

Abstract
N-methyl-d-aspartate receptor (NMDAR)-mediated excitotoxicity is implicated as a proximate cause of neurodegeneration in Huntington Disease (HD). This hypothesis has not been tested rigorously in vivo. NMDAR–NR2B subunits are a major NR2 subunit expressed by striatal medium spiny neurons that degenerate in HD. To test the excitotoxic hypothesis, we crossed a well validated murine genetic model of HD (Hdh(CAG)150) with a transgenic line overexpressing NMDAR–NR2B subunits. In the resulting double-mutant line, we show exacerbation of selective striatal neuron degeneration. This is the first direct in vivo evidence of NR2B–NMDAR-mediated excitotoxicity in the context of HD. Our results are consistent with previous suggestions that direct and/or indirect interactions of mutant huntingtin with NMDARs are a proximate cause of neurodegeneration in HD.
Introduction
Huntington disease (HD) is a dominantly inherited, incurable neurodegeneration caused by expanded CAG repeat/polyglutamine repeats within the huntingtin locus, resulting in a pathogenic huntingtin protein (htt). Degeneration of striatal medium spiny neurons (MSNs) is a hallmark of HD. Clinical manifestations include progressive cognitive decline, psychiatric disturbances, and involuntary movements. Although the molecular mechanisms underlying selective degeneration of MSNs in HD are not understood, convergent evidence supports a role for N-methyl-d-aspartate receptor (NMDAR)-mediated excitotoxicity (Coyle and Schwarcz, 1976; McGeer and McGeer, 1976; Schwarcz et al., 1983; Beal et al., 1986; Fan et al., 2007).
The striatum receives dense glutamatergic corticostriatal and thalamostriatal inputs, and MSNs are endowed heavily with glutamate receptors, including NMDARs. Coyle and Schwarcz (1976) and McGeer and McGeer (1976) developed the first models of HD by demonstrating that acute intrastriatal administration of the glutamate agonist kainic acid produced neurodegeneration with features of HD. Later work indicated that acute striatal lesions with NMDAR-selective agonists reproduced more features of HD striatal pathology (Beal et al., 1986; Bazzett et al., 1993; Ferrante et al., 1993). This is true for both NMDAR agonists and metabolic inhibitors whose indirect effects facilitate NMDAR activation (Brouillet et al., 1995; Greene and Greenamyre, 1995). Although the correlation between the effects of acutely administered NMDAR agonists and HD striatal pathology is impressive, the NMDAR excitotoxicity hypothesis of striatal degeneration in HD has not been previously tested rigorously in vivo.
NMDARs are hetero-multimeric ionotropic receptors that mediate extracellular calcium entry and consist of NR1 subunits and NR2 (A–D) or NR3 (A–C) subunits (Dingledine et al., 1999; Waxman and Lynch, 2005). Pharmacological and functional properties of NMDARs depend heavily on NR2 subunit composition (Cull-Candy et al., 2001; Loftis and Janowsky, 2003). NMDAR activation promotes both prodeath and prosurvival signaling (Papadia and Hardingham, 2007). Regulation of the balance between prodeath and prosurvival NMDAR signaling is understood incompletely. In some model systems, extrasynaptic NMDARs promote prodeath signals, whereas synaptic NMDARs promote neuronal survival (Hardingham et al., 2002; Léveillé et al., 2008; Papadia et al., 2008; Martel et al., 2009). Other data suggests that the critical factor is NMDAR subunit composition: NR2B-containing NMDARs promoting neuronal death and NR2A-containing NMDARs promoting neuronal survival (Liu et al., 2007). These hypotheses may be partially convergent. In maturer mammalian CNS synapses, NR2B-containing NMDARs tend to be extrasynaptic, whereas NR2A-containing NMDARs dominate synapses (Tovar and Westbrook, 1999). NR2B subunit containing NMDARs are highly expressed by MSNs (Monyer et al., 1992). Previous in vitro studies suggest sensitization of NR2B function by mutant htt (Chen et al., 1999; Zeron et al., 2001).
To determine whether striatal pathology and behavioral deficits of HD are caused by NMDAR-mediated excitotoxic injury in vivo, we created a modified murine model of excitotoxic injury by crossing a well validated murine genetic model of HD, Hdh(CAG)150, to a transgenic mouse overexpressing NMDAR–NR2B subunits (Tang et al., 1999; Heng et al., 2007).
