

Abstract
Substantia nigra pars reticulata (SNr) is a key basal ganglia output nucleus critical for movement control. Its GABA-containing projection neurons intermingle with nigral dopamine (DA) neuron dendrites. Here we show that SNr GABA neurons coexpress dopamine D1 and D5 receptor mRNAs and also mRNA for TRPC3 channels. Dopamine induced an inward current in these neurons and increased their firing frequency. These effects were mimicked by D1-like agonists, blocked by a D1-like antagonist. D1-like receptor blockade reduced SNr GABA neuron firing frequency and increased their firing irregularity. These D1-like effects were absent in D1 or D5 receptor knock-out mice and inhibited by intracellularly applied D1 or D5 receptor antibody. These D1-like effects were also inhibited when the tonically active TRPC3 channels were inhibited by intracellularly applied TRPC3 channel antibody. Furthermore, stimulation of DA neurons induced a direct inward current in SNr GABA neurons that was sensitive to D1-like blockade. Manipulation of DA neuron activity and DA release and inhibition of dopamine reuptake affected SNr GABA neuron activity in a D1-like receptor-dependent manner. Together, our findings indicate that dendritically released dopamine tonically excites SNr GABA neurons via D1–D5 receptor coactivation that enhances constitutively active TRPC3 channels, forming an ultra-short substantia nigra pars compacta → SNr dopamine pathway that regulates the firing intensity and pattern of these basal ganglia output neurons.
Introduction
The substantia nigra is an important component of the basal ganglia (DeLong and Wichmann, 2007). It comprises two distinct parts: pars compacta (SNc) and pars reticulata (SNr). The major cell type in SNc is the dopamine (DA) neuron whose axon projects mainly to the striatum, forming the long-distance nigrostriatal DA pathway (Parent et al., 2000; Haber, 2003). SNr is populated primarily by a majority of GABA projection neurons and a minority of DA projection neurons (Fallon and Loughlin, 1995; Nelson et al., 1996; Parent et al., 2000). The GABA projection neurons fire spontaneous, mostly regular, high-frequency spikes (Hikosaka and Wurtz, 1983; Schultz, 1986).
SNr is a key output nucleus of the basal ganglia and inhibits its targets such as brainstem motor nuclei and thalamus (Hikosaka et al., 2000; McHaffie et al., 2005). It is critical to motor control, eye, and orofacial movements in particular. SNr neuronal activity is known to be influenced by the striatal and pallidal inputs that are themselves regulated by SNc-originated DA projection, thus forming the nigro-striato-nigral loop (Haber, 2003). Loss of SNc DA neurons in animal models of Parkinson's disease (PD) and human PD patients often alters the firing frequency and/or pattern of the SNr neurons (Wichmann et al., 1999; Obeso et al., 2000; Nevet et al., 2004).
Evidence indicates that nigral DA cell somata and dendrites may release DA via vesicular exocytosis and/or reverse transport (Cheramy et al., 1981; Jaffe et al., 1998; Cragg et al., 2001; Falkenburger et al., 2001; Beckstead et al., 2004; Staal et al., 2004; Rice and Cragg, 2008). The Ca2+-dependent vesicular DA release from DA neuron dendrites may be triggered by pace-making activity-driven, L-type Ca2+ channel-mediated Ca2+ oscillation (Chan et al., 2007), potentially leading to a tonic extracellular DA level that is also regulated by reuptake (Cragg et al., 2001). Indeed, somatodendritically released DA has been indicated to induce a tonic autoinhibition of nigral DA neurons by activating the inhibitory D2 autoreceptors (Lacey et al., 1987; Pucak and Grace, 1994; Mercuri et al., 1997).
SNc DA neurons extend their dendrites deep into SNr (see Fig. 1A) (Björklund and Lindvall, 1975; González-Hernández and Rodríguez, 2000), in which tyrosine hydroxylase (TH)-positive DA dendrites and parvalbumin (PV)-positive GABA neurons intermingle closely. Such a proximity between DA dendrites and SNr GABA neurons may have important functional consequences because dendritically released DA may diffuse for a considerable distance as a result of the relatively low density of dopamine transporter (DAT) in SNr (Cragg et al., 2001).
These anatomical and physiological facts led to our hypothesis that dendritically released DA may directly activate DA receptors on SNr GABA output neurons, forming an ultra-short SNc → SNr DA pathway that provides a fast DA control over this key basal ganglia output nucleus. Because DA receptors do not directly gate any ion channel, we further hypothesized that DA receptor activation may couple to and enhance TRPC3 channels, a type of cation channel that have been shown recently to be tonically active and regulate SNr GABA neuron activity (Zhou et al., 2008).
Materials and Methods
Mice.
Wild-type 2- to 4-week-old male and female C57BL/6J mice were purchased from The Jackson Laboratory. D1 knock-out (KO) mice and D5 KO mice were generated, maintained, and genotyped according to established procedures (Xu et al., 1994; Hollon et al., 2002). All mice were housed in the animal facility on the campus of University of Tennessee Health Science Center (Memphis, TN).
Patch-clamp recording in midbrain slices.
Coronal midbrain slices containing SNc and SNr were prepared according to well established procedures (Atherton and Bevan, 2005; Zhou et al., 2006, 2008). The cutting solution contained the following (in mm): 220 sucrose, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, and 10 d-glucose. The slices were then transferred to a holding chamber containing the bathing solution (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2.5 CaCl2, 1.3 MgCl2, and 10 d-glucose. This bathing solution was also the normal perfusing solution. Drugs were applied via the bathing solution. The perfusion rate was 2 ml/min controlled by a peristaltic pump. Patch electrodes had resistances of 2–3 MΩ when filled with intracellular solution (in mm): 135 KCl, 0.5 EGTA, 10 HEPES, 2 Mg-ATP, and 0.2 Na3GTP. pH was adjusted to 7.3 with KOH. In a few cells, 125 mm K-gluconate replaced KCl on an equal molar basis. For perforated patch-clamp recording, 50 μg/ml gramicidin was added to the KCl-based intracellular solution that led to perforated whole-cell mode 10–15 min after forming tight seal.
Recordings were made at 30°C under visual guidance of a video microscope (Olympus BX51W1) equipped with Nomarski optics and 60× water-immersion lens. We targeted large cells (likely to be GABA projection neurons) for recording and selected cells with action potentials overshooting at least +20 mV for analysis. Signals were recorded with Multiclamp 700B patch-clamp amplifiers, low-pass filtered at 5 kHz by the built-in Bessel filter in the Multiclamp 700B amplifiers, digitized at 5–20 kHz, and analyzed with pClamp 9 program (Molecular Devices). For display and for reducing file size for the computer to handle, data (except action potentials) were digitally refiltered at 1 or 2 kHz and resampled at lower sampling rates. Recordings with access resistance increase of ≥15% were rejected.
The summary data in Figures 2B and 4B were from two groups of selected neurons with stable baseline firing, good response to dopaminergic drugs, good recovery, and also matching temporal patterns in their response and recovery. A total of 21 neurons with significantly no-matching temporal patterns in their response and recovery were excluded to prevent the smearing-out of the response and recovery. The excluded cells had a response magnitude similar to that of included cells such that this exclusion did not affect our data or conclusion at all. Also, the recording of the initial several minutes was often not stable and thus was not included in analysis. The starting point of stable recording was defined as time = 0 min.
Identification of substantia nigra pars reticulata GABA and DA neurons.
Reliable and unambiguous identification of SNr GABA and DA neurons by their distinct electrophysiological characteristics has been described in detail in previous studies (Atherton and Bevan, 2005; Zhou et al., 2006; Lee and Tepper, 2007; Zhou et al., 2008).
Double immunostaining of GABA neurons and DA neurons.
