Article Text

Abstract
OBJECTIVES To compare the neurological and psychometric characteristics of presymptomatic gene carriers and non-gene carriers who are at risk for developing Huntington's disease so as to characterise early signs of disease and to identify markers of neurological function that could be used to assess the impact of experimental therapies on the progression of disease, even among those who are clinically presymptomatic.
METHODS A sample of people at risk for Huntington's disease was genotyped and evaluated using subscales of the Wechsler adult intelligence scale–revised (WAIS-R), a quantified neurological rating scale, and computerised physiological measures including speed of movement and reaction time.
RESULTS Genotyping and clinical examination determined that 171 participants were presymptomatic gene carriers (PSGCs) and 414 participants were non-gene carriers (NGCs). The PSGCs performed significantly worse when compared with the NGCs on the digit symbol, picture arrangement, and arithmetic subscales of the WAIS-R (p<0.02) and for the physiological measures: button tapping, auditory reaction time, visual reaction time with decision, and movement time with and without decision (p<0.05). Although no PSGCs had sufficient neurological findings to warrant a diagnosis of Huntington's disease on clinical examination, the PSGCs had more frequent possible or definite abnormality for oculomotor function, chorea, muscle stretch reflexes, gait, and station stability, and rapid alternating movements (p⩽0.02).
CONCLUSIONS Among Huntington's disease gene carriers, subtle cognitive and motor deficits precede the onset of sufficient neurological abnormality to warrant a clinical diagnosis of Huntington's disease.
- Huntington's disease
- presymptomatic gene carriers
- early clinical signs
Statistics from Altmetric.com
Huntington's disease is a neurodegenerative disorder characterised neuropathologically by the loss of medium sized spiny neurons in the neostriatum. An autosomal dominant genetic instability syndrome, it is the result of an increased number of triplet (CAG) repeats in the huntingtin gene.1 People without Huntington's disease have a variable number of repeats, ranging in number from 11 to 28. The penetrance is repeat length dependent with those having a larger number of repeats more likely to develop disease. The clinical implication of an allele with 29 to 39 repeats is ambiguous. Those with 29–35 CAG repeats have not been found to develop the disease themselves but they may pass it to their children due to paternal meiotic instability.2 People with 36–39 CAG repeats exhibit reduced penetrance. Those with 40 or more CAG repeats exhibit 100% penetrance and are certain to develop manifest Huntington's disease if they live long enough.
Even though gene testing is available, the variability in age at disease onset, the initial symptom presentation, and the course of disease makes equivocal the determination of the exact age at symptom onset and the clinical categorisation of a person as affected or unaffected.3-5 A triad of progressive motor, cognitive, and emotional symptoms characterises adult onset Huntington's disease; however, the actual sequence of symptom presentation is variable. In a clinical setting the diagnosis of adult onset Huntington's disease in at risk persons is typically made after the onset of involuntary, choreiform movements.6 7
Motor changes that include mild chorea, abnormal extraocular movements, brisk muscle stretch reflexes, and diminished rapid alternating movements are the most consistent early findings in manifest disease.8 9 However, conflicting results have been found for motor impairment in people not experiencing choreiform movements. Oculomotor deficits including abnormal ocular saccades may be indicative of imminent manifest Huntington's disease,8 10 11 although this has not been consistently found.12 13 Other motor differences, including slower reaction time, have inconsistently been seen to differ between gene carriers and non-gene carriers.14-16 In addition to motor abnormalities, progressive cognitive decline is part of the Huntington's disease phenotype. Early changes in information processing, cognitive flexibility, and memory retrieval typically progress to more severe and widespread abnormalities later in the course of disease.17 Although cognitive decline is thought by clinicians and family members often to precede the onset of motor abnormalities, this has not been demonstrated consistently.18-26
In interim analyses, we reported subclinical motor and cognitive changes among a subset of the presymptomatic gene carriers (PSGCs).14 21 27 28 We present herein, results of analyses performed on the full data set of 657 people at risk for Huntington's disease who consider themselves presymptomatic, the largest sample of gene tested, at risk persons evaluated for signs of preclinical Huntington's disease. These analyses are intended: (1) to identify subtle subclinical motor and cognitive abnormalities in PSGCs; (2) to expand our previous studies to evaluate the progression of early signs of Huntington's disease throughout the preclinical disease process; and (3) to investigate the predictive power of these early signs to classify a person as a Huntington's disease gene or non-gene carrier. With molecular testing available, predicting gene carrier status with clinical markers is not as essential. Rather, the identification of markers of early clinical disease is necessary to better understand the very early disease process and the pathogenesis of the disease. In addition, these markers may be used as surrogate measures to evaluate the efficacy of neuroprotective agents.
