Article Text

Abstract
Objective The enteric nervous system (ENS) undergoes neuronal loss and degenerative changes with age. The cause of this neurodegeneration is poorly understood. Muscularis macrophages residing in close proximity to enteric ganglia maintain neuromuscular function via direct crosstalk with enteric neurons and have been implicated in the pathogenesis of GI motility disorders like gastroparesis and postoperative ileus. The aim of this study was to assess whether ageing causes alterations in macrophage phenotype that contributes to age-related degeneration of the ENS.
Design Longitudinal muscle and myenteric plexus from small intestine of young, mid-aged and old mice were dissected and prepared for whole mount immunostaining, flow cytometry, Luminex immunoassays, western blot analysis, enteric neural stem cell (ENSC) isolation or conditioned media. Bone marrow derived macrophages were prepared and polarised to classic (M1) or alternative (M2) activation states. Markers for macrophage phenotype were measured using quantitative RT-PCR.
Results Ageing causes a shift in macrophage polarisation from anti-inflammatory ‘M2’ to proinflammatory ‘M1’ that is associated with a rise in cytokines and immune cells in the ENS. This phenotypic shift is associated with a neural response to inflammatory signals, increase in apoptosis and loss of enteric neurons and ENSCs, and delayed intestinal transit. An age-dependent decrease in expression of the transcription factor FoxO3, a known longevity gene, contributes to the loss of anti-inflammatory behaviour in macrophages of old mice, and FoxO3-deficient mice demonstrate signs of premature ageing of the ENS.
Conclusions A shift by macrophages towards a proinflammatory phenotype with ageing causes inflammation-mediated degeneration of the ENS.
- MACROPHAGES
- AGEING
- ENTERIC NERVOUS SYSTEM
- INFLAMMATION
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Direct crosstalk between muscularis macrophages and enteric neurons controls GI neuromuscular function.
Muscularis macrophages have been implicated in disease pathogenesis of motility disorders including gastroparesis and postoperative ileus.
Ageing is associated with neurodegenerative changes to the enteric nervous system (ENS) and impairment of neuromuscular function, but the mechanism remains unclear.
What are the new findings?
A shift in macrophage polarisation from anti-inflammatory ‘M2’ to proinflammatory ‘M1’ states occurs in muscularis macrophages with ageing and is associated with chronic low-grade inflammation in the ENS microenvironment and delayed transit.
Intrinsic changes in macrophages, particularly age-dependent decrease in expression of the transcription factor FoxO3, account for some of the alterations in macrophage phenotype with ageing.
How might it impact on clinical practice in the foreseeable future?
Our findings indicate that ‘inflammaging’, a chronic low-grade inflammatory state associated with ageing, may contribute to age-dependent degeneration of the ENS and age-associated motility disorders.
This may lead to novel immunotherapies to treat common disorders such as constipation and faecal incontinence.
Introduction
Ageing causes physiological changes in GI function that contribute to many disorders including constipation and faecal incontinence which cause significant physical, emotional and financial burdens.1 ,2 Age-dependent loss and degeneration of the enteric nervous system (ENS), the intrinsic nervous system of the GI tract, likely plays a role in these disorders.1 ,3 There is evidence that oxidative stress, alterations in neurotrophic factors and calcium dysregulation may contribute, but the exact mechanism for these age-related changes remains poorly understood.3
The muscular layer of the gut wall has been found to contain numerous tissue-resident macrophages, termed muscularis macrophages (MMs), residing in close apposition to enteric ganglia.4–6 Through direct crosstalk with enteric neurons, MMs are important for GI neuromuscular function, and their depletion results in the prolongation of colonic transit times.6 Macrophages have been classified as either M1 (classically activated) or M2 (alternatively activated) based on their role in inflammatory processes.7 M1 macrophages produce proinflammatory mediators including cytokines and reactive oxygen and nitrogen species and function in host defence. In contrast, M2 macrophages play a role in dampening inflammation and promoting wound healing.7 In the intestine, macrophage phenotype differs depending on their location, with macrophages in the lamina propria layer preferentially assuming a proinflammatory M1 phenotype while MMs displaying an anti-inflammatory M2 phenotype.4 However, macrophages demonstrate tremendous plasticity and are capable of reversibly switching between M1 and M2 phenotypes.8 Perturbation of the normal balance of M1/M2 macrophages appears to be an important factor in disease pathogenesis including cancer, atherosclerosis, autoimmune disease, osteoporosis and neurodegeneration.9 Alterations in the phenotype of MMs have been implicated in GI motility disorders including gastroparesis and postoperative ileus (POI).10–14 Loss of expression of mannose receptor C type 1 (CD206), an M2 marker, in MMs of non-obese diabetic (NOD) mice has been found to correlate with the development of gastric delay11 suggesting that alterations in M1/M2 phenotype may contribute to GI neuromuscular disorders. Whether ageing causes changes in macrophage phenotype that may contribute to the degeneration of the ENS and age-related GI disorders is unknown. Here we show that ageing causes a shift in macrophage polarisation from an anti-inflammatory ‘M2’ state to a proinflammatory ‘M1’ state. This shift is partly due to decreased expression of the transcription factor FoxO3, a gene linked to longevity, and accompanies inflammation-mediated loss of enteric neurons and enteric neural stem cells (ENSCs) and delayed intestinal transit.
Materials and methods
Please see supplementary material methods for an expanded version of this section.
supplementary data
Animals
Male C57/BL6 mice of different age groups (3 months, 10–12 months and 20–24 months) acquired from the National Institute on Aging aged rodent colony, FoxO3−/− mice in FVB/N background15 and Wnt1-cre;tdTomato mice16 were used in experiments. All animal experiments were approved by Stanford University's institutional animal care and use committees.
