


Abstract
M-channel inhibitors, especially XE991, are being used increasingly in animal experiments; however, insufficient characterization of XE991 at times confounds the interpretation of results when using this compound. Here, we demonstrate that XE991 and linopirdine are state-dependent inhibitors that favor the activated-subunit of neuronal Kv7/KCNQ channels. We performed patch-clamp experiments on homomeric Kv7.2 or heteromeric Kv7.2/3 channels expressed in Chinese hamster ovary cells to characterize XE991 and linopirdine. Neither inhibitor was efficacious around the resting membrane potential of cells in physiologic conditions. Inhibition of Kv7.2 and Kv7.2/3 channels by XE991 was closely related with channel activation. When the voltage dependence of activation was left-shifted by retigabine or right-shifted by the mutation, Kv7.2(R214D), the shift in half-activation voltage proportionally coincided with the shift in the half-effective potential for XE991 inhibition. Inhibition kinetics during XE991 wash-in was facilitated at depolarized potentials. Ten-minute washout of XE991 resulted in ∼30% current recovery, most of which was attributed to surface transport of Kv7.2 channels. Linopirdine also exhibited similar inhibition characteristics, with the exception of near- complete current recovery after washout at depolarized potentials. Inhibition kinetics of both XE991 and linopirdine was not as sensitive to changes in voltage as would be predicted by open- channel inhibition. Instead, they were well explained by binding to a single activated subunit. The characteristics of XE991 and linopirdine should be taken into account when these M-channel inhibitors are used in experiments.
Introduction
Subclasses of Kv7/KCNQ subunits form tetrameric channels that underlie the M-current, a low-threshold noninactivating voltage-gated potassium current, which regulates neuronal excitability (Jentsch, 2000; Delmas and Brown, 2005; Greene and Hoshi, 2017). Activation of Gq-coupled receptors, such as muscarinic acetylcholine receptors (m1 and m3), suppresses the M-current and induces transient hyperexcitability in a wide range of neurons (Delmas and Brown, 2005). Accordingly, M-channel inhibitors were developed in an effort to ameliorate defective neuronal activity, such as in Alzheimer dementia. Linopirdine is one such prototypical compound that was found to have a cognitive enhancing effect in an animal model (Fontana et al., 1994); however, linopirdine did not pass phase 3 clinical trials (Rockwood et al., 1997). XE991 was developed as an improved compound with a similar chemical structure (Zaczek et al., 1998). Although no clinical trials have been conducted for this compound, XE991 has been used increasingly in cell culture and animal experiments to investigate physiologic (Vetter et al., 2013; Young and Thomas, 2014; Martinello et al., 2015) and pathologic roles (Mani et al., 2013; Kay et al., 2015) of the M-current.
It has been shown that KCNQ2 gene knockout is lethal (Watanabe et al., 2000; Soh et al., 2014); however, administration of linopirdine is well tolerated in humans (Pieniaszek et al., 1995; Rockwood et al., 1997), as well as in animals for linopirdine and XE991 (Fontana et al., 1994; Zaczek et al., 1998). In addition, even though XE991 is a highly potent inhibitor, it sometimes requires prolonged incubation to inhibit (Yue and Yaari, 2004), or it may have no observed effect (Romero et al., 2004). These lines of evidence suggest that there are conditions in which these Kv7 channel inhibitors are not efficacious.
Since XE991 is among the predominant choices for inhibiting Kv7 channels, inconsistencies in the literature denote a need to further characterize its mode of inhibition. To this end, we performed an electrophysiological study in a heterologous expression system. We determined the conditions under which these compounds are efficacious, as well as their mode of interaction, and addressed the past inconsistencies regarding the washout of these compounds. The determined characteristics of XE991 and linopirdine should be pertinent for experimenters using these inhibitors.
Materials and Methods
Reagents and Plasmids.
Linopirdine (1,3-dihydro-1-phenyl-3,3-bis(4-pyridinylmethyl)-2H-indol-2-one dihydrochloride), XE991 dihydrochloride (10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone dihydrochloride), and Exo1 (2-[(4-fluorobenzoyl)amino]-benzoic acid methyl ester) were purchased from Tocris (Bristol, UK). Retigabine (ethyl N-[2-amino-4-[(4-fluorophenyl)methylamino]phenyl]carbamate) was purchased from Alomone Laboratories (Jerusalem, Israel). Concanavalin A was purchased from Sigma-Aldrich (St. Louis, MO). Mammalian expression plasmids containing rat Kv7.2, Kv7.3 (Hoshi et al., 2003) and Kv7.2-monomeric citrine (mCit) (Kosenko et al., 2012) have been described. Kv7.2 mutation (R214D) was generated using QuickChange II XL site-directed mutagenesis (Agilent Technologies, San Diego, CA) and was validated by DNA sequencing.
