

Abstract
Myosin Va (MyoVa) mediates F-actin-based vesicular transport toward the plasma membrane and is found at neuronal postsynaptic densities (PSDs), but the role of MyoVa in synaptic development and function is largely unknown. Here, in studies using the dominant-negative MyoVa neurological mutant mouse Flailer, we find that MyoVa plays an essential role in activity-dependent delivery of PSD-95 and other critical PSD molecules to synapses and in endocytosis of AMPA-type glutamate receptors (AMPAR) in the dendrites of CNS neurons. MyoVa is known to carry a complex containing the major scaffolding proteins of the mature PSD, PSD-95, SAPAP1/GKAP, Shank, and Homer to dendritic spine synapses. In Flailer, neurons show abnormal dendritic shaft localization of PSD-95, stargazin, dynamin3, AMPARs and abnormal spine morphology. Flailer neurons also have abnormally high AMPAR miniature current frequencies and spontaneous AMPAR currents that are more frequent and larger than in wild-type while numbers of NMDAR containing synapses remain normal. The AMPAR abnormalities are consistent with a severely disrupted developmental regulation of long-term depression that we find in cortical Flailer neurons. Thus MyoVa plays a fundamentally important role both in localizing mature glutamate synapses to spines and in organizing the synapse for normal function. For this reason Flailer mice will be valuable in further dissecting the role of MyoVa in normal synaptic and circuit refinement and also in studies of neurological and neuropsychiatric diseases where disruptions of normal glutamate synapses are frequently observed.
Introduction
Myosin Va (MyoVA), abundant in postsynaptic densities (PSDs) of glutamatergic neuronal dendritic spines (Walikonis et al. (2000)), is a member of a family of nonconventional, processive, plus-end actin motors, consisting of a “head” region that binds to actin and hydrolyzes ATP followed by calmodulin-binding regions that endow the transport system with Ca2+ sensitivity, dimerization domains that allow stepping motion along actin filaments, and finally a “tail” cargo binding domain (Akhmanova and Hammer, 2010).
Studies of MyoVa in the brain are relatively few because MyoVa −/− (Dilute-Lethal) mice have severe seizures and die before postnatal day 21 (P21; Mercer et al., 1991). Viable MyoVa mutants are ataxic, because they lack mGluR1-dependent long-term depression (LTD) caused by the absence of dendritic spine smooth endoplasmic reticulum (SER) in cerebellar Purkinje cell dendrites (Miyata et al., 2000). This results from the absence of MyoVa's normal binding of the IP3 receptor. The motor normally pulls the receptor and its attached SER into dendritic spines (Wagner et al., 2011). MyoVa is also associated with several mRNA-binding proteins (Ohashi et al., 2002) and carries TLS/FUS to the synapse (Yoshimura et al., 2006).
MyoVa involvement in hippocampus long-term potentiation (LTP) has been examined in cultured hippocampal slices where shRNA knockdown of MyoVa eliminates NMDA receptor (NMDAR)-dependent LTP (Correia et al., 2008). This finding is surprising (Hammer and Sellers, 2012), because an earlier electron microscopic (EM) study (Petralia et al., 2001) showed AMPARs at synapses of MyoVa−/− Dilute-Lethal mice and an electrophysiological study used acute hippocampal slices from P12–P19 Dilute-Lethal and found normal NMDAR-LTP (Schnell and Nicoll, 2001). A similar study on acute P24–P26 Flailer, the mouse studied here, hippocampal slices in our laboratory was fully consistent with the Schnell and Nicoll (2001) study and also showed that hippocampal NMDAR-LTD is normal in Flailer at this age (Feinberg-Zadek et al., 2010; Pandian et al., 2011).
This work evolved from our interest in visual synapse development, the role of the membrane associated guanylate kinase (MAGUK) scaffold PSD-95 in this process, and previous findings: (1) only palmitoylated PSD-95 trafficks to synapses (El-Husseini Ael et al., 2002; Yoshii et al., 2011), implying vesicular transport; (2) MyoVa carries vesicle-bound molecules to synapses; (3) the scaffold SAPAP1/GKAP binds PSD-95 (Kim et al., 1997; Takeuchi et al., 1997) and is associated with the dynein light chain (DLC2) (Espindola et al., 2000; Naisbitt et al., 2000); (4) SAPAP1 also interacts with Shank proteins, the common synaptic scaffold for all glutamate receptors (Naisbitt et al., 1999; Tu et al., 1999); and (5) a preformed complex of PSD-95, SAPAP1, and Shank travels along dendrites and delivery of these clusters to spine synapses requires actin transport and DLC2 (Gerrow et al., 2006).
Here, we use the Flailer neurological mutant mouse that expresses a fusion protein driven by the promoter for the largely brain-specific protein Gnb-5. The flailer gene expresses the first two exons of Gnb5 fused to the cargo binding and part of the dimerization domain of MyoVa but does not code the motor's actin binding domain. Flailer also expresses normal Gnb5 and MyoVa proteins. Significantly, whenever flailer and wild-type (WT) myo5a genes are in a 1:1 ratio, for example, when Flailer is crossed with a heterozygous Dilute-Lethal mouse so that it carries one chromosome with no Myo5a gene, the flailer protein acts in brain as an intrinsic dominant-negative mutation that competes with normal MyoVa for binding of cargo (Jones et al., 2000). Flailer protein expression varies in different regions and is different from that of WT MyoVa because the flailer gene is under the control of the Gnb5 promoter while myo5a is under the control of its own promoter. Remarkably, Flailer, unlike Dilute-Lethal, survives and breeds in the homozygous state allowing this study of MyoVa's role in excitatory synapse development.
Materials and Methods
Animals.
Animal manipulations were performed in accord with the guidelines of the Massachusetts Institute of Technology Institutional Animal Care and Use Committee. For most experiments, litters of C57BL/6Awj WT and Flailer mice in the same background were kept under 12 h light/dark cycles. Both C57BL/6Awj WT and Flailer used for Figures 4D–F, 6, and 7 were crossed with thy-1 GFP mice (S-strain) (Feng et al., 2000) for at least five generations. Eyelids were glued shut with Vetbond (3M). On the day of eye opening (EO), lids were separated under photopic conditions and pups were checked every 15–30 min to ensure that they were awake until they were killed for protein fractionations.
Synaptosome fractionation.
Synaptosomes were prepared as previously reported (Dunah and Standaert, 2001). Visual cortex (VC) tissues were collected from pups of both sexes in ice-cold TEVP buffer containing the following (in mm): 10 Tris-HCl, 5 NaF, 1 Na3VO4, 1 EDTA, and 1 EGTA, pH 7.4, 320 mm sucrose, and homogenized using a tissue grinder. Homogenates were centrifuged at 1000 × g removing nuclei and large debris (P1). Supernatant (S1) was centrifuged at 10,000 × g. The pellet (P2) was resuspended in TEVP buffer and centrifuged at 25,000 × g to pellet a synaptosomal membrane fraction (LP1). After each centrifugation pellets was rinsed with ice-cold TEVP buffer to avoid contamination between each fraction.