Tissue and cell preparation
Mice were anesthetised, euthanised, and laparotomy was performed. Small intestine was removed, lavaged, and longitudinal muscle with the adherent myenteric plexus (LMMP) was dissected as previously described.16 Tissue was either freshly frozen in dry ice, fixed in paraformaldehyde, enzymatically digested with collagenase or placed in media for ex vivo organotypic cultures. Tissue digestion was performed using protocol adapted from Joseph et al17 by sequentially incubating the tissue in digestion buffer (consisting of M199 media (Invitrogen, Carlsbad, California, USA), 0.1% BSA, 1 mM CaCl2, 20 mM HEPES and 150 μM P188) containing 1.1 mg/mL collagenase (Sigma-Aldrich) for 30 min at 37°C, followed by 10 U/mL papain (Worthington Biochemicals, Lakewood, New Jersey, USA) in Hank's buffered salt solution (HBSS, Invitrogen) without calcium and magnesium, then quenched with ice-cold Digestion Buffer containing 50 U/mL DNAse I (Worthington). Tissue was mechanically dissociated by gentle trituration and filtered through a 40 μm nylon mesh cell strainer.
Flow cytometry
Dissociated cells were incubated with Zombie Aqua Viability (Biolegend, San Diego, California, USA) followed by blocking with 5% rat serum and mouse anti-CD16/CD32 (eBioscience, San Diego, California, USA) in HBSS containing 2% BCS. Samples were incubated for 30 min at 4°C with primary antibodies and isotype controls (see online supplementary table S1). After washing, cells were passed through a 40 μm nylon mesh cell strainer, and sorting was performed directly into Trizol (Qiagen, Valencia, California, USA) using a BD FACS Aria (BD Bioscience, San Jose, California, USA). Gating strategy for sorting of macrophages is shown in online supplementary figure S1. Analysis was performed using FlowJo software (Tree Star, Ashland, Oregon, USA).
supplementary tables
supplementary figures
RNA isolation and quantitative real-time PCR
RNA was extracted from cells with miRNeasy kit (Qiagen) and converted to cDNA using High-Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, California, USA). For sorted MMs, cDNA preamplification was performed with Taqman PreAmp Master Mix Kit (Applied Biosystems). Quantitative RT-PCR was performed on ABI StepOnePlus real-time instrument using the Taqman expression assays listed in online supplementary table S2. For analysis, each gene was normalised to the housekeeping gene GAPDH, and fold change in gene expression between groups was calculated using the Pfaffl method.
Statistics
Data are expressed as mean±SE of mean and was analysed using Mann-Whitney, t-test and one-way analysis of variance (ANOVA). Significance was deemed when p value was <0.05. Statistical analysis was performed with GraphPad Prism V.5 (GraphPad Software, La Jolla, California, USA).
Results
MMs demonstrate shift from anti-inflammatory to proinflammatory phenotype with ageing
Consistent with a recent report by Gabanyi et al,4 which showed that macrophages in the muscularis preferentially express M2 markers compared with those in the lamina propria, we found the majority of MMs (in both young and old mice) to express the M2 marker CD206 by immunostaining (see online supplementary figure S1A). Within the muscle layers of the intestine, MMs were found to reside primarily in the myenteric plexus layer, surrounding enteric neurons (see online supplementary figure S1B). We performed flow cytometry on dissociated cells from LMMP of mice from three different age groups, young (3 months old), mid-aged (10–12 months old) and old (20–24 months old). Macrophages, defined as CD45+F4/80+ cells, were the predominant immune cells in the muscularis layer, comprising nearly 20% of total live cells and representing between 60% and 80% of all leucocytes or CD45+ cells (figure 1A and see online supplementary figure S1C). The numbers of MMs (as a per cent of live cells) did not differ significantly with age (figure 1A). Next, we sorted MMs (see online supplementary figure S1D for gating scheme) and performed mRNA expression analysis of a variety of anti-inflammatory ‘M2’ markers including CD206, found in inflammatory zone 1 (FIZZ1) and transforming growth factor β (TGFβ), and proinflammatory ‘M1’ markers including inducible nitric oxide synthetase (iNOS), interleukin-6 (IL-6) and tumour necrosis factor α (TNFα). MMs from mid-aged and old mice revealed reduced expression of M2 markers, CD206, FIZZ1 and TGFβ compared with young mice (figure 1B). Additionally, MMs expressed higher proinflammatory M1 markers with age, although only IL-6 from old mice reached statistical significance (figure 1C). Taken together, this suggests that ageing is accompanied by a shift in macrophages from an anti-inflammatory to proinflammatory phenotype.
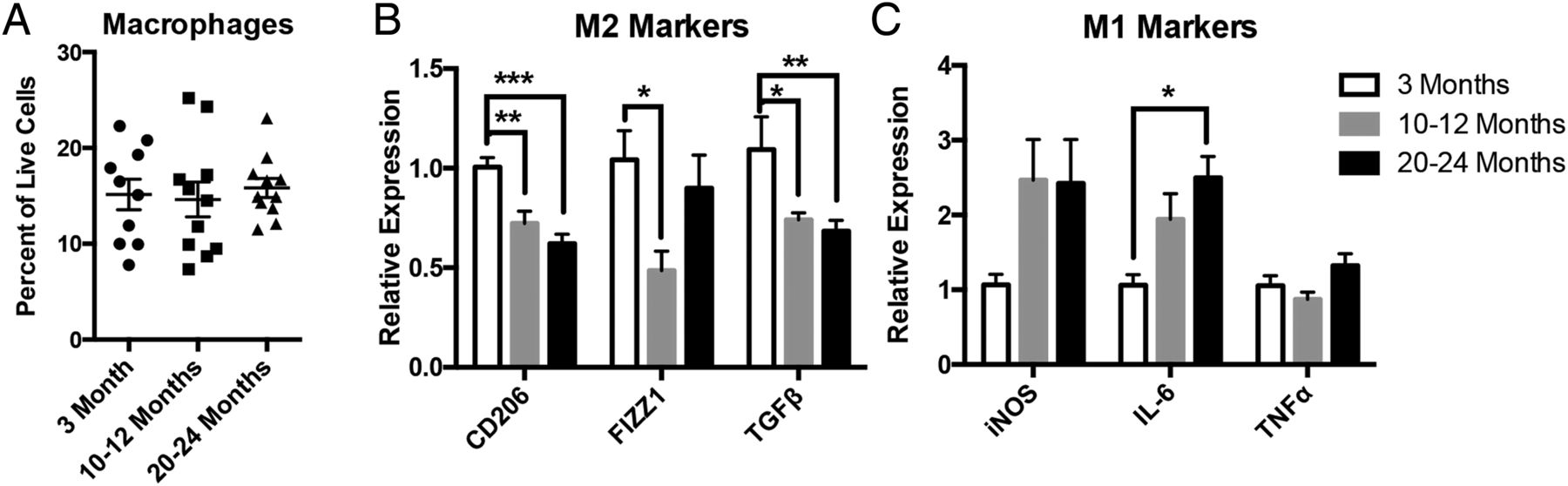
Muscularis macrophages (MMs) demonstrate a shift from anti-inflammatory to proinflammatory state with ageing. On flow cytometry, MMs (CD45+F4/80+ cells) were found to be the predominant immune cell in the muscularis layer, comprising between 15% and 20% of all live cells, but their numbers did not differ significantly with age (A). MMs sorted from old-aged and mid-aged mice revealed a decline in the expression of M2 markers CD206, found in inflammatory zone 1 (FIZZ1) and transforming growth factor β (TGFβ) (B) and a rise in expression of M1 markers, particularly interleukin-6 (IL-6), which reached statistical significance in old (C) compared with young mice by qRT-PCR. n≥10 for all groups, *p<0.05, ***p<0.001 by one-way analysis of variance, with Bonferroni's multiple comparisons test. iNOS, inducible nitric oxide synthetase; TNFα, tumour necrosis factor α.