Under lethal anesthesia, mice were intracardially perfused with the ice-cold cutting solution described above and then 4% paraformaldehyde in 0.1 m phosphate buffer (PB). The removed brains were postfixed in the same fixative for 4 h at 4°C and then infused with 30% sucrose in 0.1 m PB, pH 7.4, at 4°C over two nights. Coronal midbrain sections (50 μm in thickness) were cut on a Vibratome 1000 Plus. The sections then were processed for immunofluorescence detection of PV [a calcium-binding protein localized in GABA neurons but not in DA neurons in the SNr (González-Hernández and Rodríguez, 2000)] and TH (the rate limiting enzyme in DA biosynthesis). The free-floating sections were incubated with 2% fat-free milk, 1% bovine serum albumin (BSA), and 0.8% Triton X-100 in 0.01 m PBS for 1 h at room temperature to block nonspecific binding and permeate the cell membrane. After thorough rinsing, free-floating sections were incubated for 48 h at 4°C with the two primary antibodies (see below) and then rinsed in 0.01 m PBS three times for 10 min, followed by incubating with the two secondary antibodies (see below) for 2 h at room temperature in the dark. Both the primary and secondary antigen-antibody reactions occurred in 0.01 m PBS containing 3% normal donkey serum, 1% BSA, and 0.8% Triton X-100. The two primary antibodies were a polyclonal TH antibody raised in sheep (diluted at 1:1000; Millipore Corporation) and a monoclonal PV antibody raised in mouse (diluted at 1:2000; Sigma). The two secondary antibodies were as follows: (1) donkey anti-sheep IgG antibody, conjugated with red Alexa Fluor 594 (diluted at 1:200; Invitrogen), used for labeling TH, and (2) donkey anti-mouse IgG antibody, conjugated with green fluorescent Alexa Fluor 488 (diluted at 1:200; Invitrogen), used for labeling PV. Detection of intracellularly infused D1 antibody was achieved by using an Alexa Fluor 594-tagged secondary antibody. An identical strategy was used in the study by Zhou et al. (2008).
Single-cell reverse transcription PCR.
Our single-cell reverse transcription (scRT)-PCR procedures were described in detail by Zhou et al. (2008) and followed the principles and general methods of Surmeier et al. (1996). Patch pipettes were autoclaved to inactivate RNase. The intracellular solution (in mm: 135 KCl, 0.5 EGTA, 10 HEPES, and 1.5 MgCl2, pH adjusted to 7.3) was prepared with RNase- and DNase-free water. Brief (≤5 min) recording was done to provide electrophysiological fingerprints for SNr GABA and DA neurons. Once a stable and reliable recording was obtained, gentle suction was applied to aspirate the cytoplasm.
The aspirated cell content was expelled into a 0.2 ml PCR tube, and potential genomic DNA was digested by DNase I (5 min at room temperature). Complete removal of genomic DNA was verified by RT-minus control in which no expected amplicon was detected when the reverse transcriptase was omitted, whereas all other reaction components were exactly the same. cDNA was synthesized using SuperScript III reverse transcriptase-based Cells-Direct cDNA Synthesis kit containing RNase inhibitor (Invitrogen). RT was performed at 50°C for 50 min, yielding 30–35 μl of cDNA solution. The reaction was terminated by heating to 85°C for 5 min and then chilling on ice. The 30–35 μl of cDNA solution was used either immediately for PCR amplification or stored at −80°C for later use. All steps ware performed according to the instructions of the manufacturer.
The cDNAs were amplified using a hot start, high specificity, and high yield Platinum PCR SuperMix (Invitrogen). To increase the detection probability of D1 and D5 mRNAs in a single neuron, the two-stage, nested PCR procedure was used, as detailed by Zhou et al. (2008).
To avoid potential interference among the primers, singleplex scRT-PCR was used in the initial screening (Fig. 1B). In later experiments, to confirm the coexpression of multiple mRNAs in a single cell, the limited amount (30–35 μl) of cDNA from a single neuron was divided, and separate PCR runs were performed to detect D1, D5, TRPC3, and glutamic acid decarboxylase 1 (GAD1) mRNAs in the same SNr GABA neurons (Fig. 1C).
PCR products from the second-stage PCR amplification were separated by 2% agarose gel electrophoresis and visualized by ethidium bromide under ultraviolet light. Then the bands on the gel were cut out, and the DNA fragments were extracted and purified with QiaQuick extraction kit (Qiagen) and sequenced. The obtained sequences were positively identified at http://blast.ncbi.nlm.nih.gov/Blast.cgi.
Primers were designed according to the sequences in GenBank (supplemental Table S1, available at www.jneurosci.org as supplemental material) and were accomplished using the web-based Primer3 software (Massachusetts Institute of Technology, Cambridge, MA). Whenever possible, intron-spanning primer pairs were used that help detect genomic DNA contamination. In cases in which negative scRT-PCR results were obtained, the effectiveness of the primer pairs was verified by whole-brain mRNAs that yielded positive products (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). All primers were synthesized by Integrated DNA Technologies.
To rule out contamination of extracellular debris that may contain mRNAs, the patch pipette was lowered into the tissue without actually aspirating cytoplasm, and then the pipette content was subjected to RT-PCR, yielding no product at all.
Chemicals.
All chemicals, including d-2-amino-5-phosphonopentanoic acid (d-AP-5), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and picrotoxin, were purchased from Sigma-Aldrich or Tocris Bioscience. Fresh DA solutions were used for each experiment.
Statistics.
Descriptive statistics such as mean and SE were used. Paired t test and Kolmogorov–Smirnov (K–S) test were also used to compare measurements in the same groups/cells when applicable. ANOVA was used for comparing the measurements obtained in two groups of mice. Linear regression analysis was used to analyze the linearity of the I–V curves and determine the reversal potential. p < 0.05 was considered to be significant.
Results
SNr GABA neurons express D1 and D5 receptor mRNAs
The first question we addressed was: do SNr GABA neurons express any DA receptors? We chose scRT-PCR to answer this question because of its high sensitivity and specificity (Surmeier et al., 1996). SNr GABA neurons in brain slices were identified by their characteristic fast spiking properties (Atherton and Bevan, 2005; Zhou et al., 2008). Then the cell content was aspirated into the recording patch pipette and expelled into a PCR tube. Reverse transcription was performed, followed by two-stage nested PCR (Zhou et al., 2008). Singleplex PCR was used to avoid potential interference among primers. We also used detection of GAD1 (a key GABA synthesis enzyme) mRNA as an indicator of successful cytoplasmic aspiration and reverse transcription. If GAD1 mRNA was not detected, indicating failure in cytoplasm aspiration and/or reverse transcription, no additional detection of DA receptor mRNAs was pursued. Under these conditions, D1 and D5 mRNAs were detected in every GAD1 mRNA-positive SNr GABA neuron (n = 16) (Fig. 1B), demonstrating that most, if not all, SNr GABA neurons express D1 and D5 mRNAs (Fig. 1B). In contrast, D2, D3, and D4 receptor mRNAs were not detected (for positive control for primer effectiveness, supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The mRNA for the key enzyme for DA synthesis, TH, was not detected in any of these electrophysiologically identified SNr GABA neurons (data not shown). Furthermore, after dividing the limited amount (30–35 μl) of cDNA from a single SNr GABA neuron, separate PCR runs detected D1, D5, TRPC3, and GAD1 mRNAs in the same single SNr GABA neurons (n = 5) (Fig. 1C). These results indicate a coexpression of D1 and D5 receptors in SNr GABA neurons, providing a molecular substrate for dendritically released DA to directly influence these basal ganglia output neurons and also for a potential interaction between DA signaling and TRPC3 channel activity.

The SNc DA dendrites intermingle with SNr GABA neurons expressing D1 and D5 receptor and TRPC3 mRNAs. A, Confocal images of double-immunohistochemical staining for TH (red; a key enzyme for DA synthesis) and PV (green; expressed only in GABA neurons). Most TH-positive DA neurons are in SNc with their dendrites extending into SNr, in which most neurons are the PV-positive GABA neurons. Scale bar, 100 μm. B, To ensure maximal detection, singleplex scRT-PCR was used to detect GAD1 mRNA in electrophysiologically identified individual SNr GABA neurons (B1). Positive GAD1 mRNA detection indicated successful cell content aspiration and RT, providing the necessary condition for subsequent detection of DA receptor mRNAs from the same cell sample (B2). All amplicons were of expected sizes. No TH mRNA amplicon of 197 bp was seen in GABA neurons, but it was detected in electrophysiologically identified DA neurons. D5/RT-, RT-minus negative genomic DNA control for the intronless D5 gene. D2, D3, and D4 receptor mRNAs were not detected (for positive control for their primers, see supplemental Fig. S1, available at www.jneurosci.org as supplemental material). C, After dividing the limited amount (30–35 μl) of cDNA from a single neuron, separate PCR runs detected D1, D5, TRPC3, and GAD1 mRNAs in the same single SNr GABA neuron, confirming the coexpression of D1, D5, GAD1, and TRPC3 mRNAs in these neurons.