Gel electrophoresis, quantitative immunoblotting, and statistical analysis.
Immunoblotting was performed as previously described (Yoshii et al., 2011). Concentrations of protein sample were measured using Pierce BCA protein Assay Kit (Thermo Scientific). Equal amount of proteins (5 μg or 10 μg per lane) were run on 6% or 8% SDS-PAGE, then transferred to polyvinylidene difluoride membrane by electroblotting (Idea Scientific). Blots were blocked with blocking buffer (Sigma) diluted with Tween/0.1 m PBS (TPBS) for 30 min, then incubated in primary antibody in TPBS for 1 h at room temperature. After three 5 min rinses in TPBS, blots were incubated in secondary antibody (horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse in TPBS at 1:5000), washed several times for 5 min in TPBS, reacted with chemiluminescent substrate (Pierce), and exposed to film (Kodak). Films were scanned and band density was measured by ImageJ. Measurements were confirmed to be within the linear range of density analyses of dilution series of the samples. In Figure 2, fold change of each band was calculated by dividing its intensity with that of band labeled WL, BEO. Results are reported as averages ± SEM. All immunoblot analyses used three sets of protein samples from three different litters. Two gels were run for each sample and blotted (N = 6). Parametric Student's tests were used and *p < 0.05 and **p < 0.01.
Immunoprecipitation.
The solubilized protein samples were diluted 20-fold with immunoprecipitation (IP) buffer containing the following (in mm): 150 NaCl, 50 Na3SO4, and 2 EDTA; pH 7.2, plus 1% sodium deoxycholate and 1% Triton X-100 and incubated for 3 h at 4°C and gentle rotation with 10 μl of anti-PSD-95 or SAP102 and 40 μl of protein A-Sepharose beads slurry for each 100 μg of protein. Immunoprecipitates were washed three times with ice-cold IP buffer after brief centrifugations, and were resuspended in loading buffer for immunoblotting.
Antibodies for IP.
Anti-PSD-95 (Millipore Biotech #05-494, 1:1000), anti-SAP102 (JH 62514, a gift from Dr. J. Hell, 1:1000), anti-GKAP (c9589, a gift from Dr. M. Sheng, 1:1000), anti-MyoVa (Santa Cruz Biotechnology #sc-17706 for N terminal, 1:1000; Sigma-Aldrich #LE-18 for C terminal, 1:1000; DIL-2 for the central domain, 1:1000, a gift from Dr. J. Hammer).
Primary neuron culture, lipofection, and immunocytochemistry.
Occipital cortices of E15.5 mouse brains were dissected, and digested with a solution containing papain and DNase for 25 min. Cells were dissociated using fire-polished glass pipettes and plated to a coverslip coated with laminin and poly-d-lysine. Lipofection was performed at 8 d in vivo (DIV) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. On DIV 16–18, cultures were fixed with 4% paraformaldehyde (15 min). Except where otherwise stated, neurons were permeabilized with 0.3% Triton X-100 for 5 min and washed. Cultures were blocked with 9% bovine serum albumin (BSA; 1 h), and incubated with primary antibodies at room temperature overnight. After washing with PBS (3×), cultures were incubated with secondary antibodies (2 h), Alexa Fluor 488 and 633 (Invitrogen). Primary antibodies were PSD-95 (NeuroMab #K28/43, 1:1000), Dynamin 3 (a gift from Dr. McNiven, 1:500), Stargazin (Calbiochem #681517, 1:500), and GluA2 (Millipore Bioscience Research Reagents #MAB397, 1:1000). Images were captured using a 60× objective on a Nikon PCM 2000 confocal microscope and a 6–8 μm, Z-series, of optical sections were taken at intervals of 0.5 μm.
In utero electroporation.
Following laparotomy on timed pregnant mice, the uterus was externalized. Approximately 0.5–0.6 μl of solution containing either a PSD-95-GFP construct (2 μg/μl) or a GFP-SAP102 (2 μg/μl) was injected into the ventricle of E15.5 fetuses through the uterine wall. Using an ECM 830 apparatus (Harvard Apparatus), brains were electroporated with five 30 V, 50 ms pulses at intervals of 950 ms. After recovery, pregnancies continued and pups were delivered normally.
Electrophysiology.
Young (P14–P17) and adult (>6 months old) male WT and Flailer mice were anesthetized with isoflurane and decapitated. Slice preparation was conducted as previously described (Zhao and Constantine-Paton, 2007) except that adult mice were perfused transcardially before decapitation. Young mouse sagittal slices of midbrain were used for whole-cell spontaneous current recordings in the superficial layers of the superior colliculus (sSC) with the same recording procedures and solutions as used in cortical slices except that tetrodotoxin (TTX; Sigma) was omitted from the bath and the slices were incubated in 50 μm SYM2081 (Tocris Bioscience) a Kainate receptor (KAR) agonist for at least 15 min to desensitize and eliminate KAR currents that are prevalent in sSC at this age (van Zundert et al., 2010). Young mouse VC slices were used for whole-cell voltage-clamp recording of miniature and evoked EPSCs on L2/3 pyramidal neurons, pipettes and pipette solution were as previously described (Zhao and Constantine-Paton, 2007). Miniature AMPAR currents were recorded at −70 mV in the presence of 1 μm TTX (Sigma), 10 μm bicuculline methochloride (BMC; GABAA receptor antagonist; Sigma), 5 μm glycine, and 0 mm Mg2+ with the NMDAR antagonist 100 μm d-AP5 (100 μm) (Tocris Bioscience). For miniature NMDAR current recordings, cells were held at +40 mV with perfusion solution containing 1 μm TTX, 10 μm BMC, 5 μm glycine, and 1.5 mm Mg2+. All miniature event counts, measurements of evoked AMPAR/evoked NMDAR current ratios, and paired-pulse ratios were analyzed as previously reported (Zhao and Constantine-Paton, 2007).
For LTP and LTD experiments in mature mice, field potentials were recorded from L2/3 in response to stimulation of L4 in VC slices from male Flailer and WT mice >6 months old following the procedure of Kirkwood et al. (1997). Theta burst stimulation (four 100 Hz pulses, every 200 ms) was applied for LTP induction and 900 pulses at 1 Hz were used for inducing LTD. The mean maximum negative amplitude of the field EPSP (fEPSP) amplitude during the baseline was compared with the amplitude of the same fEPSP amplitude during an interval 30–40 min after induction.
Two-tail unpaired Student's t tests following F tests indicating normal distributions were used to compare results between WT and Flailer groups for all electrophysiology experiments. All analyses were accomplished blind to whether the animals were WT or Flailer mice.
Biotinylation assay.