Cell Culture.
Chinese hamster ovary (CHO) hm1 cells (Jiang et al., 2015) were cultured in α minimum essential media containing 5% fetal calf serum and 500 µg/ml G418 sulfate. Cells were maintained in a humidified incubator at 37°C with 5% CO2. CHO cells were grown to 30% confluence on 35-mm plates before being transfected with 1 µg of plasmid DNA and 4 µl of LT1 reagent (Mirus Bio LLC, Madison, WI).
Electrophysiology.
All patch-clamp recordings were performed at room temperature on isolated CHO cells using an Axon Multiclamp 700B patch clamp amplifier (Molecular Devices, CA). Data were acquired using pClamp software (version 10, Molecular Devices). Signals for current traces of ≤1 second duration were sampled at 2 kHz and low-pass-filtered at 1 kHz. Current recordings between 1 and 15 seconds were sampled at 500 Hz, and those longer than 15 seconds were sampled at 250 Hz. Whole-cell patch-clamp recordings on CHO hm1 cells have been described previously (Kosenko et al., 2012). Briefly, cells were constantly perfused with Q2 solution containing 144 mM NaCl, 5 mM KCl, 2 mM CaCl2, 0.5 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4). Patch pipettes (3–4 MΩ) were filled with intracellular solution containing 135 mM potassium aspartate, 2 mM MgCl2, 3 mM EGTA, 1 mM CaCl2, 4 mM ATP, 0.1 mM GTP, and 10 mM HEPES (pH 7.2). Successful patches that maintained Rs < 6 MΩ were selected for experiments. Liquid junction potential was not corrected in this study.
Live Cell Imaging.
Protocol for total internal reflection florescence (TIRF)-based assessment of surface transport of Kv7.2 channel has been described previously (Jiang et al., 2015). Briefly, 1 day after transfection, cells were replated onto 18-mm round cover glasses. On the 2nd day after transfection, cells were used for TIRF experiments. For recording, medium was replaced with Q2 solution. For Exo1 experiments, cells were pretreated with 100 µM Exo1 in Q2 solution at room temperature for 2 minutes before proceeding with experiments. Fluorescence emission was acquired using an inverted microscope IX-81 (Olympus Tokyo, Tokyo, Japan) with an ImageEM CCD camera (Hamamatsu Photonics, Hamamatsu, Shizuoka, Japan) controlled by MetaMorph 7.6.3 (Molecular Devices). For excitation in TIRF experiments, a 515-nm diode-pumped solid-state laser (Cobolt, Stockholm, Sweden) with an acousto-optic tunable filter was used with a TIRF module (Olympus). Emission images were obtained through a dual-view module (Photometrics, Tucson, AZ) with ET535/30m, ET480/40m emission filters, and a T505lpxr dichroic mirror (Chroma Technology, Bellows Falls, VT); 100-millisecond exposure time of images was taken every 10 seconds for time-lapse imaging measurements.
Statistics.
The activation curves were obtained by nonlinear regression to a Boltzmann equation, Y = (1+exp((V1/2-x)/k))-1, where x is membrane potential, V1/2 is the half activation potential, and k is the slope factor unless stated otherwise in the text. All results are expressed as the mean ± s.e.m. Statistical significance of the results was assessed by nonparametric analysis of variance (Kruskal-Wallis test) followed by Dunn’s multiple comparisons test or Mann-Whitney test. All statistical tests were performed by a computer program Prism 6 (GraphPad, La Jolla, CA). P < 0.05 is considered significant.
Results
Voltage Dependence of XE991 Inhibition is Closely Related to Activation of Kv7 Channels Rather than the Membrane Potential Per Se.
During our previous study (Kay et al., 2015), we observed that XE991 is more effective in highly active neurons compared with less active neurons. It has previously been reported that M-current inhibition by XE991 is voltage-dependent; and, under some conditions, it does not inhibit the M-current (Romero et al., 2004). We alternatively reasoned that these characteristics of XE991 are derived from state-dependent inhibition rather than voltage-dependent inhibition since the activation threshold of Kv7 channel is close to the resting membrane potential.
We characterized the voltage dependency of homomeric Kv7.2 and heteromeric Kv7.2/3 channel activation to XE991-mediated inhibition. Exposure to 10 µM XE991 for 25 seconds had no effects when cells were held at −70 mV (Fig. 1A), but it showed near-complete inhibition when XE991 was applied at potentials more positive than −30 mV (Fig. 1B). XE991-mediated inhibition of Kv7.2 or Kv7.2/3 channels showed voltage dependence with half-inhibition potentials of −51.6 ± 0.0 mV for Kv7.2 channels and −50.7 ± 0.9 mV for Kv7.2/3 channels (Fig. 1, C and D). When XE991-mediated inhibition is plotted against relative activation of Kv7.2 and Kv7.2/3 channels (Fig. 1E), it indicates that XE991 is effective only when cells are held at potentials where Kv7 channels are activated more than 1%.