Materials and methods
PARTICIPANTS
This sample of 657 people at risk for Huntington's disease was recruited through the national research roster for patients with Huntington's disease and families at Indiana University, Indianapolis, USA. Only those without a prior diagnosis of Huntington's disease and reporting themselves as asymptomatic but with a parent affected with the disease were invited to participate. Participants were informed of the risks and benefits involved in the study and informed consent was obtained (IUPUI IRB Study No 8806–02).
After the participants' visits, DNA samples were analyzed with a polymerase chain reaction based diagnostic screen to determine the number of CAG repeats29 30; therefore, the participants were unaware of their gene testing results. Of the 657 participants genotyped, 72 were eliminated before data analyses due to: lack of a molecular test result (n=33), an allele in the inconclusive, intermediate size range (32–37 CAG repeats) (n=7), or presence of sufficient neurological symptoms to warrant a clinical diagnosis of manifest Huntington's disease (n=32). The participants that remained were assigned to one of two groups: (1) presymptomatic gene carriers (PSGCs) (n=171), defined as those with an expanded Huntington's disease gene (⩾38 CAG repeats) but who did not have sufficient neurological symptoms to warrant a clinical diagnosis of manifest disease; and (2) non-gene carriers (NGCs) (n=414), defined as those with two unexpanded Huntington's disease alleles (<32 CAG repeats). Ten participants had either 38 or 39 CAG repeats. Two of these were shown to have manifest Huntington's disease on clinical examination and were not included in the analysis. The other eight were included as PSGCs as they had an affected parent and a genotype highly likely to develop manifest Huntington's disease in the future.
CLINICAL EXAMINATION
The participants' motor function was assessed with a quantified neurological examination adapted from previously described protocols.8 31 The examination encompassed extraocular movements, chorea, gait, postural stability, dystonia, parkinsonism, tremor, muscle stretch reflexes, and cerebellar function. A major portion of the neurological examination was designed to detect motor features of Huntington's disease to determine whether PSGCs had a consistent pattern of early clinical findings before the onset of the characteristic chorea. A board certified neurologist with specialisation in movement disorders (ES) performed over 90% of the examinations. The remaining examinations were completed by two neurologists trained in the use of the rating scale. The neurologists were unaware of the other test results. Ratings were 0 (normal), 1 (possibly abnormal), or 2 (abnormal) for 29 neurological tests.14 Because the purpose of the study was to detect early changes in those at risk for Huntington's disease, clinical ratings were assigned to maximise sensitivity, even at the expense of some specificity. After the clinical examination the neurologist assigned a score from 0 to 3, summarising his perception of the participant's overall risk for developing Huntington's disease based solely on the neurological examination. A score of 0 indicated a normal examination, 1 indicated minor soft signs consistent with Huntington's disease (for example, brisk reflexes), 2 indicated major soft signs consistent with Huntington's disease (for example, possible chorea), and 3 indicated manifest Huntington's disease. A score of 3 was assigned to persons who in the neurologist's perception had manifest Huntington's disease based on criteria designed to approximate those used in clinical practice. Only those persons with definite chorea, in the absence of other possible causes (for example, hyperthyroidism), or other unequivocal abnormalities consistent with Huntington's disease, were assigned a score of 3. The 32 subjects with an overall score of 3 and who, therefore, had manifest disease were not included in the statistical analyses, as these comparisons were designed to detect early clinical changes in PSGCs.
To complement the analysis of motor function assessed by the neurological examination, six physiological measures of CNS function were obtained using an automated computer driven program (H-Scan, Hoch Co, Corona Del Mar, CA, USA).32 These tests, measuring speed of movement and reaction time, included alternate button tapping, movement time with and without decision, auditory reaction time, and visual reaction time with and without decision.
To measure cognitive function, each participant was evaluated by standard administration procedures for several verbal and performance subscales of the Wechsler adult intelligence survey-revised (WAIS-R).33 Verbal subscales included digit span, vocabulary, and arithmetic. Performance subscales included digit symbol, picture arrangement, and block design.
