


Abstract
Pertussis toxin (PTX)-insensitive mutants of Gαi/o proteins expressed in C6μ cells were used to examine the hypothesis that there are agonist-specific conformational states of the μ-opioid receptor with coupling preferences to different Gαi/o subtypes, as measured by the degree of stimulation of [35S]guanosine 5′-O-(3-thio)triphosphate (GTPγS) binding. Binding of [35S]GTPγS to endogenous Gαi/o proteins stimulated by the full μ-opioid agonist [d-Ala2,MePhe4,Gly5-ol]enkephalin (DAMGO) was completely blocked by overnight treatment with 100 ng/ml PTX. Treatment for 4 h with lower concentrations led to a PTX-dependent reduction in the maximal effect of DAMGO but no alteration in the potency of DAMGO or morphine nor in the relative maximal effect (relative efficacy) of the partial agonists morphine and buprenorphine compared with the full agonist DAMGO. Using PTX-insensitive Gα mutants in which the PTX-sensitive cysteine was replaced with isoleucine, the potency for a series of μ-opioid agonists was highest in cells expressing Gαi3 and Gαo and lowest with Gαi1 and Gαi2, with no significant change in the order of potency, namely, etorphine >> endomorphin-1 = DAMGO = endomorphin-2 = fentanyl = morphine >> meperidine. The order of agonist relative efficacy, etorphine = DAMGO = endomorphin-1 = endomorphin-2 = fentanyl ≥ morphine ≥ meperidine > buprenorphine ≥ nalbuphine, was also the same across all of the PTX-insensitive Gαi/o subtypes. Highest relative efficacy to stimulate [35S]GTPγS binding was seen with Gαi3. Consequently, reported observations of agonist-directed trafficking at μ-opioid receptors most likely involve non-PTX-sensitive Gα protein mechanisms.
The μ-opioid receptor is a G protein-coupled receptor (GPCR) that can activate all members of the pertussis toxin (PTX)-sensitive Gαi/o family when occupied by agonist (Laugwitz et al., 1993; Chakrabarti et al., 1995). These early studies used the highly efficacious peptidergic agonist DAMGO. However, there is growing evidence at a variety of GPCRs for the formation or stabilization of agonist-specific conformations that have the potential to activate different downstream signaling pathways (Kenakin, 1995, 2003). For example, at the μ-opioid receptor, there is evidence for differential binding of peptide and nonpeptide agonists (Fukuda et al., 1995; Wang et al., 1995; Xue et al., 1995; Chaipatikul et al., 2003), and with the use of human μ-opioid receptor-Gα fusion protein constructs (hMOR-Gα) expressed in human embryonic kidney cells and Escherichia coli, differences have been observed between μ-opioid agonists in the ability to stimulate [35S]GTPγS binding (Stanasila et al., 2000, Massotte et al., 2002). In addition, the ability of μ-opioid agonists to cause receptor internalization does not correlate with their ability to activate other Gα protein-mediated events (Whistler and von Zastrow, 1998; Borgland et al., 2003).
Agonist-dependent activation of Gα subunits could have downstream consequences, resulting in agonist-specific trafficking of intracellular signals (Kenakin, 1995, 2003). This could occur because Gαi2 is thought to couple more efficiently to adenylyl cyclase than Gαo (McKenzie and Milligan, 1990; Moon et al., 2001), whereas Gαo may couple more effectively to inhibition of Ca2+ currents (Hescheler et al., 1987). Such differences may have behavioral outcomes. Thus, antisense knockdown of Gαi2 but not of the other Gαi/o proteins significantly attenuates μ-opioid-induced supraspinal antinociception in the mouse (Raffa et al., 1994), and Garzón and colleagues have demonstrated that knockdown of several Gα proteins differentially affects supraspinal antinociception induced by a variety of μ-agonists in the mouse (Sánchez-Blázquez et al., 2001).
