

Abstract
The presynaptic protein α-synuclein is a prime suspect for contributing to Lewy pathology and clinical aspects of diseases, including Parkinson's disease, dementia with Lewy bodies, and a Lewy body variant of Alzheimer's disease. α-Synuclein accumulates in Lewy bodies and Lewy neurites, and two missense mutations (A53T and A30P) in the α-synuclein gene are genetically linked to rare familial forms of Parkinson's disease. Under control of mouse Thy1 regulatory sequences, expression of A53T mutant human α-synuclein in the nervous system of transgenic mice generated animals with neuronal α-synucleinopathy, features strikingly similar to those observed in human brains with Lewy pathology, neuronal degeneration, and motor defects, despite a lack of transgene expression in dopaminergic neurons of the substantia nigra pars compacta. Neurons in brainstem and motor neurons appeared particularly vulnerable. Motor neuron pathology included axonal damage and denervation of neuromuscular junctions in several muscles examined, suggesting that α-synuclein interfered with a universal mechanism of synapse maintenance. Thy1 transgene expression of wild-type human α-synuclein resulted in similar pathological changes, thus supporting a central role for mutant and wild-type α-synuclein in familial and idiotypic forms of diseases with neuronal α-synucleinopathy and Lewy pathology. These mouse models provide a means to address fundamental aspects of α-synucleinopathy and test therapeutic strategies.
- transgenic mice
- α-synuclein
- wild-type
- A53T mutant
- Lewy pathology
- Parkinson's disease
- dementia with Lewy bodies
- ubiquitination
Idiopathic Parkinson's disease (PD), dementia with Lewy bodies (DLB), and a Lewy body variant of Alzheimer's disease (LBVAD) are characterized pathologically by proteinaceous inclusions in neurons commonly referred to as Lewy pathology in postmortem brain tissue samples (Forno, 1996; Ince et al., 1998; Irizarry et al., 1998; Spillantini et al., 1998; Takeda et al., 1998; Braak et al., 1999). The inclusions occur in the dystrophic (Lewy) neurites that constitute an important part of the pathology of PD and DLB (Irizarry et al., 1998; Spillantini et al., 1998; Braak et al., 1999), in neuronal perikarya (Lewy bodies) (Forno, 1996; Spillantini et al., 1997; Wakabayashi et al., 1997; Baba et al., 1998; Ince et al., 1998; Irizarry et al., 1998; Mezey et al., 1998; Spillantini et al., 1998; Takeda et al., 1998; Braak et al., 1999), and occasionally extracellularly (Den Hartog and Bethlem, 1960).
α-Synuclein is the major constituent of Lewy bodies and Lewy neurites (Spillantini et al., 1997; Wakabayashi et al., 1997; Baba et al., 1998;Ince et al., 1998; Irizarry et al., 1998; Mezey et al., 1998;Spillantini et al., 1998; Takeda et al., 1998; Arai et al., 1999; Braak et al., 1999; Giasson et al., 2000). To lesser and varying degrees, these inclusions contain ubiquitin (Gai et al., 1995), neurofilament proteins (Forno, 1996; Ince et al., 1998; Takeda et al., 1998), ubiquitin C-terminal hydrolase (Lowe et al., 1990), proteosomal subunits (Li et al., 1997), and a variety of other antigenic determinants (Pollanen et al., 1993). α-Synuclein immunoreactivity is the most sensitive and reliable diagnostic for Lewy-type pathology (Forno, 1996; Spillantini et al., 1997; Wakabayashi et al., 1997; Baba et al., 1998; Ince et al., 1998; Irizarry et al., 1998; Mezey et al., 1998; Spillantini et al., 1998; Takeda et al., 1998; Braak et al., 1999).
Lewy pathology in neurons appears to be central and may contribute mechanistically to their dysfunction and degeneration in disease. Altogether, the distribution of α-synuclein-containing inclusions in disorders with Lewy pathology, the discovery of two mutations in the α-synuclein gene linked to early-onset familial PD (Polymeropoulos et al., 1997; Krüger et al., 1998), the ability of the protein to self-aggregate (Conway et al., 1998, 2000; Giasson et al., 1999; Narhi et al., 1999; Wood et al., 1999), and recent findings in vivo in transgenic flies (Feany and Bender, 2000) and mice (Masliah et al., 2000) are supporting a central role for α-synuclein in the pathophysiology of diseases with Lewy pathology.
Here, we demonstrate pathology with neuronal degeneration and accompanying motor deficits in mice, by neuronal expression of the A53T mutant of human α-synuclein or its wild-type counterpart. α-Synuclein-encoding cDNAs were under control of mouse Thy1 regulatory sequences (Lüthi et al., 1997; Sturchler-Pierrat et al., 1997; Wiessner et al., 1999) that produced reliably neuron-specific expression and high protein levels in many central neurons of the mouse nervous system.
MATERIALS AND METHODS
Transgenic mice. Wild-type human α-synuclein cDNA (396 bp) was PCR amplified (2 min, 93°C; three cycles of 15 sec, 93°C; 30 sec, 55°C; 30 sec, 72°C; two cycles of 15 sec, 93°C; 30 sec, 60°C; 30 sec, 72°C; and 30 cycles of 15 sec, 93°C; 30 sec, 66°C; 30 sec, 72°C; oligonucleotides, cgacgacagtgtggtgtaaaggaa and tgggcacattggaactgagcactt) from 20 ng of human brain cDNA (Clontech, Palo Alto, CA) and cloned into pMOSBlue (Amersham Pharmacia Biotech, Little Chalfont, UK). The A53T mutation was introduced using PCR oligonucleotide-directed mutagenesis (Stratagene, La Jolla, CA; 30 sec, 93°C; 14 cycles of 30 sec, 93°C; 1 min, 55°C; 13 min, 68°C; oligonucleotides, agtggtgcatggtgtgacaacagtggctgaga and tctcagccactgttgtcacaccatgcaccact). The identity of wild-type and mutant cDNA was confirmed by sequencing and the corresponding blunted (Klenow) cDNAs (NdeI-SmaI) inserted into the blunted (XhoI site) Thy1 cassette (Lüthi et al., 1997;Sturchler-Pierrat et al., 1997; Wiessner et al., 1999). For microinjection, linear NotI DNA fragments comprising transgene without plasmid vector sequences were isolated. Injection was into homozygous C57BL/6 mouse eggs. Genotyping was performed by PCR (oligonucleotides were identical to those used for cloning the human α-synuclein cDNA; 5 min, 95°C; 30 cycles of 30 sec, 95°C; 1 min, 60°C; 1 min, 72°C; 10 min, 72°C) using column-purified (Qiagen, Hilden, Germany) tail DNA.