Dopamine directly excites SNr GABA projection neuron firing
If functional D1 and D5 receptors are indeed expressed in SNr GABA neurons, then DA should induce D1-like excitation as seen in other brain areas (Aosaki et al., 1998; Centonze et al., 2003). To detect potential direct D1-like excitation in SNr GABA neurons, we first blocked glutamate- and GABA-mediated fast synaptic transmission with 20 μm d-AP-5, 10 μm CNQX, and 100 μm picrotoxin to remove complications of synaptic activity. Under this condition, bath application of 10 and 30 μm DA reliably increased the firing rate of SNr GABA neurons by 16.2 ± 1.9 and 31.0 ± 2.1% from a baseline frequency of 10.7 ± 0.9 Hz, respectively (measured over the 2-min peak enhancement period; p < 0.01; n = 14) (Fig. 2A,B).

DA directly excited SNr GABA projection neurons. All recordings were made in the presence of picrotoxin, d-AP-5, and CNQX to block fast synaptic transmission. Sulpiride was present to prevent potential complications from D2 autoinhibition of DA neurons. A, Examples of spontaneous spikes under control conditions (left) and during 10 μm DA application (right). B, Group data of DA enhancement of spiking. The firing enhancement, measured over the 2 min peak enhancement period, was 16.2 ± 1.9 and 31.0 ± 2.1% for 10 and 30 μm DA, respectively. n = 14. C, An example recording showing that, after blocking spikes with 1 μm TTX, DA induced a clear depolarization. D, An example recording showing that, when voltage clamped at −70 mV, DA induced a clear inward current in the presence of 1 μm TTX. Note that SNr GABA neurons are normally depolarized and have a large holding current when clamped at −70 mV (Zhou et al., 2008). E, E1 shows the dose–response relationship of DA-induced inward current. The curve is from Hill equation fitting: EC50 was 13.3 ± 0.7 μm and Hill slope of 1.2 ± 0.1. n = 5–9 for each data point. E2 and E3 are example inward currents induced by 5 and 100 μm DA, respectively, in an SNr GABA neuron voltage clamped at −70 mV.
The question now was: how did DA induce this excitation? We found that DA did not affect action potential shape and, after adjusting membrane potential to the pre-DA level with hyperpolarizing current, it also did not alter the fast afterhyperpolarization (AHP) nor medium AHP. These results indicate that DA enhanced SNr GABA neuron firing without detectable effect on voltage-gated Na+ and K+ channels or Ca2+-activated K+ channels responsible for action potential generation and repolarization and the AHPs. Therefore, other mechanisms must be involved. We hypothesized that the primary mechanism of DA excitation could be a DA-induced direct depolarization in SNr GABA neurons.
To characterize the DA-induced depolarization, action potentials were blocked with 1 μm tetrodotoxin (TTX) to obtain stable membrane potential in SNr GABA neurons (Fig. 2C). Under this condition, bath application of 10 and 30 μm DA induced a clear depolarization of 1.8 ± 0.1 and 2.7 ± 0.2 mV from a baseline membrane potential of −49.3 ± 1.3 mV, respectively (n = 9) (Fig. 2C). These DA-induced direct depolarization amplitudes are similar to those in striatal cholinergic interneurons (Centonze et al., 2003) and medium spiny neurons (Yasumoto et al., 2002). Because SNr GABA neurons have a depolarized membrane potential close to the spike threshold, even a modest depolarization will affect neuronal firing and circuitry operation, conferring physiological significance onto this direct DA effect.
To further test our hypothesis, SNr GABA neurons were voltage clamped at −70 mV. At this holding potential, bath application of 10 and 30 μm DA induced an inward current of 17.4 ± 1.7 and 31.3 ± 2.9 pA from a baseline membrane current of −160.1 ± 16.5 pA, respectively (n = 9) (Fig. 2D) [much of the baseline holding current was attributable to the tonic TRPC3 current (Zhou et al., 2008)]. Hill equation fitting indicated an EC50 of 13.3 ± 0.7 μm with a Hill slope of 1.2 ± 0.1 (Fig. 2E). Whole-cell conductance, monitored with −10 mV, 100 ms voltage pulses, was increased from 5.28 ± 0.41 nS under control conditions to 6.55 ± 0.66 nS during 30 μm DA application (n = 8; p < 0.05), suggesting an opening or enhancement of ion channels that mediate an inward current at −70 mV. All these DA-evoked effects were prevented by 5 μm D1-like antagonist SKF83566 (8-bromo-2,3,4,5-tetrahydro-3-methyl-5-phenyl-1H-3- benzazepin-7-ol hydrobromide) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). A similar inward current was induced by 30 μm DA in SNr GABA neurons voltage clamped at −70 mV under conventional whole-cell recording mode with K-gluconate-based intracellular solution (32.5 ± 4.4 pA; n = 4) and under gramicidin-perforated recording configuration (29.7 ± 4.7 pA; n = 3).
Next, a voltage-ramp protocol was used to determine the I–V relationship of the DA-induced current. The linear ramp was between −90 and −20 mV. After obtaining a stable baseline recording, the ramp protocol was activated and 30 μm DA was bath applied. The net DA-induced current was obtained by subtracting the current generated under control condition from the current generated during DA application (Fig. 3A). Linear regression analysis indicated that the DA current I–V was linear between −90 and −20 mV with a reversal potential of −37.3 ± 1.3 mV (n = 5), suggesting a tonically active, mixed cation channel. It was not Ih because Ih has a voltage-dependent activation with a threshold at −60 mV or more negative (Harris and Constanti, 1995). In SNr GABA neurons, Ih is very small. Furthermore, the DA current I–V curve in the presence of ZD7288 (4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride), a selective Ih blocker (Harris and Constanti, 1995), was similar to that obtained without ZD7288. As will be discussed below (see Fig. 6), this current was likely mediated by TRPC3 channels that are tonically active cation channels in SNr GABA neurons (Zhou et al., 2008).

DA or a D1-like agonist induced a linear, voltage-independent inward current in SNr GABA projection neurons. A, Voltage ramp reveals the I–V relationship of DA-induced current. The ramp was between −90 and −20 mV. The 30 μm DA-induced net current (red trace) was obtained by subtracting the current generated under control conditions (black trace) from the current generated during DA application (blue trace). In other words, the red, net current trace = the blue DA trace − the black control trace. Linear regression analysis indicated a reversal potential at approximately −37 mV. B, The same voltage ramp protocol revealed that 5 μm SKF81297 (SKF) induced a similar linear current with a reversal potential also at approximately −37 mV. Identical subtraction procedure was performed. All traces were individual traces.
D1-like receptor agonists mimicked and an antagonist blocked D1-like effects in SNr GABA neurons
Based on the results described above, we reasoned that D1 and/or D5 receptors were mediating the direct excitation of DA in SNr GABA neurons. If this hypothesis was correct, then D1-like agonists should mimic the effects of DA. To test this reasoning, we investigated the effects of SKF81297 (6-chloro-2,3,4,5-tetrahydro-1-phenyl-1 H-3-benzazepine hydrobromide) and dihydrexidine, two selective, full agonists for D1-like receptors (Lovenberg et al., 1989; Aosaki et al., 1998). After a stable baseline recording, bath application of 5 μm SKF81297 had a clear excitatory effect on SNr GABA neurons (Fig. 4), similar to that induced by 30 μm DA (Fig. 2). The spontaneous spike firing rate was increased by 29.6 ± 2.5% during SKF81297 application (n = 9; p < 0.01) (Fig. 4A,B). This effect was prevented by 5 μm D1-like blocker SKF83566 (Fig. 4B), clearly indicating an involvement of D1-like receptors.