The studies discussed above did not examine Gαi3, which is reported to couple most efficiently to the DAMGO-activated μ-opioid receptor (Laugwitz et al., 1993; Chakrabarti et al., 1995), and generally, only a limited number of agonists have been studied that do not cover a wide range of agonist potency or efficacy. Here, we test the hypothesis that different agonists induce activation states of the μ-opioid receptor that differ in their ability to activate Gαi/o protein subtypes and that this can result in changes in the rank order of potency or efficacy as measured at the level of G protein activation. To study individual Gα subtypes, PTX-insensitive mutants of the Gα subtypes were expressed in C6 glioma cells stably expressing the μ-opioid receptor (C6μ), and agonist stimulation of [35S]GTPγS binding was measured in membranes prepared from PTX-treated cells to uncouple endogenous Gα proteins, which mainly comprise Gαi2 (Charpentier et al., 1993). PTX-insensitive subunits have been used successfully to study receptor activation of different Gαi/o proteins and have been shown to be able to couple to downstream effectors (Hunt et al., 1994, Senogles, 1994; Wise et al., 1997; Yamaguchi et al., 1997). The potency and efficacy of several μ-opioid agonists of differing structure, including an alkaloid (morphine), small synthetic or semisynthetic compounds (etorphine, fentanyl, meperidine, and nalbuphine), and peptides (DAMGO, endomorphin-1, and endomorphin-2) to activate the different Gαi/o subtypes were determined. The results show that μ-opioid agonists were most potent at activating Gαi3 and Gαo compared with Gαi1 and Gαi2 and had the highest efficacy, relative to the full agonist DAMGO, at Gαi3, with similar relative efficacy across the other Gα subtypes. However, there was no change in the rank order of potency or relative efficacy for the series of ligands across the different Gαi/o subunits, indicating a lack of agonist-specific activation of Gαi/o proteins.
Materials and Methods
Materials. Dulbecco's modified Eagle's medium, fetal bovine serum, trypsin-EDTA, Lipofectamine Plus Reagent, Geneticin, and Zeocin were purchased from Invitrogen (Carlsbad, CA). PTX was purchased from List Biological Laboratories Inc. (Campbell, CA), and [35S]GTPγS was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). Morphine, etorphine, fentanyl, meperidine, buprenorphine, and nalbuphine were obtained through the Narcotic Drug and Opioid Peptide Basic Research Center at the University of Michigan (Ann Arbor, MI). DAMGO, endomorphin-1, endomorphin-2, GDP, Trizma base, and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Human PTX-insensitive (PTXi) Gα protein cDNAs were purchased from UMA cDNA Resource Center (www.cDNA.org). The GF/B 4 × 6 glass fiber filtermats and filtermat bags were purchased from PerkinElmer Life and Analytical Sciences. EcoLume scintillation cocktail was obtained from ICN (Aurora, OH).
Cell Culture and Membrane Preparation. cDNA for human C351I GaoA, Gαi1, Gαi3, or C352I Gαi2 was inserted into pcDNA3.1zeo- vectors (Invitrogen). The identity of each of the Gαi/o cDNAs was confirmed by sequencing at the University of Michigan sequencing core. Transient transfection into C6 glioma cells stably expressing the rat μ-opioid receptor (C6μ) was performed using Lipofectamine Plus and 4 μg of appropriate cDNA (Clark et al., 2003). In one set of experiments, different quantities of human C351I Gαi3 cDNA (4, 6, or 8 μg) and vector to a total of 8 μg of total DNA were transfected. Cells were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum under 5% CO2. Transiently transfected cells were collected for membrane preparation 48 h after transfection. Cells were treated for 4 h or overnight with PTX before membranes were prepared as described previously (Clark et al., 2003).
[35S]GTPγS Binding. Cell membranes (10-20 μg) were incubated for 60 min in a shaking water bath at 25°C with 30 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 100 mM NaCl, 0.1 mM dithiothreitol (freshly prepared), 30 μM GDP, 0.1 nM [35S]GTPγS, and varying concentrations of ligand or double-distilled H2O as described by Traynor and Nahorski (1995). Samples were filtered through a glass-fiber filtermat mounted in a Brandel cell harvester and rinsed three times with ice-cold 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, and 100 mM NaCl. Filtermats were dried, and 0.3 ml of EcoLume scintillation cocktail was added to each sample area to soak the filter (total 7.2 ml). Each filtermat was placed in a polyethylene bag, which was heat-sealed, and radioactivity retained in each sample area was counted in a Wallac 1450 MicroBeta liquid scintillation and luminescence counter.