Biotinylation assay was performed following the method described by Ehlers (2000). Culture neurons were preincubated with TTX (2 μm) with leupeptin (200 μg/ml) for 30 min at 37°C to avoid lysosomal degradation of internalized AMPARs. Cultures were serially cooled to 10 and 4°C to stop membrane trafficking in cold DPBS plus 1 mm MgCl2 and 2.5 mm CaCl2, then treated with cleavable (disulfide-linked) biotin-containing solutions (1.5 mg/ml sulfo-NHS-biotin in DPBS) for 30 min to label surface receptors. Unbound biotin was quenched with cold DPBS plus 1 mm MgCl2, 2.5 mm CaCl2, and 50 mm glycine (rinse, 2 × 5 min). Culture cells were either immediately scraped into lysis buffer to determine total biotinylation or incubated with solution containing 20 μm NMDA (37°C, 2 min) to induce AMPAR internalization. Remaining NMDAR-treated cultures were returned to serum-free growth media containing leupeptin at 37°C for set times to allow internalized receptors to recycle; they were subsequently cooled to 4°C and incubated with glutathione cleavage buffer containing the following (in mm): 50 glutathione, 75 NaCl, 10 EDTA, 75 NaOH, and 1% BSA in H2O, pH 8.5, three times for 15 min each at 4°C to ensure complete cleavage of any newly appearing surface biotin. The buffer was quenched with two 5 min rinses of 5 mg/ml iodoacetamide in DPBS with 1 mm MgCl2, 2.5 mm CaCl2 to internalize residual biotinylated proteins. Cells were then lysed for 3 min in 0.5% SDS-RIPA buffer containing 1% Triton X-100, 0.1% SDS, 0.5% deoxycholic acid with the following (in mm)L 50 Tris, 150 NaCl, 2 EDTA, 50 NaF, 1 sodium orthovanadate, pH 7.4, and protease inhibitor mixture, scraped and the lysed solution was centrifuged at 16,000 × g for 15 min at 4°C. Protein concentration of the supernatant was determined and 300 μg of protein was adjusted to a volume of 1ml with 0.1% SDS-RIPA buffer. Biotinylated GluA1 receptor subunits were precipitated with 70 μl NeutrAvidin-agarose (Thermo Scientific) slurry overnight at 4°C, washed with the SDS-RIPA buffer, and spun down three times. Pulled down subunits were eluted into Laemmli sample buffer by boiling for 6 min. Biotinylated GluA1 was detected by immunoblot analysis of cultures and at each time interval examined the percentage of internalized subunit was calculated by normalizing the band intensity from these cultures to that of the total biotinylated GluA1 subunit measured in the WT or the Flailer cultures before adding NMDA.
Imaging of pHluorin- tagged GluA2.
pHluorin-tagged GluA2 was imaged as in (Lin and Huganir, 2007) with slight modifications. Cultured cells, lipofectamine transfected with pHluorin-GluA2 at DIV 8, were imaged at DIV 14–16 using a 60× objective on a Nikon PCM 2000 confocal microscope and multiple 6 μm Z-series of 0.5 μm optical sections were taken while neurons were perfused (∼1 ml/min) with recording buffer containing the following (in mm): 120 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 25 HEPES, pH 7.4, 30 glucose, and 1 TTX, then stimulated with the buffer containing 20 μm NMDA for 2 min. Change of average pHluorin fluorescence intensity in dendritic segments was measured using ImageJ software (National Institutes of Health, Bethesda, MD) and expressed as F0 − Ft/F0 at the intervals indicated in Figure 8E.
Results
MyoVa, SAPAP1/GKAP, and PSD-95 form a protein complex
We verified the Flailer phenotype described earlier (Jones et al., 2000) for mice in our colony and identified the presence of the truncated flailer protein with a MyoVa C-terminal antibody (97.4 kDa) in extracts from hippocampus (hippo), VC (visual cortex), sSC, and cerebellum (Clb) (Fig. 1A).

Immunblottings and immunocytochemistry show myosin Va in a complex with PSD-95 and a SAPAP1 isoform. A, The “flailer (Fl) protein” is present in the Hippo, sSC, and Clb, as well as in the VC. An antibody against the C terminal (head region; LE-18, 1:1000) but not the N terminal (tail region; sc-17706, 1:1000) or the central region recognizes the flailer protein (DIL-2, 1:1000). The black arrowhead indicates the level of flailer protein [predicted molecular weight: 85 kDa (Jones et al., 2000) and the gray arrowhead indicates the MyoVa (molecular weight: 190 kDa)]. Cerebellar protein bands are less dense than those of other regions because in this P16 tissue cerebellum is less differentiated. B, MyoVa colocalizes with PSD-95 in WT visual cortical cultures. C, MyoVa (LE-18, 10 μl) coprecipitates PSD-95 and high levels of the 95 kDa-SAPAP1 even though the input lane shows dense bands for both the 130 and 95 kDa isoforms (c9589, 1:1000). D, PSD-95 (05–494, 10 μl) coprecipitates predominantly with the 95 kDa isoform of SAPAP1 (c9589, 1:1000). E, SAP102 (JH 62514, 10 μl) coprecipitates with more 130 kDa-SAPAP1 than 95 kDa isoform (c9589, 1:1000). F, MyoVa (LE 18, 10 μl) coprecipitates PSD-95 (LE-18, 1:1000) but very low levels of SAP102 (JH 62514, 1:1000). G, PSD-95 (05–494, 10 μl) coprecipitates both MyoVa and the flailer protein while it interacts with only intact MyoVa (LE-18, 1:1000). Scale bar, 10 μm. For C–F, IP experiments were repeated three times with separate protein isolates and 10% of the solutions were loaded for inputs.
MyoVa colocalized with PSD-95 in cultured WT VC neurons (Fig. 1B). In whole lysates from WT VC at P14 (Fig. 1C), anti-MyoVa coimmunoprecipitated (co-IPed) both PSD-95 and high levels of the 95 kDa splice variant of SAPAP1/GKAP-95 but with considerably lower levels of the SAPAP1, the 130 kDa splice variant. Co-IPs from similar lysates using a PSD-95 antibody (Fig. 1D) reinforced a previous finding (Yoshii et al., 2003); namely that PSD-95 and only the 95 kDa splice variant of SAPAP1/GKAP move together to the synapse upon EO. However, co-IPs with an antibody to SAP102, the MAGUK already present at glutamate synapses in the fetus and neonate, showed more of the 130 kDa than the 95 kDa SAPAP1/GKAP splice variant (Fig. 1E). Significantly, PSD-95 but barely trace levels of SAP102 associate with MyoVa (Fig. 1F). IPs with the anti-PSD-95 antibody from cortical homogenates confirmed that PSD-95 interacts with both MyoVa and the flailer protein in Flailer while it interacts with only the intact MyoVa in WT (Fig. 1G).
Flailer neurons do not show synaptosomal enrichment of PSD-95 in VC and sSC upon EO
Controlled EO involves gluing eyelids of young rodents shut and opening them simultaneously at a prescribed time. This immediately increases activity in the visual pathway and represents an important paradigm for synchronizing activity-dependent developmental changes at visual synapses. As expected from our earlier studies in both VC and sSC, after EO (AEO) WT mouse synaptosomes, but not whole lysates, show an enrichment of PSD-95 (Fig. 2A,B,D). In Flailer VC and sSC, this EO-dependent synaptic increase in PSD-95 failed to occur (Fig. 2A,C,E) indicating that visual neurons of Flailer mice are deficient in transporting PSD-95 to synaptosome postsynaptic sites upon developmental increases in neural activity.