Increased inflammation in ENS microenvironment with age
To determine whether this age-dependent shift in macrophage phenotype affects the inflammatory milieu of the ENS microenvironment, we performed Luminex immunoassays on protein extract of LMMP from young and old mice. Expression of proinflammatory cytokines and chemokines was increased overall in the ENS microenvironment from old mice with IL-6, IL-1β and IL-18 reaching statistical significance (figure 2A). Elevation of IL-6 was confirmed in a second cohort of young and old mice using western blot analysis (figure 2B). To evaluate whether alterations in immune cells accompanied these changes, we performed immunophenotyping of LMMP using flow cytometry. A statistically significant increase in leucocytes (CD45+ cells) was found in the ENS microenvironment of mid-aged and old compared with young mice (figure 2C). Although no difference was seen in the numbers of macrophages (figure 1A) and dendritic cells (CD45+CD11c+F4/80−) as a per cent of live cells, a statistically significant rise in lymphocytes (CD45+CD3+CD19+) was found in old compared with young mice (figure 2D). A decline in macrophages and rise in lymphocytes as a per cent of CD45+ cells was also observed in the old age group (see online supplementary figure S1C). In summary, we demonstrate that the age-associated shift in macrophages towards a M1 phenotype is associated with a rise in proinflammatory cytokines and chemokines and increased infiltration of lymphocytes in the ENS.

Increased inflammation in enteric nervous system microenvironment with age. Heat map of cytokine and chemokine expression of longitudinal muscle and myenteric plexus (LMMP) lysate from young and old mice from Luminex analysis revealed higher proinflammatory cytokines and chemokines with age (A). Elevation of interleukin-6 (IL-6) was seen on a second cohort of young and old mice by western blot, which was statistically significant based on pixel intensity normalised to GAPDH (B). Flow cytometry performed on enzymatically dissociated LMMP with gating of live, single cells revealed increased numbers of CD45+ leucocytes (as per cent of live cells) in old mice compared with young (C). There was also a statistically significant rise in lymphocytes (CD3+CD19+) in old compared with young mice, but no difference in dendritic cells (CD11c+ F4/80−) as a per cent of live cells (D). n≥5 for each group; *p<0.05, **p<0.01 by Mann-Whitney for (A); **p<0.01 by t-test for (B); *p<0.05, **p<0.01 by one-way analysis of variance, with Bonferroni's multiple comparisons test for (C) and (D). TNFα, tumour necrosis factor α.
Age-associated neural response to inflammatory signals is associated with increased apoptosis, loss of enteric neurons and delayed intestinal transit
In order to evaluate whether this age-dependent inflammation causes changes to the ENS, we evaluated whether signal transducer and activator of transcription 3 (STAT3), a downstream mediator of IL-6, IL-18 and other cytokines, was preferentially activated in enteric neurons of old mice. We found a twofold increase in the per cent of neurons expressing phosphorylated STAT3 (pSTAT3) in old compared with young mice, indicating increased STAT3 activity (figure 3A). An increased ratio of pSTAT3/STAT3 was also observed in old mice by western blot analysis of LMMP protein extract (see online supplementary figure S2A). We found a statistically significant increase in the number of enteric neurons expressing cleaved caspase-3 (Asp175) (figure 3B and see online supplementary table S3). This perinuclear staining pattern of cleaved caspase-3 is similar to that observed in apoptotic neurons in the central nervous system18 and was observed in enteric neurons that were also TUNEL+ (see online supplementary figure S2B). A reduction in the density of neurons within myenteric ganglia (figure 3C) was observed in old compared with young mice, suggesting neuronal loss with ageing. This age-dependent reduction in neuronal density is similar to previous reports in human, rat and guinea pigs.3 These neurodegenerative changes were associated with prolonged total intestinal transit time in old mice compared with young (figure 3D). These findings suggest that age-dependent changes in macrophage phenotype are associated with neural response to inflammatory signals and loss of enteric neurons.

Age-dependent changes to myenteric plexus are associated with delayed intestinal transit. Immunostaining of whole mount preparations of longitudinal muscle and myenteric plexus labelled with pan-neuronal marker HuC/D (red) and phosphorylated signal transducer and activator of transcription 3 (pSTAT3) (green) revealed increased numbers of Hu+ neurons expressing pSTAT3 (arrows) in old compared with young mice (A). This difference expressed as a per cent of neurons (pSTAT3+Hu+/total Hu+) was statistically significant. Increased STAT3 activity corresponded to an increase in the number of Hu+ neurons (red) coexpressing the apoptosis marker cleaved caspase-3 (Asp175, green, arrows) in old mice compared with young (B). As a per cent of neurons (Asp175+Hu+/total Hu+), this difference was statistically significant. Old mice also demonstrated a statistically significant reduction in neuronal density (Hu+ cells/ganglia area (100 μm2)) compared with young (C). Whole intestinal transit studies performed on a separate cohort of young and old mice revealed prolonged transit times in old mice (D). **p<0.01, ***p<0.001 by t-test. Scale bars, 50 μm.