Northern blot analysis. Northern blot analysis was performed with total brain RNA (TriZol method; 10 μg loaded per gel lane). Probes were either 364 (Fig.1A, probe A) or 111 bp of the human α-synuclein cDNA (Fig. 1A,probe B; unlike probe A, probe B lacks homology to cDNAs encoding α-synuclein-related family members) (Lavedan, 1998), or 600 bp of mouse Thy1 3′ untranslated sequences (Lüthi et al., 1997) (Fig. 1A,probe C). Standard procedures were used.

Transgene expression. A, Schematic representation of the transgene. Roman numeralsrefer to exons in the endogenous murine Thy1 gene. Boxesrepresent a complete α-synuclein coding cDNA probe (A), a C-terminal 111 bp α-synuclein cDNA probe (B), and a Thy1 3′ untranslated region probe (C) (Lüthi et al., 1997).Arrows represent PCR primers for genotyping.B, Northern blot analysis of 10 μg of total brain RNA using probe A and brain RNA of a C57BL/6 nontransgenic mouse (lane 1), a line 9813 mouse (lane 2), a line 9956 mouse (lane 3), and the single transgenic male of line 9832 (lane 4). Thearrowhead refers to the transgene mRNA, and thehorizontal bar marks the position of endogenous mouse α-synuclein mRNA. C, Immunoblot analysis showing increased levels of α-synuclein protein in brain homogenates of a representative mouse of each of the lines 9956 (lane 1), 9813 (lane 2), the single F1 male of line 9832 (lane 3), and C57BL/6 (lane 4). The polyclonal rabbit antibody used (Chemicon) detects both mouse and human α-synuclein protein. D, Immunoblot using the antibody LB509 (specifically detects human but not the mouse α-synuclein protein; Zymed) and line 9956 (lane 1), line 9813 (lane 2), and C57BL/6 (lane 3).E, F, In situhybridization in sagittal brain sections of a line 9813 mouse (E) and a C57BL/6 mouse (F) using 35S-labeled cRNA corresponding to probe A (A). G, H, In situ hybridization in horizontal brain sections of a line 9813 mouse (G) and a C57BL/6 mouse (H) using 35S-labeled cRNA corresponding to probe B (A).
Immunoblot analysis. For Western blot analysis, 14,000 × g supernatant fractions were used of half-brain homogenates [homogenized in 2 ml of E-buffer (50 mm Tris-HCl, pH7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm EDTA, and a cocktail of protease inhibitors (Boehringer Mannheim, Mannheim, Germany) and left on ice for 30 min]. Fifteen, 25, or 50 μg of protein was loaded per lane and separated on 15% SDS-PAGE. After blotting and blocking nonspecific binding, membranes were incubated with rabbit anti-α-synuclein polyclonal antibody (1:1000; AB5038; Chemicon, Temecula, CA), followed by alkaline phosphatase (AP)-conjugated anti-rabbit IgG (1:50,0000; AO418; Sigma, St. Louis, MO), or the anti-human-α-synuclein-specific antibody LB509 (1:5000; Zymed, San Francisco, CA), followed by AP-conjugated anti-mouse IgG1 (1:40,000; Sigma) and chemiluminescent detection (Clontech).
In situ hybridizations. The spatial distribution pattern of transgene versus endogenous α-synuclein expression was determined byin situ hybridization (Wiessner et al., 1999). cRNA was transcribed using T7-RNA polymerase and a 364 bp cDNA template (complete human α-synuclein coding region) or a 111 bp cDNA template (corresponding to the region encoding amino acids 103–140 of human α-synuclein).
Immunocytochemistry. Mice (aged 3.8–6 months) were injected with pentobarbital (50 mg/ml Nembutal; Abbott laboratories, North Chicago, IL) and perfused transcardially with 0.01m PBS, followed by 4% paraformaldehyde in PBS. One brain hemisphere, spinal cord, and hind limb muscle were embedded in paraffin and cut as 4-μm-thick sections. Vibratomesections (25 μm) were cut from the other hemisphere for free-floating immunocytochemical staining using anti-α-synuclein mouse monoclonal antibody (1:500; S63320; Transduction Laboratories, Lexington, KY), biotinylated anti-mouse IgG (1:500; E0464; Dako, Copenhagen, Denmark), and the avidin-biotin peroxidase method (Elite standard kit SK6100; Vector Laboratories, Burlingame, CA). In addition, the following antibodies were used: AT8 monoclonal, which detects paired helical filament and hyperphosphorylated tau (1:1000; BR-003; Innogenetics, Zwijndrecht, Belgium); neurofilament 200 kDa-specific monoclonal, detecting normal and phosphorylated neurofilaments (1:100; NCL-NF200; Novocastra, Newcastle, UK); neurofilament M and H monoclonal (phophorylated forms; 1:500; MAB 1592; Chemicon). Stainings (including also Congo red and thioflavine S) were as described bySturchler-Pierrat et al. (1997). Deparaffinized sections were used for Campbell–Switzer silver staining (Campbell et al., 1987), Holmes–Luxol staining (Holmes, 1943), and immunostaining with rabbit anti-ubiquitin Ig fraction (1:200; Z0458; Dako), rabbit anti-glial fibrillary acidic protein (GFAP) Ig fraction (1:500; Z0334; Dako), anti-phosphotyrosine mouse monoclonal antibody (1:1500; P3300; Sigma), and anti-human α-synuclein-specific mouse monoclonal antibody (LB509; 1:2000; 18–0215; Zymed) (Baba et al., 1998; Jakes et al., 1999). Antigenity was enhanced by treating paraffin sections with concentrated formic acid for 5 min and microwave heating at 90°C for 60 min before incubation with anti-α-synuclein, microwave heating at 90°C for 30 min before anti-GFAP and anti-phosphotyrosine, and pronase treatment (37°C, 30 min) before anti-ubiquitin. Nonspecific binding sites were blocked using normal serum. Bound antibody was visualized using the avidin-biotin peroxidase method (Elite standard kit SK6100; Vector Laboratories) and DAB substrate (1718096; Boehringer Mannheim) or Vector Laboratories AEC substrate (SK-4200). Immunostaining and confocal analysis of neurofilament and synaptophysin at neuromuscular junctions (NMJs) were as follows. Mice were killed by anesthesia. Extensor digitorum longus (EDL) and soleus muscles were excised and stained with Alexa Fluor 488-labeled α-bungarotoxin (1:1000 in F-15 medium; Molecular Probes, Eugene, OR) for 2 hr at 4°C, followed by several rinses with PBS. Muscles then fixed with cold methanol (−20°C) and permeabilized with blocking solution (PBS containing 5% horse serum, 1% BSA, 1% Triton X-100, and 0.1% sodium azide) for 2 hr at 4°C. Tissue was subsequently incubated with primary antibodies against either neurofilament (1:500 in blocking solution; Sigma) or synaptophysin (1:200 in blocking solution; Dako) and was incubated for 40 hr at 4°C. After several rinses with PBS, muscles were incubated with Cy3-labeled goat anti-rabbit IgG (1:1000 in blocking solution; Jackson ImmunoResearch, West Grove, PA) overnight at 4°C. The muscles were thoroughly rinsed with PBS and mounted on glass slides using Citifluor (Plano). The tissue was examined with a confocal microscope (model TCS NT; Leica, Nussloch, Germany).
