Towards a Novel Class of Multitarget-Directed Ligands: Dual P2X7–NMDA Receptor Antagonists

 , ,
, ,
Abstract
:1. Introduction
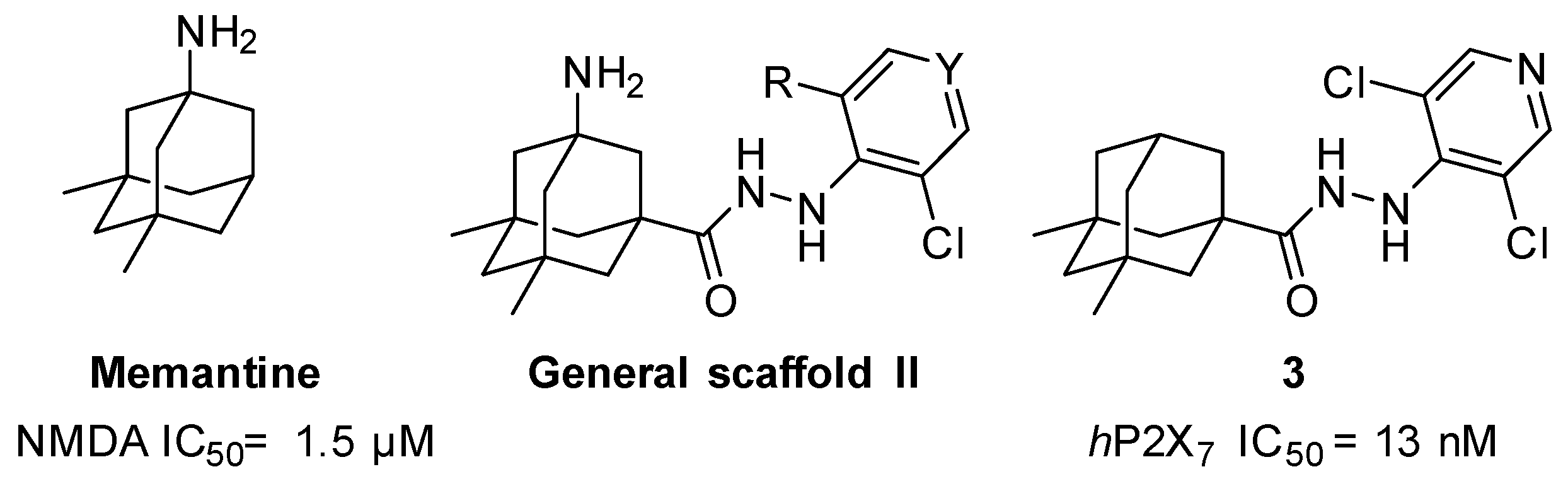
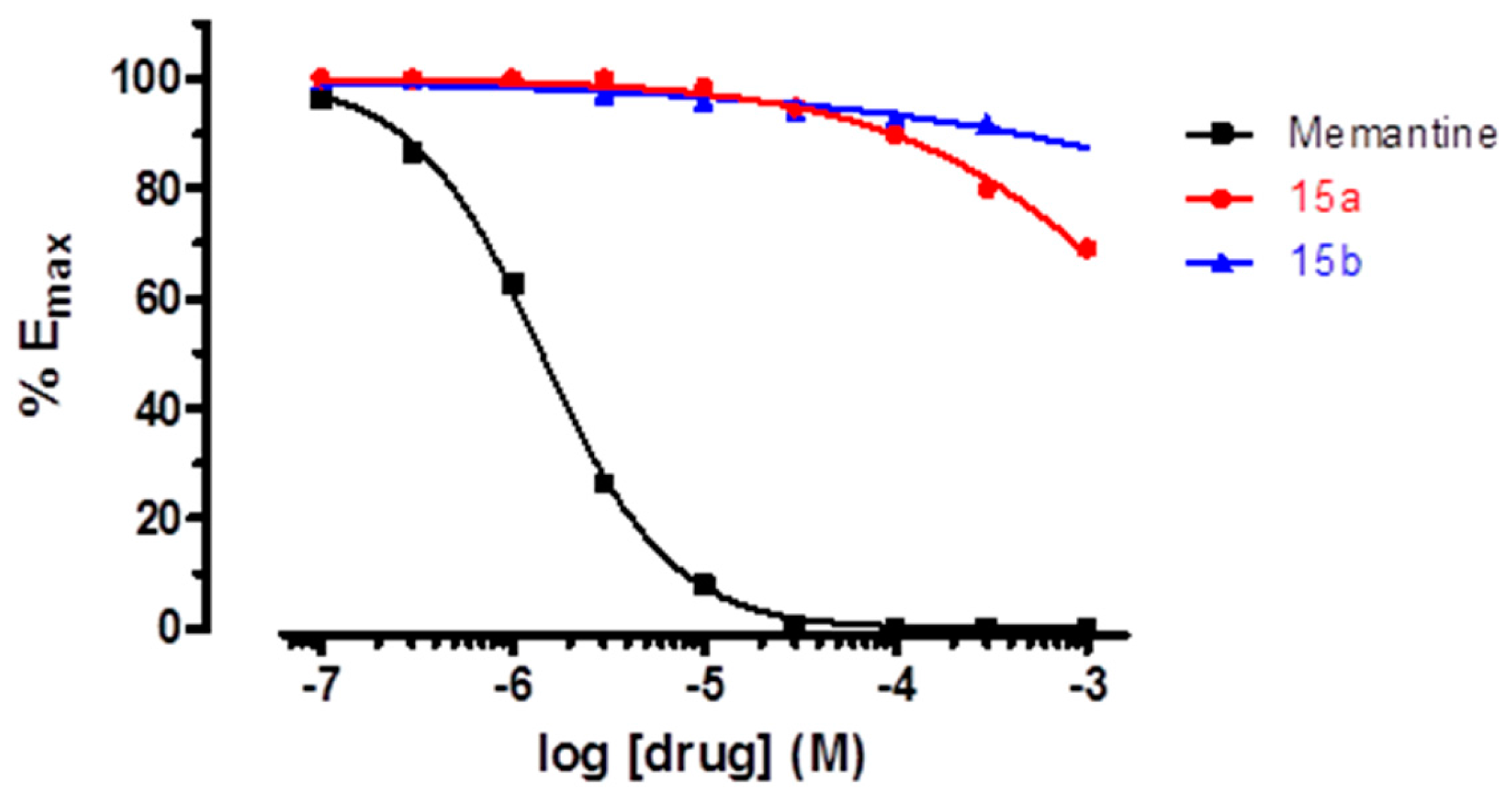
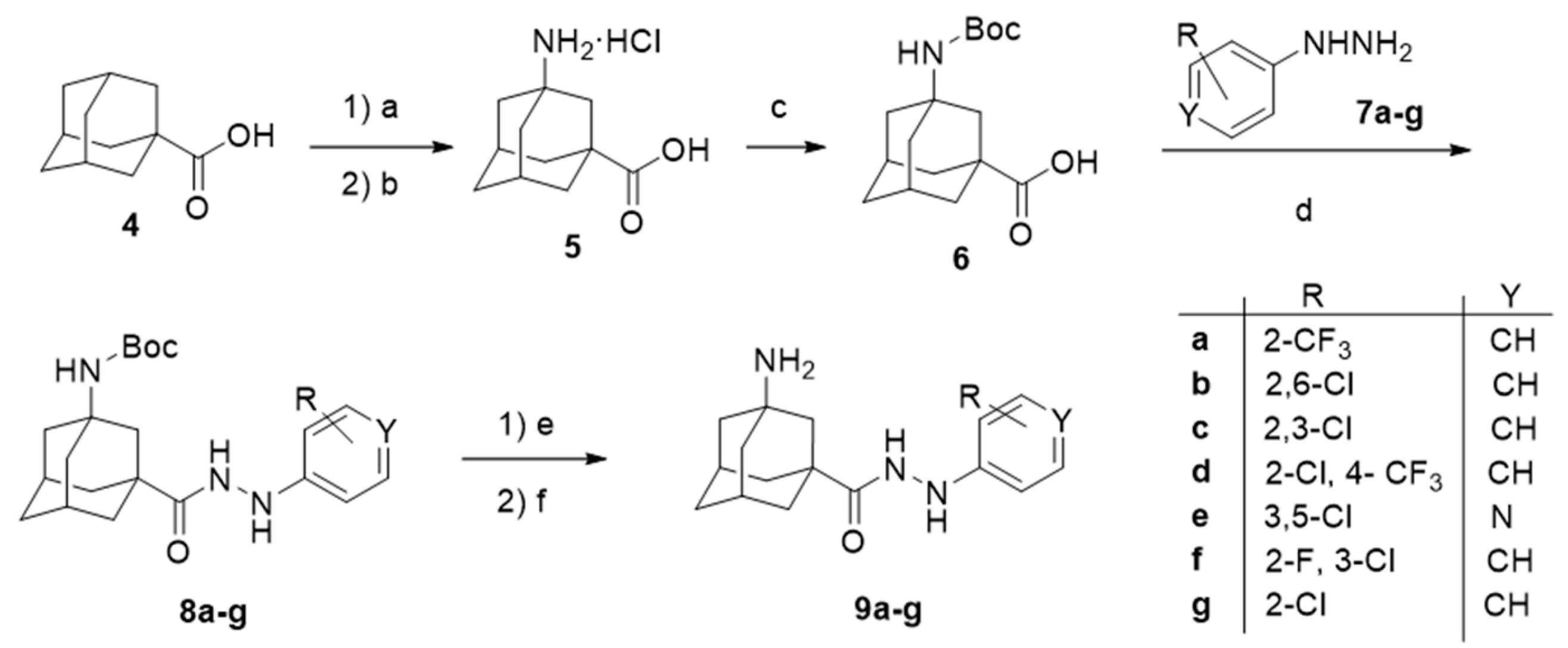
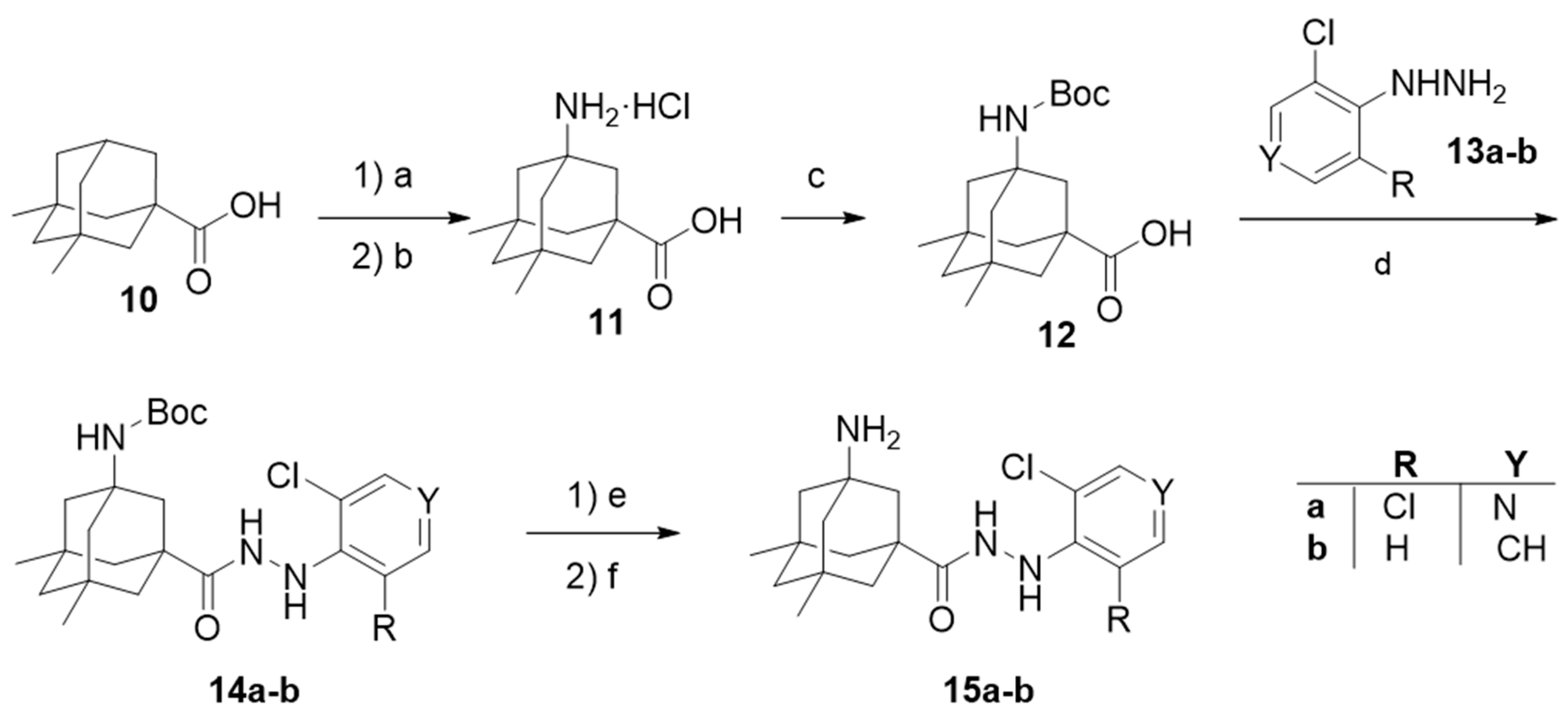
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. Chemical Synthesis
4.1.1. General Methods
4.1.2. Synthesis of the Starting Acids 6 and 12 [45]
4.1.3. General Procedures for the Synthesis of Carbohydrazides 9a–f or 15a–b.
4.1.4 Characterization Data for 2, 9a–f, and 15a–b.
4.2. P2X7 Receptor Antagonist Activity. Dye Uptake Using the Ethidium Ion Assay
4.3. NMDA Receptor Antagonist Activity
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Hickman, R.A.; Faustin, A.; Wisniewski, T. Alzheimer disease and its growing epidemic: Risk factors, biomarkers, and the urgent need for therapeutics. Neurol. Clin. 2016, 34, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Polinsky, R.J. Clinical pharmacology of rivastigmine: A new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin. Ther. 1998, 20, 634–647. [Google Scholar] [CrossRef]
- Cheewakriengkrai, L.; Gauthier, S. A 10-year perspective on donepezil. Expert Opin. Pharmacother. 2013, 14, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.; Bernabei, R.; Bullock, R.; Cruz Jentoft, A.J.; Frölich, L.; Hock, C.; Raivio, M.; Triau, E.; Vandewoude, M.; Came, E.; et al. Safety and efficacy of galantamine (Reminyl) in severe Alzheimer’s disease (the SERAD study): A randomized, placebo-controlled, double-blind trial. Lancet Neurol. 2009, 8, 39–47. [Google Scholar] [CrossRef]
- Danysz, W.; Parsons, C.G. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine—Searching for the connections. Br. J. Pharmacol. 2012, 167, 324–352. [Google Scholar] [CrossRef] [PubMed]