STATISTICAL ANALYSES
Performance differences between presymptomatic gene carriers and non-gene carriers
Initial analyses sought to identify those cognitive, physiological, and neurological variables for which the PSGCs performed worse than the NGCs. The participant's raw scores on each of the WAIS-R subscales were adjusted for age with published tables.29 Stepwise multiple linear regression was performed to identify significant (p<0.05) covariate effects of education and sex on the age adjusted WAIS-R scores. The covariate adjusted cognitive variables were then used in all subsequent analyses. Similar methods were used to adjust the physiological variables for significant effects of age and sex. One tailed multivariate analysis of variance (MANOVA) was used to examine the hypothesis that the PSGCs performed worse than the NGCs on the cognitive and physiological tests and post hoc testing using ANOVA was performed to identify those variables contributing to differences between the PSGCs and NGCs.
The Fisher exact test (one tailed) was used to compare the overall at risk score from the neurological examination for the PSGCs with that of the NGCs. Post hoc testing using Fisher's exact test was then performed to identify those specific neurological variables for which the PSGCs exhibited significantly more abnormality than the NGCs. To decrease the number of comparisons, only those variables (n=13) for which at least 10 PSGCs had a questionably abnormal (a score of 1) or definitely abnormal (a score of 2) result were used in the statistical analyses. These 13 tests were saccade accuracy, performance, smoothness, and velocity, optokinetic nystagmus, overall ocular movements, muscle stretch reflexes, chorea of the limbs, trunk, and face, gait and station stability, rapid alternating movements, and dystonia of the limbs.
Due to the possible redundancy in the measures, principal component analysis was performed on the entire data set of PSGCs and NGCs; however, only those variables for which the PSGCs scored significantly worse than the NGCs were included in the statistical analyses. The principal component analysis, completed using criteria described by Hatcher and Stepanski,34 creates multivariate measures which identify underlying structural relations between the variables. Correlation analysis was performed with these multivariate measures, the five retained principal components, to test their relation with the number of CAG repeats in the Huntington's disease gene for the PSGCs and NGCs separately. The statistical significance level was adjusted for the five comparisons.35
Predictability of gene carrier status
Logistic regression was performed to determine the predictive power of the five retained principal components to correctly classify a participant as a gene carrier or non-gene carrier. A jackknifing procedure was used that eliminated the participants one at a time and classified the participant as a gene carrier or non-gene carrier based on the model that omitted that participant. If the estimated probability of Huntington's disease carrier status was greater than or equal to 0.5, the participant was classified as a Huntington's disease gene carrier, conversely if less than 0.5, a non-gene carrier. Predictability of gene status was decreased due to the participants' wide variability in years until onset of manifest Huntington's disease. Based on the known correlation between an individual's age of onset of Huntington's disease and his or her parent's age of onset,36 a variable (diff) was computed as the difference (in years) between the participant's current age and the parent's age of onset. Logistic regression was completed both with and without the variable diff.
Comparison of principal components with overall neurological score
For the PSGCs, the five principal components were hypothesised to consist of tests measuring signs of Huntington's disease acting or progressing at different times during the disease process. The overall neurological score was hypothesised to be a marker reflecting the disease stage. To evaluate the relation between the principal components and the overall neurological score, the five principal components were entered into a MANOVA with the overall neurological score (0–2) as the independent variable. The results were then examined to determine which of the principal components contributed most to each of the stages of preclinical disease.
Results
Demographic information from the 585 study participants is summarised in table 1. Not surprisingly, the PSGCs were significantly younger than the NGCs as the sample consists only of those who reported themselves as presymptomatic and who did not have manifest Huntington's disease on clinical examination. Older persons with the expanded allele likely have already begun to experience disease symptoms and thus would not be included in our sample.