Immunoelectron and electron microscopy. For immunoelectron microscopy, transgenic and wild-type C57BL/6 mice were perfused transcardially with a mixture of 1.5% picric acid, 0.1% glutaraldehyde, and 4% paraformaldehyde in 0.1 mphosphate buffer, pH 7.4. Vibratome sections were stained free-floating with antibody to α-synuclein (LB509; 1:2000) dehydrated in ascending series of ethanol and acetone, and flat-embedded between glass slide and coverslips in Embed-812 (Electron Microscopy Sciences, Fort Washington, PA). Fragments of the spinal cord were then dissected out and ultrathin sections were cut from the tissue surface, and these were mounted on copper grids and analyzed with a Zeiss (Oberkochen, Germany) EM900 microscope. For conventional electron microscopy, mice were anesthetized and perfused transcardially with cold saline, followed by 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1m cacodylate buffer. Small tissue blocks were cut out from brainstem and spinal cord, immersion-fixed for 12 hr at 4°C in the same buffer, and epoxy-embedded, and ultrathin sections were prepared and placed on 200-mesh copper grids for staining with uranyl acetate and lead citrate.
Silver-esterase staining of neuromuscular junctions. The combined silver-esterase technique was applied on gastrocnemius muscle. This muscle was freshly dissected and, after a brief wash in 10 mm EDTA in PBS, the tissue was mounted for cryostat sectioning and cut (50 μm longitudinal sections). Slides were rinsed in 80 mm EDTA in PBS before mounting sections. Sections were dried for 30–120 min at 37°C, immersed in sodium sulfate (29%) for 3 min, washed in H2O, and incubated for 25 min at 37°C in acetylcholinesterase staining solution (Pestronk and Drachman, 1978). After washing in H2O, sections were dehydrated in 70 and 100% ethanol, respectively, for 1–2 min each. Next, sections were fixed in buffered formol saline (Pestronk and Drachman, 1978) for 30 min at room temperature, washed in H2O, and pretreated for 30 min at 37°C in 10% chloral hydrate (w/v) containing 1% pyridine (v/v). After washing in H2O, the sections were incubated in a freshly prepared solution of 20% silver nitrate containing 1% cupric sulfate. Calcium carbonate was added to each slide separately. Incubation was for 40 min at 37° before discarding the solution, washing in H2O, and incubation for 10 sec and 3–5 min, respectively, in a bath of 1% hydroquinone and 5% sodium sulfite. After washing in H2O, sections were fixed (2–5 min) in 5% sodium thiosulfate, washed (in H2O), toned for 5 min in 0.2% sodium tetrachloroaureate (after adding freshly a drop of glacial acetic acid), washed (in H2O), immersed in 1% oxalic acid for 20–120 sec, washed (in H2O), fixed again in 5% sodium thiosulfate, washed (in H2O), dehydrated in ethanol, air dried, rinsed in xylene, air dried, and mounted.
Rotating rod. The mice were trained twice daily and on two successive days to stay on a rotating rod (Technical and Scientific Equipment, Bad Homburg, Germany) for 150 sec (speed of 12 rpm). Subsequently, the animals were tested on the rotating rod once weekly, three times and at three different speeds (i.e., 12, 24, and 36 rpm). The cutoff time used for measuring the endurance performance in all of these experiments was 60 sec. The plotted mean endurance performance on a particular test day was the mean of the three performances at the given speed. Statistical evaluation of the data were using repeated measures ANOVA.
RESULTS
Six transgenic C57BL/6 lines were produced, each expressing a Thy1αSNA53T transgene to a different level. The two lines described in this report (9813 and 9956) expressed transgene mRNA levels (Fig.1B, mRNA levels were 9832 > 9813 > 9956 as shown for three lines; others not shown) that each resulted in increased levels of α-synuclein protein in brain (Fig. 1C, antibody cross-reacting with mouse and human α-synuclein; Fig.1D, antibody detecting only human α-synuclein). In both lines, transgene expression occurred in neurons throughout the telencephalon, brainstem, and spinal cord, as shown in a representative sagittal section of a mouse brain of line 9813 (Fig.1E). In situ hybridization with a cRNA probe that corresponds to the full-length coding sequence of human α-synuclein (Fig. 1A, probe A), shows the transgene mRNA expression pattern superimposed on that of endogenous mouse α-synuclein, the latter being apparent mainly in the telencephalon (Fig. 1F). Transgene expression pattern was also monitored specifically using a cRNA probe that corresponds to 111 bp encoding the C-terminal 37 amino acids of human α-synuclein (Fig. 1A, probe B). This probe is 87.5% homologous to mouse cDNA but has nucleotide differences occurring every 10–20 bp. As a result and as shown in a pair of horizontal sections, this probe detects no signal above background (as verified also by Northern blot analysis; data not shown) in the nontransgenic C57BL/6 mouse brain (Fig. 1H), whereas the signal in the transgenic brain reflects specifically the transgene expression pattern (Fig. 1G).