- Parsons, C.G.; Danysz, W.; Dekundy, A.; Pulte, I. Memantine and cholinesterase inhibitors : Complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox. Res. 2013, 24, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Mínguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Godyń, J.; Jończyk, J.; Panek, D.; Malawska, B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2016, 68, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Shakeri, A.; Rao, P.P.N. Amyloid cascade in Alzheimer’s disease: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2016, 113, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid β-protein: Synaptic and network dysfuntion. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wärmländer, S.K.; Gräslund, A.; Abrahams, J.P. Cross-interactions between the Alzheimer disease amyloid-β peptide and other amyloid proteins: A further aspect of the amyloid cascade hypothesis. J. Biol. Chem. 2016, 291, 16485–16493. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.R.; Owen, M.J. Alzheimer’s disease: The amyloid hypothesis on trial. Br. J. Phychiatry 2016, 208, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Deczkowska, A. neurological diseases as a failure of brain-immune crosstalk: The multiple faces of neuroinflammation. Trends Immunol. 2016, 37, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Sperlágh, B.; Illes, P. P2X7 receptor: An emerging target in central nervous system diseases. Trends Pharmacol. Sci. 2014, 35, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G. Physiopathological roles of P2X receptors in the central nervous system. Curr. Med. Chem. 2015, 22, 819–844. [Google Scholar] [CrossRef] [PubMed]
