

Abstract
During differentiation, neuroendocrine cells acquire highly amplified capacities to synthesize neuropeptides to overcome dilution of these signals in the general circulation. Once mature, the normal functioning of integrated physiological systems requires that neuroendocrine cells remain plastic to dramatically alter neuropeptide expression for long periods in response to hormonal and electrical cues. The mechanisms underlying the long-term regulation of neuroendocrine systems are poorly understood. Here we show that the Drosophila basic helix-loop-helix protein DIMM, a critical regulator of neuroendocrine cell differentiation, controls secretory capacity in mature neurons. DIMM expression began embryonically but persisted in adults. Through spatial and temporal manipulation of transgene expression in vivo, we defined two phases of prosecretory DIMM activity. During an embryonic critical window, DIMM controlled the differentiation of amplified expression of the neuropeptide leucokinin. At the onset of metamorphosis, levels of DIMM decreased in the insulin-producing cells (IPCs) in parallel with a marked reduction in levels of Drosophila insulin-like peptide 2 and a key neuropeptide biosynthetic enzyme peptidylglycine α-monooxygenase (PHM). Overexpression of DIMM in the IPCs prevented the decrease in PHM levels at this stage. In addition, transient overexpression of DIMM in adults produced a dramatic increase in PHM levels in numerous neurons located throughout the brain. These findings provide insights into the mechanisms controlling the maintenance of differentiated cell states, and they suggest an effective means for dynamically adjusting the strength of hormonal signals in diverse homeostatic systems.
Introduction
A fundamental question in the development and regulation of neuroendocrine systems is the control of cellular secretory capacity. Although there are many exceptions (Streit et al., 1989), neuropeptide-secreting (neuropeptidergic) cells as a rule, and neuroendocrine cells in particular, synthesize prodigious amounts of secretory peptides. This is often to support chronic secretion or signaling through the circulation (Newcomb and Scheller, 1990; Riddiford et al., 1994; Burbach et al., 2001). In addition, a critical feature of many neuropeptide systems is their inherent flexibility, which enables organisms to dramatically alter neuropeptide expression in response to internal and environmental cues. The resulting changes in the gain of neuropeptide signaling form an integral component of the neuroendocrine and physiological feedback loops that establish and regulate homeostatic mechanisms (Jequier, 2002; Meerlo et al., 2002; Plant and Shahab, 2002; Sisk and Foster, 2004). This form of regulation has been intensively investigated in a few amenable systems, and contributions of factors such as cAMP response element-binding protein (Montminy and Bilezikjian, 1987) and steroids (Burbach, 2002) to changes in neuropeptide gene expression are well documented. Nevertheless, in most cases, the heterogeneity and scattered distribution of neuropeptidergic cells has restricted progress toward a general molecular understanding of amplified secretory capacity and its regulation.
Several members of the Atonal family of basic helix–loop–helix (bHLH) transcription factors are important regulators of neuropeptide gene expression in differentiating cells. This family includes Drosophila Atonal and the vertebrate NeuroD (neurogenic differentiation), neurogenin, Mist1 (muscle, intestine, and stomach expression 1), and Olig (oligodendrocyte lineage transcription factor) proteins, all of which play critical roles in the determination and execution of cell fate decisions (Kageyama and Nakanishi, 1997; Lee, 1997; Hassan and Bellen, 2000). In addition to these developmental actions, the direct regulation of neuropeptide gene expression by Atonal-like proteins suggests that they may also regulate neuroendocrine function dynamically in the context of homeostatic signaling or postembryonic developmental changes. Consistent with this model, several members of the Atonal family are expressed in terminally differentiated cells (Morrow et al., 1999; Lee et al., 2000). In addition, recent studies have suggested a link between one Atonal family protein, NeuroD, and the neuroendocrine/endocrine signaling pathways that control energy balance (Nilaweera et al., 2002; Khoo et al., 2003), and disruptions in NeuroD are associated with the development of certain forms of type 2 diabetes (Malecki et al., 1999). Nevertheless, the importance of Atonal family proteins in the regulation or maintenance of neuropeptide and peptide hormone levels in fully differentiated cells in vivo has not been directly established.
The Drosophila dimmed (dimm) gene encodes an Atonal family bHLH protein, DIMM, that is required for the differentiation of diverse neuropeptidergic and endocrine cell lineages (Hewes et al., 2003). Reminiscent of NeuroD, expression of a dimm reporter gene continues in adult neurons and endocrine cells (Taghert et al., 2001; Hewes et al., 2003). Here, through spatiotemporal manipulation of transgenes, we show that cellular levels of neuroendocrine proteins are controlled by postembryonic changes in dimm expression. This work provides the first direct in vivo demonstration that an Atonal family protein dynamically regulates adult neuroendocrine cell properties.
Materials and Methods
Fly strains and genetic manipulations.
Fly stocks were cultured on a standard cornmeal–yeast–agar medium at 22–25°C. Larvae were collected from apple juice–agar egg-collection plates supplemented with yeast paste. Larval genotypes were distinguished by green fluorescent protein (GFP) scoring. Several of the aberrations and transposon insertions used in this paper were described by Hewes et al. (2000, 2003): dimm-Gal4 (P{GawB}crc929) (FBal0147207); P{UAS-dimm.MYC}2-A-3 (FBal0147203); dimmRev8 (FBal0147204); and Df(2L)Rev4 (FBab0029399). Other strains were as follows: P{UAS-mCD8:GFP.L}LL5 (FBti0012685) (Lee and Luo, 1999); ap-Gal4 (FBal0052383) (O’Keefe et al., 1998); dilp2-Gal4 (FBal0137378) (Rulifson et al., 2002); P{UAS-GFP.Y}B1 (FBti0002751) (Yeh et al., 1995); CyO, P{Ubi-GFP.S65T}PAD1 (FBti0012182) (CyO, Ubi-GFP); P{tubP-GAL80ts}7 (FBti0027798) and P{tubP-GAL80ts}20 (FBti0027796) (McGuire et al., 2003); and Oregon-R (wild type).
