course of these experiments (average + 4.7 ± 4.9%; n = 17).
D2 receptor desensitization is decreased by strong calcium chelation
One potential mechanism that would result in a receptor-specific inhibition of the dopamine IPSC is through desensitization of the receptors responsible for the IPSC. To test this possibility, a high concentration of dopamine (100 μm) was bath applied and the subsequent outward current was measured with control internal solution (EGTA, 0.4 mm) and with an internal solution containing BAPTA (10 mm) (Fig. 4 supplemental Fig. 4, available at www.jneurosci.org as supplemental material). One striking difference between the two internal solutions was the amplitude of the outward current induced by dopamine was larger with the BAPTA internal. Larger amplitude currents were observed when dopamine was applied via bath perfusion (control 191 ± 26 pA; BAPTA 294 ± 22 pA; n = 20–21) (Fig. 4a) (data not shown), iontophoresis (control 249 ± 19; BAPTA 413 ± 34 pA; n = 25), and synaptic release (control 56.6 ± 5.4 pA; BAPTA 80.2 ± 7.3 pA; n = 15–19) (Fig. 2a). The second observation was that the D2 receptor-dependent outward currents desensitized more slowly and less completely with the BAPTA-containing internal than with control internal (Fig. 4a,b). The results of both of these experiments have also been reproduced in dopamine neurons from Wistar rats (peak currents, BAPTA 211 ± 28 pA, control 116 ± 12 pA, p = 0.005; 5 min desensitization, BAPTA 36.0 ± 2.6%, control 63.8 ± 5.2%, p = 0.0001; n = 12–13).

Chelating calcium increases the amplitude of GIRK currents and decreases the desensitization rate of D2 receptor-mediated currents. a, b, When intracellular calcium was chelated with BAPTA (10 mm), bath perfusion of dopamine (DA; 100 μm) elicited larger D2 receptor-mediated outward currents (a) that desensitized less (a, b; p = 0.00093; F(9,144) = 3.35 for time-chelation interaction; n = 8–10). c, d, GABAB receptor-mediated outward currents elicited by baclofen (30 μm) were also larger in the presence of calcium chelation (t(40) = 2.52; p = 0.016; n = 18–24), but there was no difference in desensitization rate (c, d; p = 0.507 for time-chelator interaction; F(6,168) = 0.884; n = 13–17). Perfusion of CGP 56999a (100 nm) or sulpiride (50–200 nm) confirmed that the currents were mediated by GABAB or D2 receptors, respectively.
The receptor specificity of desensitization was examined by the activation of GABAB receptors with baclofen (30 μm). The peak amplitude of the current induced by baclofen was significantly larger with the BAPTA-containing internal (control 309 ± 27 pA; BAPTA 439 ± 48 pA; n = 18–24) (data not shown), but there was no difference in the extent of desensitization during a 7 min application of baclofen (Fig. 4c,d). Therefore, although chelating calcium with a high concentration of BAPTA increased the GIRK-mediated peak current, desensitization and the induction of LTD were D2 receptor-specific.
Desensitization of D2 receptors and LTD
To test whether D2 receptor activation and subsequent desensitization was responsible for depression of the dopamine IPSC, a high concentration of dopamine (100 μm) was bath applied. The high concentration of dopamine reliably occluded the IPSC during the superfusion and for several minutes as the dopamine washed out of the slice (Fig. 5). With BAPTA-containing internal solution, the amplitude of the IPSC returned to baseline 20–30 min after washing the dopamine (Fig. 5c,d). In experiments with the control internal solution (EGTA, 0.4 mm) the IPSC remained depressed (Fig. 5). Furthermore, in slices that had already been treated with a desensitizing concentration of dopamine, low-frequency stimulation did not induce a decrease in the IPSC. That is, previous desensitization with dopamine occluded LTD (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). This suggests that bath application of a high concentration of dopamine activated and desensitized dendritic and somatic D2 receptors, resulting in a depression of the dopamine IPSC.