XE991 inhibits homomeric Kv7.2 and heteromeric Kv7.2/3 channels at more depolarized potential than their activation thresholds. (A) Voltage protocol and representative traces showing exposure to 10 µM XE991 at −70 mV for 25 seconds, indicated as a black box, was ineffective on Kv7.2 current. Right panel shows expanded traces of indicated time points (1 and 2). (B) Voltage protocol and representative traces showing 10 µM XE991 completely blocked Kv7.2 current when exposed to XE991 at 0 mV for 25 seconds, indicated as a black box. Expanded current traces are also shown. (C) Summary graph showing voltage-XE991 inhibition relationships and activation curve for homomeric Kv7.2 channels. (D) Voltage-XE991 inhibition relationships and activation curve for heteromeric Kv7.2/3 channels. (E) Results shown in (C) and (D) replotted for relative inhibition by XE991 versus relative activation of channels. Inhibition of XE991 is closely correlated with activation of Kv7 channels in a semi-log plot (Kv7.2: r = 0.94, Kv7.2/3: r = 0.90). Data are shown as average values ± S.E.M.
To further characterize the relationships between channel activation and XE991-mediated inhibition, we shifted the activation voltage of Kv7 channels to more hyperpolarized potentials with retigabine and evaluated whether it changed the efficacy of XE991 (Fig. 2). In the presence of 10 µM retigabine, the half-activation voltage of Kv7.2 was −47.0 ± 0.8, a −24-mV shift from the control (Fig. 2B). The half-inhibition potential of XE991 also shifted −24 mV from the control (−75.2 ± 0.6 mV, n = 5) (Fig. 2, B and C). Notably, XE991 inhibited Kv7.2 current at a holding potential of −70 mV in the presence of retigabine (Fig. 2A), a potential at which XE991 did not inhibit Kv7.2 current in the control and close to the resting membrane potential in neurons (Fig. 1C).

Retigabine shifted effective potentials of XE991 to more negative potentials. (A) Voltage protocol and representative traces showing that when cells were pretreated with 10 µM retigabine, 25-second treatment with 10 µM XE991 (black box) inhibited Kv7.2 current at a holding potential of −70 mV. (B) Voltage-XE991 inhibition relationships and activation curve for Kv7.2 channels pretreated with 10 µM retigabine. (C) Activation-XE991 inhibition relationships for Kv7.2 channel from results shown in (B). Error bars show S.E.M.
An additional characterization of voltage-dependent inhibition by XE991 was assessed in a mutant Kv7.2 channel (R214D), which had a 26-mV right shift in the half-activation potential (3.4 ± 14.0 mV) (Fig. 3). Accordingly, the rightward shift in the voltage dependence of activation coincided with an equivalent rightward shift (26 mV) in the half-inhibition potential (−26.3 ± 0.27 mV) (Fig. 3B). Relationships between inhibition by XE991 and channel activation were maintained in this condition (Fig. 3C). The voltage dependence of Kv7 channel activation versus inhibition by XE991 followed a linear function with a slope of 0.97 ± 0.03 compared with wild-type, retigabine-treated, and Kv7.2(R214D) channels (Fig. 3D), suggesting a close relation between the voltage dependence of Kv7.2 channel activation and the efficacy of XE991.

Kv7.2 mutant (R214D) shifted voltage dependence of activation and XE991 inhibition to more positive potentials. (A) Voltage protocol and representative traces showing that 25-second treatment with 10 µM XE991 (black box) inhibited WT Kv7.2 current at a holding potential of −40 mV, whereas Kv7.2(R214D) mutant channel had minimal inhibition at this holding potential. (B) Voltage-XE991 inhibition relationships and activation curve for Kv7.2(R214D) channels. (C) Activation–XE991 inhibition relationships for Kv7.2(R214D) channel from results shown in (B). (D) Pooled results for V1/2 inhibition and V1/2 activation potentials of Kv7.2, retigabine-treated Kv7.2 and Kv7.2(R214D) channel. Slope of regression line is also shown. Error bars show S.E.M.
Inhibition Kinetics of XE991.
We then tested the effect of membrane potential on inhibition kinetics (Fig. 4). First, cells were held at −70 mV, a potential where XE991 has no effect, followed by a 20-second depolarization to various test potentials, with or without XE991, and then analyzed the time course of inhibition. XE991 inhibited Kv7.2 currents with time constants that decreased with increasing depolarization (Fig. 4, A–C). An interesting question would be whether such differences in inhibition kinetics change the potency of XE991 or only the speed of inhibition. To address this question, we measured the time course of Kv7.2 current inhibition by 1 µM XE991, which is close to the reported IC50 of XE991 (Wang et al., 1998), using test pulses with 20-second intervals at holding potentials of either 0 mV or −40 mV (Fig. 4D). Even though there was a difference in the speed of current inhibition, depending on holding potentials, both conditions reached almost complete block after 5 minutes.