Increased proinflammatory cytokines cause reduction in ENSCs with ageing
The ENS contains a resident population of multipotent stem cells termed ENSCs capable of regeneration following injury.16 ,19 ,20 In culture, these stem cells, which can be identified by a variety of markers including p75, CD49b and nestin, can differentiate into neurons, glia and myofibroblasts.17 We have previously shown that, similar to in vitro cultures, an ex vivo organotypic culturing system can be used to assess proliferation and differentiation of ENSCs. This ex vivo system, however, has the advantage of allowing their visualisation within intact myenteric ganglia.16 We cultured LMMP sections from young and old mice with the thymidine analogue EdU for 40 hours to assess proliferation. We found a nearly threefold reduction in the number of EdU+ cells per myenteric ganglia area in old compared with young mice (figure 4A), suggesting a decline in the number of ENSCs with age. These results are consistent with a prior report showing fewer cells expressing the ENSC marker p75 and decreased multipotent neurospheres isolated from guts of old mice compared with young.21 Next, we evaluated whether factors in the aged ENS microenvironment directly impact survival of ENSCs. Using a Wnt1-cre;tdTomato mouse line in which neural crest-derived cells are fluorescently labelled,22 we enriched for ENSCs using CD49b17 magnetic beads and cultured them in conditioned media prepared with LMMP from young (young CM) or old mice (old CM). Old CM caused a twofold increase in TUNEL+ ENSCs compared with young CM (figure 4B,C). Luminex immunoassays revealed that old CM contained twofold greater levels of IL-6 compared with young CM (see online supplementary figure S3). Since IL-6 has been implicated in impaired survival of neural progenitor cells in the central nervous system,23 ,24 we explored whether this cytokine in particular was responsible for decreased survival of ENSCs. The addition of IL-6 neutralising antibody (and not IgG control) reversed the proapoptotic effect of old CM (figure 4B, C). Additionally, we found that recombinant murine IL-6 caused increased TUNEL positivity when cultured with ENSCs but required over a 20-fold higher concentration (100 ng/mL) than that found in old CM (figure 4D and see online supplementary figure S3). Given that cells which do not express IL-6 receptor can still signal through IL-6 via soluble IL-6 receptor, termed trans-signalling,25 we wanted to evaluate whether this may explain the discrepancy. We found that adding soluble recombinant IL-6 receptor α (sIL-6Rα) caused increased TUNEL positivity at a lower concentration of IL-6 (50 ng/mL) (figure 4D), suggesting that IL-6 exerts its proapoptotic effect on ENSCs through trans-signalling.
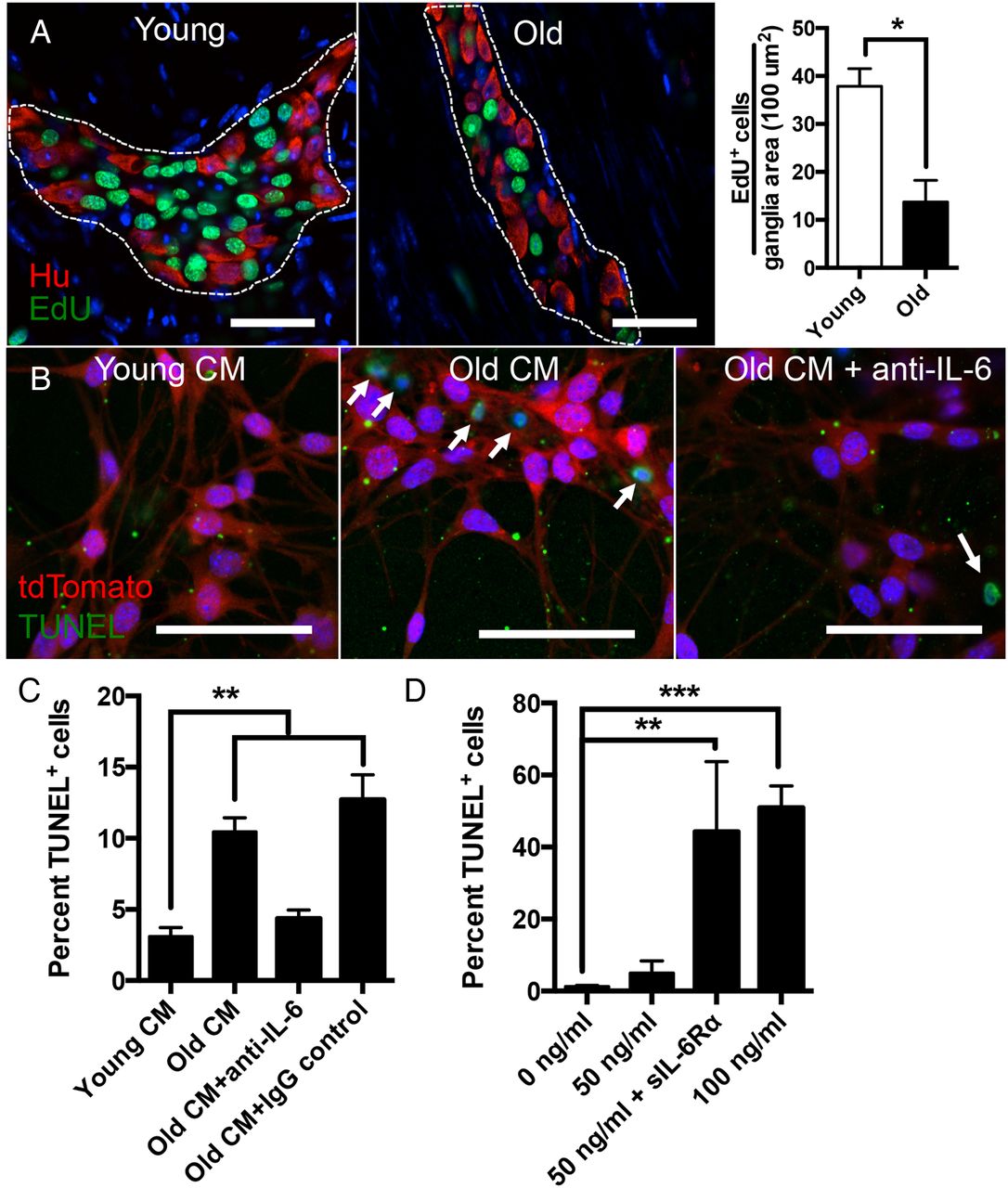
Effect of ageing and proinflammatory cytokines on enteric neural stem cells (ENSCs). Using an ex vivo organotypic culturing system to measure proliferation, a nearly threefold reduction of EdU+ cells (green) were found per ganglia area in old compared with young mice (A). Using CD49b magnetic beads, ENSCs were isolated from a Wnt1-cre;tdTomato mouse (fluorescently labelled neural crest cells) and cultured with conditioned media prepared with longitudinal muscle and myenteric plexus from young (young CM) or old (old CM) mice. Old CM was also cultured with either interleukin-6 (IL-6) neutralising antibody or an IgG control. TUNEL assay revealed over twofold increase in numbers of TUNEL+ cells (green) when ENSCs (red) were cultured with old CM compared with young CM indicating increased apoptosis due to factors in the aged enteric nervous system microenvironment. This increase in TUNEL