D1-like agonist SKF81297 mimicked DA effects in SNr GABA projection neurons that were prevented by the D1-like blocker SKF83566. All recordings were made in the presence of picrotoxin, d-AP-5 and CNQX to block fast synaptic transmission. A, Examples of spontaneous spikes under control (left) and during 5 μm SKF81297 application (right). B, Group data showing SKF81297 enhancement of firing and the recovery of the effect. The SKF81297-induced firing enhancement was 29.6 ± 2.5%, measured over the 2 min peak enhancement period. n = 9. The D1-like excitatory effect was prevented by the D1-like antagonist SKF83566. Note that SKF83566 induced an inhibition indicated by the arrowhead, suggesting an endogenous tonic D1-like activity that will be discussed later. At the end of the recording, 30 μm DA was applied to show that the neuron was still healthy. C, D, Example recordings showing that, after blocking spikes with 1 μm TTX, SKF81297 induced a clear depolarization in current-clamp recording mode (C) and a clear inward current when voltage clamped at −70 mV (D). This depolarization or inward current was prevented by the D1-like antagonist SKF83566 (5 μm). Also note that SKF83566 itself induced a hyperpolarization or outward current (arrowhead in C, D), suggesting an endogenous tonic D1-like activity (addressed later).
To further characterize the depolarization that accompanied the SKF81297-induced increase in SNr GABA neuron firing rate, action potentials were blocked with 1 μm TTX such that changes in membrane potential could be reliably detected. Under these conditions, bath application of 5 μm SKF81297 induced a clear depolarization of 2.6 ± 0.2 mV (n = 9), and this effect was prevented by 5 μm D1-like antagonist SKF83566 (Fig. 4C).
When the SNr GABA neurons were voltage clamped at −70 mV, 5 μm SKF81297 induced an inward current of 30.8 ± 3.8 pA (n = 8) (Fig. 4D), and whole-cell conductance was increased to 6.22 ± 0.34 nS from 5.12 ± 0.40 nS under control condition (n = 6), indicating an opening of ion channels. These effects were prevented by 5 μm SKF83566 (Fig. 4D). Furthermore, in the presence of 1 μm TTX, voltage ramp experiments showed that the SKF81297-induced current was linear between −90 and −20 mV with a reversal potential of −37.5 ± 1.5 mV (n = 4) (Fig. 3B). These characteristics were identical to those of the DA-induced current (Fig. 3A).
Similar current-clamp and voltage-clamp results were obtained with a second D1-like agonist, dihydrexidine, that is molecularly distinct from the phenylbenzazepines such as SKF81297 (Lovenberg et al., 1989).
Together, these data indicate that D1-like agonists mimic, and a D1-like antagonist blocks, DA-induced effects in SNr GABA neurons, supporting our idea that D1 and/or D5 receptor activation may directly depolarize these basal ganglia output neurons.
In contrast, in the presence of 5 μm D1-like antagonist SKF83566, neither D2-like receptor agonist quinpirole (Lacey et al., 1987; Mercuri et al., 1997) (1–10 μm) nor D2-like receptor antagonist sulpiride (1 μm) affected the firing rate, membrane potential, or membrane current in SNr GABA neurons (n = 2–3; data not shown). These results were consistent with our scRT-PCR data showing a lack of D2, D3, and D4 mRNA in SNr GABA neurons. Quinpirole and sulpiride had typical effects in SNc and SNr DA neurons (n = 3–6) as reported in the literature (Lacey et al., 1987).
Absence of D1-like DA effects in SNr GABA neurons in D1 or D5 receptor KO mice
D1 and D5 receptors are known to induce similar effects (Neve et al., 2004), and SNr GABA neurons express both D1 and D5 receptor mRNAs (Fig. 1B,C). Thus, the D1-like effects described above may be mediated by D1 and/or D5 receptors. There is currently no ligand selective for D1 or D5 receptors. To overcome this difficulty, we used D1 receptor KO mice and D5 KO mice (Xu et al., 1994; Hollon et al., 2002).
In contrast to wild-type mice, in SNr GABA neurons from D1 KO mice, 30 μm DA had no significant effect on the spontaneous firing rate (9.9 ± 0.8 Hz under control vs 9.7 ± 0.8 Hz during DA; p > 0.05; n = 5). In the presence of 1 μm TTX, 30 μm DA had no significant effect on the membrane potential (n = 4) (Fig. 5A) or membrane current (n = 5; data not shown) in SNr GABA neurons from D1 KO mice. SNr GABA neurons (n = 5) from wild-type littermates showed the same D1-like DA responses as in wild-type mice described previously. Similar to D1 KO mice, in SNr GABA neurons from D5 KO mice, 30 μm DA did not affect the spontaneous firing rate (9.6 ± 0.7 Hz under control vs 9.5 ± 0.7 Hz during DA; p > 0.05; n = 4), membrane potential (n = 4) (Fig. 5B), or membrane current (n = 5).

Both D1 and D5 receptors are needed for D1-like excitation in SNr GABA neurons. A, B, Exogenous 30 μm DA was without effect on the membrane potential in SNr GABA neurons in both D1 KO mice (A) and D5 KO mice (B). C, D, In wild-type mice, intracellular infusion of D1 receptor antibody caused a small but clear hyperpolarization, indicating a tonic D1-dependent depolarization. Once infused with D1 antibody, the DA-induced depolarization was greatly inhibited (C). Inset, Conventional fluorescence pictures of immunostaining detection of D1 antibody infused into a PV-positive SNr GABA neuron. In contrast, intracellular infusion of a mixture of D1 antibody and its antigen peptide had no effect by itself and did not inhibit the DA response (D). E, F, Similarly, in wild-type mice, intracellular infusion of D5 receptor antibody caused a hyperpolarization (0.6 ± 0.1 mV; n = 5), indicating a tonic D5-dependent depolarization. Once infused with D5 antibody, 30 μm DA only induced a depolarization of 0.6 ± 0.1 (E). Intracellular infusion of a mixture of D5 antibody and its antigen peptide had no effect by itself and did not inhibit the DA response with 30 μm DA inducing a depolarization of 2.5 ± 0.4 mV (F). Comparable voltage-clamp data were also obtained. G, H, Intracellular infusion of D1 receptor antibody into SNr GABA neurons in D1 KO mice had no effect (G). Similarly, infusion of D5 receptor antibody into SNr GABA neurons in D5 KO mice also had no effect (H). All traces were individual traces.
Also, in these same D1 KO mice and D5 KO mice, SNc DA neurons (n = 4–6) and SNr DA neurons (n = 3–4) displayed the typical D2 receptor-mediated autoinhibition when 30 μm DA was bath applied, indicating that the lack of D1-like responses in SNr GABA neurons in these mutant mice was not attributable to unhealthy brain slices.
In aggregate, these results indicate an involvement of an apparent D1 and D5 receptor coactivation, i.e., both D1 and D5 receptors is required for the D1-like responses.
D1 or D5 receptor antibody inhibited the D1-like effects in SNr GABA neurons
To provide additional evidence from a different approach for the D1 and D5 receptor coactivation indicated by D1 and D5 receptor knock-out mice, we used D1 and D5 receptor antibodies against an intracellular domain of the receptor proteins and applied these antibodies intracellularly via the patch pipette. Intracellular infusion of proteins (enzymes and antibodies) is a well established technique (Tauc, 1980; Aragay et al., 1995). The antibody binds to and may thus interfere with the functioning of the corresponding receptor. This strategy has been used to identify receptor subtypes when subtype-specific ligands are not available (Amaral and Pozzo-Miller, 2007; Zhao et al., 2008; Zhou et al., 2008) (Dr. Jonathan Jaggar, personal communication). The antibodies, purchased from Santa Cruz Biotechnology, were diluted at 1:100 in intracellular pipette solution. We found that intracellular application of D1 receptor antibody caused a clear hyperpolarization in current clamp (0.6 ± 0.1 mV; n = 5) (Fig. 5C). Effective infusion of the antibody into the recorded neuron was confirmed by immunohistochemical detection (Fig. 5C, inset). More importantly, in cells infused with D1 receptor antibody, 30 μm DA induced a depolarization of only 0.6 ± 0.1 mV (n = 5) (Fig. 5C), a 78% inhibition compared with that under control condition shown in Figure 2C. When voltage clamped at −70 mV, similar results that mirror the current-clamp data were obtained (data not shown). In contrast, a mixture of D1 receptor antibody and its antigenic peptide (at a weight ratio of 1:1 or 1:2) had no effect by itself and did not inhibit the 30 μm DA-induced depolarization (2.7 ± 0.4 mV; n = 3) (Fig. 5D). Thus, the effects of D1 receptor antibody were specific.