Voltage dependence of XE991 inhibition kinetics with Kv7.2 channels. (A) Voltage protocol and representative Kv7.2 current traces from 20-second test potential to −40 mV during absence, XE(–), and presence, XE(+) of 10 µM XE991 (red box). Note that in the presence of XE991, the initial activation phase of Kv7.2 current is unaffected. Kv7.2 current ratio of XE(+)/XE(–) showing that current inhibition followed a single exponential decay (lower graph, red line) with the indicated time constant. (B) Same as (A) with voltage step to 0 mV. (C) Inhibition-time constants of Kv7.2 current from wash-in experiments at indicated holding potentials measured as shown in (A) and (B). ** P < 0.01, nonparametric analysis of variance (ANOVA) (Kruskal-Wallis test), followed by Mann-Whitney test, n = 6. (D) Time courses of Kv7.2 current inhibition by 1 µM XE991 at indicated holding potentials. XE991 was applied at t = 0. Cells were held at indicated potentials, and current was measured by 500-millisecond step hyperpolarizations to −60 mV with 20-second intervals. Error bars show S.E.M.
Limited Recovery of Kv7 Current after Washout of XE991 is Predominantly due to Channel Trafficking.
Reports about recovery of Kv7 currents from XE991 after washout are conflicting; some groups consider inhibition by XE991 to be reversible (Rennie et al., 2001; Yue and Yaari, 2004; Zaika et al., 2006), whereas others consider it irreversible (Wladyka and Kunze, 2006; Brueggemann et al., 2012). To test this, we first treated CHO cells expressing Kv7.2 channels with 10 µM XE991 at 0 mV for 25 seconds, and then we held the cells at holding potentials of either −70 or −30 mV during the wash process to assess current recovery (Fig. 5). Washout experiments between these two potentials did not show significant difference (20.2% ± 3.9% and 18.2% ± 2.9% recovery after 10 minutes of washout at −70 mV and −30 mV holding potential, respectively (Fig. 5, A and B). Control experiments without XE991 showed stable Kv7.2 current in this recording condition (Fig. 5B). We then measured rate of current recovery from distinct concentrations of XE991 (0.25, 1, 10 µM), which also showed no differences (Fig. 5C). These results suggest that current recovery after washout is quite limited for XE991. We then tested heteromeric Kv7.2/3 channels, which showed similar slow and limited recovery after washout [33.4% ± 5.9% (n = 6) recovery at −70 mV, 37.8% ± 3.8% (n = 6) at −30 mV after 10 minutes’ wash].

Kv7.2 current recovery from XE991 washout is limited. (A) Representative Kv7.2 current traces showing control, 10 µM XE991 inhibited, and after 10-minute washout at indicated holding potentials. For washout at −30 mV, current was measured by 1-second test potential to −60 mV from a holding potential of −30 mV. For washout at −70 mV, cells were depolarized to 0 mV for 200 milliseconds from a holding potential of −70 mV. (B) Summary showing slow and limited recovery of Kv7.2 current after washout in cells held at −30 mV or −70 mV. Wash started at t = 0. (C) Kv7.2 current recovery from indicated concentrations of XE991 at a holding potential of 0 mV. (D) TIRF experiments showing 100 µM Exo1 suppressed constitutive exocytosis of mCit-tagged Kv7.2 (KCNQ2-mCit) by 50 µg/ml concanavalin A (black box) applied at t = 2 minutes. (E) Kv7.2 current recovery from 10 µM XE991 at −70 mV showing that 100 µM Exo1 suppressed recovery of the current. Wash started at t = 0. (F) Summary of current recovery from 10 µM XE991 after10 minutes of washout at indicated conditions. −70 mV wash and −70 mV Exo1 results are from the same data set shown in (E) **P < 0.01; NS, P > 0.05, nonparametric analysis of variance (Kruskal-Wallis test) followed by Dunn’s multiple comparisons test. Error bars show S.E.M.
