
Naturally Occurring and Experimentally Induced Rhesus Macaque Models for Polycystic Ovary Syndrome: Translational Gateways to Clinical Application


Abstract
:1. Introduction
2. PCOS and Its Potential Origins
3. The Evidence for Genetic Origins of PCOS
4. The Evidence for Developmental Origins of PCOS from Clinical Studies
5. Attributes of Indian Female Rhesus Macaques Enhance Their Use in Clinical Translational Research, with Particular Relevance to PCOS
6. Female Indian Macaque Models of PCOS
7. Infant and Peripubertal Reproduction-Related Endocrine and Ovarian Characteristics Preceding Adult Onset of PCOS-Like Traits
8. Adult PCOS-Related Traits in Female Macaque Models
8.1. Reproductive Endocrine, Ovarian, Adrenal and Infertility Traits
8.2. Neuroendocrine PCOS-Related Traits in Macaque Models
8.3. Metabolic PCOS-Related Traits in Macaque Models
8.4. Behavioral PCOS-Related Traits in Macaque Models
8.5. Gestational and Placental Contributions to Transgenerational Transmission of PCOS-Like Traits
8.6. Translational Considerations
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J. International PCOS Network. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Fertil Steril. 2018, 110, 364–379. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, R.; Avery, J.; Moore, V.; Davies, M.; Azziz, R.; Stener-Victorin, E.; Moran, L.; Robertson, S.; Stepto, N.; Norman, R.; et al. Complex diseases and co-morbidities: Polycystic ovary syndrome and type 2 diabetes mellitus. Endocr. Connect. 2019, 8, R71–R75. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Oberfield, S.E.; Stener-Victorin, E.; Marshall, J.C.; Laven, J.S.; Legro, R.S. Scientific Statement on the Diagnostic Criteria, Epidemiology, Pathophysiology, and Molecular Genetics of Polycystic Ovary Syndrome. Endocr. Rev. 2015, 36, 487–525. [Google Scholar] [CrossRef] [PubMed]
- Bozdag, G.; Mumusoglu, S.; Zengin, D.; Karabulut, E.; Yildiz, B.O. The prevalence and phenotypic features of polycystic ovary syndrome: A systematic review and meta-analysis. Hum. Reprod. 2016, 31, 2841–2855. [Google Scholar] [CrossRef] [PubMed]
- Lizneva, D.; Suturina, L.; Walker, W.; Brakta, S.; Gavrilova-Jordan, L.; Azziz, R. Criteria, prevalence, and phenotypes of polycystic ovary syndrome. Fertil. Steril. 2016, 106, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Neven, A.C.H.; Laven, J.; Teede, H.J.; Boyle, J.A. A Summary on Polycystic Ovary Syndrome: Diagnostic Criteria, Prevalence, Clinical Manifestations, and Management According to the Latest International Guidelines. Semin. Reprod. Med. 2018, 3, 5–12. [Google Scholar] [CrossRef]
- Escobar-Morreale, H.F. Polycystic ovary syndrome: Definition, aetiology, diagnosis and treatment. Nat. Rev. Endocrinol. 2018, 14, 270–284. [Google Scholar] [CrossRef]
- Azziz, R.; Marin, C.; Hoq, L.; Badamgarav, E.; Song, P. Health care-related economic burden of the polycystic ovary syndrome during the reproductive life span. J. Clin. Endocrinol. Metab. 2005, 90, 4650–4658. [Google Scholar] [CrossRef]
- Fraissinet, A.; Robin, G.; Pigny, P.; Lefebvre, T.; Catteau-Jonard, S.; Dewailly, D. Use of the serum anti-Müllerian hormone assay as a surrogate for polycystic ovarian morphology: Impact on diagnosis and phenotypic classification of polycystic ovary syndrome. Hum. Reprod. 2017, 32, 1716–1722. [Google Scholar] [CrossRef]
- Lizneva, D.; Kirubakaran, R.; Mykhalchenko, K.; Suturina, L.; Chernukha, G.; Diamond, M.P.; Azziz, R. Phenotypes and body mass in women with polycystic ovary syndrome identified in referral versus unselected populations: Systematic review and meta-analysis. Fertil. Steril. 2016, 106, 1510–1520. [Google Scholar] [CrossRef]
- Abbott, D.H.; Dumesic, D.A.; Levine, J.E. Hyperandrogenic origins of polycystic ovary syndrome—Implications for pathophysiology and therapy. Expert Rev. Endocrinol. Metab. 2019, 14, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.H.; Tarantal, A.F.; Dumesic, D.A. Fetal, infant, adolescent and adult phenotypes of polycystic ovary syndrome in prenatally androgenized female rhesus monkeys. Am. J. Primatol. 2009, 71, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.H.; Rayome, B.H.; Dumesic, D.A.; Lewis, K.C.; Edwards, A.K.; Wallen, K.; Wilson, M.E.; Appt, S.E.; Levine, J.E. Clustering of PCOS-like traits in naturally hyperandrogenic female rhesus monkeys. Hum. Reprod. 2017, 32, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Gorsic, L.K.; Kosova, G.; Werstein, B.; Sisk, R.; Legro, R.S.; Hayes, M.G.; Teixeira, J.M.; Dunaif, A.; Urbanek, M. Pathogenic Anti-Müllerian Hormone Variants in Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 2862–2872. [Google Scholar] [CrossRef] [PubMed]