Materials and Methods
Hdh150/+;NR2BTg/WT double-mutant mice.
Interbreeding the Hdh(CAG)150 and NR2B transgenic lines results in four groups of mice, Hdh+/+;NR2BWT/WT [wild-type (WT) huntingtin; no transgene], Hdh+/+;NR2BTg/WT (wild-type huntingtin; one NR2B transgene copy), Hdh150/+; NR2BWT/WT (one mutant huntingtin copy; no transgene), Hdh150/+; NR2BTg/WT (one mutant huntingtin copy; one NR2B transgene copy).
All four groups were generated from a cross of Hdh(CAG)150 mice maintained on a mixed genetic background, 129/Ola and C57BL/J6 expressing 75–90% C57BL/6; (P. J. Detloff, unpublished data), with NR2B transgenic mice maintained on a genetic CBA/C57BL/6 F1 hybrid background resulting in an F1 mixed background 129/Ola and CBA/C57BL/J6. Both male and female mice and WT littermates were used. All animals were housed in cages grouped by gender with food and water ad libitum. Animals were housed in specific pathogen-free conditions with a 12 h light/dark cycle maintained at 23°C. All procedures were conducted in compliance with the Guide for the Care and Use of Laboratory Animals and approved by the Committee on Use and Care of Animals (UCUCA), University of Michigan, and the Veterinary Medical Unit (VMU) at the Veterans Affairs Ann Arbor Health System.
Genotyping.
All mice studied were genotyped for both Hdh alleles and the NR2B transgene. The Hdh allele and NR2B transgene genotyping protocols are published (Tang et al., 1999; Heng et al., 2007). Periodic CAG sizing was performed outside our laboratory (Laragen, Los Angeles, CA) to confirm consistency of CAG repeat number, which ranged from 145 to 150 CAG repeats. For zygosity testing, NR2B transgene detection was performed outside our laboratory (Charles River Laboratories).
Euthanasia.
Animals were killed according to national guidelines. Euthanasia was performed by decapitation and approved by UCUCA and the VMU.
Western blotting.
Brains from Hdh150/+;NR2BTg/WT and Hdh150/+;NR2BWT/WT were flash-frozen. Striata were homogenized in radioimmunoprecipitation assay buffer containing proteinase inhibitor mixture (Roche) and total protein extracts were obtained. Protein concentration was determined by the Bradford Assay (Bio-Rad). Approximately 60 μg was separated by SDS-PAGE (4–12% gradient) and transferred to nitrocellulose membranes. Membranes were blocked in 5% nonfat milk in PBS with 0.1% Tween for 1 h at room temperature and probed overnight at 4°C with antibodies raised against NR2B (1:1000) or Actin (1:500) (Sigma-Aldrich), washed in PBS containing 0.1% Tween, and incubated with secondary antibodies in 5% nonfat milk in PBS with 0.1% Tween for 1 h. Membranes were washed in PBS containing 0.1% Tween followed by several washes in PBS, and protein bands were visualized by chemiluminescence (Pierce) and autoradiography.
Behavioral examination.
Six behavioral assays were used: (1) accelerating rotarod, (2) hanging wire test, (3) tail suspension test, (4) activity monitoring, (5) footprint analysis, and (6) balance beam. All tests were performed at 20, 40, 50, 70, and 100 weeks (Heng et al., 2007). Eight to fourteen animals were used per group.
Pathologic analyses.
Immunohistochemistry, stereology, and receptor autoradiography analyses were performed as described previously (Heng et al., 2007).
Stereological counts of striatal neurons were obtained from striata of animals at 70 and 100 weeks of age using StereoInvestigator software (Microbrightfield). The optical fractionator method was used to generate an estimate of NeuN immunoreactive cells. Striatal volume was reconstructed with StereoInvestigator software. Serially cut sagittal tissue sections (every fourth section) were analyzed for one entire hemisphere of animals in each genotype cohort (n = 4 per group).
Statistical analyses.
All studies were performed blind to genotype. Comparisons were performed with one-way ANOVA and post hoc comparisons with the Tukey honestly significant difference when p < 0.05. Repeated measures ANOVA was performed for behavioral tests requiring repeated tests. A critical p < 0.05 was used for statistical significance in all analyses. SPSS was used.