Data Analysis. Concentration-response data were fitted to sigmoidal curves using GraphPad Prism 4 (GraphPad Software Inc., San Diego, CA) to determine EC50 and maximal stimulation. Data from at least three separate experiments, each carried out in duplicate, are presented as mean ± S.E.M. Comparisons between drugs or across Gα subunits were made by one-way analysis of variance followed by Tukey's multiple comparison test.
Results
Efficacy and Potency of μ-Opioid Agonists in Wild-Type C6μ Cells. The C6μ cells used in this study express 3.2 ± 0.2 pmol of μ-opioid receptors/mg of protein. Basal [35S]GTPγS binding in these cells was 33 ± 5 fmol/mg protein, and the full μ-opioid agonist DAMGO stimulated [35S]GTPγS binding to 175 ± 11 fmol/mg protein. In C6μ cells expressing wild-type Gα proteins, the rank order of efficacy, as measured by the maximal ability to stimulate [35S]GTPγS binding, was DAMGO = etorphine = endomorphin-1 = endomorphin-2 = fentanyl ≥ morphine ≥ meperidine > buprenorphine ≥ nalbuphine (Table 1). Potency for the series of compounds followed the order etorphine >> endomorphin-1 = DAMGO = endomorphin-2 = fentanyl = morphine >> meperidine (Table 1). Potency data for buprenorphine and nalbuphine could not be reliably obtained due to the small degree of stimulation. DAMGO stimulation of [35S]GTPγS binding in C6μ cells expressing wild-type Gα proteins (485 ± 76%) was completely abolished (0.8 ± 2.7%) by overnight (18 h) treatment with 100 ng/ml PTX, confirming that the μ-opioid receptor was acting only through PTX-sensitive Gαi/o proteins.
Stimulation of [35S]GTPγS binding to membranes from C6μ cells by various opioid agonists Agonist-stimulation of [35S]GTPγS (100 pM) binding to membranes from C6μ cells was determined in the presence of 30 μM GDP using at least five concentrations of each agonist as described under Materials and Methods. Data are expressed as percentage maximal DAMGO stimulation and as EC50 values from concentration-effect curves determined in three separate experiments, each carried out in duplicate.
Effect of G Protein Concentration on μ-Opioid Agonist Relative Efficacy and Potency. In studies with different PTX-insensitive Gα subtypes, it is not possible to directly compare the absolute level of Gα expressed. Consequently, comparisons of agonist potency and efficacy in cells expressing different Gα subtypes can only be made if these parameters are independent of the level of Gα expression. To test this concept, the number of functional endogenous Gα proteins was reduced in wild-type C6μ cells by treatment with varying concentrations of PTX for 4 h before preparation of membranes. The maximal degree of stimulation of [35S]GTPγS binding by the highly efficacious μ-opioid agonist DAMGO was reduced by PTX treatment (Fig. 1A) in direct proportion to the amount of PTX used, resulting in a 3-fold loss of maximal stimulation after exposure to 100 ng/ml PTX for 4 h. The maximal degree of [35S]GTPγS binding stimulated by the μ-opioid partial agonists morphine and buprenorphine was similarly decreased by PTX treatment so that the maximal stimulation by the two compounds remained constant relative to DAMGO (Fig. 1B). The potencies of DAMGO and morphine, as measured by their EC50 values to stimulate [35S]GTPγS binding, were not significantly altered by reducing the number of functional Gα proteins with PTX (Fig. 1C). Therefore, μ-opioid agonist relative efficacy, as measured by maximal [35S]GTPγS stimulation relative to DAMGO, and absolute as well as relative potencies can be compared in cells expressing different expression levels of Gα subunits. In all subsequent studies that use cells expressing PTX-insensitive Gα, cells were treated with 100 ng/ml PTX for 18 h to prevent activation of endogenous G proteins.