- Gorsic, L.K.; Dapas, M.; Legro, R.S.; Hayes, M.G.; Urbanek, M. Functional Genetic Variation in the Anti-Müllerian Hormone Pathway in Women with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 2855–2874. [Google Scholar] [CrossRef] [PubMed]
- Dapas, M.; Sisk, R.; Legro, R.S.; Urbanek, M.; Dunaif, A.; Hayes, M.G. Family-based quantitative trait meta-analysis implicates rare noncoding variants in DENND1A in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 3835–3850. [Google Scholar] [CrossRef]
- Vink, J.M.; Sadrzadeh, S.; Lambalk, C.B.; Boomsma, D.I. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J. Clin. Endocrinol Metab. 2006, 91, 2100–2104. [Google Scholar] [CrossRef]
- Crisosto, N.; Ladrón de Guevara, A.; Echiburú, B.; Maliqueo, M.; Cavada, G.; Codner, E.; Paez, F.; Sir-Petermann, T. Higher luteinizing hormone levels associated with antimüllerian hormone in postmenarchal daughters of women with polycystic ovary syndrome. Fertil. Steril. 2019, 111, 381–388. [Google Scholar] [CrossRef]
- Legro, R.S.; Driscoll, D.; Strauss, J.F., III; Fox, J.; Dunaif, A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc. Natl. Acad. Sci. USA 1998, 95, 14956–14960. [Google Scholar] [CrossRef]
- Nasiri Amiri, F.; Ramezani Tehrani, F.; Esmailzadeh, S.; Tohidi, M.; Azizi, F.; Basirat, Z. Sexual function in women with polycystic ovary syndrome and their hormonal and clinical correlations. Int. J. Impot. Res. 2018, 30, 54–61. [Google Scholar] [CrossRef]
- Pastoor, H.; Timman, R.; de Klerk, C.M.; Bramer, W.; Laan, E.T.; Laven, J.S. Sexual function in women with polycystic ovary syndrome: A systematic review and meta-analysis. Reprod. Biomed. Online 2018, 37, 750–760. [Google Scholar] [CrossRef]
- Dokras, A.; Stener-Victorin, E.; Yildiz, B.O.; Li, R.; Ottey, S.; Shah, D.; Epperson, N.; Teede, H. Androgen Excess- Polycystic Ovary Syndrome Society: Position statement on depression, anxiety, quality of life, and eating disorders in polycystic ovary syndrome. Fertil. Steril. 2018, 109, 888–899. [Google Scholar] [CrossRef]
- Mykhalchenko, K.; Lizneva, D.; Trofimova, T.; Walker, W.; Suturina, L.; Diamond, M.P.; Azziz, R. Genetics of polycystic ovary syndrome. Expert Rev. Mol. Diagn. 2017, 17, 723–733. [Google Scholar] [CrossRef]
- Dunaif, A. Perspectives in Polycystic Ovary Syndrome: From Hair to Eternity. J. Clin. Endocrino.l Metab. 2016, 101, 759–768. [Google Scholar] [CrossRef]
- Burt Solorzano, C.M.; Beller, J.P.; Abshire, M.Y.; Collins, J.S.; McCartney, C.R.; Marshall, J.C. Neuroendocrine dysfunction in polycystic ovary syndrome. Steroids 2012, 77, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Phan, J.D.; Leung, K.L.; Grogan, T.R.; Ding, X.; Li, X.; Hoyos, L.R.; Abbott, D.H.; Chazenbalk, G.D. Adipose Insulin Resistance in Normal-Weight Women With Polycystic Ovary Syndrome. J. Clin. Endocrino.l Metab. 2019, 104, 2171–2183. [Google Scholar] [CrossRef] [PubMed]
- Rosenfield, R.L.; Ehrmann, D.A. The Pathogenesis of Polycystic Ovary Syndrome (PCOS): The Hypothesis of PCOS as Functional Ovarian Hyperandrogenism Revisited. Endocr. Rev. 2016, 37, 467–520. [Google Scholar] [CrossRef]
- Day, F.; Karaderi, T.; Jones, M.R.; Meun, C.; He, C.; Drong, A.; Kraft, P.; Lin, N.; Huang, H.; Broer, L.; et al. Large-scale genome-wide meta-analysis of polycystic ovary syndrome suggests shared genetic architecture for different diagnosis criteria. PLoS Genet. 2018, 14, e1007813. [Google Scholar] [CrossRef]
- Barrett, E.S.; Hoeger, K.M.; Sathyanarayana, S.; Abbott, D.H.; Redmon, J.B.; Nguyen, R.H.N.; Swan, S.H. Anogenital distance in newborn daughters of women with polycystic ovary syndrome indicates fetal testosterone exposure. J. Dev. Orig. Health. Dis. 2018, 9, 307–314. [Google Scholar] [CrossRef]
- Homburg, R.; Gudi, A.; Shah, A.; M Layton, A. A novel method to demonstrate that pregnant women with polycystic ovary syndrome hyper-expose their fetus to androgens as a possible stepping stone for the developmental theory of PCOS. A pilot study. Reprod. Biol. Endocrinol. 2017, 15, 61. [Google Scholar] [CrossRef]
- Sir-Petermann, T.; Codner, E.; Maliqueo, M.; Echiburú, B.; Hitschfeld, C.; Crisosto, N.; Pérez-Bravo, F.; Recabarren, S.E.; Cassorla, F. Increased anti-Müllerian hormone serum concentrations in prepubertal daughters of women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 3105–3109. [Google Scholar] [CrossRef]
- Detti, L.; Christiansen, M.E.; Francillon, L.; Ikuwezunma, G.; Diamond, M.P.; Mari, G.; Tobiasz, A.M. Serum Anti-Müllerian hormone (AMH) in mothers with polycystic ovary syndrome (PCOS) and their term fetuses. Syst. Biol. Reprod. Med. 2019, 65, 147–154. [Google Scholar] [CrossRef]