In all of the mice of lines 9813 and 9956, we observed an early-onset (>3 weeks of age) and a progressive decline of motor performance. A more severe motor deficit was seen in a single transgenic F1 male of a third independent founder (line 9832). This male died at an age of 5 weeks and the line was lost. Brain transgene mRNA levels in the line 9832 single F1 male (Fig. 1B, lane 4) superseded those detected in line 9813.
The progressive decline in motor performance was measured in a rotating rod experiment. A group of transgenic (n = 7) versus nontransgenic (n = 12) littermate males of line 9813 were compared, starting at age 40 d and up to age 200 d (Fig.2). The results show a progressive decline in motor performance and a dramatic reduction in endurance of the transgenic mice to remain on the rotating rod. A similar difference and effect was observed when comparing aging (40–200 d) female transgenic (n = 15) versus nontransgenic (n = 8; data not shown) mice of line 9813. The ANOVA indicated a highly significant effect of genotype (p < 0.001), as well as a significant age, speed, and genotype interaction (p < 0.005; repeated measures ANOVA for two trial factors). A comparison of the performance curves of the two groups (transgenic and wild-type) revealed that, at both speeds 12 and 36 rpm, the performance of the transgenic animals differed significantly (p < 0.001) from that of their nontransgenic littermates.

Rotating rod performance of transgenic mice. Starting at age 40 d and up to age 200 d, transgenic (n = 7) and C57BL/6 nontransgenic (n = 12) littermate male mice were tested for endurance to stay on the rotating rod. The mice were tested weekly, and their performance is shown for two different rotation speeds, speed 1 (12 rpm) and speed 3 (36 rpm). C57BL/6 mice showed maximum endurance performance (60 sec) at both speeds and as shown for speed 3 (36 rpm).
The pronounced motor phenotype found in all of the mice of both A53T transgenic lines and the documented expression of Thy1-based transgenes in motor neurons (Aigner et al., 1995) prompted us to examine more closely the pathological changes in these cells. In the anterior horns of the spinal cord, ∼80% of the motor neurons expressed the human α-synuclein A53T mutant protein, as shown by immunoreactivity for the human α-synuclein-specific antibody LB509 (Baba et al., 1998; Jakes et al., 1999). Specific to the transgenic mice, many of these cells showed diffuse perikaryal α-synuclein staining (Fig.3A) and some also showed Lewy-like pathology (Fig. 3C, D) with pronounced ubiquitin immunoreactivity (Fig. 3D). Furthermore, staining with the Campbell–Switzer pyridine silver technique (Campbell et al., 1987) (Fig. 3C), a routine and sensitive method used to detect Lewy-type changes in human brain tissue (Braak and Braak, 1999;Sandmann-Keil et al., 1999), revealed intense staining. These results indicate that (rodent) motor neurons are susceptible to Lewy-like changes when expressing the A53T mutant of human α-synuclein. Other changes associated with the development of the motor neuron pathology (and seen also in other affected brain regions; data not shown) included astrocytic gliosis (Fig. 3E) and microglial activation (Fig. 3F).

Lewy-like pathology in the transgenic mouse spinal cord. A–H correspond to sections through the anterior horn. A, Prominent perikaryal and proximal neuritic α-synuclein staining of motor neurons in a transgenic mouse spinal cord section but not in C57BL/6 (B).C, Campbell–Switzer-stained motor neurons in a transgenic mouse spinal cord, which also immunoreacted with anti-ubiquitin antibody (D). E,G, Anti-GFAP and anti-phosphotyrosine antibody stainings (F, H) in transgenic (E, F) versus nontransgenic (G, H) C57BL/6 spinal cord show evidence for astrocytic gliosis (E) and reactive microglia (F) specific to the transgenic tissue. Scale bars: A–D (in D), 20 μm;E–H (in H), 20 μm. Magnification: A–D, 250×; E–H, 200×.
Spinal roots (Fig. 4A) and nerve fiber bundles in muscles (Fig. 4C) immunostained for α-synuclein. Axonal degeneration was apparent in spinal roots, with nerve fibers showing breakdown and segmentation into ellipsoids of the myelin sheath (Fig. 4B). Also, skeletal muscles contained small angular fibers (Fig. 4D,arrowheads), indicating neurogenic muscular atrophy, consistent with the denervation and neuropathology observed to varying degrees at NMJs of gastrocnemius muscle taken from several line 9813 mice (n = 7, age 6.5 months; Fig.4E–J). Depending on the individual mouse, 10–40% of the synapses were denervated and thinning of preterminal nerves, and/or swellings were detected in at least 50% of innervated synapses. Pathological changes were also observed in two other muscles, the EDL, containing fast-twitch muscle fibers, and the soleus, which contains mostly slow-twitch muscle fibers (Fig.5). Whereas the structure of the postsynaptic apparatus, visualized by α-bungarotoxin, was similar in transgenic and wild-type mice, the presynaptic motor neurons showed clear signs of degeneration. In transgenic but not in wild-type C57BL/6 littermates, NMJs often showed a reduction in the neurofilament staining, indicating that motor neurons were about to retract from synapses (Fig. 5C, D). Moreover, neurofilament staining in more proximal regions of the motor nerve was discontinuous (data not shown). The hypothesis that motor neurons abandon synaptic sites was supported in staining for the synaptic vesicle protein synaptophysin (Fig. 5A, B). In wild-type animals, synaptophysin matches the outline of the postsynaptic acetylcholine receptors (AChRs) (Fig.5A). In transgenic animals, synaptophysin appeared punctated and thinner so that it did not cover the entire area of AChR aggregates (Fig. 5B). The finding that neurodegeneration including denervation of NMJs is observed in all of the muscles examined suggests that a universal mechanism of synapse maintenance is affected by transgene expression of human α-synuclein. Together with the neuropathological changes detected in central areas (see below), including some that are involved in motor function (e.g., globus pallidus; data not shown), these findings provide a likely explanation for the observed reduction in complex motor performance.