Results
NR2B expression
Western blot analysis confirmed that Hdh150/+;NR2BTg/WT double-mutant mice exhibited increased levels of NR2B–NMDAR subunits compared with Hdh150/+;NR2BWT/WT littermate controls (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Hdh150/+;NR2BTg/WT double-mutant mice exhibit striatal neuron loss and decreased striatal dopamine receptors
At 100 weeks of age, Hdh150/+;NR2BTg/WT double-mutant mice displayed atrophic and irregularly shaped striatal neurons compared with controls (Hdh+/+;NR2BWT/WT, Hdh+/+;NR2BTg/WT and Hdh150/+;NR2BWT/WT) (Fig. 1a–d). The Hdh150/+;NR2BTg/WT double mutants exhibited marked decreases in striatal neuron number and striatal volume with a mean 51% decrease in striatal neuron number and a mean 36% decrease in striatal volume compared with all three control groups (Hdh+/+;NR2BWT/WT, Hdh+/+;NR2BTg/WT and Hdh150/+;NR2BWT/WT) (Fig. 1e,f). At 70 weeks, there was no decrease in striatal neuron number or volume (data not shown).
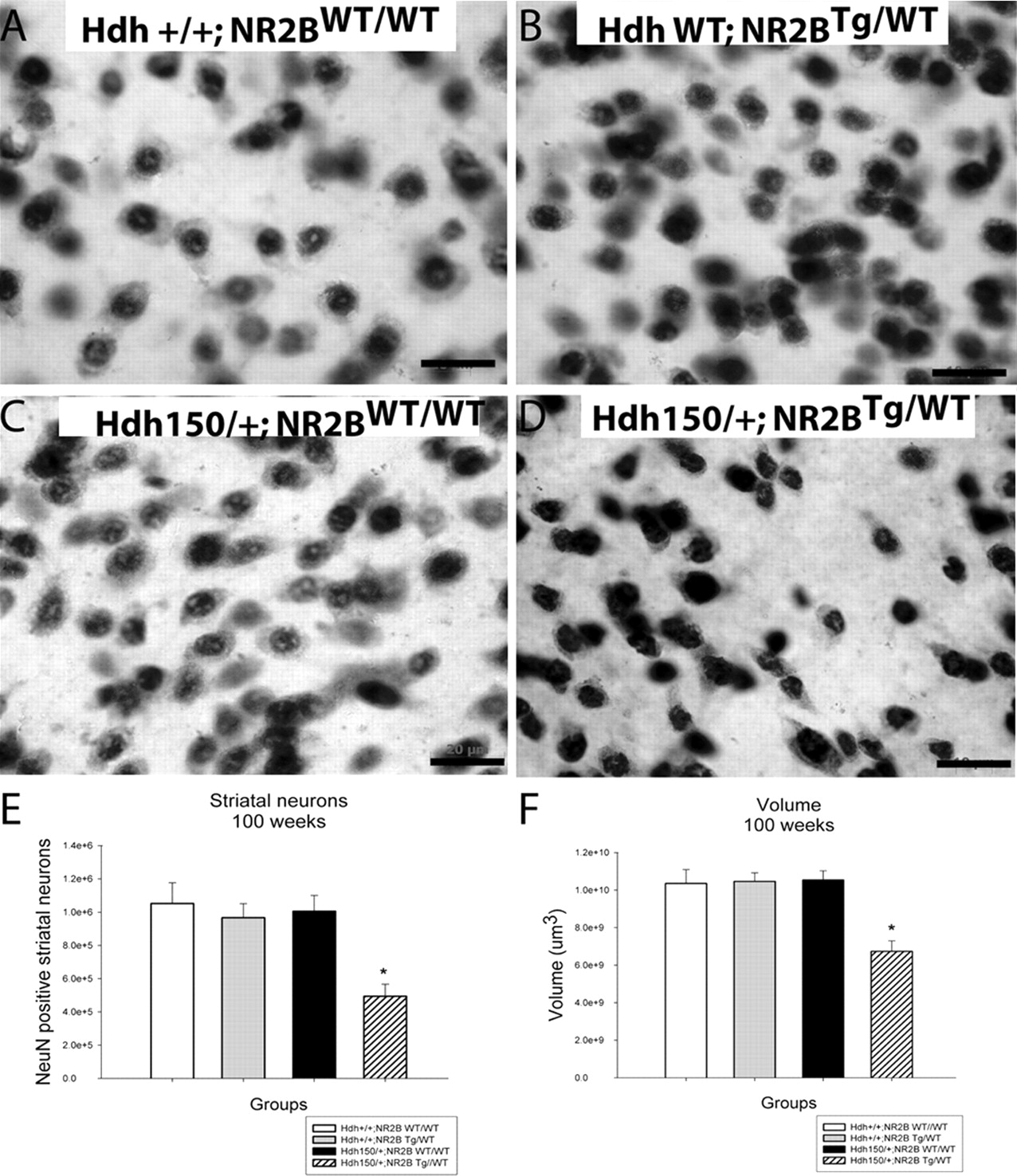
Hdh150/+;NR2BTg/WT double-mutant mice exhibit striatal neuron number and volume loss at 100 weeks. a–d, Hdh150/+;NR2BTg/WT double-mutant mice display atrophic and irregular shaped NeuN-stained striatal neurons compared with control groups (Hdh+/+; NR2BWT/WT, Hdh+/+; NR2BTg/WT, and Hdh150/+ NR2BWT/WT). e, Hdh150/+;NR2BTg/WT exhibit a 51% loss in striatal neuron number and a 36% reduction in striatal volume (f) compared with all three littermate control groups (Hdh+/+; NR2BWT/WT, Hdh+/+; NR2BTg/WT, and Hdh150/+ NR2BWT/WT). *p < 0.05. Values are expressed as mean ± SEM. n = 4 animals per group; scale bar, 20 μm.
Striatal pathology observed in double-mutant mice was confirmed by loss of striatal dopamine D1 and D2 receptors (Fig. 2; supplemental Table 1, available at www.jneurosci.org as supplemental material). The Hdh150/+;NR2BTg/WT double-mutant mice showed ∼45% decreases of D1 receptor binding in the ventral and