- Rech, J.C.; Bhattacharya, A.; Letavic, M.A.; Savall, B.M. The evolution of P2X7 antagonists with a focus on CNS indications. Bioorg. Med. Chem. Lett. 2016, 26, 3838–3845. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, D.J. Activation of microglia by amyloid β requires P2X7 receptor expression. J. Immunol. 2009, 182, 4378–4385. [Google Scholar] [CrossRef] [PubMed]
- Miras-Portugal, M.T.; Gomez-Villafuertes, R.; Gualix, J.; Diaz-Hernandez, J.I.; Artalejo, A.R.; Ortega, F.; Delicado, E.G.; Perez-Sen, R. Nucleotides in neuroregerenation and neuroprotection. Neuropharmacology 2016, 104, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.; Pérez-Sen, R.; Morente, V.; Delicado, E.G.; Miras-Portugal, M.T. P2X7, NMDA and BDNF receptors converge on GSK3 phosphorylation and cooperate to promote survival in cerebellar granule neurons. Cell. Mol. Life Sci. 2010, 67, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Delarasse, C.; Auger, R.; Gonnord, P.; Fontaine, B.; Kanellopoulos, J.M. The purinergic receptor P2X7 triggers alpha-secretase-dependent processing of the amyloid precursor protein. J. Biol. Chem. 2011, 286, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Miras-Portugal, M.T.; Diaz-Hernández, J.I.; Gomez-Villafuertes, R.; Diaz-Hernández, M.; Artalejo, A.R.; Gualix, J. Role of P2X7 and P2Y2 receptors on α-secretase-dependent APP processing: Control of amyloid plaques formation “in vivo” by P2X7 receptor. Comput. Struct. Biotechnol. J. 2015, 13, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Parvathenani, L.K.; Tertyshnikova, S.; Greco, C.R.; Roberts, S.B.; Robertson, B.; Posmantur, R.J. P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. Biol. Chem. 2003, 278, 13309–13317. [Google Scholar] [CrossRef] [PubMed]