To examine whether endogenous Gα proteins in C6μ cells would interfere with the exogenously expressed PTX-insensitive Gα subunits, we compared stable clones expressing PTX-insensitive GαoA (C351I) or Gαi3 (C351I) with and without PTX treatment. The maximal level of DAMGO-stimulated [35S]GTPγS incorporation over basal was greater in the non-PTX-treated cells (GαoA = 503 ± 29%; Gαi3 = 500 ± 53%) than the PTX-treated cells (GαoA = 205 ± 20%; Gαi3 = 63 ± 5%), suggesting an additive effect with endogenous Gα subunits. However, the potency obtained with non-PTX-treated cells expressing GαoA (205 ± 20 nM) and Gαi3 (49.4 ± 5.7 nM) was reduced compared with PTX-treated cells (GαoA = 56 ± 7 nM; Gαi3 = 7.5 ± 2.4 nM). Thus, if endogenous Gα proteins do interfere with the exogenously expressed PTX-insensitive Gα subunits, this interference is inhibited by the PTX treatment.
Comparison of Agonist Potency and Relative Efficacy of μ-Opioid Agonists at Different G Protein Subtypes. C6μ cells were transiently expressed with PTX-insensitive GαoA, Gαi1, Gαi2, or Gαi3 subunits with a C351(2)I mutation and treated overnight with 100 ng/ml PTX, such that there was no response to opioid agonists in nontransfected cells. The maximal stimulation of [35S]GTPγS binding by DAMGO was at least 6 times greater with the C351(2)I mutation for each of the Gαi/o subtypes than with the C351(2)G mutation that we have previously used (Clark et al., 2003; data not shown), in agreement with studies at the α2A-adrenoceptors (Bahia et al., 1998). The C351(2)I mutation was used in subsequent studies.
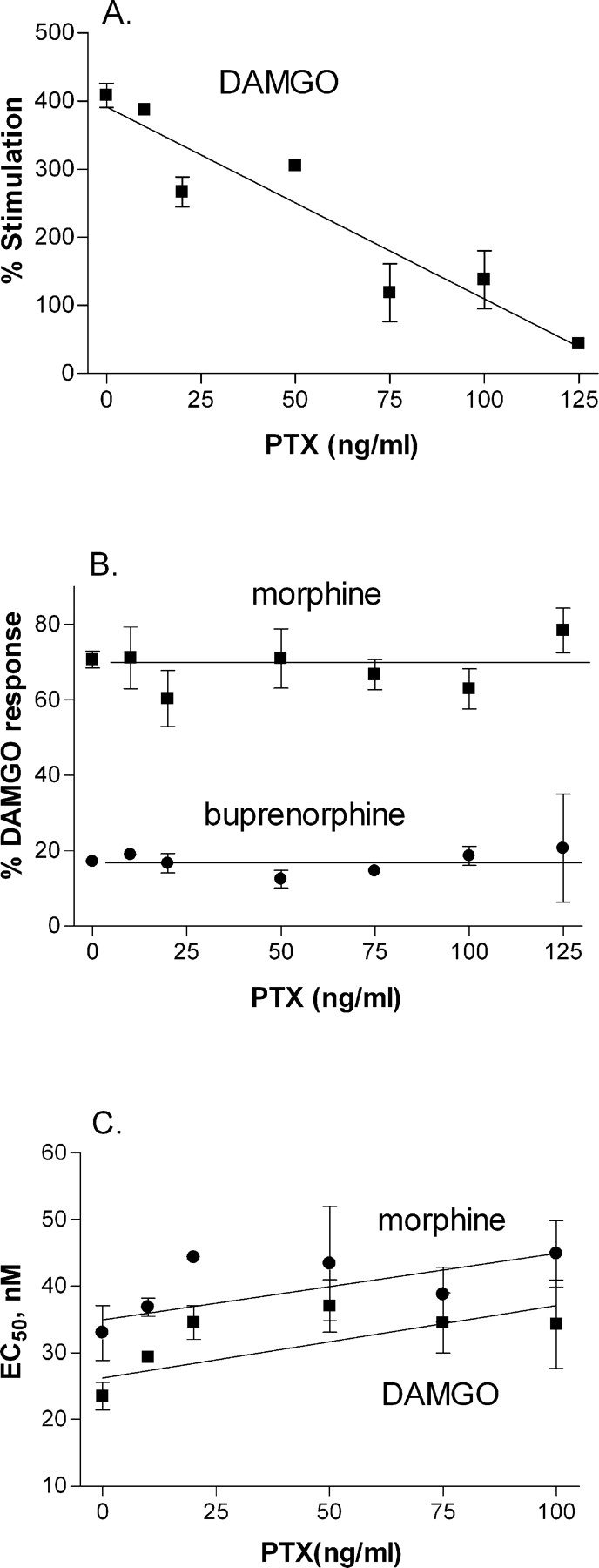
Effect of functional G protein expression on agonist efficacy. C6μ cells were treated for 4 h with 0 to 25 ng/ml PTX. Agonist-stimulation of [35S]GTPγS (100 pM) binding in membranes from these cells was determined in the presence of 30 μM GDP as described under Materials and Methods. Data are expressed as percentage stimulation over basal (A), as percentage maximal DAMGO stimulation (B), or as EC50 (C). Data points are from three separate experiments, each carried out in duplicate. PTX treatment did not have a significant effect on the EC50 value for DAMGO [F(5,22) = 2.05, p = 0.123] or morphine [F(5,20) = 1.25, p = 0.34).