- Torchen, L.C.; Legro, R.S.; Dunaif, A. Distinctive Reproductive Phenotypes in Peripubertal Girls at Risk for Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 3355–3361. [Google Scholar] [CrossRef]
- Abbott, D.H.; Vepraskas, S.H.; Horton, T.H.; Terasawa, E.; Levine, J.E. Accelerated Episodic Luteinizing Hormone Release Accompanies Blunted Progesterone Regulation in PCOS-like Female Rhesus Monkeys (Macaca Mulatta) Exposed to Testosterone during Early-to-Mid Gestation. Neuroendocrinology 2018, 107, 133–146. [Google Scholar] [CrossRef]
- McGee, W.K.; Bishop, C.V.; Pohl, C.R.; Chang, R.J.; Marshall, J.C.; Pau, F.K.; Stouffer, R.L.; Cameron, J.L. Effects of hyperandrogenemia and increased adiposity on reproductive and metabolic parameters in young adult female monkeys. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1292–E1304. [Google Scholar] [CrossRef] [Green Version]
- Torchen, L.C.; Idkowiak, J.; Fogel, N.R.; O’Neil, D.M.; Shackleton, C.H.; Arlt, W.; Dunaif, A. Evidence for Increased 5α-Reductase Activity During Early Childhood in Daughters of Women With Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2016, 101, 2069–2075. [Google Scholar] [CrossRef]
- Ibáñez, L.; Oberfield, S.E.; Witchel, S.; Auchus, R.J.; Chang, R.J.; Codner, E.; Dabadghao, P.; Darendeliler, F.; Elbarbary, N.S.; Gambineri, A.; et al. An International Consortium Update: Pathophysiology, Diagnosis, and Treatment of Polycystic Ovarian Syndrome in Adolescence. Horm. Res. Paediatr. 2017, 88, 371–395. [Google Scholar] [CrossRef]
- Witchel, S.F.; Oberfield, S.E.; Peña, A.S. Polycystic Ovary Syndrome: Pathophysiology, Presentation, and Treatment With Emphasis on Adolescent Girls. J. Endocr. Soc. 2019, 3, 1545–1573. [Google Scholar] [CrossRef]
- Jones, M.R.; Goodarzi, M.O. Genetic determinants of polycystic ovary syndrome: Progress and future directions. Fertil. Steril. 2016, 106, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.J.; Zhao, H.; He, L.; Shi, Y.H.; Qin, Y.Y.; Shi, Y.Y.; Li, Z.Q.; You, L.; Zhao, J.L.; Liu, J.Y.; et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat. Genet. 2011, 43, 55–59. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, H.; Chen, Z.J. Genome-Wide Association Studies for Polycystic Ovary Syndrome. Semin. Reprod. Med. 2016, 34, 224–229. [Google Scholar] [CrossRef]
- Jones, M.R.; Chua, A.K.; Mengesha, E.A.; Taylor, K.D.; Chen, Y.-D.I.; Li, X.H.; Krauss, R.M.; Rotter, J.I.; Legro, R.S.; Azziz, R.; et al. Metabolic and cardiovascular genes in polycystic ovary syndrome: A candidate-wide association study (CWAS). Steroids 2012, 77, 317–322. [Google Scholar] [CrossRef] [Green Version]
- McAllister, J.M.; Modi, B.; Miller, B.A.; Biegler, J.; Bruggeman, R.; Legro, R.S.; Strauss, J.F., III. Overexpression of a DENND1A isoform produces a polycystic ovary syndrome theca phenotype. Proc. Natl. Acad. Sci. USA 2014, 111, E1519–E1527. [Google Scholar] [CrossRef] [Green Version]
- Diamanti-Kandarakis, E.; Dunaif, A. Insulin resistance and the polycystic ovary syndrome revisited: An update on mechanisms and implications. Endocr. Rev. 2012, 33, 981–1030. [Google Scholar] [CrossRef]
- Sir-Petermann, T.; Angel, B.; Maliqueo, M.; Carvajal, F.; Santos, J.; Pérez-Bravo, F. Prevalence of Type II diabetes mellitus and insulin resistance in parents of women with polycystic ovary syndrome. Diabetologia 2002, 45, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-mapping T2D loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef] [Green Version]
- Meigs, J.B.; Cupples, L.A.; Wilson, P.W. Parental transmission of T2D: The Framingham Offspring Study. Diabetes 2000, 49, 2201–2207. [Google Scholar] [CrossRef] [Green Version]
- Strauss, J.F., III. Some new thoughts on the pathophysiology and genetics of polycystic ovary syndrome. Ann. N.Y. Acad. Sci. 2003, 997, 42–48. [Google Scholar] [CrossRef]
- Hayes, M.G.; Urbanek, M.; Ehrmann, D.A.; Armstrong, L.L.; Lee, J.Y.; Sisk, R.; Franks, S.; Lindgren, C.M.; Welt, C.K.; Diamanti-Kandarakis, E. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat. Commun. 2015, 6, 7502. [Google Scholar] [CrossRef] [Green Version]
- Teede, H.J.; Joham, A.E.; Paul, E.; Moran, L.J.; Loxton, D.; Jolley, D.; Lombard, C. Longitudinal weight gain in women identified with polycystic ovary syndrome: Results of an observational study in young women. Obesity 2013, 21, 1526–1532. [Google Scholar] [CrossRef]
- Lim, S.S.; Hutchison, S.K.; Van Ryswyk, E.; Norman, R.J.; Teede, H.J.; Moran, L.J. Lifestyle changes in women with polycystic ovary syndrome. Cochrane. Database Syst. Rev. 2019. [Google Scholar] [CrossRef]