LTD is induced by D2 receptor activation. a, A 4 min bath application of a high concentration of dopamine (DA, 100 μm) produced an outward current that occluded the dopamine IPSC for the first 10–20 min of washout. a–d, The amplitude of the IPSC remained depressed because of receptor desensitization, even after dopamine was washed out of the slice (1, before dopamine; 2, after 20 min washout). c, d, The amplitude of the dopamine IPSC recovered completely only when intracellular calcium was chelated with BAPTA (10 mm; t(11) = 2.58; p = 0.026; n = 6–7) (Supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Imaging of intracellular calcium was used to further investigate the synergism between D2 receptor desensitization and resting calcium required for LTD. Cells were loaded with Fluo-4 (40 μm) without other calcium chelators. As expected, depolarization of the neuron (to 0 mV for 50–500 ms) resulted in a dramatic rise in intracellular calcium, whereas iontophoresis of dopamine had no effect on calcium levels (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Application of the low-frequency stimulation protocol had no obvious effect on calcium levels. Furthermore, neither the outward current nor the rise in calcium resulting from the depolarization were changed by induction and expression of LTD (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Thus, it appears that a change in intracellular calcium is not responsible for LTD of the dopamine IPSC. Rather, resting calcium levels and the activation of D2 receptors is sufficient (and necessary) to induce LTD.
If desensitization is responsible for LTD of the dopamine IPSC, then any manipulation that inhibits calcium-dependent desensitization of D2 receptors should also inhibit the induction of LTD. Calmodulin, PKC, and β-arrestin are three potential mediators of D2 receptor signaling (Bofill-Cardona et al., 2000; Gainetdinov et al., 2004; Namkung and Sibley, 2004) that were next tested for their role in desensitization. Inhibiting calmodulin with calmidazolium (4 μm) or PKC with staurosporine (1 μm) had no effect on the desensitization induced by dopamine (100 μm) applied for 7 min (control, 69.4 ± 1.8%; calmidazolium, 71.0 ± 3.3%; staurosporine, 69.1 ± 4.9%; n = 6–8). Desensitization was also unchanged in recordings from β-arrestin 2 knock-out mice (72.9 ± 2.4%; n = 7), and LTD of the dopamine IPSC was successfully induced by low-frequency stimulation in four of five cells tested from these animals (data not shown). This suggests that none of these three mediators is singly responsible for D2 receptor desensitization or LTD. The mechanism(s) that underlie this desensitization remain unknown.
Depression of the dopamine IPSC decreases the stimulus-induced pause
The firing of dopamine neurons was examined in the cell-attached patch configuration to determine whether depression of the dopamine IPSC could affect the firing rate and/or pattern. The spontaneous firing of dopamine cells in brain slices is invariably a rhythmic, pacemaker pattern. Applying a train of five stimulations (0.5 ms, 40 Hz) produced a pause in firing that was D2 receptor-dependent and lasted for ∼one second (Beckstead et al., 2004). After the application of the low-frequency stimulation protocol used to induce LTD, there was a modest decrease in the duration the D2 receptor-mediated pause in eight of eight cells tested (Fig. 6). There was no effect on the spontaneous pacemaker firing rate (+4.9 ± 2.1%; n = 8) (data not shown). This result demonstrates that stimulation-induced depression of the dopamine IPSC can affect the pattern of firing after a burst of activity without changing the basal firing rate.

LTD of the dopamine IPSC increases neuron firing. Recordings were obtained in the loose cell-attached patch configuration, and the pacemaker firing pattern that dopamine neurons exhibit in vitro was analyzed. a, As reported previously (Beckstead et al., 2004), electrical stimulation (5 stims) produced a pause in firing that lasted ∼1 s, a result of D2 receptor activation (top trace, 13 sweeps overlaid). b, Low-frequency stimulation (0.5 ms, 2 Hz, 300 s) shortened the duration of the pause (a, bottom trace, 13 sweeps overlaid) in eight of eight cells tested (b; t(8) = 5.92; p = 0.0004). c, This change in firing produced by synaptic depression did not recover for the duration of the experiment.