Intracellularly applied D5 antibody had effects similar to those of D1 receptor antibody (n = 5) (Fig. 5E,F). Furthermore, D5 receptor antigen peptide did not neutralize the effects of the D1 antibody (n = 2), and D1 antigen peptide did not neutralize the effects of the D5 antibody (n = 2), also indicating that the effects of D1 and D5 antibodies were specific. As shown in Figure 5, C and E, intracellularly applied D1 or D5 antibody, respectively, also induced a hyperpolarization, indicating a tonic D1 and D5 receptor-dependent depolarization that was inhibited by either one of these two antibodies. Finally, none of these antibody-induced effects was seen in SNr GABA neurons in D1 KO mice (n = 3) and D5 KO mice (n = 3) (Fig. 5G,H).
Together, our data clearly suggest that an apparent coactivation of D1 and D5 receptors was required to cause an inward current or depolarization in SNr GABA neurons. Inhibition of one of these two receptors led to the inhibition of the entire D1-like excitation.
Next we turned to this question: because D1 and D5 receptors do not directly gate any ion channel, what effector ion channel mediates the D1-like excitation? The experiments below were designed to identify the effector ion channel(s).
TRPC3 channels mediate the D1-like effects in SNr GABA neurons
Our data presented in Figure 3 indicate that the DA current I–V curve was linear between −90 and −20 mV with a reversal potential at approximately −37 mV, suggesting a constitutively active, mixed cation channel, similar to the I–V relationship of the TRPC3 channel-mediated current in SNr GABA neurons (Zhou et al., 2008). Our scRT-PCR data also showed that D1, D5, and TRPC3 mRNAs were coexpressed in the same SNr GABA neurons (Fig. 1C). Together, these data indicate that the apparent D1–D5 receptor coactivation opened a type of depolarizing cation channels, consistent with the idea that DA may enhance the tonically active TRPC3 channels. To test this hypothesis, we investigated whether blocking TRPC3 channels also blocked the D1-like excitation (Fig. 6A).

TRPC3 channels are the likely effector of D1–D5 receptor coactivation in SNr GABA neurons. A, Diagrammatic representation of the hypothesized D1–D5 receptor coactivation coupled to TRPC3 channels that may be inhibited by intracellularly infused TRPC3 antibody. B, Intracellular infusion of TRPC3 channel antibody revealed a tonic depolarization. Once infused with TRPC3 antibody, the DA-induced depolarization was greatly reduced. The open arrow points to the gradual process by which TRPC3 antibody exerts its effect. C, Premixing of TRPC3 antibody with TRPC3 antigenic peptide neutralized TRPC3 antibody's effects (compare with B), indicating that TRPC3 antibody effects were specific. Comparable voltage-clamp data were also obtained. The traces in B and C were individual traces.
Because of the lack of selective agonists and antagonists, we applied a TRPC3 antibody intracellularly via the recording pipette as illustrated in Figure 6A. The TRPC3 antibody (purchased from Alomone Labs) is known to interfere with or inhibit the functioning of TRPC3 channels (Albert et al., 2006; Amaral and Pozzo-Miller, 2007; Zhou et al., 2008). Intracellular infusion of this antibody (diluted at 1:100 in the pipette solution) caused a hyperpolarization of 6.9 ± 0.6 mV (n = 6), probably by inhibiting the tonic TRPC3-mediated depolarization (Fig. 6B, open arrow) (Amaral and Pozzo-Miller, 2007; Zhou et al., 2008). In these TRPC3 antibody-infused cells, the DA-induced depolarization was substantially inhibited with the residual amplitude being only 0.6 ± 0.1 mV (n = 6) (Fig. 6B). The effects of TRPC3 antibody were neutralized by its antigenic peptide (antibody/peptide ratio was 1:1 or 1:2 by weight) with an intact 30 DA μm-induced depolarization (2.8 ± 0.4 mV; n = 3) (Fig. 6C), indicating that the inhibitory effects of TRPC3 antibody were specific. Together, these results strongly suggest that TRPC3 channels are the effector channel of D1–D5 receptor coactivation in SNr GABA neurons such that an inhibition of TRPC3 channels leads to an inhibition of D1-like excitation.
The next question was: how does D1–D5 receptor coactivation lead to enhanced TRPC3 channel activity? TRPC3 channels are known to be stimulated by lipid-mediated signaling mechanisms (Clapham, 2003; Albert and Large, 2006; Hardie, 2007; Venkatachalam and Montell, 2007; Nilius et al., 2008). Whereas D1 and D5 receptors are known to use the classical Gs– or GOlf–cAMP signaling pathway (Neve et al., 2004), D1–D5 coactivation may stimulate Gq/11 and phospholipase C (PLC), thereby triggering lipid signaling mechanisms (Rashid et al., 2007). If these lipid signaling mechanisms are involved, then inhibition of PLC should inhibit the DA response. Indeed, in SNr GABA neurons from slices incubated with a PLC inhibitor U73122 (1-[6[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione) (5 μm) (Horowitz et al., 2005), 30 μm DA induced a depolarization of 0.5 ± 0.1 mV from their resting potential at approximately −50 mV in the presence of 1 μm TTX (n = 9) (Fig. 7A,C). In contrast, in SNr GABA neurons from slices incubated with an adenylyl cyclase inhibitor 2′,5′-dd-3′-AMP-bis(t-Bu-SATE) (5 μm) (Laux et al., 2004), 30 μm DA induced a depolarization of 2.6 ± 0.3 mV (n = 8) from their resting potential at approximately −50 mV in the presence of 1 μm TTX (Fig. 7B,C). Under control condition, in SNr GABA neurons, 30 μm DA induced a depolarization of 2.8 ± 0.2 mV from their resting potential at approximately −50 mV in the presence of 1 μm TTX (n = 13) (Fig. 7C) (see also Fig. 2C). Thus, inhibition of PLC, but not adenylyl cyclase, caused a substantial inhibition of the DA response.

Inhibition of PLC, but not adenylyl cyclase, causes a substantial inhibition of the DA response. A, In SNr GABA neurons from slices incubated for at least 20 min with a PLC inhibitor U73122 (5 μm), 30 μm DA induced a depolarization of 0.5 ± 0.1 mV (n = 9) from their resting potential at around −50 mV in the presence of 1 μm TTX. B, In SNr GABA neurons from slices incubated for at least 20 min with an adenylyl cyclase inhibitor 2′,5′-dd-3′-AMP-bis(t-Bu-SATE) (5 μm), 30 μm DA induced a depolarization of 2.6 ± 0.3 mV (n = 8) from their resting potential at approximately −50 mV in the presence of 1 μm TTX. C, Summary graph showing the 30 μm DA-induced depolarization under control condition and after incubation with U73122 or 2′,5′-dd-3′-AMP-bis(t-Bu-SATE). The depolarization in U73122-treated cells was significantly smaller than other two groups (p < 0.01), whereas there was no significant difference between control and treatment with 2′,5′-dd-3′-AMP-bis(t-Bu-SATE).
D1-like receptor blockade decreased SNr GABA neuron firing frequency and increased their firing irregularity: evidence for tonic D1–D5 receptor activity
Thus far, our data demonstrated unambiguously that exogenously applied DA- or selective D1-like agonists induced D1-like excitation in SNr GABA neurons. Now we turn to this critical question: can endogenous DA released from DA dendrites induce a similar D1-like excitation? Data addressing this question were already shown in Figures 4 and 5 and also in supplemental Figure S2 (available at www.jneurosci.org as supplemental material) and will be discussed in combination with additional data.