- Lambertini, L.; Saul, S.R.; Copperman, A.B.; Hammerstad, S.S.; Yi, Z.; Zhang, W.; Tomer, Y.; Kase, N. Intrauterine Reprogramming of the Polycystic Ovary Syndrome: Evidence from a Pilot Study of Cord Blood Global Methylation Analysis. Front. Endocrinol. 2017, 8, 352. [Google Scholar] [CrossRef] [Green Version]
- Hiam, D.; Simar, D.; Laker, R.; Altıntaş, A.; Gibson-Helm, M.; Fletcher, E.; Moreno-Asso, A.; Trewin, A.J.; Barres, R.; Stepto, N.K. Epigenetic reprogramming of immune cells in women with PCOS impact genes controlling reproductive function. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef]
- Sagvekar, P.; Kumar, P.; Mangoli, V.; Desai, S.; Mukherjee, S. DNA methylome profiling of granulosa cells reveals altered methylation in genes regulating vital ovarian functions in polycystic ovary syndrome. Clin. Epigenetics. 2019, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Palomba, S.; Marotta, R.; Di Cello, A.; Russo, T.; Falbo, A.; Orio, F.; Tolino, A.; Zullo, F.; Esposito, R.; La Sala, G.B. Pervasive developmental disorders in children of hyperandrogenic women with polycystic ovary syndrome: A longitudinal case-control study. Clin. Endocrinol. 2012, 77, 898–904. [Google Scholar] [CrossRef]
- Schindler, A.E. Hormones in human amniotic fluid. Monogr. Endocrinol. 1982, 21, 1–158. [Google Scholar]
- Wang, R.; Hartmann, M.F.; Tiosano, D.; Wudy, S.A. Characterizing the steroidal milieu in amniotic fluid of mid-gestation: A GC-MS study. J. Steroid Biochem. Mol. Biol. 2019, 193, 105412. [Google Scholar] [CrossRef]
- Berenbaum, S.A.; Beltz, A.M. Sexual differentiation of human behavior: Effects of prenatal and pubertal organizational hormones. Front Neuroendocrinol. 2011, 32, 183–200. [Google Scholar] [CrossRef]
- Wu, Y.; Zhong, G.; Chen, S.; Zheng, C.; Liao, D.; Xie, M. Polycystic ovary syndrome is associated with anogenital distance, a marker of prenatal androgen exposure. Hum. Reprod. 2017, 32, 937–943. [Google Scholar] [CrossRef]
- Sánchez-Ferrer, M.L.; Mendiola, J.; Hernández-Peñalver, A.I.; Corbalán-Biyang, S.; Carmona-Barnosi, A.; Prieto-Sánchez, M.T.; Nieto, A.; Torres-Cantero, A.M. Presence of polycystic ovary syndrome is associated with longer anogenital distance in adult Mediterranean women. Hum. Reprod. 2017, 32, 2315–2323. [Google Scholar] [CrossRef] [Green Version]
- Simsir, C.; Kuru Pekcan, M.; Aksoy, R.T.; Ecemis, T.; Coskun, B.; Hamdemir Kilic, S.; Tokmak, A. The ratio of anterior anogenital distance to posterior anogenital distance: A novel biomarker for polycystic ovary syndrome. J. Chin. Med. Assoc. 2019. [Google Scholar] [CrossRef]
- VÁzquez-martÍnez, E.R.; Gómez-Viais, Y.I.; García-Gómez, E.; Reyes-Mayoral, C.; Reyes-Muñoz, E.; Camacho-Arroyo, I.; Cerbón, M.A. DNA Methylation in the Pathogenesis of Polycystic Ovary Syndrome. Reproduction 2019. [Google Scholar] [CrossRef]
- Jacobsen, V.M.; Li, S.; Wang, A.; Zhu, D.; Liu, M.; Thomassen, M.; Kruse, T.; Tan, Q. Epigenetic association analysis of clinical sub-phenotypes in patients with polycystic ovary syndrome (PCOS). Gynecol. Endocrinol. 2019, 35, 691–694. [Google Scholar] [CrossRef]
- Weiss, P.A.; Scholz, H.S.; Haas, J.; Tamussino, K.F.; Seissler, J.; Borkenstein, M.H. Long-term follow-up of infants of mothers with type 1 diabetes: Evidence for hereditary and nonhereditary transmission of diabetes and precursors. Diabetes Care. 2000, 23, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Sacks, D.A. The global burden of hyperglycemia in pregnancy—Trends from studies in the last decade. Diabetes Res. Clin. Pract. 2018, 145, 17–19. [Google Scholar] [CrossRef]
- Lowe, W.L., Jr.; Scholtens, D.M.; Kuang, A.; Linder, B.; Lawrence, J.M.; Lebenthal, Y.; McCance, D.; Hamilton, J.; Nodzenski, M.; Talbot, O.; et al. HAPO Follow-up Study Cooperative Research Group. Hyperglycemia and Adverse Pregnancy Outcome Follow-up Study (HAPO FUS): Maternal Gestational Diabetes Mellitus and Childhood Glucose Metabolism. Diabetes Care. 2019, 42, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Kelley, A.S.; Smith, Y.R.; Padmanabhan, V. A narrative review of placental contribution to adverse pregnancy outcomes in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef]
- Allvin, K.; Ankarberg-Lindgren, C.; Niklasson, A.; Jacobsson, B.; Dahlgren, J. Altered umbilical sex steroids in preterm infants born small for gestational age. J. Matern. Fetal. Neonatal. Med. 2019, 18, 1–7. [Google Scholar] [CrossRef] [Green Version]
- de Zegher, F.; López-Bermejo, A.; Ibáñez, L. Central Obesity, Faster Maturation, and ‘PCOS’ in Girls. Trends Endocrinol. Metab. 2018, 29, 815–818. [Google Scholar] [CrossRef]