Demographic data (mean(SD)) for the 585 study participants with unequivocal DNA results and without manifest Huntington's disease
PERFORMANCE DIFFERENCES BETWEEN PRESYMPTOMATIC GENE CARRIERS AND NON-GENE CARRIERS
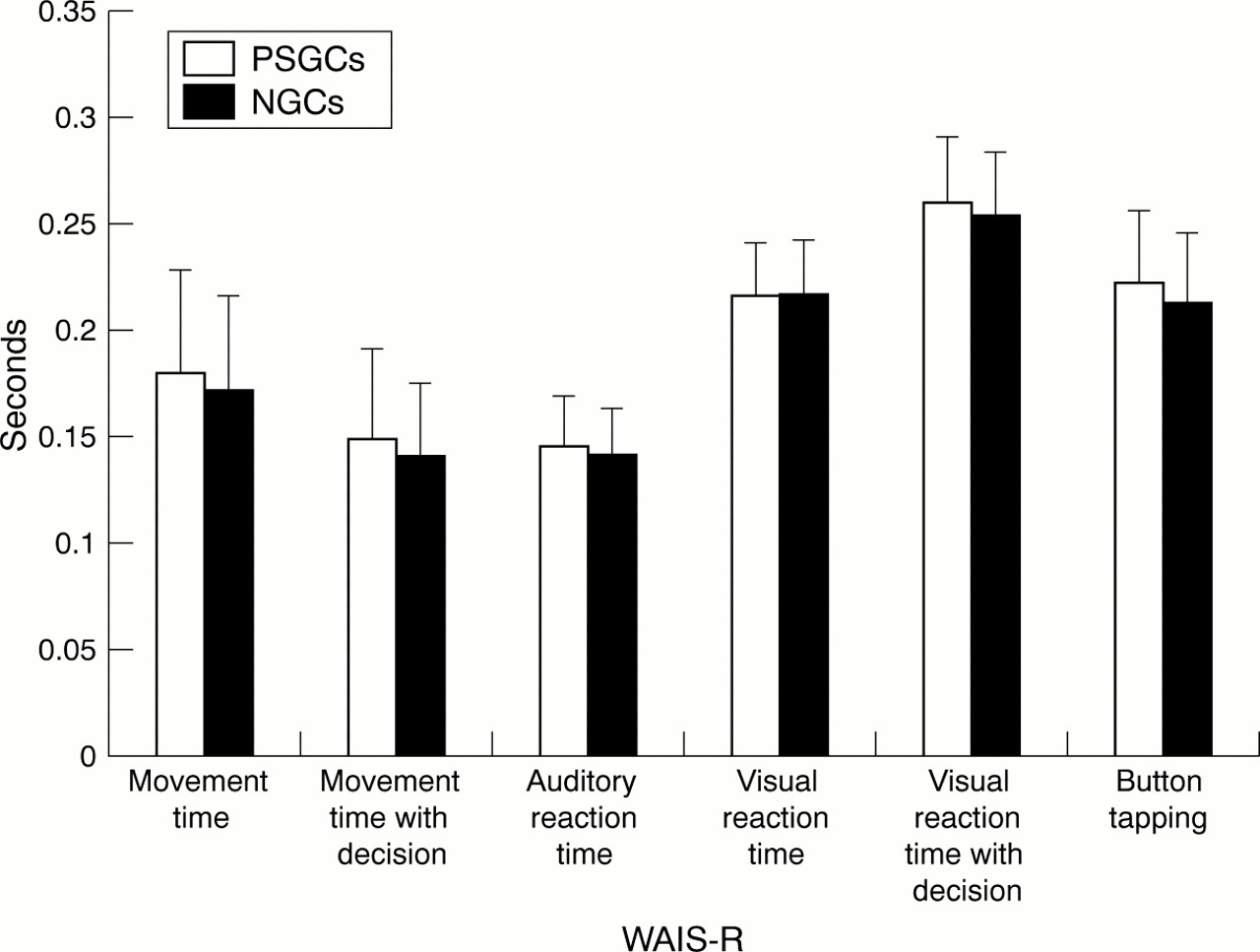
The PSGCs were found to have subtle cognitive, physiological, and motor signs of Huntington's disease when their performance was compared with that of the NGCs. The PSGCs performed significantly worse than the NGCs on the WAIS-R (Wilks' Λ=0.97,F=2.68, p=0.014). The measures found to contribute to this significantly lower cognitive performance were the digit symbol, picture arrangement, and arithmetic subscales (p<0.02, fig 1). The PSGCs also performed significantly worse on the physiological variables (Wilks' Λ=0.95,F=4.40, p=0.0002). The performance of the PSGCs was significantly slower than the NGCs for button tapping, auditory reaction time, visual reaction time with decision, movement time, and movement time with decision (p<0.05, fig2).

Comparison of WAIS-R mean scores for the presymptomatic gene carriers (PSGCs) and non-gene carriers (NGCs) using ANOVA. Significantly worse WAIS-R scores for the PSGCs denoted by * p<0.02; ** p = 0.001.

Comparison of H-scan speed of movement and reaction time mean scores for the presymptomatic gene carriers (PSGCs) and non-gene carriers (NGCs) using ANOVA. Significantly slower performance for the PSGCs denoted by * p<0.05; ** p<0.005.