Synaptosome fractions of Flailer fails to show enrichment of PSD-95 and its activity-dependent upregulation after EO. A–E, PSD-95 is enriched in synaptosomes (Syn) compared with whole lysates (WL) and its level increases 1 d after controlled eye opening (AEO: P14) as compared with 1 d before eye opening (BEO: P12) in both VC (B) and sSC (D) of WT. Flailer VC does not show an enrichment of PSD-95 in synaptosome or an increase AEO as compared with BEO (C, E). Histone 2B (H2B) is concentrated only in the WL as a measure of the purity of the synaptosomes. Immunoblotting experiments were repeated six times (twice from 3 litters) and equal amounts of proteins were loaded (10 μg) for both WL and Syn lanes. For quantification, band intensities of PSD-95 in B and D were normalized to BEO WL. *p < 0.05; for B and D. Error bars indicate SEM.
Impaired MyoVa-mediated transport selectively decreases localization of PSD-95 but not of SAP102 on VC dendritic protrusions
We examined endogenous PSD-95 distribution in dissociated neuron cultures from VC of WT and Flailer mice (DIV 14–16). In Flailer most endogenous PSD-95 immunolabeling was limited to dendritic shafts and the number of distinct PSD-95 synaptic puncta located away from the shaft were significantly reduced compared with WT neurons (Fig. 3A,B,G). While Flailer neurons had more immunolabeling of the presynaptic protein synaptophysin than WT (Fig. 3G), most overlap of synaptophysin and PSD-95 occurred on dendritic shafts where PSD-95 was distributed. Available antibodies to SAP102 were inadequate for immunohistochemistry. However, we used lipofectamine transfection in cultured VC neurons (Fig. 3C,D), or in utero electroporation to introduce a GFP-tagged version of either PSD-95 or SAP102 into layer 2/3 VC cortical neurons (Fig. 3H,I), to quantify the effectiveness of each scaffold's transport to synaptic puncta. GFP-tagged PSD-95 distribution in culture was consistent with the endogenous PSD-95 data, except that somewhat higher densities of puncta were found in both mouse strains, due to overexpression (Fig. 3G). Cultured WT neurons also showed increased PSD-95-GFP puncta on dendritic protrusions compared with PSD-95 puncta in Flailer cultures where most PSD-95 was localized on dendritic shafts (Fig. 3C,D). In contrast, neurons transfected with SAP102-GFP showed similar puncta numbers in Flailer and WT cells and also much less expression of SAP102-GFP in dendritic shafts (Fig. 3E–G). SAP102-GFP or PSD-95-GFP in VC layer 2/3 neurons in vivo imaged in fixed slices from P14 pups (1 day AEO) showed distributions similar to those in VC cultures and quantification of puncta verified the culture results (Fig. 3H,I). We previously reported (Yoshii et al., 2011) that PSD-95-GFP puncta in WT mouse VC layer 2/3 neurons in vivo significantly increased 1 d after EO. Figure 3, H and I, indicate that this same increase in PSD-95 does not occur in Flailer whereas SAP102 puncta are not altered between WT and mutant.

In Flailer VC neurons, both endogenous and GFP-tagged PSD-95 protein is present mainly along dendritic shafts with fewer PSD-95 puncta. A, B, G, WT and Flailer neurons dissected from embryonic VC were immunostained with PSD-95 and synaptophysin at DIV 14–16. Three different dendritic segments are shown from a WT and a Flailer neuron. While PSD-95 shows a distinct punctate distribution pattern in WT, it is present mainly in dendritic shafts with significantly fewer discernible puncta in Flailer. More synaptophysin labels are present along dendrites in Flailer as compared with WT. C, D, G, Despite overexpression, the numbers of PSD-95-GFP-positive puncta were significantly lower in Flailer neurons than in WT neurons. E–G, Flailer and WT neurons show similar punctate distributions of SAP102-GFP. G, Also plots endogenous PSD-95 and synaptophysin label and both PSD-95-GFP puncta and SAP102-GFP puncta on the same axis, a result consistent with the electrophysiological data showing more functional synapses in Flailer. H, I, In utero electroporation of a PSD-95-GFP DNA at E15.5 specifically labels layer 2/3 visual cortical pyramidal neurons. When examined at P14, 1 d after EO, Flailer neurons show reductions in the density of PSD-95-GFP-positive protrusions as compared with WT neurons in vivo. The densities of SAP102-GFP-positive protrusions are comparable between WT and Flailer. I, Shows representative images of dendritic segments in WT and Flailer neurons. Scale bars: (in A–F, I) 10 μm. Magnified dendritic branches are sampled from areas indicated by dashed boxes (A–F). For each condition, N = 30 branches in 10 neurons. **p < 0.01 for G. Error bars indicate SEM. ***p < 0.001 for H. Error bars indicate SEM.