Animals with the ap-Gal4, UAS-dimm::MYC genotype die as larvae, and this lethality was prevented by culturing y* w*; ap-Gal4, UAS-dimm::MYC, tubP-Gal80ts/CyO, Ubi-GFP stocks at 18°C. Developmental rates were slower at 18–22°C than at 25–30°C. However, at 27.5–30°C, larvae expressing the UAS-dimm::MYC transgene [e.g., under the control of apterous-Gal4 (ap-Gal4)] developed at rates approximately equal to control larvae at 18–22°C.
Immunostaining.
Immunostaining was performed as described previously (Benveniste et al., 1998; Hewes et al., 2003). For each experiment, larvae from the various control and test groups were dissected and stained in parallel, or, when necessary, antisera and other reagents were aliquoted in advance to ensure consistency. Tissues were fixed for 1 h in 4% paraformaldehyde (PFA), 4% paraformaldehyde/7% picric acid (PFA-PA), or Bouin’s fixative. Polyclonal and monoclonal antisera were used overnight at 4°C to detect the following proteins: Drosophila insulin-like peptide 2 (dILP2) (affinity purified, 1:50, PFA-PA) (Rulifson et al., 2002); DIMM (affinity purified, 1:200, PFA-PA) (Allan et al., 2005); leucokinin (LK) (1:750, preabsorbed on 0–12 h wild-type embryos, PFA-PA) (Nässel and Lundquist, 1991); MYC (monoclonal antibody 9E 10, 1:100, PFA-PA; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) (Evan et al., 1985); and peptidylglycine α-monooxygenase (PHM) (affinity purified, 1:100, Bouin’s fixative) (Jiang et al., 2000). For imaging of CD8::GFP, tissues were fixed in PFA. Cyanine 3- and Alexa 488-conjugated goat or donkey secondary antisera from Jackson ImmunoResearch (West Grove, PA) and Invitrogen (Carlsbad, CA) were used at a 1:500 dilution. Confocal z-series projections were obtained using an Olympus Optical (Melville, NY) FluoView FV500 microscope.
In situ hybridization. Whole-mount in situ hybridization was performed on wild-type tissue using a digoxigenin-labeled DNA probe (Hewes et al., 2003). Image stacks, at constant focus increments, were taken with an Olympus Optical CV12 CCD camera and BX61WI microscope. Two-dimensional projections were obtained using Olympus Optical MicroSuite Extended Focal Imaging software.
Staining quantification.
Quantification was performed as described previously (Hewes et al., 2003) on confocal images, and the images shown in the figures are representative of the mean staining intensity values. The same confocal scanning settings, which were optimized to avoid image saturation, were used for all preparations within each experiment. Intensity indices are not directly comparable between figures, because different scanning settings were used for each experiment. For cell counts, the range of pixel intensities for each image was compressed using the adjust levels function in Adobe Photoshop (Adobe Systems, San Jose, CA), and automated counts (with manual corrections for missed or merged cells) were performed using the analyze particles function in NIH ImageJ (version 1.32j). All of the software settings used for cell counts were identical for each image. Tests for normality, omnibus tests [multivariate ANOVA (MANOVA) and general linear model (GLM) ANOVA], and parametric (GLM ANOVA and Tukey–Kramer multiple comparison post hoc tests) and nonparametric (Kruskal–Wallis one-way ANOVA on ranks and Kruskal–Wallis multiple comparison Z-value post hoc tests) ANOVA were performed using NCSS-2001 (NCSS, Kaysville, UT); we used sequential Bonferroni corrections of test α to control for type I errors with multiple comparisons (Rice, 1989). All values for n refer to the number of CNS sampled (which varied because of differences in fecundity and survival for each genotype and treatment), and values for bilaterally paired neurons were averaged to yield a single value per CNS. Reference means for the post hoc comparisons in each figure are shown as dotted lines.
Results
Maintenance of DIMM expression in mature neurons
To detect dimm mRNA expression in postembryonic cells, we performed in situ hybridization with an antisense dimm probe on the CNS and associated endocrine glands (ring glands) of third-instar larvae (Fig. 1A). Within the CNS, we observed primarily cytoplasmic dimm mRNA in >100 neurons and in the peptidergic endocrine cells of the corpora cardiaca portion of the ring gland. This distribution matched the expression pattern for a dimm reporter gene (dimm-Gal4), containing the yeast transcriptional activator Gal4 expressed under the control of dimm gene regulatory regions (Fig. 1B) (Hewes et al., 2003). Strong expression of dimm-Gal4 is found in ∼200 neurons and endocrine cells in larvae (Hewes et al., 2003) and adults (Taghert et al., 2001). Many of these cells are identifiable in late embryos and are fully differentiated by the time of larval hatching, judging by their high-level expression of mature neuropeptides, full neuritic arbors, and involvement in homeostatic and developmental neuroendocrine signaling pathways (Hewes et al., 2003). Thus, the distribution of dimm mRNAs, and of cells with active dimm enhancer activity, remains essentially unchanged throughout larval development and includes many fully differentiated cells.

DIMM protein and dimm mRNA are expressed in terminally differentiated neurons. A, In situ hybridization with a dimm antisense probe in third-instar larvae. B, Colocalization of dimm reporter expression (CD8::GFP, green; expression of UAS-mCD8::GFP was directed by dimm-Gal4) and DIMM protein (α-DIMM, purple) in third-instar larvae. Areas of merged green and purple signals are white. Arrows indicate clusters of lateral abdominal neurons that express dimm mRNA, DIMM protein, and the dimm-Gal4 reporter gene. Arrowheads (in B) indicate selected neurons that express the DIMM protein but not the dimm reporter. CC, Intrinsic endocrine cells of the corpora cardiaca; DC, dorsal chain Ap-let neurons (Park et al., 2004); LP1, MP2, SP1, Tv, Tvb, other morphologically distinguishable dimm-positive neurons (Hewes et al., 2003). Scale bars, 50 μm.