The potency of DAMGO and other μ-opioid agonists to stimulate [35S]GTPγS binding to the C351(2)I Gα subtypes decreased in the order Gαi3 ≥ GαoA > Gαi1 > Gαi2 (Table 2). When compared as potencies relative to DAMGO, there were no differences confirming that absolute values can be used. The overall agonist potency in membranes expressing Gαi3 was 4.6 ± 0.4-fold higher than in membranes from cells expressing Gαi2 across all seven agonists examined (Table 2). In contrast, treatment of wild-type cells with 100 ng/ml PTX for 4 h, which reduced maximal DAMGO stimulation of [35S]GTPγS binding and, presumably, functional Gα subunits by at least 3-fold (Remmers et al., 2000) (Fig. 1) only reduced the potency of DAMGO and morphine potency by a nonsignificant 1.4-fold (Fig. 1C).
Potency of μ-opioid agonists to stimulate binding of [35S]GTPγS to different PTX-insensitive Gα proteins C6μ cells transiently transfected with C351(2)I-PTXi Gα subunits were treated overnight with 100 ng/ml PTX. Agonist stimulation of [35S]GTPγS (100 pM) binding in membranes from these cells was measured with at least five concentrations of each agonist in the presence of 30 μM GDP as described under Materials and Methods. Shown are the mean EC50 ± S.E.M. from at least three separate experiments, each performed in duplicate, and the relative potency compared with DAMGO (in parentheses).
The degree of maximal stimulation of [35S]GTPγS binding by DAMGO and the other μ-opioid agonists varied with the different Gα subtypes in the decreasing order GαoA > Gαi2 > Gαi1 > Gαi3 (Fig. 2), possibly as a consequence of differential expression. Consequently, the maximal stimulation for the different agonists was compared with that obtained with DAMGO. The rank order of maximal stimulation of [35S]GTPγS binding across the C351(2)I mutant Gα subtypes was constant in the order: DAMGO = etorphine = endomorphin-1 = endomorphin-2 = fentanyl ≥ morphine ≥ meperidine > buprenorphine ≥ nalbuphine (Fig. 3). The same rank order was obtained in membranes from cells stably expressing the less efficient C351(2)G Gα mutants (data not shown). Although the rank order of μ-agonist-mediated [35S]GTPγS-stimulated binding was unchanged across the various Gαi/o subunits, the maximal [35S]GTPγS stimulation relative to DAMGO was generally greatest with Gαi3. However, this difference was only significant for endomorphin-1, endomorphin-2, morphine, and buprenorphine (Fig. 3).

DAMGO-stimulated [35S]GTPγS binding to C351(2)I-PTXi mutant Gα subunits. C6μ cells transiently transfected with appropriate PTXi Gα subunits were treated overnight with 100 ng/ml PTX. Membranes from these cells were incubated with 30 μM GDP, 100 pM [35S]GTPγS, and 0 to 10 μM DAMGO as described under Materials and Methods. DAMGO stimulation of [35S]GTPγS binding was expressed as percentage stimulation over basal. Shown are the combined results from three separate experiments, each carried out in duplicate. Maximal stimulation and potency values obtained for C351(2)I mutants were: GαoA (maximum = 595 ± 89%, EC50 = 44 ± 5 nM), Gαi1 (maximum = 130 ± 28%, EC50 = 84 ± 18 nM), Gαi2 (maximum = 265 ± 96%, EC50 = 154 ± 47 nM), and Gαi3 (maximum = 61 ± 10%, EC50 = 38 ± 7 nM).