positivity was reversed with the addition of IL-6 neutralising antibody but not IgG control (B and C). When cultured with recombinant IL-6 at increasing concentrations, ENSCs demonstrated a statistically significant increase in TUNEL+ cells (D) at the highest concentration (100 ng/mL). The addition of soluble IL-6 Rα (200 ng/mL) augmented the number of TUNEL+ cells when added to IL-6 at a lower concentration (50 ng/mL). *p<0.05 by t-test for (A); *p<0.05, **p<0.005 by one-way analysis of variance, with Bonferroni's multiple comparisons test for (C) and (D). Scale bars, 50 μm.
Age-dependent loss of anti-inflammatory phenotype is in part intrinsic to macrophages
To assess whether the shift in phenotype is intrinsic to macrophages or related to extrinsic influences in aged microenvironment, we generated congenic bone marrow (BM) chimaeras by transplanting BM from young or old mice into lethally irradiated young or old mice (figure 5A). This technique has been shown to be an effective way to deplete host MMs and replace them with macrophages derived from BM graft.13 Similar to old mice, sorted MMs from O→Y revealed reduction in expression of M2 markers CD206 (p<0.05), FIZZ1 and TGFβ (p=0.06) compared with Y→Y mice (figure 5B), suggesting that cell intrinsic factors contribute to the age-dependent loss of suppressive phenotype. While not statistically significant, we also found a trend towards increased leucocytes in O→Y compared with Y→Y (see online supplementary figure S4A). MMs from chimaeras transplanted with old BM (O→O and O→Y) showed a trend towards increased iNOS expression, suggesting a more proinflammatory phenotype (figure 5C). We also found a modest but statistically significant preservation of neuronal density in enteric ganglia from Y→O chimaeras compared with O→O (see online supplementary figure S4B), supporting the possibility that young BM confers some protection against age-related neuronal loss.
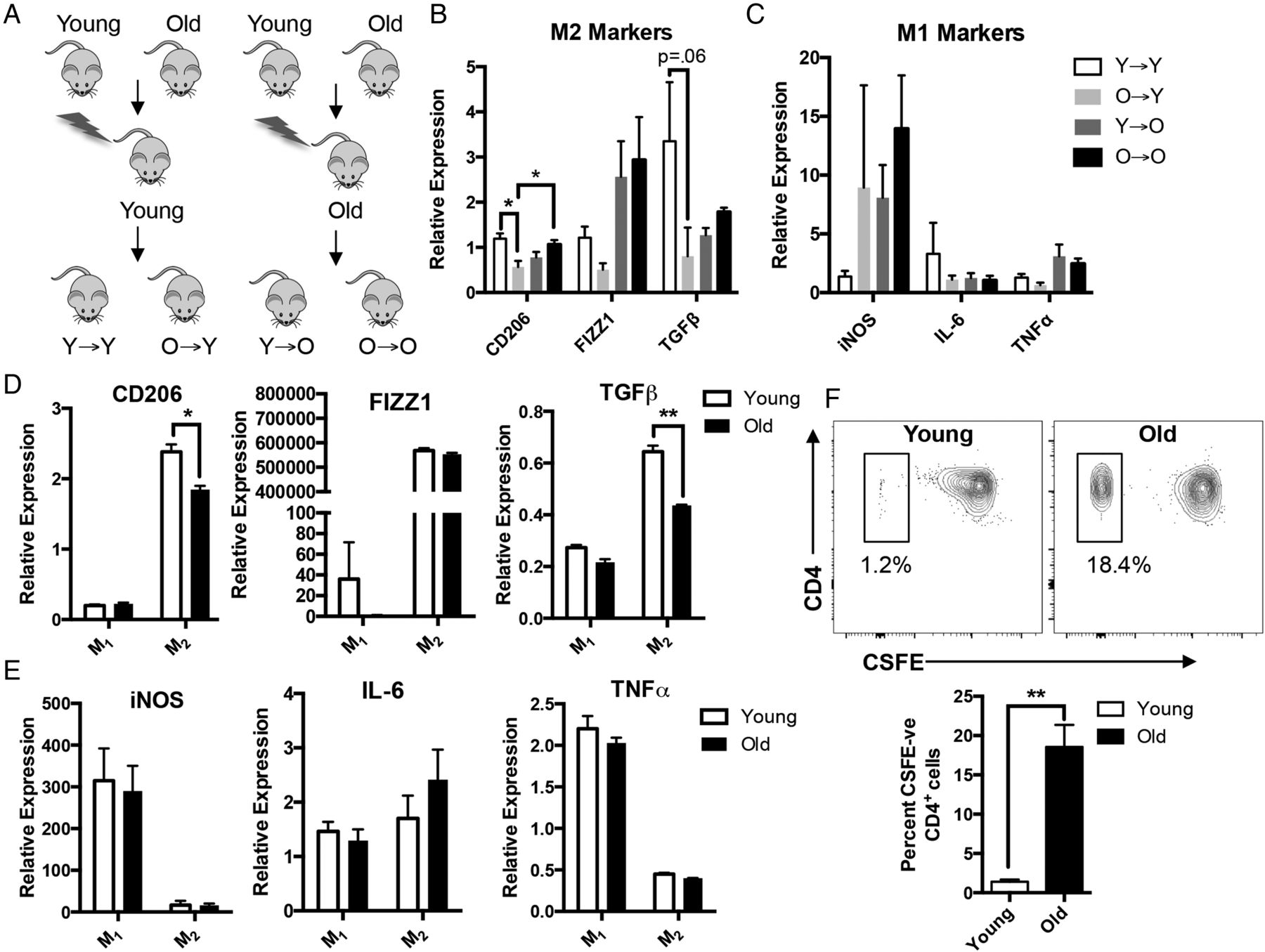
Macrophages from old mice have intrinsic differences that make them less anti-inflammatory. Congeneic bone marrow (BM) chimaeras were generated by injecting BM harvested from young or old C57BL/6 mice into lethally irradiated young or old mice (A). Muscularis macrophages sorted from O→Y expressed decreased M2 markers compared with Y→Y, suggesting that the age-dependent loss of anti-inflammatory phenotype is intrinsic to the macrophage (B). Although not statistically significant, mice transplanted with old BM (O→Y and O→O) had a trend towards increased expression of inducible nitric oxide synthetase (iNOS) (C). BM-derived macrophages (BMDMs) prepared from old mice and treated with interleukin (IL)-4 and IL-13 to polarise to M2 activation state (M2) exhibited reduced expression of M2 markers, particularly CD206 and transforming growth factor β (TGFβ) compared with young (D). Interferon γ (IFNγ)-treated BMDMs (M1) from young and old mice did not demonstrate significant differences in the expression of proinflammatory M1 markers (E). When incubated with CD4+ T cells activated with anti-CD3 and anti-CD28 and labelled with CSFE, IL-4/IL-13-treated BMDMs from old mice demonstrated reduced suppression of lymphocyte proliferation (higher per cent of CSFE-negative CD4+ cells) compared with those from young (F). For (D) and (E), RNA expression was normalised to untreated (M0) macrophages from young mice. *p<0.05 by one-way analysis of variance, with Bonferroni's multiple comparisons test and t-test; **p<0.01 by t-test. n≥5 for chimaeric mouse groups. FIZZ1, found in inflammatory zone 1; TNFα, tumour necrosis factor α.