Neuromuscular degeneration. A, Longitudinal section of a spinal root with human α-synuclein-immunoreactive (LB509 antibody) nerve fibers that are specific to the transgenic mice (C57BL/6 not shown). B, Holmes–Luxol-stained spinal root showing axonal degeneration with breakdown and segmentation of myelin into ellipsoids (“digestive chambers”). C, Cross-sectioned bundle of nerve fibers in a muscle showing strongly human α-synuclein-specific (antibody LB509) immunoreactive axons. D, Cross-sectioned muscle fiber bundle containing small angular fibers consistent with denervation (arrowheads), indicating neurogenic muscle atrophy (Holmes–Luxol stain). Scale bars:A–D, 20 μm. E–J, Medial gastrocnemius NMJs from C57BL/6 (E) and two different line 9813 mice (mouse 1, F–H; mouse 2, I,J), aged 6.5 months. The combined silver-esterase reaction reveals nerves in black and the synaptic acetylcholine esterase reaction product (asterisks) inblue. NMJs in the transgenic mice show neuropathological changes ranging from swellings of preterminal nerves (F, arrow), thinning out of the preterminal nerve (G), retraction of the nerve from the synaptic region (H), to complete denervation (I, arrows). A thin and characteristically twisted nerve sprout (arrows) has regrown to a denervated synapse shown in J. Dark ovals (in I and J) are attributable to background labeling of nuclei. Scale bar, 40 μm. Magnification: A, 100×; B,C, 400×; D, 200×; E–J, 400×.

Neurodegeneration of soleus neuromuscular junctions. A–D, The structure of the postsynaptic apparatus is visualized by α-bungarotoxin staining of AChRs (green), which was similar in C57BL/6 (A, C) and transgenic (B,D) mice. Costainings are shown for α-bungarotoxin (green) and synaptophysin (A,B), and α-bungarotoxin and neurofilament (C, D). Scale bar, 3 μm. Magnification, 700×.
To investigate pathological changes in the brain more closely, animals of both lines, aged 12 weeks and older (n = 19), were examined by applying immunohistochemical techniques routinely used to assess Lewy pathology in human brain (Gai et al., 1995; Forno, 1996;Spillantini et al., 1997; Wakabayashi et al., 1997; Baba et al., 1998;Ince et al., 1998; Mezey et al., 1998; Takeda et al., 1998). In transgenic mouse brains, many neurons in the telencephalon (Fig.6A,B,D,E) and brainstem (Fig.7B,C) showed intense α-synuclein staining in cell bodies and dendrites. This pattern was seen when using an antibody that specifically detects human but not mouse α-synuclein (Fig.6A,D; for negative control, see Fig. 3B). It was also seen when using an antibody that detects both mouse and human α-synuclein (Figs.6B,E,7B,C) and in sharp contrast to the axonal and presynaptic distribution of endogenous mouse α-synuclein in the nervous system of nontransgenic mice (Fig. 6C, F).

α-Synuclein protein expression in the transgenic mouse brain. A–F, α-Synuclein staining in the hippocampal CA1 region (A, B) and neocortex (D, E) of a transgenic mouse compared with the respective regions in a nontransgenic C57BL/6 mouse (C, F). Unlike in the transgenic brain, the C57BL/6 hippocampus lacks α-synuclein immunoreactivity in CA1 pyramidal cell bodies in stratum pyramidale (st.p.). In both mice, immunostaining is prominent in stratum radiatum (st.r.). Strongly immunoreactive neurites (i.e., CA1 cell dendrites) in stratum radiatum are specific to the transgenic brain. Similar dendritic structures in the C57BL/6 brain appear pale and without α-synuclein. Immunostaining was in paraffin (A, D; human α-synuclein-specific antibody LB509) and free-floating brain sections (B,C, E, F; antibody detects both mouse and human α-synuclein). Scale bars: A,D (in D), 20 μm (250× magnification);B, C, E, F (in F), 20 μm (200× magnification).

α-Synuclein and ubiquitin in transgenic mouse and human PD brain. A–F, Sections are shown of a human PD substantia nigra (A, D) and a transgenic mouse brain pontine reticular nucleus (B,C, E, F) immunostained for α-synuclein (A–C) and ubiquitin (D–F). Note the prominent somatodendritic staining for α-synuclein and the neuritic staining for ubiquitin in both human and mouse neurons. Lewy-like neurites are indicated by arrowheads. A cell of the substantia nigra in the human PD brain shows a Lewy body inclusion (A,arrow). G, H, Two consecutive 3 μm paraffin sections of the same group of cells in a cerebellar nucleus, stained for α-synuclein (LB509 antibody;G) and ubiquitin (H).G, Several neurons showing similar degrees of perikaryal α-synuclein immunoreactivity. Many cross-sectioned neurites are also stained. H, Abundant ubiquitin immunoreactivity is seen in only one of the three grouped cells and a nearby neurite (arrowheads in H and Gindicate the same cell and neurite showing α-synuclein immunoreactivity). In addition, compared with the number of α-synuclein-positive neurites in G, inH a smaller number of these structures (dark and punctate) show immunoreactivity for ubiquitin. Scale bars:A, D, 20 μm (magnification:A, 320×; D, 630×); B, C, E–H (in H), 20 μm (magnification, 400×).
Increased perikaryal and neuritic staining of neurons by α-synuclein antibodies is one characteristic feature in the diseased human brain (Spillantini et al., 1997; Wakabayashi et al., 1997; Baba et al., 1998;Irizarry et al., 1998; Mezey et al., 1998; Spillantini et al., 1998;Braak et al., 1999). Compared with affected neurons in human PD brain (Fig. 7A), α-synuclein staining in transgenic mouse brains showed heterogeneous changes in neurites, very similar to those observed in human brain (Forno, 1996; Wakabayashi et al., 1997;Spillantini et al., 1997; Baba et al., 1998; Ince et al., 1998;Irizarry et al., 1998; Mezey et al., 1998; Spillantini et al., 1998;Takeda et al., 1998; Braak et al., 1999). Frequently observed changes included sausage-like enlargements of proximal and distal neuritic segments, thick or fine thread-like inclusions, as well as beaded or spindle-shaped neurites (Fig.7B,C). They were most prominent and frequent in areas including the nucleus centralis oralis pontis, the nucleus vestibularis lateralis, the deep cerebellar nuclei, the deep aspects of the tectal plate, and as shown above (Fig. 3), motor nuclei in the spinal cord.