We hypothesized that spontaneously released DA from DA neuron dendrites may induce a tonic activation of D1 and D5 receptors and exert a tonic influence on SNr GABA neurons. Consequently, blocking these D1 and D5 receptors may reveal this potential tonic excitation. To test this idea, we first blocked fast GABA- and glutamate-mediated synaptic transmission with picrotoxin, d-AP-5, and CNQX. After establishing stable baseline recording, 5 μm SKF83566 (a D1-like blocker) was bath applied to probe the potential tonic D1/D5 receptor activity. Indeed, SKF83566 significantly decreased SNr GABA neuron firing frequency by 8.6 ± 1.7% (from 10.8 ± 0.9 Hz under control conditions to 9.8 ± 0.7 Hz during antagonist application; n = 10; p < 0.01) (Fig. 8A–D) (see also Fig. 4B). These results suggest that there is indeed a tonic D1-like receptor activity that normally increases SNr GABA neuron firing rate.
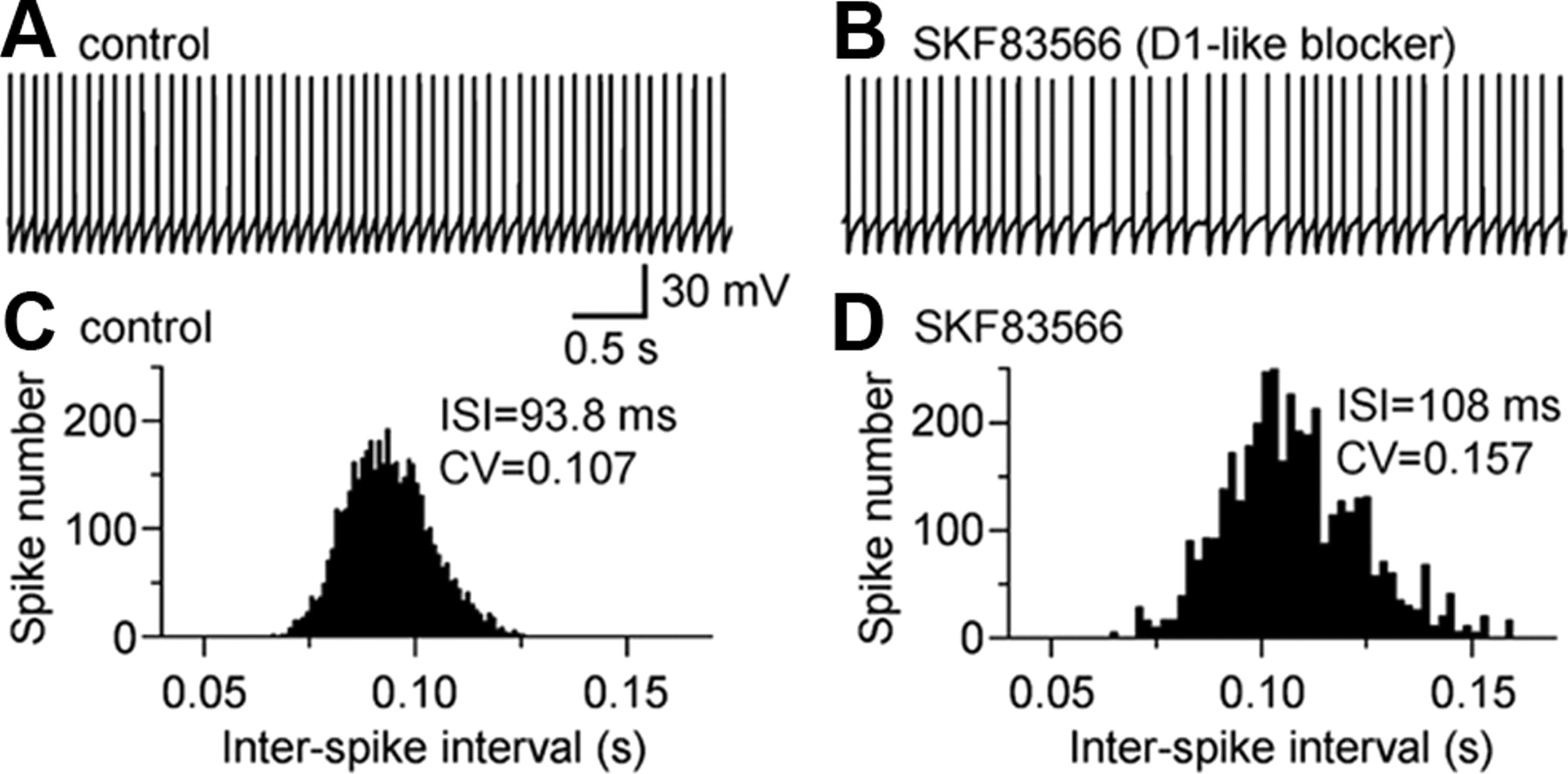
D1-like receptor blockade increased SNr GABA neuron firing irregularity. A, Spontaneous spikes recorded in an SNr GABA neuron under control conditions. B, During exposure to D1-like blocker SKF83566 (5 μm), spiking became slower in frequency and more irregular in interval. C, Control ISI distribution was narrow and smooth. D, Distribution of ISI under SKF83566 became wider and more variable. Mean ISI and its CV are listed in the graphs.
After blocking action potentials with 1 μm TTX and obtaining stable membrane potential, bath application of 5 μm SKF83566 induced a clear hyperpolarization of 0.7 ± 0.2 mV (n = 9) in SNr GABA neurons (Fig. 4C). When the neurons were voltage clamped at −70 mV, bath application of 5 μm SKF83566 induced a clear outward current (8.4 ± 1.3 pA; n = 8) (Fig. 4D) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Whole-cell conductance was also decreased from 5.08 ± 0.37 nS under control condition to 4.72 ± 0.35 nS during SKF83566 application (n = 4). These effects were not seen in SNr GABA neurons in D1 KO mice (n = 2) and D5 KO mice (n = 2). Intracellular infusion of D1 or D5 receptor antibody also revealed a tonic D1 and D5 receptor-dependent excitation (Fig. 5C–H). These data clearly indicate that, in SNr GABA neurons, inhibition of D1 and D5 receptors induced an outward current, most likely by downregulating the tonically active TRPC3 channels.
The SNr GABA projection neuron firing pattern is often altered in Parkinson's disease (Wichmann et al., 1999; Tang et al., 2005). Therefore, a question arises: can D1 and D5 receptor activation in SNr GABA projection neurons contribute to the regulation of the firing pattern in these neurons? If it does, then DA neuron degeneration will likely lead to degradation of this regulation, potentially contributing to the symptoms of Parkinson's disease. We noticed that, during SKF83566 exposure, the decrease in SNr GABA neuron firing frequency or the increase in interspike interval (ISI) was accompanied by an increase in the irregularity of ISI. To quantify this change, the coefficient of variation (CV) of ISI under control and during SKF83566 was calculated. CV was computed by dividing the SD of ISI by the mean ISI (Bennett and Wilson, 1999; Lee et al., 2007; Zhou et al., 2008). Indeed, bath application of 5 μm SKF83566 increased the CV of ISI by 27.5 ± 4.5% (p < 0.05; n = 8) (Fig. 8C,D). These results indicated that tonic activation of D1 and D5 receptors arising from endogenous DA release may help keep SNr GABA neurons sufficiently depolarized and spiking reliably at a regular interval.
Additional evidence for DA neurons directly influencing SNr GABA neurons
To provide more evidence for our hypothesis that SNc DA neurons can directly affect SNr GABA neurons, we did the following three additional sets of experiments.