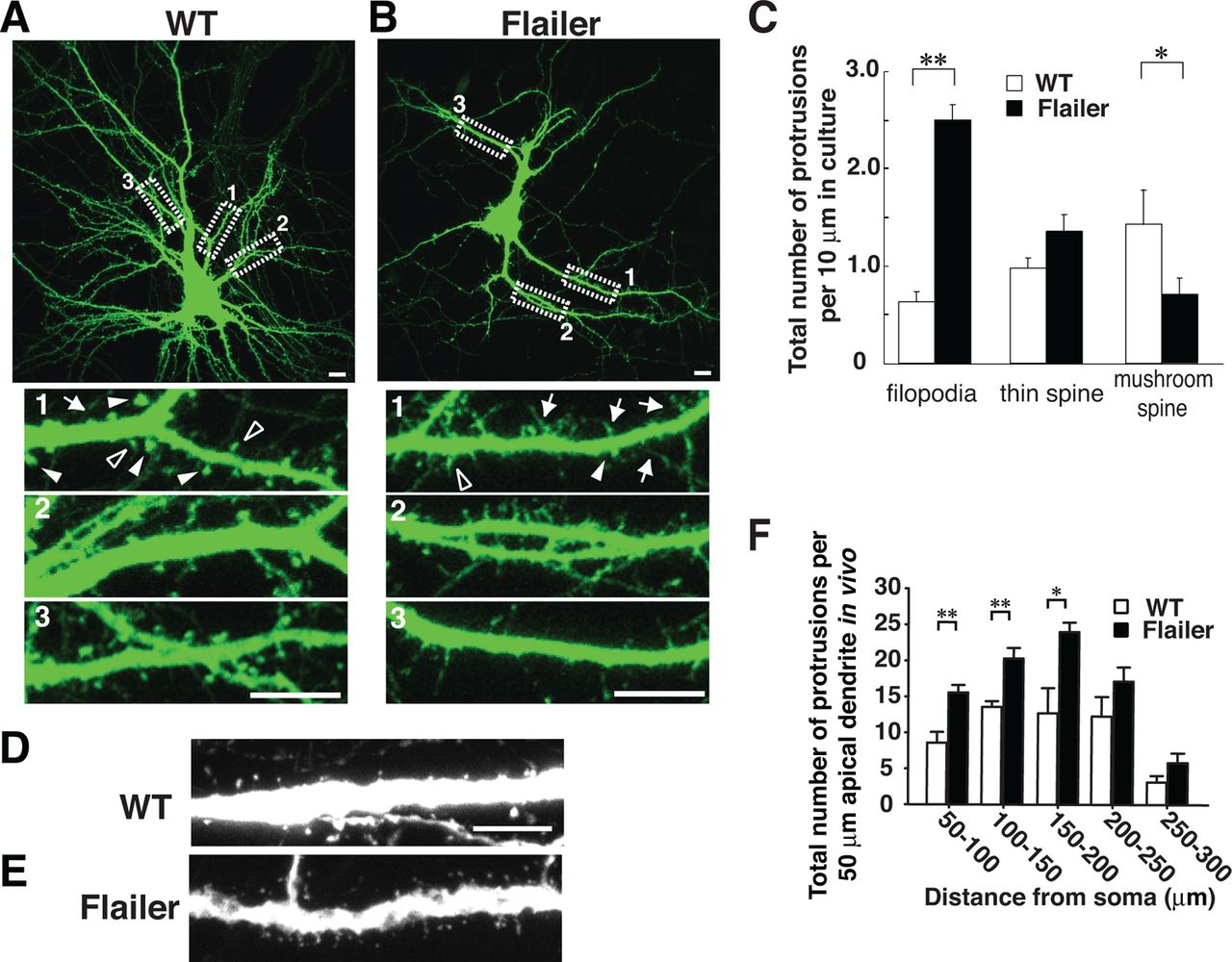
Flailer neurons show reductions in dendritic spines and increases in filopodia
GFP transfected into WT and Flailer-cultured VC neurons allowed analysis of dendritic morphology in secondary branches (Fig. 4A,B). Classification of spines and protrusion is based on Harris et al. (1992). We defined that thin spines had heads ≥2 and ≤3 times the spine neck diameter and mushroom spines had heads >3 times the spine neck diameter. Compared with WT, Flailer neurons had higher densities of dendritic protrusions with the majority showing filopodia-like morphology. Mushroom spines were few in Flailer neurons, but thin spines were present at approximately equal densities in both Flailer and WT (Fig. 4A–C). We confirmed these alterations in dendritic protrusions in vivo by crossing Flailer with Thy-1 EGFP mice (Feng et al., 2000) and breeding to homozygosity at the flailer locus. We compared apical dendrites of P16 VC layer 5 pyramidal neurons in Flailer/EGFP and WT/EGFP mice (secondary dendrites were difficult to discern because of the high density of EGFP-filled layer V pyramidal neurons and consequent dendritic overlap). These developing pyramidal neurons had higher densities of thin, long dendritic protrusions in Flailer, and when spine heads were present, they were qualitatively smaller than WT (Fig. 4D,E). The density difference between WT and Flailer was most pronounced near the cell body (Fig. 4F). In these young animals in vivo distal dendritic differences in WT versus Flailer protrusion density were not present. The finding probably results because the cortex is getting thicker and apical dendrites grow at distal tips. In such immature regions SAP102 is still the dominant MAGUK in both genotypes and SAP102 is not mislocated in Flailer.

Flailer neurons have fewer mature spines in vitro and in vivo. A–C, WT (A) and Flailer (B) neurons dissected from embryonic VC, transfected with eGFP at DIV 8 and imaged at DIV 14–16. A, Filled triangles show examples of mushroom spines and outlined triangles indicate examples of thin spines. B, Arrows indicate examples of filopodia and solid arrows indicate mushroom spines. As compared with WT, Flailer neurons have fewer mushroom spines and more filopodia (C). For each strain, N = 30 segments in 10 neurons. Magnified dendritic branches are sampled from areas indicated by dashed boxes. D–F, Apical dendrites of WT (D) and Flailer (E) layer 5 VC neurons in vivo at P16. Both strains were crossed with thy-1 GFP transgenic mice. As compared with WT, Flailer neurons have higher densities of thinner and longer protrusions (F). For each strain, N = 10 branches in 10 neurons. For C and F, *p < 0.05; **p < 0.01. Error bars indicate SEM. Scale bars: (in A, B, D, E) 10 μm.
Flailer visual neurons have increased spontaneous and miniature AMPAR currents
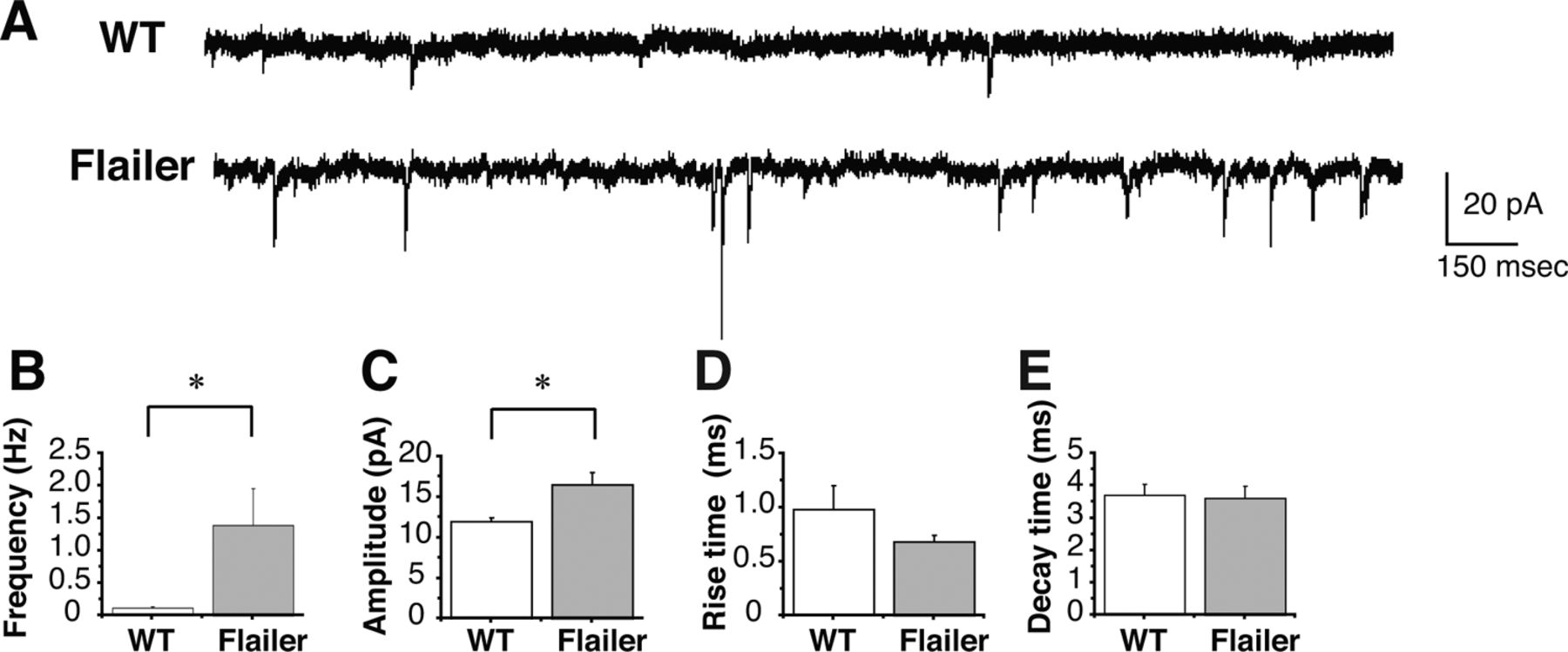
Initial exploratory whole-cell voltage-clamp studies using acute slices of the sSC demonstrated higher frequencies and amplitudes of spontaneous AMPAR currents in Flailer as compared with WT sSC neurons (Fig. 5). Consequently, we performed detailed electrophysiological synaptic analyses of Flailer and WT on specific layer 2/3 neurons in acute VC slices. At P16 (2 d after EO), Flailer cells had significantly higher frequencies of pharmacologically isolated miniature AMPAR currents than WT neurons (Fig. 6A,B), without changes in amplitude, rise time, or decay time (Fig. 6A,C,D,E). This indicated abnormally high AMPAR-containing functional synaptic contacts in VC layer 2/3 Flailer pyramidal neurons than in WT. In contrast, miniature NMDAR currents recorded at +40 mV to include any NMDAR-only contacts (silent synapses) were not significantly different in frequency, amplitude, rise time, or decay time between Flailer and WT (Fig. 6F–J). We also found no change in spontaneous NMDAR currents at +40 mV (data not shown). Consistent with the miniature current recordings indicating more AMPAR- than NMDAR-containing synapses, Flailer neurons showed higher evoked AMPAR/NMDAR amplitude ratios than WT after stimulating layer 2/3 from layer 4 (Fig. 6K). Paired-pulse analyses of AMPAR currents were not significantly different between WT and Flailer neurons (Fig. 6L) indicating no changes in presynaptic glutamate release.