A 30% turnover rate of Kv7.2 channel in 10 minutes is comparable to surface transport of Kv7.2 subunit that we described previously in CHO cells (Jiang et al., 2015). To evaluate the contribution of Kv7.2 channel trafficking to the current recovery, we tested whether an exocytosis inhibitor, 100 µM Exo1 (Feng et al., 2003), can suppress current recovery after washout. We first confirmed the effects of 100 µM Exo1 on the surface transport of the Kv7.2 channel by total internal reflection fluorescence (TIRF) measurements using monomeric citrine-tagged Kv7.2 channel, Kv7.2-mCit. TIRF selectively illuminates <100 nm from the cover glass, which can be used to monitor Kv7.2 surface transport at the bottom surface of cells (Jiang et al., 2015). To confirm Exo1 ability to suppress exocytosis, we inhibited endocytosis by 50 µg/ml concanavalin A, which induced a gradual increase of TIRF signal of Kv7.2-mCit owing to constitutive exocytosis (Fig. 5D) (Jiang et al., 2015). A 2-minute pretreatment with 100 µM Exo1 resulted in a 75% ± 5% reduction in concanavalin A-induced increase in TIRF signals from Kv7.2-mCit (Fig. 5D), confirming that most exocytosis is inhibited in this condition. Using this condition, we examined whether Exo1 prevents recovery of Kv7.2 current after 10 µM XE991 washout; 100 µM Exo1 application alone did not inhibit Kv7.2 current (0.98 ± 0.06, n = 6). In the control washout without Exo1, Kv7.2 current recovered 32.3% ± 2.1% after a 10-minute washout at a holding potential of −70 mV (Fig. 5E). In the presence of 100 µM Exo1, current recovery was reduced to 12.1% ± 2.1% (Fig. 5, E and F). To further assess whether open probability of Kv7.2 affects current recovery, we tested washout in the presence of 10 µM retigabine at −30 mV, as well as washout at a holding potential of 0 mV. These conditions did not alter current recovery (Fig. 5F). We concluded that current recovery after washout of XE991 derives mostly from new Kv7 channels surfaced to the plasma membrane rather than dissociation of XE991 from Kv7.2 channel.
Linopirdine Shares Basic Features with XE991.
Since current recovery after washout of XE991 did not accurately reflect dissociation of XE991, we examined linopirdine to evaluate inhibition kinetics of this type of Kv7 channel inhibitor. Linopirdine is a prototypical M-channel inhibitor that was developed before and considered structurally similar to XE991 (Zaczek et al., 1998). As summarized in Fig. 6, 30 µM linopirdine showed voltage-dependent inhibition of Kv7.2 current with a half-inhibition potential at −55.7 ± 0.4 mV (Fig. 6, A and B), which is close to that of XE991. We then assessed the inhibition kinetics of linopirdine (30 µM) at various test potentials using the same voltage protocols as used in XE991 (Fig. 6, D–F). Similar to XE991, inhibition time constants of linopirdine were decreased with more depolarized potentials (Fig. 6F).

Linopirdine replicated most of XE991 effects. (A) Voltage protocols and representative Kv7.2 current traces showing voltage-dependent inhibition of 30 µM linopirdine (black box). Right traces show expanded currents from indicated time points 1 and 2. (B) Pooled results for voltage-inhibition relationships of linopirdine and activation curve for Kv7.2 channels as measured in A. (C) Results shown in (B) replotted as relative activation against relative inhibition of Kv7.2 by linopirdine. Results of Kv7.2/3 are also included. (D) Voltage protocol and representative Kv7.2 current traces with a test potential to −40 mV during absence, Lin(–), and presence, Lin(+), of 30 µM linopirdine (red box). Kv7.2 current ratio Lin(+)/Lin(–) showing that current inhibition followed a single exponential decay with the indicated time constant (bottom graph, red line). (E) Same as (D) with a test potential to 0 mV. (F) Inhibition time constants of wash-in from Kv7.2 current measured as shown in (D) and (E). As with XE991, time constants decreased with depolarizing potentials. **P < 0.01, nonparametric analysis of variance (Kruskal-Wallis test) followed by the Mann-Whitney test, n = 5. Error bars show S.E.M.
Although wash-in inhibition was similar to XE991, there was a difference in current recovery during washout. When linopirdine-treated CHO cells were washed at a holding potential of −70 mV, Kv7.2 currents showed 24.2% ± 4% recovery after 10 minutes (n = 6) (Fig. 7, A and B), comparable to that of XE991. When cells were held at −30 mV, however, Kv7.2 currents showed almost full recovery after a 10-minute wash (93.4% ± 4.2%, n = 5 (Fig. 7, A and B). A similar recovery profile was observed with heteromeric Kv7.2/3 channels (Fig. 7C). To further characterize current recovery at depolarized potentials, the Kv7.2 current was monitored by 500-millisecond hyperpolarized steps with 20-second intervals. Time courses of current recovery (Fig. 7D) were best fit by a single exponential function with time constants as summarized in Fig. 7E, which was facilitated as holding potentials were depolarized (Fig. 7, D and E).