- Ficker, C.; Rozmer, K.; Kató, E.; Andó, R.D.; Schumann, L.; Krügel, U.; Franke, H.; Sperlágh, B.; Riedel, T.; Illes, P. Astrocyte-neuron interaction in the substancia gelatinosa of the spinal cord dorsal horn via P2X7 receptor-mediated release of glutamate and reactive oxygen species. Glia 2014, 62, 1671–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervetto, C.; Alloisio, S.; Frattaroli, D.; Mazzotta, M.C.; Milanese, M.; Gavazzo, P.; Passalacqua, M.; Nobile, M.; Maura, G.; Marcoli, M. The P2X7 receptor as a route for non-exocytotic glutamate release: Dependence on the carboxyl tail. J. Neurochem. 2013, 124, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Marcoli, M.; Cervetto, C.; Paluzzi, P.; Guarnieri, S.; Alloisio, S.; Thellung, S.; Nobile, M.; Maura, G. P2X7 pre-synaptioc receptors in adult rat cerebrocortical nerve terminals: A role in ATP-induced glutamate release. J. Neurochem. 2008, 105, 2330–2342. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, F.J.; Bustos, F.J.; Inostroza, E.; Zúñiga, F.E.; Neve, R.L.; Montecino, M.; van Zundert, B. Differential roles of NMDA receptor subtypes NR2A and NR2B in dendritic branch development and requirement of RasGRF1. J. Neurophysiol. 2010, 103, 1758–1770. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Tomé, P.; Brera, B.; Arévalo, M.; de Ceballos, M.L. β–Amyloid25-35 inhibits glutamate uptake in cultured neurons and astrocytes: Modulation of uptake as a survival mechanism. Neurobiol. Dis. 2004, 15, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases—What is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.W.; Kotermanski, S.E. Mechanism of action of memantine. Curr. Opin. Pharmacol. 2006, 6, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Rauw, G.; Baker, G.B.; Kar, S. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur. J. Neurosci. 