Flailer sSC neurons have higher frequency spontaneous AMPAR but not NMDAR currents. A–E, Spontaneous isolated AMPAR currents in the sSC recorded in acute slices prepared from P14 of WT and Flailer (A). Frequency (B) and amplitude (C), but neither rise time (D) nor decay time (E), are significantly different between WT and Flailer. Six cells for each genotype.

Flailer VC neurons, compared with WT VC neurons, show higher frequencies of miniature AMPAR currents, no change in miniature NMDAR currents, and increased evoked AMPAR/NMDAR ratios. A, Miniature AMPAR currents of layer 2/3 pyramidal neurons recorded in acute slices prepared from P16 VC of WT and Flailer mice. B–E, Miniature AMPAR current frequency (B), but not amplitude (C), rise time (D) or decay time (E), was significantly different between WT and Flailer. F, Miniature NMDAR currents of layer 2/3 pyramidal neurons recorded in acute slices prepared from P16 VC of WT and Flailer. G–J, Miniature NMDAR current frequency (G), amplitude (H), rise time (I), and decay time (J) were not significantly different between WT and Flailer. K, Ratio of evoked AMPAR/NMDAR current amplitude was significantly higher in Flailer neurons than in WT neurons. L, Paired-pulse ratios were not significantly different between WT and Flailer neurons indicating no change in probability of neurotransmitter release. *p < 0.05 for B, ***p < 0.001 for K. Error bars indicate SEM. Number of cell and animals are indicated as (n = number of cells/number of animals) in B–E and G–L.
The finding of more AMPAR synapses than NMDAR synapses is novel in cortex and suggests that the increase in AMPAR spontaneous currents seen in the sSC and AMPAR miniature currents (synaptic contacts) seen in the cortex could represent constitutive (NMDAR independent) AMPAR synaptic expression. Constitutive AMPAR currents have been documented in acute hippocampus slices and slice cultures (Li et al., 1994; Zhu and Malinow, 2002) and in acute slices of sSC (Colonnese et al., 2003) after long-term APV blockade of NMDARs. They might also reflect reduced endocytosis of AMPARs.
Adult Flailer neurons show abnormally enhanced plasticity in response to low frequency stimulation
Our finding of increased miniature AMPAR current frequency and evoked AMPAR/NMDAR amplitude ratios in Flailer mice motivated an examination of LTP and LTD in their neurons. We studied these forms of plasticity in the same layer 2/3 neurons examined in the young mice using a protocol for stimulation of VC layer 4 and field recording of NMDAR-dependent LTP and LTD in layer 2/3 of mature mouse VC (Kirkwood et al., 1997). VC neurons from older than 6-month-old WT and Flailer mice showed an LTP in layer 2/3 neurons (Fig. 7A). In both the earlier study and our WT NMDAR-dependent LTD stimulation of layer 4 at 1 Hz for 15 min failed to produce layer 2/3 LTD in these older neurons (Fig. 7B). However, in marked contrast to WT, the same LTD stimulation protocol produced a significant LTP in mature Flailer VC slices (Fig. 7B). The finding suggested that the skewed AMPAR and NMDAR ratios seen in the young Flailer were due to a failure of activity-dependent AMPAR endocytosis: specifically, in Flailer NMDAR activity was producing normal increases in AMPAR insertion without triggering a balanced endocytosis of AMPARs in response to low frequency, LTD producing activity.

Adult Flailer neurons show normal LTP but abnormally enhanced plasticity in response to low frequency stimulation. A, Theta burst stimulation (TBS) induced a similar LTP in both WT (filled diamonds: 1.14 ± 0.04) and Flailer (open diamonds: 1.13 ± 0.04). B, No LTD was observed (filled diamonds: 1 ± 0.04) in WT, but LTD stimulation induced a significant increase in fEPSP amplitude in Flailer (LTP) (open diamonds: 1.13 ± 0.04). *p < 0.05 for B. Error bars indicate SEM. Number of cell and animals are indicated as (n = number of slices/number of animals) in A and B.
Flailer neurons show perturbed internalization of AMPAR subunits due to the mislocalization of endocytic machinery
To test this defective AMPAR endocytosis hypothesis directly we quantified the relative amounts of AMPARs on the neuronal cell surfaces of Flailer and WT neurons using biotinylation assays. In cultured VC neurons we found that Flailer neurons had significantly higher surface levels of both the GluA1 and GluA2 AMPAR subunits (Fig. 8A,B).