Immunostaining for ubiquitin is also frequently used to visualize Lewy pathology in human brain (Gai et al., 1995; Forno, 1996; Spillantini et al., 1997; Wakabayashi et al., 1997; Baba et al., 1998; Ince et al., 1998; Irizarry et al., 1998; Mezey et al., 1998; Spillantini et al., 1998; Takeda et al., 1998; Braak et al., 1999; Gómez-Tortosa et al., 2000b). In the transgenic mice, dystrophic neurites and cell bodies occasionally stained intensely for ubiquitin (Fig.7E,F). The stained neurites displayed morphological features (Fig.7E,F) similar to those seen in human brains (Fig. 7D) with Lewy pathology (Gai et al., 1995; Forno, 1996; Spillantini et al., 1997; Wakabayashi et al., 1997;Baba et al., 1998; Ince et al., 1998; Irizarry et al., 1998; Mezey et al., 1998; Spillantini et al., 1998; Takeda et al., 1998; Braak et al., 1999). However, both neuronal cell bodies and neurites with ubiquitin immunostaining were less frequent when compared with α-synuclein-positive cells and neurites (Fig.7G,H) and were restricted primarily to those regions that showed the most pronounced α-synuclein immunopathology in cells and neurites, including the nucleus centralis oralis pontis, the nucleus vestibularis lateralis, the deep cerebellar nuclei, the deep aspects of the tectal plate, and motor nuclei in the spinal cord. Within each of these regions, neurons and neurites showed no significant ubiquitin immunostaining, weak staining (with variable degrees of punctate cytoplasmic and/or perinuclear immunolabeling), or an intense ubiquitin immunostaining, as illustrated in two consecutive 3 μm paraffin sections (Fig. 7G,H). These sections show the same set of cells and neuritic structures in a cerebellar nucleus, stained for α-synuclein (Fig. 7G) and ubiquitin (Fig. 7H).
Ubiquitin compared with α-synuclein immunopathology in human brains with Lewy bodies and Lewy neurites is also less frequent (Forno, 1996;Ince et al., 1998; Irizarry et al., 1998; Spillantini et al., 1998;Takeda et al., 1998; Braak et al., 1999; Gómez-Tortosa et al., 2000b), suggesting that ubiquitination might be a late event in the development of the pathology in mouse and human. Curiously, ubiquitination was evident in some but not all of the mice with α-synuclein immunopathology, and the phenomenon was observed in both lines (9813 and 9956) and sexes. For example, in one group of five 6-month-old transgenic mice of line 9813 that were processed simultaneously for pathology, only two of the five animals showed a prominent ubiquitin immunopathology in the restricted brain regions. The stochastic manner in which ubiquitination appeared led us to compare transgene mRNA levels between different mice and sexes within each line but no differences between either individuals or sexes were detected (data not shown). Apparently, factors other than these or strain genetic background (all mice are C57BL/6) play a role in this phenomenon.
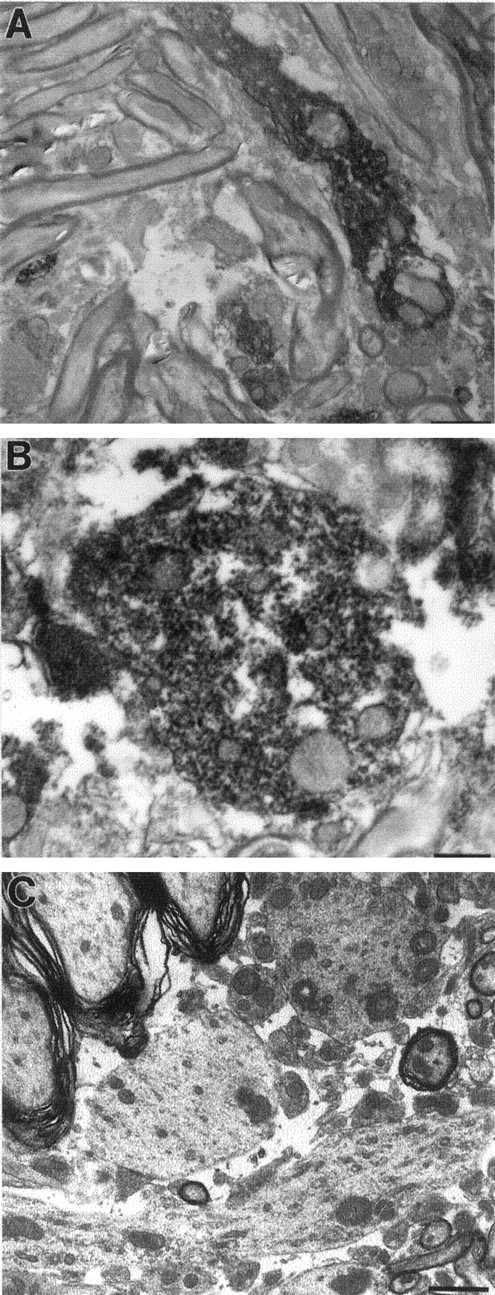
Finally, ultrastructural features of the human α-synuclein-positive neuronal inclusions were characterized by immunoelectron and conventional electron microscopy. In contrast to nontransgenic C57BL/6 mice, which showed no staining with the LB509 human α-synuclein-specific antibody (data not shown; for reference, see Fig. 3B), neurons of transgenic mice showed α-synuclein-containing granular deposits (Fig.8A,B). Using conventionally stained ultrathin sections, brainstem and spinal cord tissue of transgenic but not nontransgenic mice showed dendrites (often dilated) containing electron-dense finely structured granular material (Fig. 8C). α-Synuclein-immunoreactive fibrillar components as seen in human brains with Lewy pathology (Spillantini et al., 1997; Wakabayashi et al., 1997; Goedert et al., 1998; Takeda et al., 1998) were not seen.

Ultrastructural features of neurons in Thy1αSNA53T mice. Shown are immunoelectron (A,B) and a conventional micrograph (C) of spinal cord gray matter. A,B, Five-month-old Thy1αSNA53T mouse of line 9813. The immunoelectron micrographs are specifically stained for human but not mouse α-synuclein (LB509 antibody). A, Longitudinal section through a small dendritic appendage showing prominent staining of elements in the dendritic cytoplasm. B, Immunolabeled cross-sectioned dendrite, showing the fine granular composition of the α-synuclein-containing structures, which occasionally appeared attached to the endoplasmic reticulum and mitochondria, for example.C, Conventionally stained electron micrograph of a brainstem section of a 4-month-old transgenic mouse showing fine granular electron-dense material in dendritic profiles, most evident in the cross-sectioned dendrite located in the top rightpart between the three larger and a smaller myelinated axon. Scale bars: A, 1.7 μm (4500× magnification);B, 0.6 μm (12,000× magnification); C, 1.9 μm (5000× magnification).