Discussion
The results demonstrate that low-frequency electrical stimulation produces a robust and persistent decrease in transmission at dendrodendritic dopamine synapses. The blockade of LTD with intracellular application of a high concentration of BAPTA suggests a postsynaptic site of action. This intracellular solution also increased GIRK-dependent currents and reduced D2-receptor dependent desensitization. Thus, it appears that desensitization of D2 receptors may underlie LTD. Another way that is commonly used to identify the site of action is with a paired-pulse protocol. Although the paired-pulse ratio was not affected after the induction of LTD, which would suggest a postsynaptic change, it was considered to be a very insensitive measure because it was not changed even by experiments where presynaptic release was drastically decreased (our unpublished observation).
Although the IPSC is thought to result from dendrodendritic release of dopamine, several observations indicate that the IPSC is not the result of dopamine released from the neuron that is being recorded. (1) All experiments were done in voltage clamp making it unlikely that there would be sufficient calcium entry during the stimulus to evoke transmitter release. (2) There is no change in the outward current after a depolarization following the induction of LTD (supplemental Fig. 2, top traces, available at www.jneurosci.org as supplemental material). (3) Depolarizing the postsynaptic neuron repeatedly to 0 mV at 2 Hz for 300 s did not decrease in the IPSC either by causing an increase in intracellular calcium or by evoking dopamine release. (4) The release of dopamine from a cell filled with BAPTA (10 mm) should be blocked from the chelation of calcium. Larger IPSCs were observed with the BAPTA internal solution. Together, these results indicate that there was insufficient dopamine release from the postsynaptic neuron to affect the IPSC.
Long-term synaptic depression
LTD is often described experimentally as a persistent decrease in synaptic strength that occurs in response to low-frequency electrical stimulation (1–5 Hz, 5–15 min). It has been demonstrated extensively at glutamate synapses (Malenka and Bear, 2004), a few central GABAA and glycinergic synapses (Gaiarsa et al., 2002) and at a kainate synapse in the perirhinal cortex (Park et al., 2006). The depression of dopamine IPSCs described here was blocked by buffering intracellular calcium to a very low level with BAPTA, indicating that LTD is mediated by a postsynaptic mechanism. Unlike some forms of LTD, the depression of the dopamine IPSC does not require a rise in intracellular calcium (Malenka and Bear, 2004). The LTD observed in the present study is similar to that described at excitatory AMPA receptor-dependent synapses on dopamine neurons, where LTD is blocked by chelating intracellular calcium and is not NMDA receptor-dependent (Jones et al., 2000; Thomas et al., 2000; Thomas and Malenka, 2003). A recent report suggests that mGluR activation depresses excitatory transmission in the VTA in a manner distinct from low-frequency stimulation-induced LTD (Bellone and Luscher, 2005). Regardless of mechanistic differences, the present findings demonstrate that dopamine synapses can undergo use-dependent decreases in synaptic strength in a manner similar to previously reported forms of synaptic plasticity. A comparison to other forms of LTD at Gi/o-coupled inhibitory synapses is not possible, as no others have been reported.
LTD was induced by prolonged activation of D2 receptors, either by evoking synaptic release of endogenous dopamine or by perfusion of a high concentration of dopamine. Simply increasing intracellular calcium with glutamate receptor activation or repeated depolarizations did not induce LTD. Furthermore, activation of D2 receptors when intracellular calcium was lowered with BAPTA also did not induce LTD. Thus, induction of LTD required cooperation between a resting calcium level and D2 receptor desensitization. The calcium-imaging experiments confirmed that a rise in intracellular calcium was not required for depression of the dopamine IPSC.