- Mas-Parés, B.; Xargay-Torrent, S.; Bonmatí, A.; Lizarraga-Mollinedo, E.; Martínez-Calcerrada, J.M.; Carreras-Badosa, G.; Prats-Puig, A.; de Zegher, F.; Ibáñez, L.; López-Bermejo, A.; et al. Umbilical cord microRNA in small-for-gestational age children and association with catch-up growth: A pilot study. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef]
- Li, S.; Zhu, D.; Duan, H.; Tan, Q. The epigenomics of polycystic ovarian syndrome: From pathogenesis to clinical manifestations. Gynecol. Endocrinol. 2016, 32, 942–946. [Google Scholar] [CrossRef]
- Barbour, L.A.; Scifres, C.; Valent, A.M.; Friedman, J.E.; Buchanan, T.A.; Coustan, D.; Aagaard, K.; Thornburg, K.L.; Catalano, P.M.; Galan, H.L.; et al. A cautionary response to SMFM statement: Pharmacological treatment of gestational diabetes. Am. J. Obstet Gynecol. 2018, 219, 367.e1–367.e7. [Google Scholar] [CrossRef]
- Zheng, W.; Huang, W.; Zhang, L.; Tian, Z.; Yan, Q.; Wang, T.; Zhang, L.; Li, G. Early pregnancy metabolic factors associated with gestational diabetes mellitus in normal-weight women with polycystic ovary syndrome: A two-phase cohort study. Diabetol. Metab. Syndr. 2019, 11, 71. [Google Scholar] [CrossRef] [Green Version]
- Kent, J.; Dodson, W.C.; Kunselman, A.; Pauli, J.; Stone, A.; Diamond, M.P.; Coutifaris, C.; Schlaff, W.D.; Alvero, R.; Casson, P.; et al. Reproductive Medicine Network. Gestational Weight Gain in Women With Polycystic Ovary Syndrome: A Controlled Study. J. Clin. Endocrinol. Metab. 2018, 103, 4315–4323. [Google Scholar] [CrossRef]
- Abbott, D.H.; Bruns, C.R.; Barnett, D.K.; Dunaif, A.; Goodfriend, T.L.; Dumesic, D.A.; Tarantal, A.F. Experimentally induced gestational androgen excess disrupts glucoregulation in rhesus monkey dams and their female offspring. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E741–E751. [Google Scholar] [CrossRef] [Green Version]
- Villarroel, C.; Salinas, A.; López, P.; Kohen, P.; Rencoret, G.; Devoto, L.; Codner, E. Pregestational type 2 diabetes and gestational diabetes exhibit different sexual steroid profiles during pregnancy. Gynecol. Endocrinol. 2017, 33, 212–217. [Google Scholar] [CrossRef]
- Bimber, B.N.; Yan, M.Y.; Peterson, S.M.; Ferguson, B. mGAP: The macaque genotype and phenotype resource, a framework for accessing and interpreting macaque variant data, and identifying new models of human disease. BMC Genomics. 2019, 20, 176. [Google Scholar] [CrossRef] [Green Version]
- Phillips, K.A.; Bales, K.L.; Capitanio, J.P.; Conley, A.; Czoty, P.W.; ‘t Hart, B.A.; Hopkins, W.D.; Hu, S.; Miller, L.A.; Nader, M.A.; et al. Why Primate Models Matter. Am. J. Primatol. 2014, 76, 801–827. [Google Scholar] [CrossRef] [Green Version]
- Arifin, E.; Shively, C.A.; Register, T.C.; Cline, J.M. Polycystic ovary syndrome with endometrial hyperplasia in a cynomolgus monkey (Macaca fascicularis). Vet. Pathol. 2008, 45, 512–515. [Google Scholar] [CrossRef] [Green Version]
- Francis, P.J.; Appukuttan, B.; Simmons, E.; Landauer, N.; Stoddard, J.; Hamon, S.; Ott, J.; Ferguson, B.; Klein, M.; Stout, J.T.; et al. Rhesus monkeys and humans share common susceptibility genes for age-related macular disease. Hum. Mol. Genet. 2008, 17, 2673–2680. [Google Scholar] [CrossRef] [Green Version]
- Rogers, J.; Raveendran, M.; Fawcett, G.L.; Fox, A.S.; Shelton, S.E.; Oler, J.A.; Cheverud, J.; Muzny, D.M.; Gibbs, R.A.; Davidson, R.J.; et al. CRHR1 genotypes, neural circuits and the diathesis for anxiety and depression. Mol. Psychiatry 2013, 18, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Oler, J.A.; Fox, S.A.; Shelton, S.E.; Rogers, J.; Dyer, T.D.; Davidson, R.J.; Shelledy, W.; Oakes, T.R.; Blangero, J.; Kalin, N.H. Amygdalar and hippocampal substrates of anxious temperament differ in their heritability. Nature 2010, 129, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Dray, B.K.; Raveendran, M.; Harris, R.A.; Benavides, F.; Gray, S.B.; Carlos, J.; Perez, C.J.; McArthur, M.J.; Williams, L.E.; Baze, W.B.; et al. Mismatch repair gene mutations lead to lynch syndrome colorectal cancer in rhesus macaques. Genes Cancer 2018, 9, 142–152. [Google Scholar] [PubMed] [Green Version]
- Moshiri, A.; Chen, R.; Kim, S.; Harris, R.A.; Li, Y.; Raveendran, M.; Davis, S.; Liang, Q.; Pomerantz, O.; Wang, J.; et al. A nonhuman primate model of inherited retinal disease. J. Clin. Invest. 2019, 129, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Fawcett, G.L.; Dettmer, A.M.; Kay, D.; Raveendran, M.; Higley, J.D.; Ryan, N.D.; Cameron, J.L.; Rogers, J. Quantitative Genetics of Response to Novelty and Other Stimuli by Infant Rhesus Macaques (Macaca mulatta) Across Three Behavioral Assessments. Int. J. Primatol. 2014, 35, 325–339. [Google Scholar] [CrossRef] [Green Version]