The vast majority of human cases with Lewy pathology are idiopathic, and α-synuclein mutations in the coding region, other than A53T and A30P, have not been found in cohorts of familial or sporadic PD (Chan et al., 1998; Farrer et al., 1998; The French Parkinson's Disease Study Group, 1998; Vaughan et al., 1998). To test whether the pathology seen in the mice is unique for the A53T mutant of α-synuclein, in which case it could bear little on idiopathic forms of the diseases, we also generated transgenic lines (n = 5) expressing wild-type human α-synuclein. Wild-type human α-synuclein (αSNwt) expression was under control of the mouse Thy1 regulatory sequences as for the A53T mutant (Fig. 1A). Mice of one of these lines (S969) showed a motor phenotype and expressed brain transgene mRNA and protein levels similar to those in seen in Thy1αSNA53T mice of line 9813 (data not shown). Brain and spinal cord contained many neurons with pronounced α-synuclein immunostaining in cell perikarya and dendrites (Fig. 9). Cells and neurites with Lewy-like features were prominent and frequent mainly in those areas of the CNS as reported for the Thy1αSNA53T mice. Two representative examples shown are from the deep mesencephalic nucleus (Fig. 9D) and the spinal cord (Fig.9E,F). Gastrocnemius neuromuscular junctions showed in some regions over 50% denervation, and sprouting was often seen at the remaining endplates like in the A53T mice (data not shown). In conclusion, like its A53T mutant, expression of wild-type human α-synuclein can cause pathogenicity in central neurons.

Immunopathology in Thy1αSNwt.A, Cerebral cortex showing prominent somatic and axodendritic α-synuclein immunostaining on neurons of the deep cortical layers. B, CA1 region of the hippocampus showing prominent α-synuclein immunostaining in principal cell somata and dendrites. C, CA3 sector of the hippocampus. Note some pyramidal cells with prominent cytoplasmic staining next to unstained cells and the immunostaining of the mossy fiber terminals.D, Deep mesencephalic nucleus with a neuron showing a strong α-synuclein immunostaining in soma and proximal dendrites. Note the presence of unstained axons in the vicinity.E, Spinal cord longitudinal section with α-synuclein-immunostained motor neurons and enlarged proximal dendrites (arrow). F, Same area as inE showing ubiquitin immunostaining in the cytoplasm of some neurons. Scale bars: A, B (inB), 20 μm; C, D (inD), 20 μm; E, F (inF), 20 μm. Magnification: A,B, 100×; C, D, 250×;E, F, 320×.
DISCUSSION
The transgenic mice described here cover a remarkable spectrum of the pathological changes that are seen in postmortem brain tissue of DLB, PD, and LBVAD patients. Our results identify the A53T mutant of human α-synuclein and its wild-type counterpart as pathogenic agents that can drive the formation of extranigral pathology in rodent central neurons. The A53T mutation may enhance but is not required for pathogenicity of the human protein. This finding is in agreement with recent reports describing its pathogenicity in transgenic mice (Masliah et al., 2000) and Drosophila(Feany and Bender, 2000). Expression of the human α-synuclein A30P mutant has also revealed changes in neurons that are reminiscent of Lewy-like pathology in both the mouse (Kahle et al., 2000a,b) and the fly (Feany and Bender, 2000). Therefore, all three human α-synuclein proteins can be pathogenic in neurons in vivo. In each case, their expression resulted in aberrant perikaryal and dendritic accumulations of α-synuclein in neurons, suggesting that this could play a central role in the development of the pathology. In our mice, human α-synuclein expression also induced motor neuron degeneration, with profound effects on axonal integrity, and denervation of NMJs in several muscles examined. These observations suggest that α-synuclein (over)expression affects a (perhaps universal) mechanism of synapse maintenance. Whether the three human α-synuclein proteins affect one or different (for example, axonal transport; Jenssen et al., 1999) mechanisms and with similar or different thresholds for pathogenicity remains to be determined. However, because the α-synuclein gene is infrequently mutated in its coding sequence, it will be key to identify and study cellular processes that facilitate α-synuclein protein accumulation, such as after insult (Kowall et al., 2000; Vila et al., 2000), or its degradation, which likely involves the proteasome–ubiquitin pathway (Leroy et al., 1998; Bennett et al., 1999).
An interesting feature of the pathology in our transgenic mice and in human diseases with Lewy pathology (Braak et al., 1996; Forno, 1996;Ince et al., 1998; Takeda et al., 1998; Braak et al., 2000) is a selective and topographically confined vulnerability of neurons to develop Lewy (human) or Lewy-like (mouse) pathology. In the mice, prominently affected areas included the nucleus centralis oralis pontis, the nucleus vestibularis lateralis, the deep cerebellar nuclei, the deep aspects of the tectal plate, and motor nuclei in the spinal cord. Of course, the pattern in the mouse is dictated a priori by the properties of the Thy1 gene-based expression cassette. This cassette yielded widespread neuronal expression of α-synuclein. However, pathology was most pronounced in certain brain regions, suggesting that some neurons are more vulnerable. Perhaps modulators of α-synucleinopathy vary regionally in the brain. The Thy1 cassette generally fails to express in dopaminergic neurons of the substantia nigra pars compacta (S. Bischoff and H. van der Putten, unpublished observations), and we confirmed this to be the case also in the A53T transgenic mice (data not shown). In the human brain, the dopaminergic cells of the substantia nigra pars compacta appear most sensitive to develop Lewy pathology, and the resulting nigral lesions are primarily involved in the clinical symptoms of PD (Forno, 1996;Spillantini et al., 1997; Wakabayashi et al., 1997; Baba et al., 1998;Ince et al., 1998; Irizarry et al., 1998; Mezey et al., 1998;Spillantini et al., 1998; Takeda et al., 1998; Braak et al., 1999). Furthermore, when using transgene cassettes that allow for expression in dopaminergic neurons, these cells seem highly prone to develop Lewy-like pathology in both mouse (Masliah et al., 2000) and fly (Feany and Bender, 2000). However, extranigral Lewy pathology is very common in both PD and DLB brains (Gai et al., 1995; Braak et al., 1996, 2000;Forno, 1996; Gómez-Tortosa et al., 2000a), and it is suspected to contribute to the variable cognitive and neuropsychiatric features in these diseases. By excluding transgene effects in dopaminergic neurons of the substantia nigra pars compacta, Thy1αSN mice seem particularly useful to address and model extranigral aspects of the pathology as it is seen in PD and DLB.