dorsal striatum compared with both Hdh+/+;NR2BWT/WT and Hdh+/+;NR2BTg/WT wild-type mice (Fig. 2a). Although not significantly different, Hdh150/+;NR2BWT/WT mice exhibit a decrease of D1 receptor binding intermediate between the Hdh+/+;NR2BWT/WT, Hdh+/+;NR2BTg/WT wild-type controls and Hdh150/+;NR2BTg/WT double-mutant mice. Hdh150/+;NR2BTg/WT double mutants exhibited an ∼50% reduction in the ventral and dorsal striatal D2 receptor binding compared with Hdh+/+;NR2BWT/WT wild-type controls. Hdh150/+;NR2BWT/WT again displayed an intermediate effect (Fig. 2b). GABAA/benzodiazepine receptor binding was used to survey multiple extrastriatal brain regions and revealed no differences between groups at 100 weeks, indicating little or no neuronal loss in these brain regions (Fig. 2c). Decreased D1 and D2 receptor binding was observed in double mutants at 70 weeks (Fig. 2d,e). The Hdh150/+;NR2BTg/WT double-mutant mice and Hdh150/+;NR2BWT/WT mice displayed similar average reductions in D1- and D2-binding sites in both dorsal and ventral striatum. Both D1- and D2-binding decreases approximated 30% (Fig. 2d,e).

Hdh150/+;NR2BTg/WT double-mutant mice exhibit reductions in striatal D1 and D2 receptor binding, whereas Hdh150/+;NR2BWT/WT show an intermediate reduction at 100 weeks. Pseudocolor images of receptor binding. Autoradiographs were analyzed by quantitative densitometry using an MCID-M2 image analysis system. Histograms show results of densitometric analysis of film images converted to pCi of 3H-ligand bound per gram of protein. Regions analyzed are the substantia nigra (SN), ventral striatum (ventral Str), dorsal striatum (dorsal Str), whole striatum (STR), cerebellum (Cer), hippocampus (HIP), thalamus (THAL), frontal cortex (FRCT), and whole brain (Whole). a–c, D1, D2, and GABAA receptor binding at 100 weeks. d, e, D1 and D2 receptor binding at 70 weeks. *p < 0.05. Values are expressed as mean pCi/g ± SEM. n = 4 animals per group.