To help confirm that ageing causes intrinsic changes to macrophages, we prepared BM-derived macrophages (BMDMs) from young and old mice. BMDMs were treated with either interferon γ (IFNγ) or IL-4 and IL-13 to induce M1 or M2 polarisation states, respectively. When treated with IL-4 and IL-13, cells of old mice showed decreased anti-inflammatory M2 markers CD206 and TGFβ compared with young (figure 5D) by RT-PCR. There was no difference in expression of proinflammatory markers iNOS, IL-6 and TNFα in IFNγ-treated BMDMs from young and old mice (figure 5E). To assess whether this difference might be due to an age-related alteration in the ability to polarise, we performed flow cytometry on the BMDMs. The majority of CD45+ cells (>96%) from both young and old expressed the macrophage marker F4/80. Furthermore, we found similar expression of CD206 in BMDMs treated with either IL-4/IL-13 (see online supplementary figure S5A) or IFNγ (see online supplementary figure S5B) from young and old, suggesting the differences we observed in mRNA gene expression were not due to altered polarisation. To determine whether reduction in ‘M2’ markers corresponds to diminished anti-inflammatory properties, we performed a lymphocyte suppression assay. CD4+ T cells isolated from spleen of a young mouse were labelled with CSFE, activated with anti-CD3 and anti-CD28, then co-cultured with IL-4/IL-13-treated BMDMs from young or old mice for 72 hours. Co-culturing with BMDMs from young mice yielded dramatically fewer proliferated (CSFE-negative) lymphocytes (1.2% vs 18.4% of CD4+ cells) compared with old (figure 5F), indicating diminished suppressive behaviour in macrophages from old mice. Taken together, these findings suggest that intrinsic changes in macrophages contribute to their loss of anti-inflammatory properties.
Reduction in FoxO3 expression is associated with decreased anti-inflammatory phenotype in macrophages
FoxO3, a member of the family of Forkhead transcription factors, is one of a few genes in which genetic variants have been consistently linked to longevity in humans.26 In addition to regulating metabolism, apoptosis, autophagy and stem cell homeostasis, FoxO3 plays a role in modulating the immune response.26 FOXO3 genetic variants have been associated with more aggressive disease courses for patients with Crohn's disease and rheumatoid arthritis, and FoxO3-deficient mice demonstrate an exaggerated inflammatory response to experimental colitis.27 Since FoxO3 appears to regulate the immune response through myeloid cells,27 ,28 we evaluated whether alterations in its expression may account for age-related changes to MMs. We found a 20% reduction in FoxO3 expression in LMMP from old compared with young mice (figure 6A), which is similar to the reduction seen in skeletal muscle of old rats.29 This age-dependent reduction was also seen in sorted MMs (figure 6B) and BMDMs by both qRT-PCR (figure 6C) and western blot analysis (see online supplementary figure S5C). To determine whether this reduction in FoxO3 expression may account for changes in macrophage phenotype, we generated BMDMs from FoxO3−/− and wild type (WT) mice. As seen in old mice, BMDMs from FoxO3−/− mice treated with IL-4 and IL-13 demonstrated reduced expression of CD206 and TGFβ compared with WT (figure 6D). IFNγ-treated BMDMs from FoxO3−/− mice demonstrated decreased expression of proinflammatory markers (figure 6E). Despite this decline in M1 markers, we found that administration of LPS to similar IFNγ-treated BMDMs caused increased elevation of proinflammatory markers relative to WT, suggesting a more proinflammatory phenotype (see online supplementary figure S5D). These results suggest that an age-dependent reduction of FoxO3 accounts for the loss of suppressive phenotype of macrophages with ageing.