The PSGCs had higher rates of minor or major soft signs of Huntington's disease as measured by their overall at risk score for Huntington's disease (p<0.0001). However, no PSGCs had sufficient neurological findings to warrant a diagnosis of Huntington's disease on clinical examination and 78% of the PSGCs had either a completely normal examination or only minor soft signs of Huntington's disease. The PSGCs were found to have significantly (p<0.05) higher rates of probable or definite abnormality (a neurological score of 1 or 2) compared with the NGC for 10 of the 13 neurological tests including saccade accuracy and velocity, optokinetic nystagmus, overall ocular function, chorea of the limbs, trunk, and face, muscle stretch reflexes, gait and station stability, and rapid alternating movements (table 2). Although two PSGCs exhibited definite chorea of the face and one definite chorea of the trunk, no definite chorea of the limbs was seen for the PSGCs; the positive ratings for chorea of the limbs were only for possible abnormality (a score of 1). The percentages of possible chorea of the limbs exhibited by the NGCs (22%) (table 2) is consistent with the strategy of attempting to maximise sensitivity during the neurological examination even at the expense of some specificity.
Neurological examination results: percentage of possible, definite, and overall (possible or definite) abnormal findings for the PSGC and NGC
Principal component analysis using the cognitive, physiological, and motor variables for which the PSGCs performed significantly worse than the NGCs resulted in five multivariate measures or principal components (table 3). Variables that loaded on the first component were primarily oculomotor measures, those loading on the second component were primarily movement time measures from the H scan, those on the third component were functional neurological motor measures, those on the fourth component were cognitive measures, and the measures loading on the fifth component were reaction time measures.
Principal components analysis
Further analyses of the five principal components identified a significant correlation (r=−0.29, p=0.0002) between the number of CAG repeats in the disease producing allele of the PSGCs and the fourth component (table 4). Similar results were not seen for the NGCs (r=0.09, p=0.086). When the three WAIS-R subscales picture arrangement, digit symbol, and arithmetic, which loaded onto the fourth component, were examined individually for the PSGCs, poorer performance on each was correlated with a larger number of CAG repeats (p⩽0.005, table 4).
Correlation between the retained principal components and the number of trinucleotide repeats on the disease producing allele for PSGCs only
PREDICTABILITY OF GENE CARRIER STATUS
The ability of the principal components to predict gene carrier status was evaluated by logistic regression. The specificity was high (95.1%), but as would be expected with a presymptomatic group of gene carriers, the sensitivity was low (12.6%). When the difference (in years) between the participants' age and their affected parents' age of onset of symptoms was included in the logistic regression, the sensitivity improved, with 24.0% of PSGCs correctly classified as gene carriers.
COMPARISON OF PRINCIPAL COMPONENTS WITH OVERALL NEUROLOGICAL SCORE
The five principal components were found to differ significantly (p=0.0001) for the three groups classified based on the overall at risk score. The components found to contribute significantly to this finding were components 1 (p<0.0001) and 3 (p<0.0001). The other three components did not vary significantly with the overall neurological at risk score (fig 3).

{kind=link}
{kind=link}
{kind=link}
Comparison of principal components with overall neurological score. Components 1 (p<0.0001) and 3 (p<0.0001) varied with overall neurological score.
Discussion
Analyses of this sample, the largest reported cohort of those at risk for Huntington's disease with confirmed molecular test results (PSGCs n=171, NGCs n=414), were aimed at further delineating the range of early clinical findings with Huntington's disease and at identifying markers of neurological function that could be used to assess the impact of experimental therapies on the progression of disease, even among people who are clinically presymptomatic. The categorisation of participants as gene carriers or non-gene carriers was only made after unambiguous molecular test results. In addition, all participants were adult presymptomatic persons and after molecular testing were found to have fewer than 50 CAG repeats. Therefore, our sample is representative of persons with adult onset Huntington's disease and the detected deficits are consistent with the typical Huntington's disease prodrome.
Subtle cognitive and motor deficits preceded the onset of unequivocal neurological abnormality among the PSGCs. Cognitive functioning, as determined by six subscales of the WAIS-R, showed statistically significant poorer performance for the PSGCs in three measures, the arithmetic, digit symbol, and picture arrangement subscales. Given the writing necessary for the digit symbol subscale, a potential confounding factor in the interpretation of this test is subtle changes in motor function. However, principal component analysis demonstrated that the digit symbol subscale loaded with the other cognitive measures, suggesting that the deficit in digit symbol function is correlated with cognitive performance as measured by the other subscales, and is not simply a matter of early motor function abnormality. Executive functions such as the picture arrangement test are thought to be mediated by striatocortical circuits, in particular connections between the caudate and prefrontal cortex. Thus, abnormalities in executive functions in PSGCs is consistent with the head of the caudate being the site of the earliest and most profound pathology in Huntington's disease. In addition, slower speed of movement and reaction time were seen for the PSGCs. On neurological examination, significantly more possible or definite abnormalities of oculomotor function (saccade accuracy and velocity, optokinetic nystagmus, and overall ocular movements), chorea, postural instability, and deficits in rapid alternating movements were also present. Whereas the statistical presence of these abnormalities could suggest that the PSGCs are in fact symptomatic, it is important to note that no participants classified as PSGCs exhibited definite chorea or other signs sufficient to warrant a diagnosis of manifest Huntington's disease.