2008, 28, 1989–2002. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Minarini, A.; Melchiorre, C. Multi-target design strategies in the context of Alzheimer’s disease: Acetylcholinesterase inhibition and NMDA receptor antagonism as the driving forces. Neurochem. Res. 2014, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaur, M.; Chadha, N.; Silakari, O. Hybrids: A new paradigm to treat Alzheimer’s disease. Mol. Divers. 2016, 20, 271–297. [Google Scholar] [CrossRef] [PubMed]
- Blanpied, T.A.; Clarke, R.J.; Johnson, J.W. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neuroscience. 2005, 25, 3312–3322. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.W.; Sarris, K.; Kalvin, D.M.; Namovic, M.T.; Grayson, G.; Donnelly-Roberts, D.L.; Harris, R.; Honore, P.; Jarvis, M.F.; Faltynek, C.R.; et al. Structure-activity relationship studies on N’-aryl carbohydrazide P2X7 antagonists. J. Med. Chem. 2008, 51, 3030–3034. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.G.; Lee, S.D.; Cho, J.H.; Jung, Y.; Kim, J.H.; Hien, T.T.; Kang, K.W.; Ko, H.; Kim, Y.C. Structure-activity relationships and optimization of 3,5-dichloropyridine derivatives as novel P2X(7) receptor antagonists. J. Med. Chem. 2012, 55, 3687–3698. [Google Scholar] [CrossRef] [PubMed]
- Wanka, L.; Cabrelle, C.; Vanejews, M.; Schreiner, P.R. γ-Aminoadamantanecarboylic acids through direct C-H bond amidations. Eur. J. Org. Chem. 2007, 9, 1474–1490. [Google Scholar] [CrossRef]
- Keystone, E.C.; Wang, M.M.; Layton, M.; Hollis, S.; McInnes, I.B. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann. Rheum. Dis. 2012, 71, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Canudas, A.M.; Pubill, D.; Sureda, F.X.; Verdaguer, E.; Camps, P.; Muñoz-Torrero, D.; Jiménez, A.; Camins, A.; Pallàs, M. Neuroprotective effects of (±)-huprine Y on in vitro and in vivo models of excitotoxicity damage. Exp. Neurol. 2003, 180, 123–130. [Google Scholar] [CrossRef]