- Xue, C.; Raveendran, M.; Harris, R.A.; Fawcett, G.L.; Liu, X.; White, S.; Dahdouli, M.; Deiros, D.R.; Below, J.E.; Salerno, W.; et al. The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences. Genome Res. 2016, 26, 1651–1662. [Google Scholar] [CrossRef] [Green Version]
- Barnett, D.K.; Abbott, D.H. Reproductive adaptations to a large-brained fetus open a vulnerability to anovulation similar to polycystic ovary syndrome. Am. J. Hum. Biol. 2003, 15, 296–319. [Google Scholar] [CrossRef]
- Kenealy, B.P.; Terasawa, E. Hypothalamic control of female reproduction. In Oxford Research Encyclopedias, Neuroscience, Neuroendocrine and Autonomic Systems; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- Terasawa, E. Control of luteinizing hormone-releasing hormone pulse generation in nonhuman primates. Cell Mol. Neurobiol. 1995, 15, 141–164. [Google Scholar] [CrossRef]
- Knobil, E. The GnRH pulse generator. Am. J. Obstet Gynecol. 1990, 163, 1721–1727. [Google Scholar] [CrossRef]
- Krey, L.C.; Butler, W.R.; Knobil, E. Surgical disconnection of the medial basal hypothalamus and pituitary function in the rhesus monkey. I. Gonadotropin secretion. Endocrinology 1975, 96, 1073. [Google Scholar] [CrossRef]
- Plant, T.M.; Krey, L.C.; Moossy, J.; McCormack, J.T.; Hess, D.L.; Knobil, E. The arcuate nucleus and the control of gonadotropin and prolactin secretion in the female rhesus monkey (Macaca mulatta). Endocrinology 1978, 102, 52–62. [Google Scholar] [CrossRef]
- Chaffin, C.L.; Vandevoort, C.A. Follicle growth, ovulation, and luteal formation in primates and rodents: A comparative perspective. Exp. Biol. Med. 2013, 238, 539–548. [Google Scholar] [CrossRef]
- Stouffer, R.L.; Chandrasekher, Y.A.; Slayden, O.D.; Zelinski-Wooten, M.B. Gonadotrophic and local control of the developing corpus luteum in rhesus monkeys. Hum. Reprod. 1993, 8 (Suppl. 2), 107–111. [Google Scholar] [CrossRef] [Green Version]
- Zeleznik, A.J.; Somers, J.P. Regulation of the Primate Corpus Luteum: Cellular and Molecular Perspectives. Trends Endocrinol. Metab. 1999, 10, 189–193. [Google Scholar] [CrossRef]
- Zeleznik, A.J. In vivo responses of the primate corpus luteum to luteinizing hormone and chorionic gonadotropin. Proc. Natl. Acad. Sci USA 1998, 95, 11002–11007. [Google Scholar] [CrossRef] [Green Version]
- Michael, A.E.; Abayasekara, D.R.; Webley, G.E. Cellular mechanisms of luteolysis. Mol. Cell. Endocrinol. 1994, 99, R1–R9. [Google Scholar] [CrossRef]
- Nio-Kobayashi, J.; Kudo, M.; Sakuragi, N.; Iwanaga, T.; Duncan, W.C. Loss of luteotropic prostaglandin E plays an important role in the regulation of luteolysis in women. Mol. Hum. Reprod. 2017, 23, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Castracane, V.D.; Moore, G.T.; Shaikh, A.A. Ovarian function of hysterectomized Macaca fascicularis. Biol. Reprod. 1979, 20, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Abbott, D.H.; Barnett, D.K.; Levine, J.E.; Padmanabhan, V.; Dumesic, D.A.; Jacoris, S.; Tarantal, A.F. Endocrine antecedents of polycystic ovary syndrome in fetal and infant prenatally androgenized female rhesus monkeys. Biol. Reprod. 2008, 79, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Koster, M.P.; de Wilde, M.A.; Veltman-Verhulst, S.M.; Houben, M.L.; Nikkels, P.G.; van Rijn, B.B.; Fauser, B.C. Placental characteristics in women with polycystic ovary syndrome. Hum. Reprod. 2015, 30, 2829–2837. [Google Scholar] [CrossRef] [Green Version]
- Palomba, S.; de Wilde, M.A.; Falbo, A.; Koster, M.P.; La Sala, G.B.; Fauser, B.C. Pregnancy complications in women with polycystic ovary syndrome. Hum. Reprod. Update 2015, 21, 575–592. [Google Scholar] [CrossRef] [Green Version]
- Maliqueo, M.; Lara, H.E.; Sánchez, F.; Echiburú, B.; Crisosto, N.; Sir-Petermann, T. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 166, 151–155. [Google Scholar] [CrossRef]
- Ibáñez, L.; Del Río, L.; Díaz, M.; Sebastiani, G.; Pozo, Ó.J.; López-Bermejo, A.; de Zegher, F. Normalizing Ovulation Rate by Preferential Reduction of Hepato-Visceral Fat in Adolescent Girls With Polycystic Ovary Syndrome. J. Adolesc. Health. 2017, 61, 446–453. [Google Scholar] [CrossRef]
- Plant, T.M. Neuroendocrine control of the onset of puberty. Front. Neuroendocrinol. 2015, 38, 73–88. [Google Scholar] [CrossRef] [Green Version]
- Terasawa, E.; Fernandez, D.L. Neurobiological mechanisms of the onset of puberty in primates. Endocr. Rev. 2001, 22, 111–151. [Google Scholar] [CrossRef]
- Foster, D.L.; Ryan, K.D. Endocrine mechanisms governing transition into adulthood: A marked decrease in inhibitory feedback action of estradiol on tonic secretion of luteinizing hormone in the lamb during puberty. Endocrinology 1979, 105, 896–904. [Google Scholar] [CrossRef]