D2 receptor-dependent desensitization
Desensitization of G-protein coupled receptors has been extensively studied in neurons and is thought to involve phosphorylation by G-protein-coupled receptor kinases and subsequent binding of β-arrestin (for review, see Gainetdinov et al., 2004). This desensitization mechanism is well established and explains many of the pharmacological effects of exogenous compounds on G-protein-coupled receptor signaling in central neurons. The demonstration of a decreasing response of photoreceptors to a prolonged exposure to light suggests a physiological role for desensitization of G-protein-coupled receptors (Calvert and Makino, 2002). The present work expands this evidence by demonstrating that G-protein-coupled receptor desensitization can produce synaptic plasticity and cause a robust physiological adaptation in response to the release of endogenous dopamine. Repetitive release of dopamine in dendritic areas may produce a transient high local concentration of dopamine at postsynaptic receptors resulting in desensitization. The localized action of endogenous dopamine was demonstrated by experiments showing that low-frequency stimulation did not decrease the current elicited by dopamine applied at the soma by iontophoresis. This suggests that different receptors may mediate the local action of vesicular dopamine and the exogenously applied dopamine. Desensitization of synaptically activated receptors produced depression of the IPSC whereas receptors that were activated by the iontophoretic application of dopamine applied at the cell body remained unaffected.
Inhibitory autoreceptors such as the D2 receptor are thought to provide a negative cellular feedback mechanism. The depression of the dopamine IPSC would diminish autoreceptor signaling in dopamine neurons, effectively removing one mechanism that “brakes” cell excitability. Thus, autoreceptor desensitization is itself counterintuitive and seems more likely to be responsible for the creation of disease states rather than serving a homeostatic physiological role. Autoreceptor desensitization in the cell body region could also be responsible for some of the effects of the dopamine precursor l-DOPA in Parkinson's disease and other hypodopaminergic syndromes. If l-DOPA also induced sustained depression of D2 receptors in the substantia nigra, it could directly increase cell excitability and firing. This would increase terminal dopamine release and perhaps contribute to therapeutic efficacy.
LTD increases dopamine neuron firing
In vivo, dopamine neurons switch between a single pulse, random firing pattern and a more active bursting mode consisting of series of 2–8 closely spaced action potentials (Grace and Bunney, 1984). Bursting facilitates release of dopamine in terminal limbic fields, overwhelming uptake transporters and producing a supra-additive phasic response (Chergui et al., 1994; Floresco et al., 2003). This transition from tonic to phasic levels of extracellular dopamine is hypothesized to be ultimately responsible for many of the behavioral consequences of dopamine release (Phillips et al., 2003). The excitatory and inhibitory inputs that shape the pattern of activity are severed in acutely prepared brain slices and, consequently, dopamine neurons exhibit a rhythmic, pacemaker-like spontaneous firing pattern in vitro (Grace and Onn, 1989). The low-frequency stimulation-induced depression of the dopamine IPSC did not significantly increase the dopamine neuron pacemaker firing rate. However, when a burst of action potentials was mimicked with a train of five stimulations, the duration of the postburst pause was decreased. Although it is difficult to predict the precise influence on firing in vivo, these results suggests that plasticity of the dopamine IPSC would more prominently affect active bursting cells than quiescent or slowly firing ones.
In summary, applying low-frequency stimulation to dopamine neurons in the ventral midbrain induces a novel form of synaptic plasticity: long-term depression of dendrodendritic dopamine-mediated IPSCs. The mechanism for this plasticity is a calcium-dependent, agonist-induced postsynaptic desensitization of D2 receptors.
Footnotes
-
This work was supported by National Institute on Drug Abuse Grants F32 DA16467 (M.J.B.) and R01 DA4523 (J.T.W.). J.T.W. was a NARSAD Ritter Foundation Investigator.
- Correspondence should be addressed to John T. Williams, Vollum Institute, L474, Oregon Health and Science University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239. williamj{at}ohsu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}