The remarkable spectrum of pathological changes seen in the mice included subsets of central neurons showing α-synuclein-stained perikarya, dendrites, Lewy-like neurites, and a smaller subset of these cells staining for ubiquitin. In contrast, no pathological staining was observed when using antibodies that detect paired helical filament and hyperphosphorylated tau, neurofilaments, or when using stains to detect Aβ plaque-like pathology (Sturchler-Pierrat et al., 1997; data not shown). Ubiquitination was evident in some but not in all aged-matched female and male mice, and this phenomenon was observed in both A53T lines. Because all of the mice were C57BL/6 and no variation in transgene expression was detected between individuals or sexes within a line, factors other than these seem to account for ubiquitination occurring in a stochastic manner.
Like in human PD and DLB brains (Forno, 1996; Ince et al., 1998;Irizarry et al., 1998; Spillantini et al., 1998; Takeda et al., 1998;Braak et al., 1999; Gómez-Tortosa et al., 2000b), ubiquitination in transgenic mouse neurons appears to be a late modification in cells with α-synuclein pathology. It was evident in only a subset of human α-synuclein-immunopositive mouse neurons and neurites (Fig.7G, H) and found mainly in those brain regions that showed the most pronounced α-synuclein immunopathology. A quantification of the number of perikarya and neurites showing ubiquitin versus α-synuclein immunopathology was not attempted because, unlike in human brains with Lewy pathology, the pattern and the intensity of ubiquitin immunolabeling was very heterogeneous. In DLB, percentages of α-synuclein-positive but ubiquitin-negative structures ranged only from 2 to 10% depending on brain region (Gómez-Tortosa et al., 2000b). Morphologically, and compared with classical Lewy bodies, these structures corresponded to less aggregated and less compact α-synuclein inclusions, such as pale bodies (Dale et al., 1992). α-Synuclein compared with ubiquitin antibodies thus showed higher sensitivity for identifying less aggregated and less compacted inclusions but little differences with respect to identifying classical Lewy bodies. In the mice, we have not seen classical Lewy body-like structures. Perikarya and neurites contained less compact and more diffuse α-synuclein-immunopositive material. A significant proportion of α-synuclein-immunopositive perikarya and neurites failed to show or had different degrees of punctate ubiquitin staining. Only occasional cells and neurites showed very intense ubiquitin immunostaining (Fig. 7G,H). Therefore, compared with human brains with Lewy pathology, the majority of α-synuclein-positive inclusions in the mice may represent less compact α-synuclein conglomerates associated with pre-ubiquitination or early stages of ubiquitination. In human brains, ubiquitinated structures represent mainly classical Lewy bodies and Lewy neurites, and punctate ubiquitin immunopathology is less frequent (Gómez-Tortosa et al., 2000b). Perhaps aging and/or species differences account for these observations and also for the prominent, diffuse, ubiquitin staining seen in the cytoplasm of occasional mouse but not human neuronal cell bodies (Fig.6E,F) in which it is usually confined to Lewy bodies (Gai et al., 1995; Forno, 1996; Ince et al., 1998; Irizarry et al., 1998; Spillantini et al., 1998; Takeda et al., 1998; Braak et al., 1999; Gómez-Tortosa et al., 2000b). Morphologically, ubiquitin staining of dystrophic neurites is remarkably similar in both species (Fig. 7D–F,H).
Using transmission and immunoelectron microscopy, human α-synuclein A53T-immunoreactive granular material was detected in cytoplasm, dendrites, and occasionally in axons of A53T mice. Granular material with α-synuclein has been seen in human tissue with Lewy pathology and in partially purified human Lewy bodies (Arima et al., 1998; Baba et al., 1998; Goedert et al., 1998; Wakabayashi et al., 1998). We did not detect the typical α-synuclein decorated Lewy body filaments as seen in material extracted from human DLB cingulate cortex (Spillantini et al., 1998), in tissue sections from human PD and DLB brain (Arima et al., 1998; Baba et al., 1998; Wakabayashi et al., 1998), and in synthetic α-synuclein filaments (Conway et al., 1998; Crowther et al., 1998; El-Agnaf et al., 1998; Giasson et al., 1999; Narhi et al., 1999; Wood et al., 1999). This could relate to insufficient aging of the mice and/or species differences. Others recently reported granular but no fibrillar material in mice expressing wild-type human α-synuclein (Masliah et al., 2000), whereas in Drosophilafibrillar material was seen (Feany and Bender, 2000). These different findings are intriguing because the identity of the pathogenic species in diseases with Lewy pathology and its relationship to the α-synuclein-containing fibril has not been elucidated, and it is not known whether Lewy bodies in human diseases are cytotoxic, harmless side products, or markers of cell damage. Irrespective, and likely more important, are the several other striking similarities seen between the pathology in human and the transgenic mice. All three (wild-type, A30P, and A53T) α-synuclein proteins produce nonfibrillar oligomers (Conway et al., 2000) that perhaps are critical in pathogenesis, whereas fibrils and/or Lewy bodies could represent harmless epiphenomena.
In conclusion, the changes in the transgenic mice correlate with neurodegeneration, confirming that such mouse models provide a useful means to address fundamental aspects of disorders with α-synucleinopathy and novel animal models for testing therapeutic strategies.
Footnotes
We thank Hanny Richener, Claudia Falk, Dr. Edgar Kaeslin, Corina Schneider and Areejittra Soontornmalai, Rita Meyerhofer, and Dr. Reinhard Bergmann for assistance with cloning, DNA microinjections, histology, behavioral analysis, and statistical advice, respectively.
Correspondence should be addressed to Herman van der Putten at the above address. E-mail: p_herman.van_der_putten{at}pharma.novartis.com.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}