To determine whether DIMM protein also persists in mature neurons, we performed anti-DIMM immunocytochemistry on CNS tissue in which expression of a fluorescently tagged, integral membrane fusion protein mCD8::GFP (a fusion of the mouse lymphocyte marker CD8 and GFP) (Lee and Luo, 1999) was driven by dimm-Gal4. The anti-DIMM antiserum was confirmed to be specific for DIMM protein by staining CNS tissue in dimm gain-of-function backgrounds (see below). In larvae, 90% of all mCD8::GFP-positive neurons also expressed DIMM (Fig. 1B and data not shown). A few additional neurons expressed DIMM but not the dimm-Gal4 reporter. In the adult brain, there also was clear overlap in the two patterns, but the distribution of DIMM protein expression was substantially greater (Fig. 2). Expression of the dimm-Gal4 reporter is also tightly correlated with high-level expression of the neuropeptide biosynthetic enzyme PHM and with diverse neuropeptides in various subsets of cells in the dimm-Gal4 pattern (Hewes et al., 2003). Thus, DIMM is present at high levels in numerous mature neuroendocrine cells and other strongly neuropeptidergic interneurons.

Widespread DIMM protein expression in adult brain neurons. Colocalization of UAS-mCD8::GFP (grayscale in top left inset and green in the right panel) and DIMM (grayscale in bottom left inset and purple in the right panel) in many neurons (e.g., arrows) located in the adult brain. Expression of UAS-mCD8::GFP was directed by dimm-Gal4. Arrowheads indicate selected neurons that expressed DIMM but not mCD8::GFP. Scale bar, 50 μm.
Secretory protein levels depend on dimm gene dosage
Changes in dimm expression produce corresponding changes in levels of secretory proteins. Mutations that substantially reduce both embryonic and also postembryonic dimm mRNA levels result in markedly reduced immunostaining throughout the CNS for diverse neuropeptides and neuropeptide biosynthetic enzymes (Hewes et al., 2003). These effects are scaled based on the severity of the molecular defects caused by the different loss-of-function alleles. Conversely, tonic dimm overexpression produces elevated levels of secretory proteins, including the neuropeptide LK and PHM (Hewes et al., 2003; Allan et al., 2005). Do the effects of dimm overexpression on secretory protein levels require changes in cell fate? To test this hypothesis, we drove expression of a UAS-dimm::MYC transgene, containing the wild-type dimm coding sequence in frame with a MYC epitope tag (Hewes et al., 2003), under the control of an ap-Gal4 driver. The ap-Gal4 line produces widespread, heterogeneous transgene expression in the brain lobes and relatively limited expression, including several neuropeptidergic neurons, in the ventral nerve cord (VNC) (Benveniste et al., 1998; O’Keefe et al., 1998; Hewes et al., 2003). Overexpression of DIMM under these conditions resulted in markedly increased levels of anti-PHM immunostaining in numerous cells throughout the brain lobes and in a much smaller population of cells in the VNC (Fig. 3A). We confirmed the expression of UAS-dimm::MYC in the affected cells through anti-MYC and anti-DIMM immunostaining (Fig. 3B–D).

DIMM overexpression results in elevated PHM levels in many CNS neurons. A, Anti-PHM immunostaining (α-PHM) of second-instar larval CNS after UAS-dimm::MYC expression with (left) or without (right) the ap-Gal4 driver. The top panels show ventral views of the brain lobes and ventral nerve cord. The bottom panels show dorsal views of the brain lobes. The confocal imaging settings were adjusted to avoid excessive signal saturation (left), and areas of substantial PHM immunostaining in the controls (right) are therefore only faintly visible. B, Anti-PHM and anti-MYC (α-MYC) coimmunostaining of second-instar larval CNS after UAS-dimm::MYC expression under the control of the ap-Gal4 driver in the brain lobe (top) and ventral nerve cord (bottom). C, Percentage of T cells (±SEM) double labeled for PHM and MYC (n=10). p=0.64, one-way ANOVA. D, Anti-PHM and anti-DIMM (α-DIMM) coimmunostaining of second-instar larval CNS after UAS-dimm::MYC expression under the control of ap-Gal4. The feathered arrows indicate strongly MYC- or DIMM-positive but weakly PHM-positive cells. The arrows indicate weakly MYC- or DIMM-positive but strongly PHM-positive cells, and the arrowheads indicate dorsal chain Ap-let neurons. NP, Synaptic neuropils; T cells, thoracic ventral apterous-expressing neurons. Scale bars: A, 100 μm; B, 50 μm.
Within the VNC, we observed elevated levels of PHM in two classes of cells, the thoracic ventral ap-positive cell clusters (T cells) and the dorsal chain of ap-positive neurons. Both of these groups of cells normally express several neuropeptidergic markers (Hewes et al., 2003; Park et al., 2004). In wild-type animals, the T-cell clusters in thoracic segments 2 and 3 consisted of four ap-Gal4-positive cells, including the neuropeptidergic thoracic ventral (Tv) and Tvb neurons, which expressed PHM and DIMM (data not shown) (Park et al., 2004). The T-cell cluster in thoracic segment 1 usually contains a fifth ap-Gal4-positive cell. After dimm overexpression with the ap-Gal4 driver, ∼90% of the ap-Gal4-positive cells in T1–T3 expressed high levels of PHM (4.6 ± 0.1, 4.0 ± 0.1, and 4.0 ± 0.0 cells in T1, T2, and T3, respectively; n = 14) (Fig. 3B). Hence, PHM expression was elevated in Tv and Tvb. We observed a similar increase in the dorsal chain ap-positive cells. Both of these groups of neurons express DIMM and PHM in wild-type animals, and the effects of dimm expression therefore did not require changes in cell identities.
To determine whether other cells in the normal dimm expression pattern displayed similar upregulation of PHM, we overexpressed UAS-dimm::MYC under the control of the dimm-Gal4 driver. Cells throughout the dimm-Gal4 pattern displayed substantially higher levels of PHM after DIMM overexpression (Fig. 4). The morphologies of the affected cells, including the neuritic arborizations within the CNS, appeared normal. Therefore, a quantitative increase in DIMM expression (overexpression) was sufficient to drive increased expression of PHM in most, if not all, normally DIMM-expressing cells. These effects did not require a switch in cell fate.