- Conley, A.J.; Bird, I.M. The role of cytochrome P450 17 alpha-hydroxylase and 3 beta-hydroxysteroid dehydrogenase in the integration of gonadal and adrenal steroidogenesis via the delta 5 and delta 4 pathways of steroidogenesis in mammals. Biol. Reprod. 1997, 56, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Conley, A.J.; Pattison, J.C.; Bird, I.M. Variations in adrenal androgen production among (nonhuman) primates. Semin. Reprod. Med. 2004, 22, 311–326. [Google Scholar] [CrossRef]
- Dunaif, A.; Chang, R.J.; Franks, S.; Legro, R.S. Polycystic Ovary Syndrome: Current Controversies, from The Ovary to The Pancreas; Humana Press: Totowa, NJ, USA, 2008. [Google Scholar]
- Herman, R.A.; Jones, B.; Mann, D.R.; Wallen, K. Timing of prenatal androgen exposure: Anatomical and endocrine effects on juvenile male and female rhesus monkeys. Horm. Behav. 2000, 38, 52–66. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Abbott, D.H.; Eisner, J.R.; Goy, R.W. Prenatal exposure of female rhesus monkeys to testosterone propionate increases serum luteinizing hormone levels in adulthood. Fertil. Steril. 1997, 67, 155–163. [Google Scholar] [CrossRef]
- Treloar, O.L.; Wolf, R.C.; Meyer, R.K. Failure of a single neonatal dose of testosterone to alter ovarian function in the rhesus monkey. Endocrinology 1972, 90, 281–284. [Google Scholar] [CrossRef]
- Bishop, C.V.; Mishler, E.C.; Takahashi, D.L.; Reiter, T.E.; Bond, K.R.; True, C.A.; Slayden, O.D.; Stouffer, R.L. Chronic hyperandrogenemia in the presence and absence of a western-style diet impairs ovarian and uterine structure/function in young adult rhesus monkeys. Hum. Reprod. 2018, 33, 128–139. [Google Scholar] [CrossRef]
- Vendola, K.A.; Zhou, J.; Adesanya, O.O.; Weil, S.J.; Bondy, C.A. Androgens stimulate early stages of follicular growth in the primate ovary. J. Clin. Invest. 1998, 101, 2622–2629. [Google Scholar] [CrossRef] [Green Version]
- Vendola, K.; Zhou, J.; Wang, J.; Bondy, C.A. Androgens promote insulin-like growth factor-I and insulin-like growth factor-I receptor gene expression in the primate ovary. Hum. Reprod. 1999, 14, 2328–2332. [Google Scholar] [CrossRef]
- Zeleznik, A.J.; Little-Ihrig, L.; Ramasawamy, S. Administration of dihydrotestosterone to rhesus monkeys inhibits gonadotropin-stimulated ovarian steroidogenesis. J. Clin. Endocrinol. Metab. 2004, 89, 860–866. [Google Scholar] [CrossRef] [Green Version]
- Faiman, C.; Reyes, F.I.; Dent, D.W.; Fuller, G.B.; Hobson, W.C.; Thliveris, J.A. Effects of long-term testosterone exposure on ovarian function and morphology in the rhesus monkey. Anat. Rec. 1988, 222, 245–251. [Google Scholar] [CrossRef]
- Billiar, R.B.; Richardson, D.; Anderson, E.; Mahajan, D.; Little, B. The effect of chronic and acyclic elevation of circulating androstenedione or estrone concentrations on ovarian function in the rhesus monkey. Endocrinology 1985, 116, 2209–2220. [Google Scholar] [CrossRef]
- Billiar, R.B.; Richardson, D.; Schwartz, R.; Posner, B.; Little, B. Effect of chronically elevated androgen or estrogen on the glucose tolerance test and insulin response in female rhesus monkeys. Am. J. Obstet. Gynecol. 1987, 157, 1297–1302. [Google Scholar] [CrossRef]
- Abbott, A.D.; Colman, R.J.; Tiefenthaler, R.; Dumesic, D.A.; Abbott, D.H. Early-to-mid gestation fetal testosterone increases right hand 2D:4D finger length ratio in polycystic ovary syndrome-like monkeys. PLoS ONE 2012, 7, e42372. [Google Scholar] [CrossRef] [Green Version]
- Goy, R.W.; Robinson, J.A. Prenatal exposure of rhesus monkeys to potent androgens: Morphological, behavioral and physiological consequences. In Banbury Report 11: Environmental Effects on Maturation; Cold Spring Harbor. Press: Cold Spring Harbor, NY, USA, 1982. [Google Scholar]
- Dean, A.; Sharpe, R.M. Clinical review: Anogenital distance or digit length ratio as measures of fetal androgen exposure: Relationship to male reproductive development and its disorders. J. Clin. Endocrinol. Metab. 2013, 98, 2230–2238. [Google Scholar] [CrossRef] [Green Version]
- Daan, N.M.; Koster, M.P.; Steegers-Theunissen, R.P.; Eijkemans, M.J.; Fauser, B.C. Endocrine and cardiometabolic cord blood characteristics of offspring born to mothers with and without polycystic ovary syndrome. Fertil. Steril. 2017, 107, 261–268.e3. [Google Scholar] [CrossRef] [Green Version]
- Apter, D.; Vihko, R. Endocrine determinants of fertility: Serum androgen concentrations during follow-up of adolescents into the third decade of life. J. Clin. Endocrinol. Metab. 1990, 71, 970–974. [Google Scholar] [CrossRef]