Hdh150/+;NR2BTg/WT double-mutant mice exhibit no change in distribution of neuronal intranuclear inclusions
We determined the distribution of htt aggregates. Neuronal intranuclear inclusions (NIIs) saturated striatal neurons in double-mutant and Hdh150/+;NR2BWT/WT mice at 100 weeks (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). There were only sparse NIIs in other brain regions. This result is consistent with our previous observations in the Hdh(CAG)150 line (Tallaksen-Greene et al., 2005). With quantitative real-time-PCR, we determined that the NR2B transgene did not affect huntingtin mRNA expression (data not shown).
Hdh150/+;NR2BTg/WT double-mutant mice exhibit weight loss and motor deficits
Hdh150/+;NR2BTg/WT double-mutant mice exhibited an 18.4% decrease in body weight compared with other groups [Hdh150/+;NR2BTg/WT, 29.4 ± 3.1 g (mean ± SEM); Hdh+/+;NR2BWT/WT,35.7 ± 1.4 g; Hdh+/+;NR2BTg/WT, 36.7 ± 4.5 g; Hdh150/+;NR2BWT/WT, 35.6 ± 3.9 g; p < 0.05]. No motor abnormalities were detected by rotarod with all groups exhibiting similar latencies to fall. Double-mutant mice exhibited significantly reduced general locomotion and exploratory behavior at 100 weeks (supplemental Fig. S3a, available at www.jneurosci.org as supplemental material) [Hdh150/+;NR2BTg/WT, 76.8 ± 17.7 (mean ± SEM) crossovers/2 h; Hdh+/+;NR2BWT/WT, 251.7 ± 30.7; Hdh+/+;NR2BTg/WT, 199.1 ± 43.7; Hdh150/+; NR2BWT/WT, 102.8 ± 5.1; p < 0.01]. All groups displayed similar latencies to fall on the hanging wire test, indicating that muscle strength was not compromised and no abnormalities found with muscle histology (data not shown). Both Hdh150/+;NR2BTg/WT and Hdh150/+;NR2BWT/WT require three times as long to traverse a balance beam compared with wild-type littermate controls (supplemental Fig. S3b, available at www.jneurosci.org as supplemental material) [Hdh150/+;NR2BTg/WT, 5.8 ± 0.4 s (mean ± SEM); Hdh150/+;NR2BWT/WT, 6.5 ± 0.6 s; Hdh+/+;NR2BWT/WT, 2.0 ± 0.2; p < 0.05]. Footprint analysis revealed that both Hdh150/+;NR2BTg/WT and Hdh150/+;NR2BWT/WT display a shortening of stride length and loss of normal gait pattern compared with controls (supplemental Fig. S4, available at www.jneurosci.org as supplemental material).
Discussion
We tested the hypothesis that selective striatal degeneration observed in HD is mediated by NR2B subunits of the NMDAR in vivo. We found that double-mutant mice exhibited a significant decrease in striatal neuron number and striatal volume. The striatal degeneration observed in these mice was associated with decreases in D1 and D2 receptor binding and with motor deficits. These data demonstrate synergistic effects of mutant htt and overexpression of NR2B subunits with exacerbation of the HD-like phenotype characteristic of Hdh(CAG)150 mice. The NR2B transgenic line has previously been shown to exhibit increased NMDAR activation resulting in longer EPSPs and increased calcium flux (Tang et al., 1999). In situ hybridization reveals increased expression of the NR2B transgene in the cortex, striatum, hippocampus, and amygdala with little increase of expression in thalamus, brainstem, and cerebellum (Tang et al., 1999). Within areas of increased NR2B expression, the total number of NR2B-containing NMDARs is increased in individual synapses. NR2B transgenic mice display normal growth, body weight, and survival. No anatomic abnormalities are documented in this line, and NR2B transgenic mice maintain superior performance in learning and memory tasks up to at least 20 months of age (Tang et al., 1999; Cao et al., 2007). NR2B subunit expression declines with normal aging (Magnusson et al., 2002). This is seen also in NR2B transgenic mice, but NR2B expression continues to be higher in transgenic mice than in wild-type controls (Cao et al., 2007).