DIMM overexpression boosts PHM levels in normally Phm- and dimm-expressing cells. A, Anti-PHM immunostaining (α-PHM) in the CNS of a third-instar larva after dimm overexpression in normally dimm-positive cells (dimm-Gal4/UAS-dimm::MYC) and in the CNS from a sibling control (dimm-Gal4/+). B, Intensity of anti-PHM immunostaining (±SEM) for the cell types indicated in A. Note that the Tv and Tvb neurons are repeated in each of the three thoracic ganglia (Tv1–Tv3 and Tvb1–Tvb3), but only one pair is indicated in A. d1, d4, d7, d11, Selected dorsal chain Ap-let neurons (Park et al., 2004); MP2, SP1, Tv, Tvb, other morphologically distinguishable dimm-positive neurons (Hewes et al., 2003). ∗p < 0.05, ∗∗p < 0.01 (MANOVA, p=0.000000; post hoc testing by Kruskal–Wallis one-way ANOVAs on ranks with sequential Bonferroni correction; n=10). Scale bar, 50 μm.
Although most if not all neurons respond to transgenic expression of DIMM by overexpressing PHM (Fig. 3) (Allan et al., 2005), the strength of this effect was not uniform. Some cells with high levels of DIMM::MYC overexpression, as judged by levels of anti-MYC (Fig. 3B) and anti-DIMM (Fig. 3D) immunostaining, displayed weak induction of PHM. In contrast, we also observed cells with relatively low DIMM::MYC levels and very strong anti-PHM immunostaining (Fig. 3B,D). Therefore, although DIMM is clearly a key regulator of PHM expression, other unidentified factors must also contribute in a cell-type-specific manner to define the relative levels of this biosynthetic enzyme.
Late embryonic critical window for differentiation of leucokinin expression
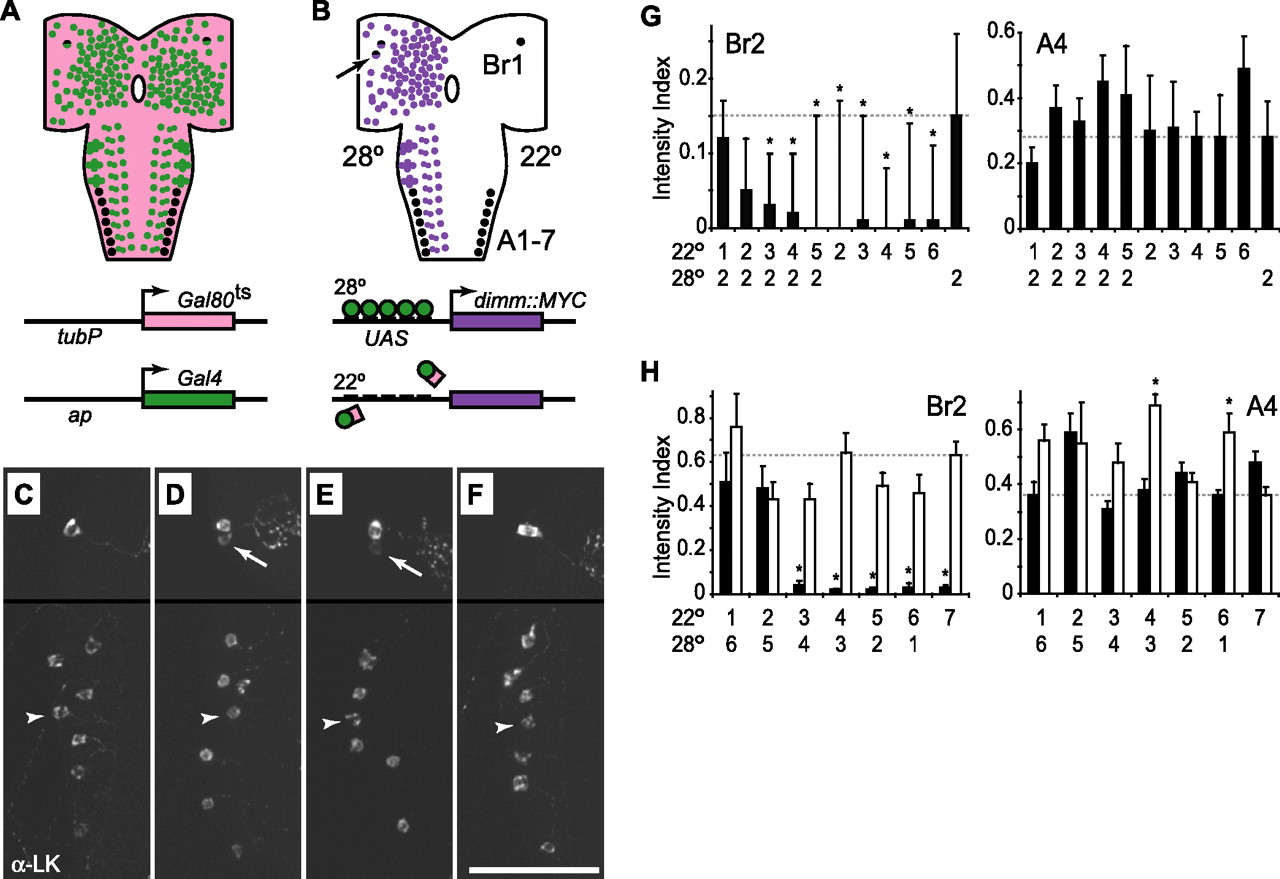
To determine the developmental stage(s) during which changes in dimm activity can drive elevated accumulation of secretory proteins in mature cells, we used the TARGET system (McGuire et al., 2003) to direct spatially and temporally restricted dimm overexpression. We obtained spatially restricted CNS expression of UAS-dimm::MYC with the ap-Gal4 driver. We used a third transgene to direct ubiquitous expression of a temperature-sensitive Gal4 repressor protein, Gal80ts, under the control of the tubulinP (tubP) promoter (Fig. 5A). At permissive temperatures (18–22°C), the Gal80ts protein binds to Gal4 and represses its transcriptional activity. At restrictive temperatures (27.5–30°C), Gal4 is released to bind to available upstream activating sequence (UAS) sites, and Gal4-dependent transcription proceeds normally (Fig. 5B). With this system, we controlled the timing of transgenic dimm expression through temperature-shift protocols. Expression of UAS-dimm::MYC was confirmed by anti-MYC and anti-DIMM immunostaining (Fig. 3).