The individual measures comprising the principal components were oculomotor function (component 1), speed of movement (component 2), motor function (component 3), cognitive function (component 4), and reaction time (component 5). Logistic regression analysis with these principal components demonstrated that the predictability of these measures to determine whether an at risk person is a Huntington's disease gene carrier is low. Possible reasons for this low predictability are: (1) the gene carriers in this sample are indeed presymptomatic and consequently, the differences between PSGCs and NGCs are subtle; (2) the specific signs experienced presymptomatically are not consistent for all gene carriers and thus, result in greater variability in the data; or (3) the signs loading on the different components act at different times or progress at different rates in the preclinical disease progression. Because greater than 98% of those carrying the mutated Huntington's disease gene are certain to develop disease, therapies that slow disease progression or delay or even prevent the onset of clinical disease have enormous potential importance. The concept of treating patients with asymptomatic conditions after identifying them as at high risk for developing clinical disease has been addressed in other medical disorders such as hypercholesterolaemia and hypertension. Identification of neuroprotective agents for presymptomatic treatment may have application for neurodegenerative diseases other than Huntington's disease, such as amyotrophic lateral sclerosis, Alzheimer's disease, or Parkinson's disease
Critical for the determination of when to administer the neuroprotective agents is the progression of signs leading to clinically symptomatic disease. In an attempt to delineate this progression, the principal components were evaluated in relation to the overall neurological score. Because our sample was cross sectional rather than longitudinal, a comparison of those with overall scores of 0, 1, or 2 is not truly a study of progression of preclinical signs. The actual progression of abnormalities can only be known with certainty from a longitudinal study. However, from our cross sectional data it seems that the oculomotor abnormalities comprising component 1 begin to worsen earlier in the disease process than the functional motor deficits such as chorea, rapid alternating movements, and gait and station stability. However, later in the disease process, when the overall at risk score is 2 (major soft signs of Huntington's disease), deficits in the functional motor component surpass the oculomotor component. This suggests that oculomotor deficits may precede the onset of equivocal functional motor deficits and supports the finding of a Venezuelan study that oculomotor deficits may be indicative of imminent manifest Huntington's disease.8 Interestingly, although the speed of movement and reaction times are slower for the PSGCs than the NGCs, they do not seem to worsen early in the disease process. The WAIS-R subscales for which the PSGCs scored significantly worse than the NGCs also did not seem to progress before overt disease onset. These findings suggest that there may be abnormalities present from early in life that do not progress until manifest disease develops.
The performance on the WAIS-R did not change with increasing abnormality in the overall at risk score, but was correlated with the length of CAG repeat for PSGCs even in this restricted repeat range (38–49 repeats). This suggests that Huntington's disease gene carriers with a larger number of CAG repeats experience deficits in cognitive performance before the onset of motor signs of Huntington's disease. Whether this implies that those with longer repeat lengths also experience more rapid decline in cognitive function cannot be determined with this cross sectional study. Although an understanding of the rate of progression of clinical symptomotology has the most obvious implications for the treatment of patients with clinically manifest Huntington's disease, further efforts to understand disease progression in presymptomatic people is clearly warranted. Longitudinal studies are essential to further characterise the subclinical markers of disease that we have identified to better define the disease process, allow for earlier diagnosis, and utilise the markers in the evaluation of agents designed to slow progression and delay the onset of clinical Huntington's disease and other neurodegenerative diseases.
Acknowledgments
This study was supported in part by grants R01-AG-08918, M01-RR-750, and PHS N01-NS-2326 from the Public Health Service, Washington, DC and by a Medical Genetics Training Grant Fellowship NICHD-t3HD07373 from the National Institute of Child Health and Human Development, Bethesda, MD (SCK). We thank all participants and members of the Huntington's disease roster for their efforts, and Dawn Kleindorfer and Cherie Bond for their assistance (Indiana University School of Medicine, Indianapolis).