In the first set of experiments, we stimulated SNc DA neurons and monitored responses in SNr GABA neurons. To evoke the maximal somatodendritic DA release and hence the maximal DA response, we adopted the published methods of burst-stimulating DA neurons (Chen and Rice, 2001; John et al., 2006). Each stimulation consisted of a burst of 10 50-Hz pulses delivered at every 5 min that prevents DA release depletion (Chen and Rice, 2001; John et al., 2006). Each pulse was 0.1 ms at 80–120 μA. The bipolar stimulating electrode was placed in the SNc. The recorded SNr GABA neurons were voltage clamped at −80 mV to increase driving force and prevent spontaneous spikes. The bathing solution was a blocking mixture (for details, see the legend of Fig. 9A). Under these conditions, burst stimulation of DA neurons induced a postsynaptic-like response in SNr GABA neurons. The peak amplitude was 7.1 ± 0.8 pA (n = 4) (Fig. 9A). The rise time was ∼0.5 s and the decay time was ∼8 s, although the small amplitude and consequently the interference from noise prevented precise measurement of these two parameters. This postsynaptic-like inward current was blocked by 2 μm D1-like antagonist SKF83566 (Fig. 9A), indicating that somatodendritically released DA may activate D1-like receptors in SNr GABA neurons. In contrast, endogenous DA induced a postsynaptic-like, G-protein-gated inwardly rectifying K+ channel-mediated, outward current in DA neurons by activating D2-like receptors in DA neurons (Beckstead et al., 2004).

More evidence for DA neurons to directly influence SNr GABA neurons. A, Burst stimulation of SNc DA neurons induced a postsynaptic-like response in SNr GABA neurons that was sensitive to 2 μm D1-like blocker SKF83566. See Results for the stimulating protocol. The bathing solution was a mixture containing the following receptor blockers: 1 μm sulpiride to prevent D2 receptor-mediated autoinhibition, 50 μm d-APV, and 20 μm CNQX to block ionotropic glutamate receptors, 500 μm (S)-α-methyl-4-carboxyphenylglycine to block metabotropic glutamate receptors, 50 μm picrotoxin to block GABA receptors, 5 μm CGP 52432 (3-[[(3,4-dichlorophenyl)-methyl]amino]propyl](diethoxymethyl)phosphinic acid) to block GABAB, 5 μm RS 102221 (8-[5-(2,4-dimethoxy-5-(4-trifluoromethylphenylsulfon-amido)phenyl-5-oxopentyl)]-1,3,8-triazaspiro[4.5]decane-2,4-dione hydrochloride) to block 5-HT2C receptors, and 200 nm prazosin to block α1 adrenoceptors. B, D2-like agonist quinpirole (Quin; 1 μm) decreased SNr GABA neuron firing rate (white bar), whereas D2-like antagonist sulpiride (Sulp; 1 μm) increased the firing rate (gray bar). K–S tests on the distributions of firing rates indicate that the inhibition was significant (p < 0.05). DAT inhibitor GBR12909 (GBR; 2 μm) together with sulpiride also increased the firing rate of SNr GABA neurons (p < 0.01; black bar). See Results for more details. C, In the presence of 1 μm TTX, in SNr GABA neurons, D2-like agonist quinpirole (1 μm) induced a hyperpolarization, whereas D2-like antagonist sulpiride (1 μm) induced a depolarization (gray bar). DAT inhibitor GBR12909 (2 μm) together with sulpiride (1 μm) also induced a depolarization (black bar). See Results for detailed description. D, An example recording showing that GBR12909 (2 μm), in the presence of 1 μm TTX and 1 μm sulpiride, induced a clear depolarization sensitive to D1-like blocker SKF83566 (5 μm). Note that the membrane potential during SKF83566 went below the initial baseline because SKF83566 blocked the tonic D1-like activity. Traces in A were digitized at 10 kHz in which the trace in D was digitized at 100 Hz. All traces are individual traces.
In the second set of experiments, we examined the effects of D2-like agonist quinpirole that is known to inhibit DA neuron firing and presynaptic DA release via D2 autoinhibition (Lacey et al., 1987; Cragg and Greenfield, 1997; Pothos et al., 1998). We found that quinpirole (1 μm) inhibited SNr GABA neuron firing rate by 5.9 ± 1.0% (n = 11) (Fig. 9B). K–S tests on the distributions of firing rates in individual SNr GABA neurons indicate that the inhibition was significant (p < 0.05). We also tested the effects of D2-like antagonist sulpiride that is known to increase DA neuron firing and presynaptic DA release (Pucak and Grace, 1994; Cragg and Greenfield, 1997). We found that sulpiride (1 μm) increased SNr GABA neuron firing rate by 6.2 ± 1.1% (n = 10; p < 0.05) (Fig. 9B).
In the third set of experiments, we bath applied a DA uptake inhibitor GBR12909 (1-[2-[bis(4-fluorophenyl)-methoxy]ethyl]-4-[3-phenylpropyl]piperazine) that is known to increase the extracellular DA level (Rice and Cragg, 2008). To prevent the complication of DA-induced autoinhibition, 1 μm sulpiride was applied first before GBR12909. As shown in Figure 9B, 2 μm GBR12909 increased the firing rate of SNr GABA neurons by 19.3 ± 2.4% above drug-free control and 13.2 ± 1.5% above that during 1 μm sulpiride alone (n = 7; p < 0.01).
In the presence of 1 μm TTX, in SNr GABA neurons, D2-like agonist quinpirole (1 μm) induced a −0.4 ± 0.0 mV hyperpolarization (n = 3) (Fig. 9C), whereas D2-like antagonist sulpiride (1 μm) induced a 0.5 ± 0.1 mV depolarization (n = 4) (Fig. 9C). DAT inhibitor GBR12909 (2 μm) together with sulpiride (1 μm) induced a 1.3 ± 0.1 mV depolarization over the drug-free baseline (n = 4) (Fig. 9C). In the presence of D1-like blocker SKF83566 (5 μm), none of the effects of quinpirole and sulpiride described above was seen. GBR12909-induced depolarization was also blocked by D1-like antagonist SKF83566 (Fig. 9D), suggesting that D1-like receptors were the common signaling pathway mediating the effects induced by D2-like ligands and DAT inhibitor GBR12909.
These results indicate that manipulation of DA neuron activity and DA release can affect SNr GABA neuron activity. The relatively weak effects are consistent with the established fact that DA neuron dendrites have Na+-spike-independent, pace-making activity-driven, L-type Ca2+ channel-mediated Ca2+ oscillation (Chan et al., 2007) that may be essential for the dendritic DA release. Certainly, Ca2+ release from internal stores may also contribute (Patel et al., 2009). Another factor is that the responses were recorded in 300-μm-thick brain slices in which substantial numbers of DA neurons and GABA neurons and their dendrites were killed or injured during the tissue sectioning procedure, leading to a reduced magnitude of the endogenous DA-mediated interactions. Thus, the endogenous DA-mediated interactions in intact animals are likely to be substantially larger.
Discussion
Based on a series of experiments using multiple different approaches, the major finding of this study is that nigral DA neurons form a novel ultra-short SNc → SNr DA pathway, in addition to the established long-distance nigro-striato-nigral loop (Fig. 9). In this ultra-short DA pathway, dendritically released DA exerts direct excitatory effects on SNr GABA projection neurons by an apparent coactivation of D1 and D5 receptors that subsequently enhances constitutively active TRPC3 channels in these neurons, leading to enhanced and more regular basal ganglia output.
Dendritically released dopamine induces tonic D1 and D5 receptor activity in SNr GABA neurons
SNr GABA projection neurons intermingle with DA dendrites that release DA (Björklund and Lindvall, 1975, Geffen et al., 1976; Cheramy et al., 1981; Cragg et al., 2001; Falkenburger et al., 2001). Endogenously released DA has been suggested to activate D1-like receptors on GABA and glutamate afferent terminals in the nigral areas and, hence, facilitate GABA and glutamate release (Cameron and Williams, 1993; Ibañez-Sandoval et al., 2006). Cheramy et al. (1981) also indicated that DA may directly affect SNr GABA projection neurons. Iontophoretic studies on DA effects on SNr GABA neurons in intact animals have produced conflicting results. For example, early studies (Ruffieux and Schultz, 1980; Waszcak and Walters, 1983) found that DA exerted an excitatory effect on SNr neurons, whereas a recent study (Windels and Kiyatkin, 2006) reported that DA had no consistent effects. Another recent study (Kliem et al., 2007) indicated that local infusion of a D1-like agonist generally reduced SNr GABA neuron firing rate via presynaptic mechanisms. However, these studies used extracellular recordings and consequently could not conclusively determine whether the effects were a direct excitation in SNr GABA neurons or were local circuit-mediated effects. These studies might also have been complicated by the fact that DA may affect multiple, potentially opposing synaptic inputs (such as glutamate and GABA) that may mask each other's effects on SNr GABA neuron firing.