dimm overexpression boosts LK levels during an embryonic critical period. A, tubP-Gal80ts (pink), ap-Gal4 (green), and LK neuropeptide (black) expression patterns. Schematics of the two transgenes are shown below the drawing of the CNS. B, Pattern of temperature-dependent UAS-dimm::MYC expression (purple). Schematics below the drawing of the CNS show the interaction between Gal4 and Gal80ts at 22°C (bottom) and between Gal4 and the UAS sites in the UAS-dimm::MYC transgene at 28°C (top). At 22°C, LK (right side of CNS drawing) is expressed in only one pair of brain neurons (Br1), and these cells also express ap-Gal4 (black and purple cell). At 28°C, LK expression (left side of CNS drawing) remains the same in cells A1–A7, which are ap-Gal4 negative, whereas greater LK expression is observed in Br1 and Br2 (arrow), which express ap-Gal4. C–F, LK immunostaining (α-LK) after 6 d at 22°C (C), 2 d at 28°C (D), 1 d at 22°C followed by 2 d at 28°C (E), and 5 d at 22°C followed by 2 d at 28°C (F). Arrows and arrowheads indicate Br2 and A4, respectively. G, H, LK immunostaining intensity (±SEM) in ap-Gal4/UAS-dimm::MYC larvae with (black bars) and without (white bars) tubP-Gal80ts. The labels on the x-axis indicate the duration (in days) that each group was maintained first at 22°C and then at 28°C. Eggs were collected during the first 24 h, and the stages of development at the time of each successive shift were as follows: day 1, embryos; day 2, embryos and hatchling larvae; day 3, first-instar larvae; day 4, first-and second-instar larvae; day 5, second-instar larvae; day 6, mostly second-instar larvae with a few (<10%) third-instar larvae; day 7 (no temperature shift), second- and third-instar larvae. A1–A7, Abdominal LK cells; ap, apterous promoter; Br1, brain LK cell; (tubP) tubulin 1α promoter. Results of the one-way ANOVAs and n values were as follows: G, Br2, p=0.000000 (n = 14–18) and A4, p=0.0094 (n=5–16); H, white bars, Br2, p=0.11 (n=9–26) and A4, p=0.00010 (n=9–24); H, black bars, Br2, p=0.000000 (n=6–26) and A4, p=0.00092 (n=6–25). ∗p < 0.05 (Tukey–Kramer test). Scale bar, 50 μm.
In the absence of Gal80ts, overexpression of dimm under the control of ap-Gal4 produced ectopic expression of LK-related neuropeptides in one pair of brain neurons, Br2, in young larvae. At older larval stages, we observed additional ectopic LK expression in up to seven neurons in each brain lobe. However, levels of LK in the ventral nerve cord neurons A1–A7, which show little or no ap-Gal4 expression, were unchanged (Hewes et al., 2003). In larvae bearing the tubP-Gal80ts transgene, ectopic LK expression in Br2 was dependent on temperature, whereas native LK expression in A1–A7 was not (Fig. 5C–F). After 6–7 d at 22°C, LK expression in Br2 was low or undetectable, and we observed weak LK expression in only three Br2 neurons in 24 brain lobes (12 CNS) (Fig. 5C). In contrast, LK expression was strongly induced in Br2 after only 2 d at 28°C (Fig. 5D).
To determine the timing of dimm action in Br2, we used ap-Gal4 to drive UAS-dimm::MYC in the presence or absence of tubP-Gal80ts while performing temperature shifts from 22 to 28°C (shift ups) at daily intervals during larval development. Because of the temperature dependence of larval development rates (see Materials and Methods), we used two different shift-up protocols. In the first experiment, we held the duration of the 28°C treatment constant (2 d), and we varied the stage of larval development at the time of dissection (Fig. 5G). In the second experiment, we varied the duration of the 28°C treatment, and we dissected all larvae at the same stage of development (mid-second instar) (Fig. 5H). Independent of the larval stage at dissection and the total length of time spent at the restrictive temperature, we detected increased expression of LK in Br2 when the larvae were shifted up to 28°C within the first 2 d, when animals were late embryos or young (hatchling) first-instar larvae. After more than 2 d of development at 22°C, we observed significantly less LK expression. This effect required the presence of the Gal80ts protein; LK levels were high in Br2 in all temperature-shift groups when tubP-Gal80ts was omitted (Fig. 5H, white bars). In contrast, LK levels in the control A4 cell were essentially unchanged in both experiments (Fig. 5G,H), although the levels were elevated, in the absence of Gal80ts, in two of seven temperature treatment groups in the second experiment (Fig. 5H). This effect may have been attributable to low-level and previously undetected expression of the transgene in A4. However, there was no consistent relationship between A4 LK levels and the temperature regimen. Taken together, these results show that there is a critical window of dimm expression that closes at approximately the time of larval hatching, for induction of high levels of ectopic LK in Br2. Thus, DIMM functions in some cells as a neuropeptidergic differentiation factor.
Adult-specific DIMM overexpression elevates levels of the neuropeptide biosynthetic enzyme PHM
In contrast to the spatially restricted effects of DIMM overexpression on LK, we (Figs. 3, 4) and others (Allan et al., 2005) have observed marked DIMM-induced increases in the intensity of PHM immunostaining in essentially all CNS neurons. These results are a mirror image of the effects of reduced dimm expression on PHM levels (Hewes et al., 2003; Allan et al., 2005), suggesting a quantitative relationship between dimm and PHM expression. Notably, LK expression depends on the LIM (Lin–11, Isl-1, and mec-3) homeodomain protein Apterous (Herrero et al., 2003), a regulator of neuropeptidergic cell differentiation, whereas PHM expression does not (Allan et al., 2005).