Linopirdine interaction kinetics with Kv7.2 channels in relation to membrane potential. (A) Representative current traces showing Kv7.2 current recovery after washout of 30 µM linopirdine from cells held at −30 mV or −70 mV. For −30 mV holding potential during washout, Kv7.2 currents were monitored by 1-second-step hyperpolarizations to −60 mV. For −70 mV holding potential, cells received a 200-millisecond step depolarization to 0 mV. (B) Summary showing almost full recovery of Kv7.2 current when cells were washed at −30 mV while showing slow and limited recovery at −70 mV. (C) Similar full recovery was observed with heteromeric Kv7.2/3 channels at −30 mV. (D) Current recovery during washout from 30 µM linopirdine at indicated holding potentials. T = 0 indicates beginning of washout. Current was measured by a 500-millisecond-step hyperpolarization to −60 mV with 20-second intervals from indicated holding potentials. (E) Summary of current recovery-time constants at indicated holding potentials during washout. **P < 0.01, nonparametric analysis of variance (Kruskal-Wallis test), followed by the Mann-Whitney test, n = 5. Error bars show S.E.M.
Quantitative Model for XE991 and Linopirdine.
Our results so far suggest that inhibition kinetics of XE991 and linopirdine are closely related to the activation state of Kv7.2 channel. A common mechanism of state-dependent ion channel inhibitors is open-channel inhibition. Therefore, we first assessed this mechanism. If XE991 and linopirdine interact with the channel at the open state, then wash-in and washout time constants should show close correlation with channel activation; Namely, if inhibitors interact only with open channels, then the rate of association at the half-activation potential for Kv7.2 channel (−21 mV, Fig. 8) should be nearly double the rate constant at maximal activation since duration of single channel opening at V1/2 is half of that at maximal activation.
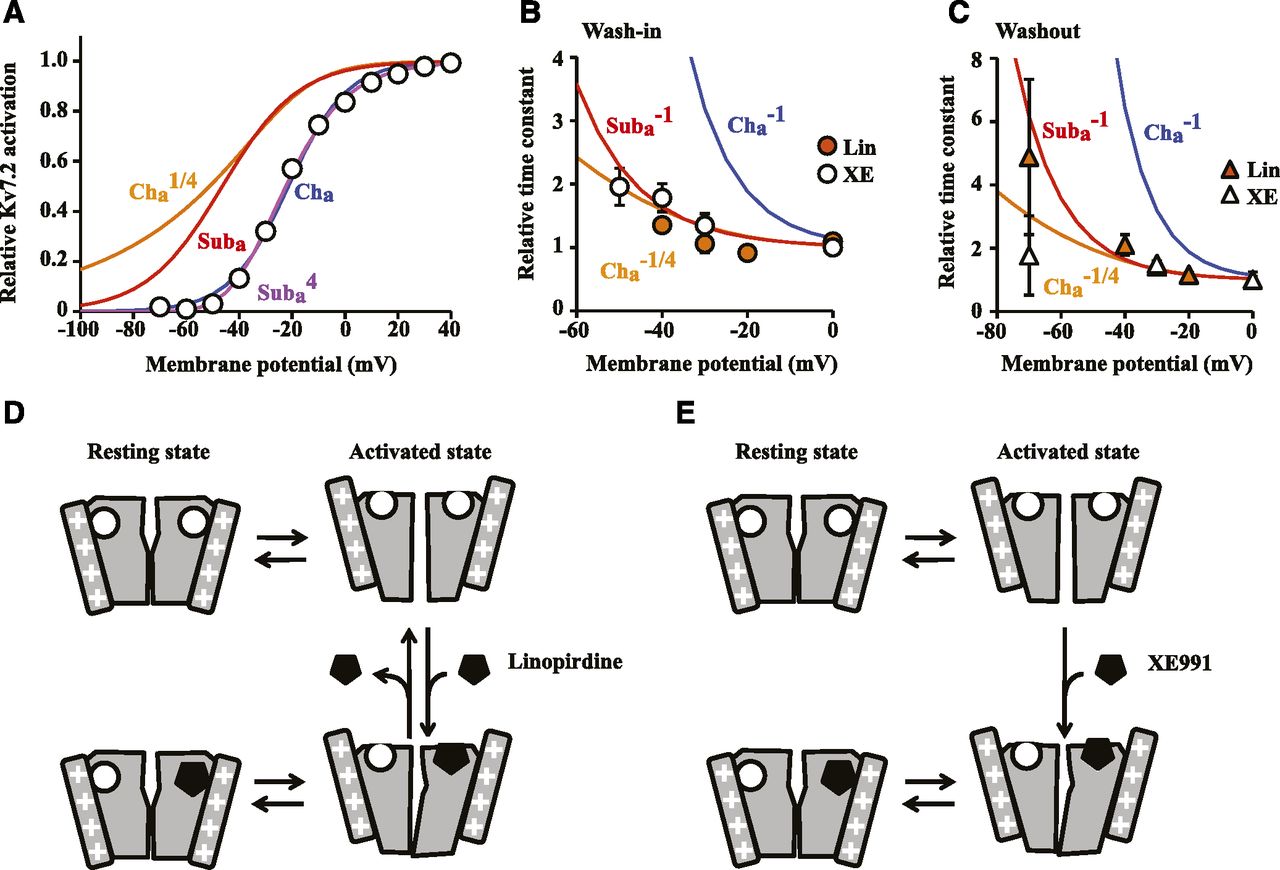
XE991 and linopirdine interaction with Kv7.2 relates with activation of single subunit. (A) Activation curve for Kv7.2 channel (blue curve, Cha), and one-quarter power of its Boltzmann function (orange curve, Cha1/4). Activation curve for a single subunit (red curve, Suba) and its fourth power function (purple curve, Suba4) are also shown (see text for details). (B) Relative inhibition time constants calculated from Fig. 4C (XE991, open circle) and Fig. 7C (linopirdine, orange circle) normalized to those from 0 mV plotted as a function of voltage. The reciprocal of the activation curves of Cha, Cha1/4 and Suba shown in (A) are also shown. (C) Relative recovery time constants from experiments shown in Fig. 4 and Fig. 5B (XE991, open triangle) and Fig. 6E and 7E (linopirdine, orange triangle), overlaid with the reciprocal of the activation curves of Cha, Cha 1/4, and Suba. (D) Schematic model for interaction between linopirdine and Kv7.2 channel. (E) Schematic model for interaction between XE991 and Kv7.2 channel. Error bars show S.E.M.
We assumed a single-step binding reaction for XE991 or linopirdine (inhibitors, INH), which can be described as follows in eq. 1: (1)where kon (M-1s−1) is the second-order association rate constant and koff (s−1) is the first-order dissociation rate constant. Thus, this reaction can be described as follows in eq. 2 and eq. 3:
(1)where kon (M-1s−1) is the second-order association rate constant and koff (s−1) is the first-order dissociation rate constant. Thus, this reaction can be described as follows in eq. 2 and eq. 3: (2)
(2) (3)using the inhibition time constant (τin) and concentration of inhibitors, as described by Goldstein and Miller (1993) and Hille (2001). In addition, koff can be measured directly from the rate of washout as described in eq. 3. As eq. 2 predicts, inhibition time constants are proportional to the reciprocal of the rate constant. Thus, we plotted normalized wash-in time constants to reciprocals of the activation curve (Cha−1, Fig. 8B). To our surprise, neither wash-in nor washout time constants overlapped with Cha-1 (Fig. 8, B and C). Instead, best-fit powers for wash-in were 0.19 ± 0.06 for linopirdine and 0.26 ± 0.03 for XE991, which were close to one-fourth power of the activation curve (Fig. 8B, Cha1/4). A similar trend was observed for washout of linopirdine (0.36 ± 0.006 for linopirdine washout, Fig. 8C). We did not perform this analysis for XE991 washout since it was not voltage-dependent. Considering that a single channel is composed of four subunits, a best fit to the one-fourth power of channel activation suggests that binding interactions are closely related to activation of a single subunit. To evaluate this, we estimated single-subunit activation by applying the fourth-power Boltzmann equation to Kv7.2 channel activation, as shown in eq. 4:
(3)using the inhibition time constant (τin) and concentration of inhibitors, as described by Goldstein and Miller (1993) and Hille (2001). In addition, koff can be measured directly from the rate of washout as described in eq. 3. As eq. 2 predicts, inhibition time constants are proportional to the reciprocal of the rate constant. Thus, we plotted normalized wash-in time constants to reciprocals of the activation curve (Cha−1, Fig. 8B). To our surprise, neither wash-in nor washout time constants overlapped with Cha-1 (Fig. 8, B and C). Instead, best-fit powers for wash-in were 0.19 ± 0.06 for linopirdine and 0.26 ± 0.03 for XE991, which were close to one-fourth power of the activation curve (Fig. 8B, Cha1/4). A similar trend was observed for washout of linopirdine (0.36 ± 0.006 for linopirdine washout, Fig. 8C). We did not perform this analysis for XE991 washout since it was not voltage-dependent. Considering that a single channel is composed of four subunits, a best fit to the one-fourth power of channel activation suggests that binding interactions are closely related to activation of a single subunit. To evaluate this, we estimated single-subunit activation by applying the fourth-power Boltzmann equation to Kv7.2 channel activation, as shown in eq. 4: (4)where x is the membrane potential, V1/2 is the half-activation potential of a single subunit, and k is the slope factor. Best-fit curve for channel activation is shown as a purple curve in Fig. 8A (Suba4), and the red curve shows a derived activation curve for a single subunit (Suba). Reciprocals of predicted single-subunit activation curves were also in good agreement with the inhibition kinetics of XE991 and linopirdine (Fig. 8, B and C, red curve). Furthermore, recalculation of relationships between half-inhibition potentials of XE991 and half-activation potentials of single-subunit of Kv7.2 channel using the results shown in Fig. 3D showed equivalent relationships (Supplemental Fig. 1).