Reduction of FoxO3 expression with ageing contributes to decrease in anti-inflammatory phenotype. Western blot revealed a modest but statistically significant reduction of FoxO3 expression in protein extract of longitudinal muscle and myenteric plexus from old compared with young mice based on pixel intensity normalised to GAPDH (A). A decline in FoxO3 expression was also found in sorted muscularis macrophages (B) and interleukin (IL)-4/IL-13-treated bone marrow derived macrophages (BMDMs) (C) from old mice. To evaluate whether decreased FoxO3 expression might be responsible for alterations in macrophage phenotype, BMDMs were derived from FoxO3−/− and WT mice and treated with either interferon γ (IFNγ) (M1) or IL-4 and IL-13 (M2). M2 BMDMs from FoxO3−/− mice demonstrated reduced expression of M2 markers, particularly CD206, compared with WT (D). Decreased expression of proinflammatory markers inducible nitric oxide synthetase (iNOS) and tumour necrosis factor α (TNFα) was observed in M1 BMDMs from FoxO3−/− mice (E), although higher expression of M1 markers was seen following LPS stimulation (see online supplementary figure S5D). For (C–E), RNA expression was normalised to untreated (M0) macrophages from WT mice. *p<0.05, **p<0.01 by t-test. TGFβ, transforming growth factor β.
Foxo3 deficiency mirrors age-associated neural activation, apoptosis and enteric neuronal loss
Similar to C57/BL6 mice, we found an age-dependent increase in the numbers of enteric neurons immunoreactive for pSTAT3 in the FVB mouse strain (p<0.01 by two-way ANOVA). FoxO3 deficiency caused a statistically significant increase in the per cent of pSTAT3+ neurons compared with WT in the mid-age and old age groups (figure 7A). We also found a statistically significant increase in the numbers of enteric neurons expressing the apoptosis marker Asp 175 in FoxO3−/− mice compared with WT in all age groups (figure 7B and see online supplementary table S3). These findings corresponded to a decrease in neuronal density in FoxO3−/− mice compared with WT in young and mid-aged groups but not old (figure 7C). On a separate cohort of male mice between 14 and 17 months of age, FoxO3−/− mice demonstrated prolonged total intestinal transit times compared with age-matched controls (figure 7D). In summary, these findings suggest that FoxO3 deficiency mimics a premature ageing phenotype in the ENS.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Degenerative changes to myenteric plexus and delayed intestinal transit in FoxO3−/− mice. Immunostaining of longitudinal muscle and myenteric plexus from FoxO3−/− and WT FVB mice from three age groups (3 months, 12 months and 20 months) with pan-neuronal marker HuC/D (red) and phosphorylated signal transducer and activator of transcription 3 (pSTAT3) (green) revealed increased numbers of Hu+ neurons expressing pSTAT3 (arrows) in FoxO3−/− compared with WT mice (A). Expressed as per cent of neurons (pSTAT3+Hu+/total Hu+), this difference was statistically significant for the mid-age and old age groups. An increased number of Hu+ neurons (red) coexpressing cleaved caspase-3 (Asp175, green, arrow) was also found in FoxO3−/− compared with WT mice and was statistically significant in all age groups (B). A statistically significant decline in neuronal density (Hu+ cells/ganglia area (100 μm2)) in FoxO3−/− compared with WT mice was observed in the age groups of 3 and 12 months, but not 20 months (C). Prolonged intestinal transit times were found in FoxO3−/− mice compared with WT (performed on a separate cohort of male mice between the age of 14 and 17 months). Representative images are from 12-month age group. *p<0.05, **p<0.01 and ***p<0.001 by multiple t-test. Scale bars, 50 μm.
Discussion
A growing body of evidence suggests that MMs play an important role in maintenance of GI neuromuscular function and that perturbations in their phenotype may underlie motility disorders.4 ,6 ,10–14 Here, we show that MMs are the predominant immune cells within the muscularis layer of intestine, comprising between 60% and 80% of all leucocytes. Although ageing does not affect their abundance, it causes a shift in macrophage polarisation from anti-inflammatory M2 to proinflammatory M1 (figure 1). This age-dependent shift in macrophage phenotype parallels inflammatory changes in the ENS microenvironment including elevated proinflammatory cytokines and infiltration of immune cells, particularly lymphocytes (figure 2), and functional delay in intestinal transit (figure 3D). Our findings mirror those seen following acute injury using animal models for POI where macrophages have been shown to be responsible for driving inflammation and disrupting GI neuromuscular function.13 ,14 Notably, chronic low-grade inflammation, termed ‘inflammaging’, has been observed in other tissues and organs with ageing,30 and macrophages have been speculated to play a critical role.31 In addition to macrophages, other cells may also contribute to the observed inflammation. We noted an increase in the numbers of lymphocytes with age (figure 2D). While this may be secondary to proinflammatory factors released by macrophages, intrinsic age-dependent alterations in lymphocyte populations, particularly regulatory and effector T cells, may contribute. Additionally, non-immune cells, particularly enteric glia and senescent cells, may have a role in the inflammation. Enteric glia have been implicated in the production of IL-6 and MCP-1 following injury.32 Senescent cells, which have been detected in myenteric ganglia of old mice,33 can secrete proinflammatory cytokines and chemokines termed senescence-associated secretory phenotype.34 Further experiments aimed at elucidating the contributions of various cell types on age-dependent inflammation are clearly needed.
Our analysis was restricted to the muscularis layer of the small intestine, but we believe our findings are generalisable to other regions of the gut including the colon and submucosal layer. While local embryonic precursor cells of yolk sac origin maintain some macrophage populations, mucosal macrophages from intestine have been shown to derive from circulating monocytes from BM.35 Given that we observed a loss of suppressive behaviour in BMDMs from old mice (figure 5E), it is likely that macrophages from other regions of the gut derived from circulating monocytes will have a similar phenotype. Nonetheless, future experiments definitively showing that MMs from adult mice are derived from circulating monocytes are clearly warranted.