Therefore, to determine whether DIMM performs two roles, both as a differentiation factor for expression of some neuropeptide genes and also as a maintenance or regulatory factor for expression of other secretory proteins, we examined the effects of postembryonic manipulation of DIMM levels on PHM expression. We reared animals bearing one copy each of the ap-Gal4 and UAS-dimm::MYC transgenes and either one or two copies of tubP-Gal80ts at 18°C throughout development to maintain minimal expression of UAS-dimm::MYC into the adult stage. After 2 d, adults were then shifted to 28°C for an additional 6 d to turn on DIMM::MYC expression. The temperature-shifted adults displayed robust increases in levels of PHM in numerous brain regions (Fig. 6), with the most prominent increases occurring in cells located throughout the optic lobes (Fig. 6A,E), dorsal protocerebrum (Fig. 6C,F), and subesophageal ganglion. PHM expression was not intrinsically age- or temperature-dependent, because we did not observe any increase in PHM levels in adults maintained at 18°C throughout the test period or in adults lacking the UAS-dimm::MYC and ap-Gal4 transgenes (Fig. 6B,D–F). Thus, PHM expression remains plastic and responsive to changes in levels of DIMM in fully differentiated neurons.

Transient adult dimm overexpression drives strong PHM overexpression. A–D, Anti-PHM immunostaining (α-PHM) in optic lobe (A, B) and dorsal protocerebrum (C, D). dimm overexpression (A, C) was induced (28°C for 6 d) in 2-d-old heterozygous ap-Gal4, UAS-dimm::MYC, tubP-Gal80ts adults. Controls (B, D) remained at the 18°C rearing temperature. E, F, Counts (±SEM) of PHM-positive optic lobe (E) and protocerebral (F) neurons. The labels on the x-axes indicate the genotype and adult shift temperature for each group. °C, Adult shift temperature; arrowheads, large ventral lateral neurons (LNvs) (Taghert et al., 2001); arrows, neurons at optic lobe–brain border; asterisks, optic lobes; GC, Gal80ts copy number; T, presence (+) or absence (−) of ap-Gal4 and UAS-dimm::MYC. Results of Kruskal–Wallis one-way ANOVAs on ranks and n values were as follows: E, p=0.000000 (n=8–12); F, p=0.000000 (n=8–13). ∗p < 0.05 (Kruskal–Wallis test). Scale bar, 50 μm.
Reduced DIMM expression results in decreased PHM levels in the Drosophila insulin-producing cells at the onset of metamorphosis
The preceding experiments demonstrated that secretory protein levels can be regulated by DIMM in mature cells, but they did not test whether these markers are regulated by DIMM under normal conditions. Therefore, we looked for cells in which changes in levels of DIMM expression are correlated with naturally occurring regulation of neuropeptide expression in the Drosophila insulin-producing cells (IPCs). We focused on these cells for three reasons. First, the IPCs are mature, fully functioning neuroendocrine neurons with important roles in the regulation of larval growth and metabolism (Rulifson et al., 2002). Second, they express multiple independently regulated dILP genes (Brogiolo et al., 2001; Ikeya et al., 2002). Third, dILP gene expression is regulated physiologically and developmentally (Ikeya et al., 2002; Li and White, 2003).
To determine whether DIMM is expressed in the IPCs, we performed double-labeling experiments on larval CNS with anti-DIMM, anti-dILP2, and reporter genes for dimm and dILP2. All four markers were coexpressed in the IPCs (Fig. 7A and data not shown). All of the dILP2-positive cells also expressed DIMM, although there were two to three additional DIMM-positive cells in each midline protocerebral cluster that lacked dILP2 expression.

Levels of DIMM and dILP2 both decrease in the insulin-producing cells at the onset of metamorphosis. A, Double labeling with anti-dILP2 (α-dILP2) and anti-DIMM (α-DIMM) in the midline protocerebral IPCs in a wild-type third-instar larva. The immunostaining for each marker is shown separately in the top and together as a merge in the bottom. The arrow indicates one of the double-labeled cells. Arrowheads indicate selected DIMM-positive cells in each midline protocerebral cluster that did not express dILP2. B, Intensity of anti-dILP2 and anti-DIMM immunostaining in the IPCs of wild-type animals in the third-larval instar and during the onset of metamorphosis. s3, Small third-instar larvae, shortly after ecdysis; m3, medium-sized third instar larvae; W, wandering stage third-instar larvae; WP, white prepupae. Results of one-way ANOVAs and n values were as follows: dILP2, p = 0.000001 (n = 6–8); DIMM, p = 0.000000 (n = 6–8). ∗p < 0.05 (Tukey–Kramer test). Scale bar, 20 μm.
In a survey of gene expression in the CNS at the onset of metamorphosis, Li and White (2003) found a 72% reduction in the level of dilp2 mRNA at pupariation (the white puparium stage) relative to 18 h earlier. To test whether this change in transcript levels was accompanied by a corresponding reduction in protein, we performed anti-dILP2 and anti-DIMM immunostaining on CNS tissue from animals at different stages relative to the onset of metamorphosis. In small- and medium-sized third-instar larvae, before the onset of metamorphosis, levels of both markers were high. However, at the wandering stage (∼18 h or less before pupariation) and at pupariation, levels of dILP2 and DIMM immunostaining were low (Fig. 7B). Thus, levels of dilp2 mRNA, dILP2 protein, and DIMM protein all were reduced in the IPCs during a brief (<18 h) period at the onset of metamorphosis.
Although the amounts of dILP2 and DIMM in the IPCs were correlated, dILP2 expression was not dependent on DIMM. The levels of anti-dILP2 immunostaining in dimm loss-of-function mutant larvae were normal or reduced only slightly (data not shown). Moreover, anti-dILP2 immunostaining and expression of the dilp2 reporter gene were both unaffected in wandering third-instar larvae that expressed UAS-dimm::MYC under control of the dimm-Gal4 driver. In contrast, anti-DIMM immunostaining was strongly elevated by this treatment (data not shown). Therefore, we turned our attention to PHM as a potential DIMM target in metamorphic IPCs.