Relative agonist efficacy in membranes from C6μ cells expressing C351(2)I-PTXi Gα subunits. Cells were transiently transfected with appropriate Gα and then treated overnight with 100 ng/ml PTX. Membranes were incubated with 30 μM GDP, 100 pM [35S]GTPγS, and varying concentrations (0-10 μM) of opioid agonist as described under Materials and Methods. Relative efficacy for each agonist was expressed as percentage maximal DAMGO stimulation of [35S]GTPγS binding. Shown are the means ± S.E.M. from at least three assays performed in duplicate. *, Gαi3 relative stimulation significantly different from that of GαoA (p < 0.05), Gαi1 (p < 0.01), and Gαi2 (p = 0.001). ≠, Gαi3 relative stimulation significantly different from that of Gαi2 (morphine, p = 0.01; buprenorphine, p < 0.05). #, Gαi3 relative stimulation significantly different from that of Gαi1 (p < 0.05). †, Gαi2 relative stimulation significantly different from that of GαoA (p < 0.05).
To test whether the low degree of [35S]GTPγS incorporation into Gαi3 was responsible for the higher relative agonist efficacy and increased potency of agonists at Gαi3, stimulation of [35S]GTPγS binding was examined in C6μ cells transfected with increasing amounts of Gαi3 cDNA. The results showed that the maximal effect of DAMGO increased with increasing cDNA, but neither the potency of DAMGO nor the relative efficacy of morphine and buprenorphine was altered (Table 3).
Effect of concentration of Gαi3 cDNA used for transfection on agonist-stimulated binding of [35S]GTPγS in C6μ cells C6μ cells were transiently transfected with C35I-PTXi Gαi3 subunits and treated overnight with 100 ng/ml PTX. DAMGO stimulation of [35S]GTPγS (100 pM) binding in membranes from these cells was measured with at least five concentrations of agonist in the presence of 30 μM GDP as described under Materials and Methods. For morphine and buprenorphine, a single maximal (10 μM) concentration was used. Values are means ± S.E.M. from at least three separate experiments, each performed in duplicate.
Discussion
The purpose of this study was to use PTX-insensitive Gα subunits and C6 cells stably expressing a μ-opioid receptor to test the hypothesis that structurally dissimilar μ-opioid agonists stabilize distinct activation states of the receptor, which differentially activate Gαi/o subtypes. Following overnight treatment with a high concentration of PTX, the ability of μ-opioid agonists to stimulate [35S]GTPγS binding was completely lost but could be rescued by transfection with PTX-insensitive mutants of Gαi/o subunits, although agonist potency to stimulate [35S]GTPγS binding varied in the order Gαi3 ≥ Gαo > Gαi1 ≥ Gαi2. Across a wide range of μ-opioid agonists, there was no change in the rank order of potency or relative maximal ability to stimulate [35S]GTPγS binding to these different PTX-insensitive Gα subunits.
Because the PTX-insensitive [C351(2)I] mutation is in a region of Gα involved in receptor coupling, it was necessary to show that functional coupling to the μ-opioid receptor is retained and that there is no interference by endogenous Gα proteins. The ability of DAMGO to stimulate [35S]GTPγS binding was much greater in cells expressing both wild-type and PTX-insensitive Gα protein before treatment with PTX, suggesting that both endogenous and exogenous Gα subunits were activated. This result agrees with findings that the C351I mutant of Gαi1 couples extremely well to the α2A-adrenergic receptor (Bahia et al., 1998). Moreover, as in other studies (Wise et al., 1997; Clark et al., 2003) additional Gβγ subunits were not required, suggesting that the Gα subunits expressed are able to use endogenous Gβγ. In contrast, agonist potency was higher in PTX-treated cells compared with non-PTX-treated cells, suggesting that competition between endogenous and exogenous Gα subunits occurred, but this was removed by PTX-mediated ADP-ribosylation of the endogenous Gα. Finally, it is possible that the relatively high-receptor expression level used in the study (Clark et al., 2003) may mask differences in relative agonist efficacies and potencies of the different ligands. Receptor number does affect relative agonist efficacy (MacEwan et al., 1995; Selley et al., 2000), and differences in rank order of efficacy and potency have been demonstrated with different receptor numbers (Brink et al., 2000; Cordeaux et al., 2000). However, this should not be a concern because many of the compounds tested, namely morphine, meperidine, buprenorphine, and nalbuphine, are confirmed to be partial agonists, indicating that a response ceiling had not been reached.