AMPARs accumulate on cell surface in Flailer (Fl) neurons. A, B, Surface proteins in cultured VC neurons were biotinylated at DIV 14–16 and pulled down using NeutrAvidin-agarose slurry. Flailer neurons have a higher amount of total surface GluA1 and GluA2 in Flailer neurons as compared with WT on baseline while transferrin receptors (TfR) levels, used as a control, were comparable. Experiments were repeated four times. C, D, Application of NMDA at 20 μm induced transient endocytosis of biotinylated GluA1 in WT but not in Flailer neurons. Biotin of the reinserted GluA1 receptors to the cell surface were stripped with glutathione. Therefore, all of biotin-tagged GluA1 proteins that were pulled down with NeutrAvidin-agarose were internalized receptors. The graph in D shows the amount ratios of internalized receptors to the total receptors before the induction of endocytosis. Internalized GluA1 was significantly lower in Flailer than WT at 5, 10, and 15 min. Experiments were repeated four times. E, Application of NMDA at 20 μm induced transient endocytosis of pHluorin-tagged GluA2 in WT but not in Flailer neurons (N = 6 neurons). The graph shows the reduced internalization of GluA2 in Flailer as compared with the WT cultures. F, I, Cultured neurons stained with an antibody to the N terminal of the GluA2 AMPAR subunit without permeabilization show surface receptor distributions. The number of surface GluA2-positive protrusions is significantly increased in Flailer neurons as compared with WT (I). Magnified dendritic branches are sampled from areas indicated by dashed boxes, and show that dendritic shafts (outlined with white dashed curves) have high signals in Flailer neurons indicating diffuse distribution of surface GluA2 along dendrites. G, I, Dynamin 3 (Dyn 3), a critical component of endocytosis at synapse, colocalizes with punctate distributions of PSD-95 in WT neurons while both proteins are localized to dendritic shafts in Flailer. Number of dynamin 3-positive puncta is significantly reduced in Flailer neurons as compared with WT (I), suggesting disrupted distribution of endocytic machinery. H, I, Stargazin colocalizes with punctate distributions of PSD-95 in WT neurons while both proteins are localized to dendritic shafts in Flailer. The number of stargazin-positive puncta is significantly reduced in Flailer neurons as compared with WT (I), correlating with an increase in extrasynaptic distribution of surface GluA2 subunit. J, Dynamin 3 is reduced in VC homogenates prepared from Flailer as compared with WT. *p < 0.05; **p < 0.01 in D, E, and I. Error bars indicate SEM in D and E. Scale bars: (in F–H) 10 μm.
We subsequently examined surface turnover of AMPARs using two different culture approaches. First, biotinylation of GluA1 was combined with NMDA application (20 μm, 2 min) to a number of equally dense cultures and different sets of cultures were fixed at different times. NMDA stimulation normally induces internalization of AMPARs (Ehlers, 2000). Testing solution included leupeptin, to prevent lysosomal degradation. To detect only internalized receptors, neurons were treated with glutathione. This cleaves the disulfide linker and strips biotin from newly appearing (reinserted) surface receptors after the induction of AMPAR endocytosis. Data were normalized by calculating the ratios of internalized receptors at each time interval to total surface receptor in cultures without induction. WT neurons showed the expected transient increases in internalized GluA1. However, in Flailer neurons the level of internalized GluA1 remained low and stable for the duration of the assay (Fig. 8C,D). In the second experiment, NMDA treatment was combined with an imaging assay using GluA2 tagged with pHluorin in the cultured VC neurons (Lin and Huganir, 2007). PHluorin is a GFP variant with significantly reduced fluorescence when it is endocytosed thereby providing an assay of changes in tagged surface molecule endocytosis in the same cultures over time. Both Flailer and WT neurons initially showed GluA2-pHluorin on their surface. In WT neurons, this fluorescence rapidly decreased with NMDA application to induce AMPAR internalization. However, the initial decrease of surface GluA2 in Flailer neurons was significantly smaller than that of WT neurons shown in the F0 − Ft/F0 plot against time (Fig. 8E). Also, Flailer neurons maintained this higher level of surface fluorescence for the 30 min duration of the experiment while WT neurons gradually reinserted GluA2 to the level of Flailer in the last 10 min of the experiment. Both sets of data document decreased internalization of AMPARs in Flailer relative to WT VC neurons.
Surface staining of Flailer VC cultures with anti-GluA2 antibody also revealed a higher density of generally larger and sometimes fused AMPAR puncta as compared with WT neurons (Fig. 8F,I). In addition surface GluA2 receptors in Flailer were also more diffusely distributed on dendritic shafts. We also found AMPAR regulatory proteins, stargazins, molecules necessary for binding AMPARs to the MAGUK scaffolds, diffusely distributed in dendritic shafts with fewer small puncta than WT and frequent fused blob-like clusters, which proved impossible to count as puncta with high accuracy (Fig. 8G,I).
Normally activity-dependent AMPAR endocytosis and recycling of AMPARs occurs at a specialized endocytic zone located adjacent to one side of the PSD in dendritic spines (Lu et al., 2007). Dynamin 3 is a postsynaptic dynamin critical for this endocytosis of AMPARs and it is held by the scaffold Homer in the endocytic zone (Gray et al., 2003). In Flailer neurons, most dynamin 3 was distributed in the dendritic shaft, as can be seen in the overlay of Flailer dendritic segments (Fig. 8H) where the shafts show an orange tint, indicating overlap of dynamin 3 with the high levels of immunostained anti-PSD-95 in the shaft. The figure also demonstrates that there was little evidence of overlap between the few discernible dynamin 3 puncta and the small number of PSD-95 puncta present in Flailer. In addition, Western blots of VC whole lysates probed with the antibody recognizing the cargo-binding N-terminal domain of MyoVa present in both WT and Flailer, as the control, revealed substantially less dynamin 3 in Flailer compared with WT (Fig. 8I). These results suggest that Flailer neurons have defects in distribution and net production of dynamin 3, the dynamin unique to activity-dependent endocytosis at glutamate synapses (Gray et al., 2003). This result is consistent with the disrupted endocytosis of AMPARs, and the abnormal accumulation of AMPARs on Flailer cell surfaces.
Discussion
In the visual pathway the activity associated with EO and the onset of pattern vision causes rapid PSD-95 trafficking to synapses. This stimulates BDNF/TrkB signaling to palmitoylate PSD-95 (Yoshii et al., 2011) and allows membrane-associated, vesicular transport of this predominant MAGUK to the maturing PSD on the spines of excitatory neurons. Here we show that the plus-end actin filament motor protein MyoVa plays an essential role in this last stage of transport. MyoVa associates with DLCs (Espindola et al., 2000) and through DLC2 (Benashski et al., 1997; Naisbitt et al., 2000) holds SAPAP1 and all of the associated excitatory scaffolding complex and transports it up the spine to the PSD. This scaffolding complex includes PSD-95 and Shank that holds Homer (Gerrow et al., 2006). The latter probably also carries dynamin 3 to the endocytic zone because we found that in Flailer hippocampus Homer1C, which holds dynamin 3, is significantly reduced in synaptosomes (Pandian et al., 2011). The scaffold complex delivery replaces the initially appearing MAGUK SAP102 with the PSD-95 MAGUK during development (Sans et al., 2000; Yoshii et al., 2003; van Zundert et al., 2004): A step in initial synapse stabilization is also likely to occur during learning and memory in the mature brain regions where SAP102 remains present (Allen Brain Atlas) and new excitatory synapses are formed in maturity. Significantly the interaction between MyoVa-DLC2 and SAPAP1 depends on sustained synaptic activity (Moutin et al., 2012). This is fully consistent with our initial finding that pattern vision initiates the increase in PSD-95 at visual synapses (Yoshii et al., 2003, 2011) and also with the activity-driven processes of learning and memory.