Through double labeling, we confirmed that PHM is coexpressed with DIMM in the IPCs (Fig. 8A). In addition, levels of PHM in the IPCs decreased dramatically as animals initiated metamorphosis. We observed decreased PHM expression in the IPCs at the wandering stage, and PHM levels in the IPCs were even lower at pupariation (Fig. 8B). To determine whether these changes were dependent on the concomitant decrease in DIMM levels (Fig. 7B) or on other factors, we drove DIMM overexpression in the IPCs and then performed immunocytochemistry for PHM and DIMM at the wandering stage. IPC levels of PHM and DIMM were high in larvae bearing both dilp2-Gal4 and UAS-dimm::MYC, but they were low in control larvae bearing either one of these transgenes alone (Fig. 8C). Therefore, the reduction in PHM expression in the IPCs is dependent on the decrease in DIMM expression that occurs in these cells at the onset of metamorphosis. These changes in transmitter properties likely contribute to the regulation of IPC function as the animal transitions from a period of feeding and dramatic growth to an extended period in which the animal stops feeding and depends entirely on stored energy reserves.

A decrease in levels of PHM in the IPCs at the onset of metamorphosis is reversed by overexpression of DIMM. A, Double labeling with anti-PHM (α-PHM) and anti-DIMM (α-DIMM) in the midline protocerebral IPCs in a wandering third-instar larva (bearing UAS-dimm::MYC but no Gal4 driver). The immunostaining for each marker is shown separately in the top and together as a merge in the bottom. The arrow indicates one of the double-labeled cells. B, Intensity of anti-PHM immunostaining in the IPCs of wild-type animals in third-instar larvae and during the onset of metamorphosis. C, Overexpression of DIMM produced increased anti-PHM and anti-DIMM immunostaining in the IPCs of wild-type animals in wandering stage third-instar larvae. The labels on the x-axes indicate the presence (+) or absence (−) of the dilp2-Gal4 and UAS-dimm::MYC transgenes in each genotype. Bouin’s fixative was used to optimize anti-PHM immunostaining in B, and the fixative PFA-PA was used to optimize for double labeling in A and C. Therefore, intensity index values for PHM in B and C cannot be directly compared. s3, Small third-instar larvae, shortly after ecdysis; W, wandering stage third-instar larvae; WP, white prepupae. Results of the statistical analysis were as follows: in B, ∗p < 0.05 [n=8–11; compared with the reference mean (dashed line) after a one-way ANOVA (p=0.000000) with the Tukey–Kramer multiple comparison test]; in C, ∗p < 0.05, Kruskal–Wallis post hoc analysis following Kruskal–Wallis one-way ANOVAs on ranks [PHM, p=0.0014 (n=3–9); DIMM, p=0.00087 (n=3–9)]. Scale bar, 20 μm.
Discussion
Through spatial and temporal manipulation of transgene expression, we tested whether the bHLH protein DIMM controls neuropeptide expression as either a differentiation factor or as a regulator of differentiated cell phenotypes. Depending on the neuropeptide or secretory proteins that we examined, we found that DIMM can play either role. In some cells, DIMM contributed to the differentiation of LK neuropeptide expression. Then, in mature cells, DIMM continued to be expressed to effect changes in the levels of the peptide biosynthetic enzyme PHM. The postembryonically regulated cells included numerous neurons located throughout the adult brain (Fig. 6) as well as larval (but fully differentiated) insulin-producing neurons (Fig. 8), which function in the regulation of metabolism and growth (Rulifson et al., 2002). Thus, our results reveal a novel mechanism for the regulation of neuroendocrine protein expression in mature animals.
DIMM is both a differentiation factor and a regulatory factor
DIMM is required for the embryonic development of secretory peptide expression in diverse neuronal and endocrine cell lineages (Allan et al., 2003, 2005; Hewes et al., 2003; Gauthier and Hewes, 2006). In the current study, we extended these findings through spatiotemporal manipulation of dimm transgene expression. We found a critical window, which closed at the end of embryogenesis, during which DIMM must be present to induce full expression of the neuropeptide leucokinin (Fig. 5). Thus, under some conditions, DIMM is a differentiation factor.
If DIMM also regulates mature neuronal cell phenotypes, then it should satisfy five conditions. First, it must be present in terminally differentiated cells. Through analysis of dimm reporter gene expression, dimm in situ hybridization, and anti-DIMM immunocytochemistry, we showed that DIMM is expressed in mature neurons (Figs. 1, 2). Second, levels of DIMM must be positively correlated with levels of secretory proteins, and this should occur without significant changes in the cell fates of the affected neurons. Consistent with this prediction, reduced dimm expression results in lower secretory protein levels, elevated expression of dimm results in higher secretory protein levels, and neither effect was accompanied by gross changes in cell morphology or transmitter identity (Figs. 3⇑–5) (Hewes et al., 2003). Third, acute changes in DIMM expression in mature cells should produce corresponding changes in the abundance of secretory proteins: PHM levels were markedly increased after transient DIMM overexpression in the adult brain (Fig. 6). Fourth, levels of both DIMM and secretory proteins should fluctuate in tandem in some cells under native conditions. We (Fig. 8) and others (Park et al., 2004) have observed positively correlated changes in cellular expression of secretory proteins and DIMM in the context of normal physiological regulation or postembryonic developmental transitions. Fifth, these natural changes in neuropeptide levels should be sensitive to experimental manipulation of DIMM. Overexpression of DIMM in the IPCs prevented the decrease in PHM levels that normally occurs in these cells at the onset of metamorphosis (Fig. 8). Together, these results provide the first direct evidence for the postembryonic regulation of differentiated cell properties by an Atonal family protein in living animals.
In a previous study, Park et al. (2004) showed that the induction of PHM and l-phenylalanyl-l-methionyl-l-arginyl-l-phenylalanine amide (FMRFamide)-related neuropeptide expression in the Tva neurons during Drosophila metamorphosis was accompanied by increased expression of a dimm reporter gene. These results are consistent with a role for DIMM in the postembryonic regulation of both Phm and FMRFamide-related (Fmrf) expression in these neurons, because both genes are regulated embryonically by dimm (Allan et al., 2005). Early expression of DIMM in the Tva neurons produced early PHM expression (Fig. 3), although Fmrf expression was not affected (Allan et al., 2005) (data not shown). However, because the Tva neurons are born in the embryo, and their larval function (if any) is unknown, it is not clear whether the correlated changes in DIMM and PHM in these cells reflect cell regulation or delayed differentiation. In contrast, the IPCs are fully functional neurons that control growth rates and circulating carbohydrate levels in larvae (Rulifson et al., 2002). Thus, the regulation of PHM in the IPCs is a clear example of DIMM activity in terminally differentiated cells.