In C6μ cells expressing endogenous Gα subunits, the ability of the efficacious agonist DAMGO to stimulate [35S]GTPγS was completely lost by overnight treatment with 100 ng/ml PTX, confirming that the response was due to coupling to PTX-sensitive Gαi/o proteins. This effect was rescued by transfection with each of the PTX-insensitive [C352(2)I] Gα subunits, confirming the expression of these subunits. However, the degree of DAMGO-mediated stimulation varied across the subtypes, possibly due to differential expression. Therefore, to be able to study agonists across Gα subtypes, we compared relative agonist efficacy as maximal stimulation of [35S]GTPγS binding relative to DAMGO as well as agonist potency. To do this, we first confirmed that different levels of Gα expression had no effect on these parameters. This confirmation was achieved in two ways. First, reduction in the number of functional Gαi/o proteins by partial treatment with PTX lessened the maximal DAMGO stimulation of [35S]GTPγS binding. Importantly, this treatment did not alter the potency of DAMGO or morphine or the efficacy of morphine and buprenorphine relative to DAMGO. The result agrees with data using α2-adrenergic receptors expressed in NIH-3T3 fibroblasts in which no change in the relative efficacy of clonidine in relation to epinephrine was observed with progressive PTX treatment (Yang and Lanier, 1999). Second, increasing the amount of Gαi3 cDNA transfected resulted in an increased ability of DAMGO to stimulate [35S]GTPγS binding, with no change in its potency and no change in the relative efficacy of morphine and buprenorphine. Together, these experiments show that comparisons of both absolute and relative potency and relative efficacy across μ-opioid agonists can be made between cells expressing PTX-insensitive Gα subtypes at different levels.
The highest potency and relative efficacy of the agonists were seen with Gαi3 ≥ GαoA = Gαi1 ≥ Gαi2, suggesting that μ-opioid receptors are most efficiently coupled to Gαi3 and GαoA and least efficiently to Gαi1 and Gαi2. This observation confirms work showing that DAMGO-stimulated incorporation of [α-32P]azidoanilide into Gα subunits was more potent for Gαi3 than for Gαi1 in SH-SY5Y cells (Laugwitz et al., 1993) and with potency in the order Gαi3 > Gαo > Gαi2 in Chinese hamster ovary cells expressing a μ-opioid receptor (Chakrabarti et al., 1995). However, the same rank order of agonist relative efficacy and potency was maintained across cells expressing GαoA,Gαi1,Gαi2,orGαi3. This contrasts with agonist-specific differences observed in systems expressing receptor-Gα fusion proteins. For example, in human embryonic kidney cells expressing a hMOR-Gαo fusion protein, morphine showed similar efficiency to stimulate [35S]GTPγS incorporation with both hMOR-Gαi1 and hMOR-Gαi2, yet endomorphin-1 and -2 promoted [35S]GTPγS binding to hMOR-Gαi1 but very poorly, if at all, to hMOR-Gαi2 (Massotte et al., 2002). The endomorphins, together with morphine and the synthetic peptide agonist DAMGO, were equipotent in stimulating [35S]GTPγS binding in E. coli expressing a hMOR-Gαo fusion protein, but morphine and endomorphin-1 were more potent than DAMGO or endomorphin-2 in cells expressing hMOR-Gαi2 (Stanasila et al., 2000). The differences between our findings and those of the fusion systems may be accounted for by the very different systems used. However, the differences may not be so definite because, even in the present system, morphine is equipotent at Gαi1 and Gαi2, whereas endomorphin-1 is more potent than endomorphin-2. Consequently, subtle differences between ligands may be apparent, but these are not sufficient to alter agonist rank order.