To ensure that the observed responses were direct effects in the recorded SNr GABA neurons, our present study was performed after blocking these fast glutamate and GABA synaptic inputs. Under this isolated condition, we found that DA or D1-like agonists (SKF81297 and dihydrexidine) induced similar D1-like excitatory effects (depolarization or inward current accompanied by increased whole-cell conductance) in SNr GABA neurons. All these effects were prevented by a D1-like receptor antagonist SKF83566. These effects were also absent in D1 KO mice and D5 KO mice and inhibited by intracellular application of D1 or D5 antibody. These results plus the data that SNr GABA neurons express both D1 and D5 mRNAs lead to the compelling conclusion that coactivation of D1 and D5 receptors induces a direct excitation in these basal ganglia output neurons.
In the present study, we also found that, after blocking fast GABA and glutamate receptors, D1-like antagonist SKF83566 induced a hyperpolarization in SNr GABA neurons and decreased their firing frequency accompanied by an increase in firing irregularity. These effects were absent in D1 KO mice and also in D5 KO mice, strongly suggesting that spontaneously released DA may induce a tonic coactivation of D1 and D5 receptors that influences SNr GABA neuron activity. Direct stimulation of DA neurons induced a direct inward current in SNr GABA neurons that was sensitive to D1-like blockade. Furthermore, manipulation of DA neuron activity and DA release with D2-like receptor ligands, and blockade of DA reuptake in particular, affected SNr GABA neuron firing. These data provide additional evidence that SNc DA neurons can directly influence SNr GABA neurons.
SNr GABA neurons may also feedback onto SNc DA neurons, as indicated by D1-like agonist SKF81297-induced increase in spontaneous IPSCs (sIPSCs) in DA neurons (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). This result is consistent with our data that SKF81297 increased SNr GABA neuron firing, although the sIPSCs in DA neurons might have originated in SNr GABA neurons and also other GABA afferents (Tepper et al., 1995; Mailly et al., 2003; Misgeld, 2004).
D1–D5 receptor coactivation enhances TRPC3 channel activity in SNr GABA neurons
Our scRT-PCR consistently detected both D1 and D5 receptor mRNAs in SNr GABA neurons, although previous studies detected D1 receptors only on striatonigral axon terminals but not in SNr GABA neurons (Mansour et al., 1992; Levey et al., 1993; Ariano, 1997). D5 receptor expression in SNr was implied, although the cell type(s) expressing this receptor was not known (Choi et al., 1995; Khan et al., 2000). The use of the highly sensitive and specific scRT-PCR techniques in the present study may explain this difference in detection.
D1 and D2 receptor coactivation and the underlying molecular mechanisms are relatively well established (Hopf et al., 2003; Rashid et al., 2007). Our functional demonstration of the requirement of both D1 and D5 receptors or coactivation of D1 and D5 receptors for the D1-like excitation is a novel finding. Despite a lack of direct molecular evidence, a recent review (Rashid et al., 2007) implied a D1–D5 receptor coactivation that can stimulate Gq/11 activation and lipid signaling mechanism. Our present study provides the first data supporting this implied novel signaling pathway associated with these DA receptors. The apparent D1 and D5 receptor coactivation we observed may arise from receptor dimerization and/or downstream signaling mechanisms. Future studies are needed to elucidate the molecular mechanisms underlying this apparent D1 and D5 receptor coactivation.
Because D1 and D5 receptors do not directly gate any ion channel, to what ion channel does this D1–D5 coactivation couple? Our results show that the biophysical characteristics of the DA-induced inward current are almost identical to those of TRPC3 channel-mediated current: both are linear and voltage independent with reversal potential at approximately −37 mV (Fig. 3) (Zhou et al., 2008). Furthermore, intracellular application of TRPC3 channel antibody inhibited the D1-like effect in SNr GABA neurons (Fig. 6). These results strongly suggest that the D1–D5 receptor coactivation may ultimately couple to and enhance the constitutively active TRPC3 channels (Fig. 6A) that in turn regulate SNr GABA neurons. A DA regulation of TRPC3 channel activity is entirely possible because D1–D5 coactivation may activate Gq/11 and phospholipase C, thereby triggering lipids-mediated signaling mechanisms (Rashid et al., 2007) that are known to stimulate TRPC3 channels (Clapham, 2003; Hardie, 2007; Nilius et al., 2008).
Functional implications
Based primarily on the fact that DA dendrites may release DA in SNr, Cheramy et al. (1981) suggested that DA may directly affect SNr GABA projection neurons. Two pioneering studies indicated that iontophoretically applied DA exerts an excitatory effect on SNr neuron firing in intact animals (Ruffieux and Schultz, 1980; Waszcak and Walters, 1983). However, these studies used extracellular recordings, did not remove synaptic inputs, and consequently could not determine whether the effect was a direct excitation in SNr neurons or local circuit-mediated effect. Because of technical limitations at these early dates, these studies also could not determine the DA receptor subtype(s) responsible for the effect. Two recent studies revisited this topic but led to conflicting conclusions on DA regulation of SNr GABA neurons (Windels and Kiyatkin, 2006; Kliem et al., 2007). Our present results are consistent with those of the two early in vivo studies (Ruffieux and Schultz, 1980; Waszcak and Walters, 1983). More importantly, our data provide cellular and molecular mechanisms for these early in vivo observations.
The nigral DA neurons are commonly described to regulate basal ganglia output through the long distance nigro-striato-nigral pathway (Parent et al., 2000; Haber, 2003). However, DA dendrites and SNr GABA neurons intermingle with each other. As illustrated in Figure 10, our study shows that there is also an ultra-short SNc → SNr DA pathway that directly regulates SNr GABA neuron output. Besides the well known D2 receptor-mediated autoinhibition of nigral DA neurons, our data indicate that dendritically released DA induces a tonic D1 and D5 receptor coactivation-mediated hetero-excitation in SNr GABA projection neurons. This D1 and D5 receptor coactivation couples to and enhances tonically active TRPC3 channels, leading to the depolarized membrane potential, high firing rate, and regular firing pattern in SNr GABA projection neurons. Loss of DA neurons in Parkinson's disease may impair this novel ultra-short DA pathway, contributing to the complex neuropathophysiology and, hence, movement deficits. Indeed, a recent study found movement control deficits in TRPC3 channel knock-out mice (Hartmann et al., 2008). Although loss of TRPC3 channel in the cerebellum may be primarily responsible for the motor deficits, TRPC3 channel loss in SNr may also be involved.

Diagrammatic representation of the novel ultra-short SNc → SNr DA pathway identified and characterized by our present study. DA dendrites are known to release DA and induce D2-mediated autoinhibition in DA neurons. There is also the established long-distance nigro-striato-nigral loop. We have demonstrated that SNr GABA neurons express D1 and D5 receptors that ultimately couple to tonically active TRPC3 channels. Dendritically released DA also directly acts on SNr GABA output neurons, forming an ultra-short DA pathway that contributes to the regulation of the firing frequency and pattern of these basal ganglia output neurons. GPe, Globus pallidus external part; motor N, brainstem motor nuclei; BG, basal ganglia; STN, subthalamic nucleus; SC, superior colliculus.
Footnotes
-
This work was supported by a grant from the American Parkinson Disease Association and a Young Investigator Award from National Alliance for Research on Schizophrenia and Depression (F.-M.Z.) and National Institutes of Health Grants R01DA021194, R01MH067119, and R01NS058850 (F.-M.Z.), DA015525 (S.G.M.), and DA017323 and DA025088 (M.X.). We thank Dr. David Sibley for generously donating dopamine D5 knock-out mice. We also thank Drs. Shengyuan Ding and Xingjun Wu for their help and Dr. Jonathan Jaggar for discussion.
- Correspondence should be addressed to Fu-Ming Zhou, Department of Pharmacology, University of Tennessee College of Medicine, Memphis, TN 38163. fzhou3{at}utmem.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}