The control of PHM expression by DIMM may serve to regulate the capacity of neurons to produce amidated neuropeptides. In Drosophila, PHM is essential for neuropeptide amidation (Jiang et al., 2000). Most insect neuropeptides (>90%) are amidated at the C terminus (Hewes and Taghert, 2001), and amidation is often required for neuropeptide signaling (Jiang et al., 2000). In addition, many secretory proteins, including the vertebrate ortholog of PHM, peptidylglycine α-amidating monooxygenase, may play indirect roles in the sorting of coexpressed neuropeptides into secretory granules (Arvan and Castle, 1998; Brakch et al., 2002). Because dILP2 is not an amidated peptide, the role of the PHM in the IPCs is unclear. It will be interesting to determine whether PHM is required for dILP packaging and sorting into secretory granules or whether other amidated neuropeptides contribute to signaling by the IPCs. Nevertheless, because DIMM regulates PHM levels pan-neuronally (Fig. 3) (Allan et al., 2005), it likely effects dynamic changes in levels of bioactive, secretion-competent neuropeptides in diverse neurons in addition to the IPCs. In turn, these changes may alter the gain of neuropeptide signaling in the context of homeostatic and developmental regulation of neuroendocrine systems.
The differential effects of dimm on expression of Phm versus Lk and Fmrf may reflect differences in the combinatorial transcription factor codes that control the expression of these secretory genes. PHM can be induced (or elevated) in most if not all neurons by expression of a wild-type dimm transgene (Allan et al., 2005), although other factors likely contribute secondarily to the fine-tuning of PHM expression, because the responses to dimm overexpression were not linear (Fig. 3). Therefore, the code for Phm expression is primarily binary and depends on whether or not DIMM is expressed and generally not on developmental stage. In contrast, the overexpression of dimm alone is not sufficient in most cells to drive Lk and Fmrf expression (Hewes et al., 2003), and other factors, such as the LIM homeodomain gene apterous and the zinc finger gene squeeze, are also required (Allan et al., 2003, 2005; Herrero et al., 2003). Thus, if some elements of these combinatorial codes are only present in differentiating cells, then the induction of Lk, Fmrf, and other similarly regulated genes may only be possible during differentiation. After this stage, other unknown mechanisms would be needed to maintain Lk and Fmrf expression.
Potential dual functionality of other Atonal family members
Atonal-related proteins operate in transcriptional hierarchies, with proteins such as the neurogenins involved in selection of cell precursors, and later acting factors such as the NeuroD proteins regulating terminal differentiation (Ma et al., 1996; Sommer et al., 1996; Schwab et al., 2000). NeuroD1/BETA2, for example, is a member of the latter class, and it is expressed in endocrine cells of the pancreas, intestine, and pituitary and in several classes of neurons (Lee et al., 1995; Naya et al., 1995; Mutoh et al., 1997; Poulin et al., 1997). It is essential for the complete differentiation of several neuronal and endocrine cell types (Naya et al., 1997; Miyata et al., 1999; Liu et al., 2000). Moreover, NeuroD1/BETA2 has been shown to control neurite outgrowth, cell excitability, and the expression of several peptide hormone genes, including insulin, secretin, and proopiomelanocortin (Naya et al., 1995; Mutoh et al., 1997; Poulin et al., 1997; Hassan et al., 2000; Schwab et al., 2000).
Interestingly, hypothalamic NeuroD mRNA levels are reduced in obese ob/ob and food-deprived mice, suggesting a functional relationship in mature neurons between NeuroD and the neuroendocrine/endocrine signaling pathways that control energy balance (Nilaweera et al., 2002). NeuroD is also required for activity-dependent granule neuron dendritic growth in the intact rat cerebellar cortex (Gaudilliere et al., 2004), and Gal4-NeuroD chimeras can activate insulin promoter elements in response to glucose stimulation of cultured pancreatic beta cells (Khoo et al., 2003). In addition, mutations in NeuroD are associated with the development of certain forms of type 2 diabetes mellitus in young people (Malecki et al., 1999). Together, these findings provide strong, albeit indirect, support for roles of other Atonal family proteins in the regulation or maintenance of neuropeptide and peptide hormone levels in fully differentiated cells. Our current results on the dual functionality of DIMM provide additional indirect evidence for this model and suggest that regulation by Atonal proteins is a conserved and important feature of many neuroendocrine systems.
In summary, this study demonstrates that DIMM controls neuropeptide expression in developing and mature neurons. This is the first direct evidence, in situ, for continued function of an Atonal family transcription factor in differentiated cells. Our findings provide insights into the general mechanisms for maintenance of terminally differentiated cells after the induction signals are gone (Baker, 2004). In addition, they suggest an effective means for the regulation of the gain of neuropeptide signaling in mature animals.
Footnotes
-
This work was supported by National Science Foundation (NSF) Grant IBN0344018 and NSF Experimental Program To Stimulate Competitive Research Grant 0132534 (R.S.H.), Oklahoma Center for the Advancement of Science and Technology Grant HR03-048S, and the Oklahoma State Regents for Higher Education. We thank Ronald Davis, Eric Rulifson, and Paul Taghert for stocks and antisera, Chad Hargrave, Audrey Kennedy, Kendal Milam, Matthew Mote, Elizabeth Pearsall, Kimberly Roark, and Jeremiah Smith for technical assistance, and Troy Littleton, Joseph Bastian, and Sebastien Gauthier for comments on this manuscript.
- Correspondence should be addressed to Randall Hewes, Department of Zoology, Stephenson Research and Technology Center, 101 David L. Boren Boulevard, University of Oklahoma, Norman, OK 73019. Email: hewes{at}ou.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}