- Valverde, E.; Sureda, F.X.; Vázquez, S. Novel benzopolycyclic amines with NMDA receptor antagonist activity. Bioorg. Med. Chem. 2014, 22, 2678–2683. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound |  | % Inhibition or IC50 a,b | |
|---|---|---|---|
| R | R’ | ||
| no BzATP | - | - | 0% |
| only BzATP | - | - | 100% |
| AZD9056 c | - | - | 4.1 ± 0.7 nM d |
| 2 |  | H | NA e |
| 9a |  | H | 48%/37% |
| 9b |  | H | 23%/16% |
| 9c |  | H | 42%/17% |
| 9d |  | H | 24%/21% |
| 9e |  | H | 53%/27% |
| 9f |  | H | 15%/17% |
| 9g |  | H | 6.0 ± 0.6 μM d |
| 15a |  | CH3 | 9.9 ± 0.7 μM d |
| 15b |  | CH3 | 16.1 ± 1.6 μM d |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karoutzou, O.; Kwak, S.-H.; Lee, S.-D.; Martínez-Falguera, D.; Sureda, F.X.; Vázquez, S.; Kim, Y.-C.; Barniol-Xicota, M. Towards a Novel Class of Multitarget-Directed Ligands: Dual P2X7–NMDA Receptor Antagonists. Molecules 2018, 23, 230. https://doi.org/10.3390/molecules23010230
Karoutzou O, Kwak S-H, Lee S-D, Martínez-Falguera D, Sureda FX, Vázquez S, Kim Y-C, Barniol-Xicota M. Towards a Novel Class of Multitarget-Directed Ligands: Dual P2X7–NMDA Receptor Antagonists. Molecules. 2018; 23(1):230. https://doi.org/10.3390/molecules23010230
Chicago/Turabian StyleKaroutzou, Olga, Seung-Hwa Kwak, So-Deok Lee, Daina Martínez-Falguera, Francesc X. Sureda, Santiago Vázquez, Yong-Chul Kim, and Marta Barniol-Xicota. 2018. "Towards a Novel Class of Multitarget-Directed Ligands: Dual P2X7–NMDA Receptor Antagonists" Molecules 23, no. 1: 230. https://doi.org/10.3390/molecules23010230