In this context the present finding that SAP102 appears at dendritic protrusions despite the disrupted MyoVa function in Flailer mice is not surprising. SAP102 is the MAGUK of new synapses. EM studies show most early synapses on dendritic shafts although a large number are also present on filopodia (Fiala et al., 1998). In differentiating neurons the SAP102/GluN2B complex travels in the dendritic shaft moving in and out of the plasma membrane (Washbourne et al., 2004) and EphB clustering of GluN2B containing NMDARs is likely the first step in synapse formation (Dalva et al., 2000). Whether SAP102 localization on dendritic protrusions reflects its initial presence on membrane where protrusions form, or whether the MAGUK actually travels to protrusion tips, remains unknown, but the process is independent of MyoVa. SAP102 is necessary to initiate spine maturation (Chen et al., 2011; Murata and Constantine-Paton, 2013) as may be indicated by similar levels of thin spines on WT and Flailer neurons (Fig. 4C). However, maturation of mushroom spines involves additional molecules and interactions, such as myosin IIb and normal positioning of PSD-95 and Shank molecules in spines (Korobova and Svitkina, 2010; Hodges et al., 2011). The latter requirement is supported by the abnormal morphology and reductions of mature spines in Flailer (Fig. 4).
AMPARs and NMDARs differ in prevalence on Flailer neurons
In addition to disruptions of MAGUK transport, whole-cell recordings of NMDAR and AMPAR miniature currents on Flailer and WT VC layer 2/3 neurons reveal significant differences between the prevalence of NMDAR and AMPAR synapses. In Flailer, layer 2/3 neurons show significant increases over WT in miniature AMPAR current frequency and therefore in AMPAR-containing synapses (Fig. 6A,B). However, the same neurons have normal NMDAR miniature current frequencies implying normal numbers of NMDAR-containing synapses (Fig. 6F,G). The data are consistent with the significantly increased AMPAR/NMDAR ratios present in Flailer VC (Figs. 6K). This excess is also suggested by recordings in the sSC (Fig. 5) and therefore is not restricted to a particular cell type or to neocortical neurons. The electrophysiological data are generally consistent with our immunohistochemical staining and MAGUK-EGFP in VC neurons. PSD-95 directly binds NMDARs it is largely in and on dendritic shafts, as is our staining for AMPARs, and the molecules dynamin 3 and stargazin, which maintain AMPARs near the PDZ domains of MAGUKs. Relatively few of these molecules are on dendritic protrusions or the rare mature spines in Flailer. Petralia et al. (2001) examined PSDs of MyoVa−/− mice. They found AMPARs at synapses with EM immunogold staining, but it is not clear if these PSDs are on shafts or spines or filopodia (Petralia et al., 2001).
There are several likely causes for the increase in AMPARs while electrophysiological properties of NMDARs are normal. One possibility, predicated from the literature on cerebellum in other MyoVa rodent mutants (Takagishi et al., 1996; Miyata et al., 2000), is that Group I metabotropic receptor (mGluR)-dependent LTD, which depends on Ca2+ release from intracellular stores, is defective due to the absence of the IP3 receptor and SER in Purkinje cell spines. SER is also absent in Flailer Purkinje cells (Jones et al., 2000) and we find that mGluR5-LTD (also a Group I mGluR) is absent in hippocampus CA1 (Feinberg-Zadek et al., 2010; Pandian et al., 2011). Layer 2/3 VC neurons do not show mGluR-dependent LTD. However, they do show NMDAR-dependent LTD. This LTD is present at layer 4 to layer 2/3 VC synapses in young mice but absent at these same synapses in mature mice (Kirkwood et al., 1997). Consequently, we examined NMDAR-dependent LTP and LTD in mature Flailer. In contrast to older WT, in Flailer older than 6 months both NMDAR-dependent LTP-inducing stimulation and NMDAR-dependent LTD-inducing stimulation produce LTP at the layer 4 to layer 2/3 synapses (Fig. 7A,B) and, as shown in Fig. 8, A–E, NMDAR-dependent AMPAR endocytosis is significantly disrupted in Flailer VC.
In fact the entire AMPAR scaffolding and recycling system normally clustered at the PSD of spine synapses is disorganized in Flailer. Stargazin family molecules that bind AMPARs to MAGUKS show a diffuse distribution in VC neurons instead of their normal distribution largely overlapping with PSD-95 puncta (Fig. 8G). In addition, Flailer cortex has decreased levels of dynamin 3 (Fig. 8J), which is localized adjacent to the PSD (Gray et al., 2003), interacts with Homer, and is specifically involved in activity-dependent AMPAR endocytosis and recycling. Although NMDARs can be endocytosed, there is no evidence that dynamin 3 is involved in this process (Ehlers, 2000; Lu et al., 2007). Dynamin 3 is diffusely distributed in Flailer dendritic shafts (Fig. 8H). These data suggest that impaired transport of the PSD complex also results in disrupted localization and function of endocytic zone, an interpretation consistent with our electrophysiological data.
A final factor contributing to AMPAR activity increases in Flailer is that AMPAR subunit trafficking to spines in dendrites involves a different non-muscle myosin, Myosin Vb (Lisé et al., 2006; Wang et al., 2008). Thus normal Myosin Vb function, combined with decreases in MyoVa function and consequent disruption of the normal PSD, probably increases surface AMPAR expression due to disrupted endocytosis. AMPAR removal or addition to synapses is a major means of glutamate synaptic strength regulation through the activity-dependent processes of LTP and LTD. This critical process is significantly disrupted in Flailer mice and probably contributes to their early seizures and abnormal behavior.
This report describes a pronounced disruption of the major excitatory synapse control system in the vertebrate brain. Indeed the severe convulsions shown by Flailer pups are readily understood in this context. Although our initial behavioral work suggests intriguing abnormalities in Flailer behavior, neither the compensations that mitigate this strain's early seizures, nor its behaviors, have been studied in depth (Jones et al., 2000; Pandian et al., 2011). Thus further work on Flailer combined with genetic spatial and temporal manipulation of the Flailer protein may prove exceptionally powerful. Disruption of synaptic weakening and the hyperconnectivity that likely results in particular brain regions or cells when the Flailer protein is selectively expressed could facilitate understanding of the molecular mechanisms underlying normal circuit development. It could also prove vital for investigations of complex neurological and psychiatric disorders that clearly arise from circuit and synapse disruption in frequently unlocalizable regions of the brain.
Footnotes
This work was supported by R01-EY014074–17 and EY014420 to M.C.-P.. We thank Dr. John Hammer for the “Central” MyoVa antibody (Figure 1A), Dr. Johannes Hell for the SAP102 antibody, Dr. McNiven for the dynamin3 antibody, Dr. Morgan Sheng for the GKAP/SAPAP1 antibody, Dr. Natalie Sans for the GFP-tagged SAP102construct, Dr. Richard Huganir for the pHluorin-tagged GluA2 construct, and Dr. Miriam Meisler for the flailer mice. We also thank Dr. Mark Mooseker for suggesting that we study flailer.
- Dr. Akira Yoshii, McGovern Institute for Brain Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Building 46-4165, Cambridge, MA 02139. ayoshii{at}mit.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}