Although our double-mutant mice exhibited exacerbation of striatal neuron degeneration, the time course of their disease was not altered by NR2B transgene addition. As in Hdh(CAG)150 mice, neuronal loss was a late event. Our D1/D2 binding data, with declines at 70 weeks, are consistent with a prolonged period of neuronal dysfunction before death of striatal neurons (Heng et al., 2007). Similar results are reported in human PET imaging studies (Weeks et al., 1996).
Rotorod performance was preserved in Hdh150/+;NR2BTg/WTmice with substantial striatal atrophy and neuronal loss. This result is consistent with our previous data in Hdh(CAG)150 mice, indicating that the rotorod is an insensitive measure of striatal dysfunction (Heng et al., 2007). The balance beam task and footprint analysis were more sensitive to striatal injury/dysfunction. These measures did not discriminate between Hdh150/+;NR2BTg/WT and Hdh150/+;NR2BWT/WTmice. The latter did exhibit evidence of striatal neuron dysfunction as shown by diminished D1/D2 dopamine receptor binding (Fig. 2; supplemental Table 1, available at www.jneurosci.org as supplemental material). Relatively sensitive measures like the balance beam task and footprint analysis may exhibit “floor” effects that made it difficult to discriminate be-tween Hdh150/+;NR2BTg/WT and Hdh150/+;NR2BWT/WT mice.
Our results are consistent with previous work in other HD murine models. Studies in transgenic mouse models of HD reveal evidence of increased NMDAR activation. Cultured striatal neurons from YAC transgenic mice expressing a full-length human HD construct exhibit increased NMDAR activation followed by increased Ca2+ levels and mitochondrial membrane depolarization in MSNs compared with wild-type controls (Zeron et al., 2003, 2004; Shehadeh et al., 2006). Striatal neurons in these transgenic models appear more susceptible to NMDA agonist-mediated toxicity compared with wild-type mice, and NMDA induced cell death is abolished by a specific NR2B antagonist (Levine et al., 1999; Zeron et al., 2002). Consistent with these results, the R6/2 transgenic mouse model of HD-expressing exon 1 of the human HD exhibits selectively increased NMDA-evoked current and intracellular calcium (Levine et al., 1999). Recent work shows amelioration of behavioral and neuropathological deficits, and increased survival of R6/2 transgenic mice with decortication, an anti-excitotoxic intervention (Stack et al., 2007). These observations support an important role for NMDAR action, particularly the NR2B subunit, in striatal neuron degeneration in HD.
Comparative studies confirm that the pharmacological and functional properties of NMDARs depend heavily on NR2 subunit composition (Cull-Candy et al., 2001; Li et al., 2003; Loftis and Janowsky, 2003). NR2B subunit presence influences Mg2+ sensitivity and confers high Ca+2 permeability (Hollmann and Heinemann, 1994). NMDAR activation may either promote neuronal survival or cause excitotoxic injury (Papadia and Hardingham, 2007). In some model systems, NR2B-containing NMDARs promote prodeath signaling, whereas NR2A-containing NMDARs promote survival (Liu et al., 2007). Considerable other work suggests that NMDAR localization is the salient factor with extrasynaptic NMDARs promoting neuronal death, and synaptic NMDARs promote survival (Hardingham et al., 2002; Léveillé et al., 2008; Papadia et al., 2008; Martel et al., 2009). In mature mammalian neurons, however, NR2B subunit containing NMDARs tend to be preferentially localized extrasynaptically (Tovar and Westbrook, 1999). NR2B subunit containing NMDARs are highly expressed by MSNs (Monyer et al., 1992). In vitro coexpression studies of NMDARs and htt in non-neuronal cells indicate sensitization of NR2B function by mutant htt (Chen et al., 1999; Zeron et al., 2001).
Recent in vitro work directly links sensitization of NMDAR–NR2B dysfunction mediated by mutant htt to deranged intracellular Ca2+ homeostasis and apoptosis in HD (Bezprozvanny and Hayden, 2004; Fan and Raymond, 2007). Coexpression of NMDARs with expanded polyglutamine htt in non-neuronal-cultured cells enhances excitotoxic cell death, an effect maximized by NR2B subunit expression (Zeron et al., 2001; Li et al., 2004). Similarly, cultured MSNs from a mouse model of HD exhibit increased glutamate-evoked peak currents and enhanced Ca2+ levels mediated by the NR2B subunit (Zeron et al., 2002, 2004; Shehadeh et al., 2006). These findings support the hypothesis that mutant htt enhances neuronal susceptibility to NMDAR-mediated excitotoxic insult, particularly in neurons expressing NR2B-containing NMDARs (Cepeda et al., 2001; Zeron et al., 2002; Li et al., 2003; Tang et al., 2003).