Overall, studies suggest that Gαi3 is the Gαi/o protein most efficiently coupled to the μ-opioid receptor. The preference of activated μ-opioid receptor for Gαi3 potentially provides for agonist-directed selectivity of second messenger signaling. There is evidence that Gαi3 is a better coupling partner for Gβγ in inwardly rectifying potassium channel activation than other Gαi/o subunits in Xenopus oocytes (Ivanina et al., 2004), and Gαo is involved in inhibition of voltage-gated Ca 2+ channels, at least in neuronal cells (Hescheler et al., 1987). In contrast, Gαi1, Gαi2, and Gαi3 are all able to mediate inhibition of adenylyl cyclase (Gerhardt and Neubig, 1991). However, maximal DAMGO-stimulated [35S]GTPγS binding was much lower in Gαi3-expressing cells than in Gαo-expressing cells, and the potency difference for agonist stimulation of [35S]GTPγS incorporation into the different Gα subunits is not large. Therefore, the relative concentration of Gα proteins may be more influential in governing downstream signaling. Indeed, Gαo is expressed at very high levels in brain and is considered to be the most important protein for transducing opioid signals (Jiang et al., 2001).
The present findings do not provide evidence for agonist-specific conformations of the μ-receptor that activate specific Gαi/o proteins and so potentially lead to agonist-directed trafficking of intracellular signaling. Instead, differential activation of second-messenger pathways could occur by a strength of a signaling mechanism (Kenakin, 1995) dependent on the degree of agonist efficacy. A full agonist, such as DAMGO, might activate all Gαi/o proteins expressed in a cell and the downstream pathways to which they are coupled, but a partial agonist may only be able to stimulate enough of the most preferred or abundant Gα protein and thus activate signaling through a lesser number of downstream pathways. In contrast, there are reports of agonist-directed trafficking of intracellular signal pathways via the μ-opioid receptor. In particular, there appears to be no relationship between the ability of μ-opioids to mediate receptor internalization and down-regulation compared with other events, including inhibition of Ca2+ currents (Borgland et al., 2003), inhibition of adenylyl cyclase (Keith et al., 1996; Whistler and von Zastrow, 1998), and activation of inwardly rectifying K+ channels (Celver et al., 2004). However, differences between μ-opioid ligands in mediating internalization and down-regulation may be due to agonist-dependent differences in the degree of phosphorylation of the μ-opioid receptor, leading to differences in the ability to recruit β-arrestin 1 and β-arrestin 2 for internalization (Bohn et al., 2004). Such actions may be independent of G protein. For example, down-regulation has a PTX-insensitive component (Yabaluri and Medzihradsky, 1997) and the ability of DAMGO but not of morphine to stimulate phospholipase D2 by a mechanism involving small G proteins may contribute to internalization (Koch et al., 2003).
In summary, the findings confirm that the agonist-activated μ-opioid receptor couples to Gαi3 more efficiently than to other Gαi/o subtypes. However, there was no change in the rank order of relative agonist efficacy or potency among the different Gαi/o subtypes to indicate that different agonists generate specific activation states of the receptor. These results imply that reported examples of agonist-specific signaling may involve the ability of μ-opioid receptors to activate downstream signaling through a non-PTX-sensitive G protein mechanism rather than by differential activation of the structurally similar Gαi/o proteins.
Acknowledgments
We thank Dr. Huda Akil for the μ-opioid receptor cDNA.
Footnotes
-
This work was supported by National Institute on Drug Abuse Grant DA 04087. T.D.G. was supported by the American Society for Pharmacology and Experimental Therapeutics Gerald J. Dalton/Vincent G. Zannoni Summer Undergraduate Research Fellowship.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.105.096818.
-
ABBREVIATIONS: GPCR, G-protein-coupled receptor; PTX, pertussis toxin; DAMGO, [d-Ala2,MePhe4,Gly5-ol]enkephalin; C6μ, rat C6 glioma cells stably expressing the μ-opioid receptor; [35S]GTPγS, guanosine-5′-O-(3-[35S]thio)triphosphate; PTXi, PTX-insensitive; C351(2), 351Cys or 352Cys in Gαi/o proteins.
- Received October 7, 2005.
- Accepted January 23, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}