While the pathogenesis of GI neuromuscular disorders including gastroparesis, functional dyspepsia and irritable bowel syndrome remains poorly understood, inflammation appears to play a role.36–38 In animal models of colitis, inflammation has been shown to cause a loss of myenteric neurons.39 ,40 Our findings support a role for inflammation in the pathogenesis of age-related GI disorders such as constipation. We demonstrate that inflammation in the ENS microenvironment of old mice correlates with a neural response (increased STAT3 activity in myenteric neurons), neuronal loss and delayed intestinal transit (figure 3). These findings suggest that immunotherapies may have a role in the treatment of age-related GI disorders. Future studies that replicate our findings in humans and manipulate the immune response to dampen age-dependent inflammation are likely to determine the potential of such therapies.
A decline in ENSCs has previously been noted in old mice,21 and our finding of reduced EdU uptake in ganglia of old mice seems to support this (figure 4A). We show that factors released from the aged ENS microenvironment, particularly IL-6, cause increased apoptosis of ENSCs (figure 4B–D). While ENSCs remain quiescent in adult mice at steady state,16 ,17 ,19 they appear capable of neurogenesis after injury.19 Thus, it suggests that elderly individuals may have more lasting damage to their ENS following acute insults such as gastroenteritis or toxin ingestion than young.
Our findings suggest that age-intrinsic differences in macrophages play a role in the observed shift in macrophage phenotype. Despite an inherent limitation in our chimaera experiment, the use of congenic mice which prevented us from determining the efficiency of tissue engraftment, we observed a reduction in M2 markers in O→Y compared with Y→Y (figure 5B), suggesting both successful engraftment and intrinsic differences in macrophage phenotype. Similar age-dependent reduction of M2 markers (FIZZ1, Arg1 and SOC1) has been observed in IL-4-treated macrophages derived from human peripheral blood monocytes and mouse adherent splenocytes,41 ,42 arguing that this difference is common to humans and other populations of macrophages. It is notable that despite the loss of anti-inflammatory phenotype, MMs from O→Y did not demonstrate a significant rise in proinflammatory M1 markers compared with Y→Y (figure 5C). Additionally, M1-polarised BMDMs from old mice did not demonstrate increased expression of proinflammatory markers compared with young either (figure 5F). This suggests that while macrophages from old mice may have less suppressive properties, extrinsic factors in the aged microenvironment are likely responsible for their assuming a proinflammatory phenotype. A recent study found a preference for M2 phenotype in mice deficient of IL-18 receptor43; thus, it is possible that the elevation of IL-18 we observed in the aged microenvironment by Luminex (figure 2A) may be one such extrinsic factor that promotes a proinflammatory phenotype.
Our findings implicate a role for FoxO3 in the intrinsic differences observed in macrophages with age. Although one explanation for the lower M1 and M2 markers seen in IFNγ and IL-4/IL-13-treated BMDMs from FoxO3−/− mice (figure 6D, E) is a diminished ability to polarise, we do not believe this to be the case. We found substantial differences in particular M1 and M2 markers (over fivefold elevation of iNOS for IFNγ-treated FoxO3−/− BMDMs and >100 000-fold elevation of FIZZ1 for IL-4/IL-13-treated FoxO3−/− BMDMs; data not shown) compared with untreated ‘M0’ BMDMs from both WT and FoxO3−/−, suggesting no lack in the ability to polarise. Therefore, our findings suggest that FoxO3 is necessary for maintaining suppressive and homeostatic properties of alternatively activated macrophages (figure 6D). A similar immune modulatory role for FoxO3 has been observed in human monocytes. A similar immune modulatory role for FoxO3 has been observed in human monocytes. A genetic variant of FOXO3 (rs12212067) that is associated with more aggressive Crohn's disease and rheumatoid arthritis was found to result in decreased expression of FoxO3 and increased production of proinflammatory cytokines in human monocytes following stimulation with LPS.27 While this genetic variant has not been linked to longevity, others found in close proximity have been.26 While the mechanism by which these FOXO3 genetic variants extend life is unclear, our results suggest that these polymorphisms may maintain higher expression of FoxO3 in macrophages with age thus dampening inflammation.
FoxO3 is expressed in a variety of tissues and cells and regulates diverse biological processes including metabolism, autophagy, apoptosis and stem cell homeostasis.26 Conditional deletion of FoxO3 in neural stem cells causes their premature depletion within the central nervous system with ageing.15 ,44 The importance of FoxO3 is highlighted by the resultant delay in intestinal transit (figure 7D). We believe this is due to the role of FoxO3 in immune modulation based on the increased STAT3 activity and neuronal loss observed in the ENS of FoxO3−/− mice (figure 7A–C); however, we cannot exclude the possibility that the effect of FoxO3 deficiency on non-immune cells such as neurons and glia may be involved. Thus, further studies including BM chimaeras and cell-type specific knockouts will be necessary to further elucidate the relative importance of FoxO3 deficiency in different cell types on the ENS. It is unclear why we did not observe a difference in neuronal density in FoxO3−/− and WT mice from the old age group, but a possible explanation is selection bias, given that only 26% of FoxO3−/− mice survived to 20 months of age (data not shown).
Based on the findings of our study, we propose a model for age-dependent motility disorders (see online supplementary figure S6). An age-related decline in FoxO3 expression in MMs causes loss of their suppressive properties and results in a low-grade chronic inflammatory state. With time, this proinflammatory state results in tissue damage, a loss of regenerative capacity and ultimately a decline in neuromuscular function.
Acknowledgments
The authors thank Jing Xue, Marisol Chang, Vishal Sharma and Sidhartha Sinha for technical assistance. They thank Anne Brunet for providing resources to conduct experiments. They thank Vanda Lennon (Mayo Clinic, Rochester) for providing the HuC/D antibody.
References
Footnotes
Contributors LB: designing research studies, conducting experiments, acquiring data, analysing data and writing manuscript; LN and JG: conducting experiments; SK and PJP: experimental design; AH: designing research studies, analysing data, overall guidance and writing manuscript.
Funding Supported by NIH grants AG045098 (LB), DK103966 (LB) and AG049622 (AH).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.