(4)where x is the membrane potential, V1/2 is the half-activation potential of a single subunit, and k is the slope factor. Best-fit curve for channel activation is shown as a purple curve in Fig. 8A (Suba4), and the red curve shows a derived activation curve for a single subunit (Suba). Reciprocals of predicted single-subunit activation curves were also in good agreement with the inhibition kinetics of XE991 and linopirdine (Fig. 8, B and C, red curve). Furthermore, recalculation of relationships between half-inhibition potentials of XE991 and half-activation potentials of single-subunit of Kv7.2 channel using the results shown in Fig. 3D showed equivalent relationships (Supplemental Fig. 1).
Discussion
It has been previously reported that efficacy of linopirdine and XE991 is voltage-dependent (Romero et al., 2004). We confirmed similar voltage-dependent inhibition by these inhibitors. On the other hand, we also found that shifts in the half-inhibition potential corresponded with equivalent shifts in the half-activation voltage by retigabine or Kv7.2(R214D), which suggests that such changes in efficacy are derived from conformational changes of Kv7.2 channel subunits rather than from voltage differences across the plasma membrane. The most common class of state-dependent ion-channel inhibitor is open-channel inhibitors. Therefore, we first suspected this mechanism. In fact, transient channel activation in the presence of inhibitors, as shown in Fig. 4 and Fig. 7, is a signature profile for open-channel inhibition (Zagotta et al., 1990); however, our inhibition kinetic analysis suggests that these inhibitors bind to a single activated subunit rather than an open channel (Fig. 8, B and C).
Although the results from linopirdine mirror most of the findings from XE991, a key difference was that inhibition by linopirdine was reversible at depolarized potentials. Together with our finding from voltage-dependent wash-in kinetics, this finding suggests that linopirdine can reversibly interact with Kv7.2 channel when subunits are at the active conformation but is trapped within Kv7.2 channels when subunits are in the resting conformation after binding (Fig. 8D). Unlike linopirdine, we did not observe full recovery for XE991. Furthermore, most of the current recovery after XE991 washout was due to new channel insertion to the plasma membrane. These findings suggest that binding of XE991 to the Kv7.2 channel is apparently irreversible (Fig. 8E). On the other hand, we would like to emphasize that the Kv7.2 current can be restored from XE991-mediated inhibition as a result of channel trafficking, which explains previous observations (Rennie et al., 2001; Yue and Yaari, 2004; Zaika et al., 2006). Our results suggest ∼30% recovery per 10 minutes, but it may depend on cell types and experimental conditions.
Our findings in this report suggest that XE991 might not be efficacious for cells staying at resting membrane potentials, such as in silent or scarcely firing neurons. Coadministration of XE991 and retigabine would be helpful to remove such biased efficacy in animal experiments. For cultured neurons, holding at depolarized potentials during voltage-clamp experiments or coadministration of XE991 and retigabine or high potassium would be effective.
Activated subunit inhibition and slow binding kinetics of these inhibitors may explain why these compounds are well tolerated in animals (Mani et al., 2013; Vetter et al., 2013; Young and Thomas, 2014; Kay et al., 2015) without causing lethal seizures, as seen in KCNQ2 gene knockout mice (Watanabe et al., 2000; Soh et al., 2014). In addition, preferential M-channel inhibition in highly active neurons by these inhibitors would exaggerate neurotransmitter-mediated M-current suppression, which may underlie the cognitive enhancing action of these compounds.
Authorship Contributions
Participated in research design: Greene, Hoshi.
Conducted experiments: Greene, Hoshi.
Contributed new reagents: Kang.
Performed data analysis: Greene, Hoshi.
Wrote or contributed to the writing of the manuscript: Greene, Kang, Hoshi.
Footnotes
- Received March 24, 2017.
- Accepted May 5, 2017.
↵1 Current affiliation: Department of Anesthesiology, Pharmacology and Physiology, Rutigers, The State University of New Jersey, New Jersey Medical School, Newark, New Jersey.
This work was supported by the National Institutes of Health National Institutes of Neurologic Disorders and Stroke [Grant R01NS067288] to N.H.
↵
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.

Abbreviations
- CHO
- Chinese hamster ovary
- Kd
- inhibition constant
- kon
- association rate constant
- koff
- dissociation rate constant
- τ
- time constant
- mCit
- monomeric citrine
- TIRF
- total internal reflection florescence
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}