Other mechanistic hypotheses of HD pathogenesis have been suggested, such as transcriptional dysregulation, mitochondrial dysfunction, axonal transport defects, brain-derived neurotropic factor depletion, and proteosomal dysfunction. All these hypotheses have some support (Imarisio et al., 2007). It is possible that expanded polyQ htt is neurotoxic because it has multiple deleterious effects (Aronin et al., 1999). Our results support NMDAR excitotoxicity as a proximate cause of neurodegeneration in HD, but there may be an overlapping network of expanded polyQ effects causing neurodegeneration. Although our double mutants exhibit exacerbation of the phenotype, they do not show any apparent change in the time course of the phenotype and exhibit similar pathological and behavioral changes to Hdh(CAG)150 mice up to 70 weeks. In other murine genetic models, enhanced NMDAR Ca2+ currents are found in embryonic neurons, suggesting that pathologic dysregulation of NMDAR signaling is an early event in HD (Zeron et al., 2004). It is plausible that later occurring, pathologic effects of expanded polyQ htt could interact with the potentiation of NR2B–NMDAR signaling in synergistic or additive manner to produce neurodegeneration. Our in vivo findings are consistent with previous reports that subunit-specific interactions between NR2B-containing NMDARs and expanded mutant htt potentiate NMDAR-initiated neuronal death and are important mediators of neurodegeneration in HD. NMDAR antagonists are viable candidates as interventions in HD, but it seems likely that highly selective NR2B antagonists will be required.
Footnotes
-
This work was supported by a Veterans Affairs Merit Review grant and the Huntington Disease Society of America. We acknowledge Lauren Cline, Kevin Duong, Chad Green, Erin Katz, Alex Martusiewicz, Adam Myrold, and Rebecca York for their technical assistance. We thank Dr. Tom Morrow for use of his MCID system and Dr. Kirk Frey for use of his stereology apparatus. We thank the anonymous reviewers for careful evaluation of a previous version of this manuscript and several constructive criticisms.
- Correspondence should be addressed to Dr. Roger L. Albin, 5023 Biomedical Science Research Building, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, MI 48109-2200. ralbin{at}umich.edu
References
- Aronin et al., 1999.↵
- Bazzett et al., 1993.↵
- Beal et al., 1986.↵
- Bezprozvanny and Hayden, 2004.↵
- Brouillet et al., 1995.↵
- Cao et al., 2007.↵
- Cepeda et al., 2001.↵
- Chen et al., 1999.↵
- Coyle and Schwarcz, 1976.↵
- Cull-Candy et al., 2001.↵
- Dingledine et al., 1999.↵
- Fan and Raymond, 2007.↵
- Ferrante et al., 1993.↵
- Greene and Greenamyre, 1995.↵
- Hardingham et al., 2002.↵
- Heng et al., 2007.↵
- Hollmann and Heinemann, 1994.↵
- Imarisio et al., 2007.↵
- Léveillé et al., 2008.↵
- Levine et al., 1999.↵
- Li et al., 2003.↵
- Li et al., 2004.↵
- Liu et al., 2007.↵
- Loftis and Janowsky, 2003.↵
- Magnusson et al., 2002.↵
- Martel et al., 2009.↵
- McGeer and McGeer, 1976.↵
- Monyer et al., 1992.↵
- Papadia and Hardingham, 2007.↵
- Papadia et al., 2008.↵
- Schwarcz et al., 1983.↵
- Shehadeh et al., 2006.↵
- Stack et al., 2007.↵
- Tallaksen-Greene et al., 2005.↵
- Tang et al., 2003.↵
- Tang et al., 1999.↵
- Tovar and Westbrook, 1999.↵
- Waxman and Lynch, 2005.↵
- Weeks et al., 1996.↵
- Zeron et al., 2001.↵
- Zeron et al., 2002.↵
- Zeron et al., 2004.↵





{kind=link}
{kind=link}