- Wiksten-Almströmer, M.; Hirschberg, A.L.; Hagenfeldt, K. Prospective follow-up of menstrual disorders in adolescence and prognostic factors. Acta Obstet. Gynecol. Scand. 2008, 87, 1162–1168. [Google Scholar] [CrossRef]
- Ibáñez, L.; Valls, C.; Potau, N.; Marcos, M.V.; de Zegher, F. Polycystic ovary syndrome after precocious pubarche: Ontogeny of the low-birthweight effect. Clin. Endocrinol. 2001, 55, 667–672. [Google Scholar] [CrossRef]
- Koivuaho, E.; Laru, J.; Ojaniemi, M.; Puukka, K.; Kettunen, J.; Tapanainen, J.S.; Franks, S.; Järvelin, M.R.; Morin-Papunen, L.; Sebert, S.; et al. Age at adiposity rebound in childhood is associated with PCOS diagnosis and obesity in adulthood-longitudinal analysis of BMI data from birth to age 46 in cases of PCOS. Int. J. Obes. 2019, 43, 1370–1379. [Google Scholar] [CrossRef] [Green Version]
- Sadrzadeh, S.; Painter, R.C.; Lambalk, C.B. Developmental origins of polycystic ovary syndrome (PCOS), a case control study comparing birth weight in women with PCOS and control group. Gynecol. Endocrinol. 2016, 32, 856–859. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.; Saxena, R.; Welt, C.K. Environmental and genetic factors influence age at menarche in women with polycystic ovary syndrome. J. Pediatr. Endocrinol. Metab. 2012, 25, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Bird, I.M.; Dumesic, D.A.; Abbott, D.H. Adrenal hyperandrogenism is induced by fetal androgen excess in a rhesus monkey model of polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 6630–6637. [Google Scholar] [CrossRef] [Green Version]
- Bishop, C.V.; Stouffer, R.L.; Takahashi, D.L.; Mishler, E.C.; Wilcox, M.C.; Slayden, O.D.; True, C.A. Chronic hyperandrogenemia and western-style diet beginning at puberty reduces fertility and increases metabolic dysfunction during pregnancy in young adult, female macaques. Hum. Reprod. 2018, 33, 694–705. [Google Scholar] [CrossRef]
- Wood, J.R.; Dumesic, D.A.; Abbott, D.H.; Strauss, J.F., III. Molecular abnormalities in oocytes from women with polycystic ovary syndrome revealed by microarray analysis. J. Clin. Endocrinol. Metab. 2007, 92, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Dumesic, D.A.; Meldrum, D.R.; Katz-Jaffe, M.G.; Krisher, R.L.; Schoolcraft, W.B. Oocyte environment: Follicular fluid and cumulus cells are critical for oocyte health. Fertil. Steril. 2015, 103, 303–316. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Schramm, R.D.; Peterson, E.; Paprocki, A.M.; Zhou, R.; Abbott, D.H. Impaired developmental competence of oocytes in adult prenatally androgenized female rhesus monkeys undergoing gonadotropin stimulation for in vitro fertilization. J. Clin. Endocrinol. Metab. 2002, 87, 1111–1119. [Google Scholar] [CrossRef]
- Waldstreicher, J.; Santoro, N.F.; Hall, J.E.; Filicori, M.; Crowley, W.F., Jr. Hyperfunction of the hypothalamic-pituitary axis in women with polycystic ovarian disease: Indirect evidence for partial gonadotroph desensitization. J. Clin. Endocrinol. Metab. 1988, 66, 165–172. [Google Scholar] [CrossRef]
- Daniels, T.L.; Berga, S.L. Resistance of gonadotropin releasing hormone drive to sex steroid-induced suppression in hyperandrogenic anovulation. J. Clin. Endocrinol. Metab. 1997, 82, 4179–4183. [Google Scholar] [CrossRef]
- Pastor, C.L.; Griffin-Korf, M.L.; Aloi, J.A.; Evans, W.S.; Marshall, J.C. Polycystic ovary syndrome: Evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J. Clin. Endocrinol. Metab. 1998, 83, 582–590. [Google Scholar] [CrossRef]
- Foecking, E.M.; McDevitt, M.A.; Acosta-Martínez, M.; Horton, T.H.; Levine, J.E. Neuroendocrine consequences of androgen excess in female rodents. Horm. Behav. 2008, 53, 673–692. [Google Scholar] [CrossRef] [Green Version]
- Park, C.J.; Zhao, Z.; Glidewell-Kenney, C.; Lazic, M.; Chambon, P.; Krust, A.; Weiss, J.; Clegg, D.J.; Dunaif, A.; Jameson, J.L.; et al. Genetic rescue of nonclassical ERalpha signaling normalizes energy balance in obese ERalpha-null mutant mice. J. Clin. Invest. 2011, 121, 604–612. [Google Scholar] [CrossRef] [Green Version]
- Musatov, S.; Chen, W.; Pfaff, D.W.; Mobbs, C.V.; Yang, X.J.; Clegg, D.J.; Kaplitt, M.G.; Ogawa, S. Silencing of estrogen receptor alpha in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 2501–2506. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; He, Y.; Yang, Y.; Zhu, L.; Wang, C.; Xu, P.; Hinton, A.O.; Yan, X.; Zhao, J.; Fukuda, M.; et al. PI3K in the ventromedial hypothalamic nucleus mediates estrogenic actions on energy expenditure in female mice. Sci. Rep. 2016, 6, 23459. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Nedungadi, T.P.; Zhu, L.; Sobhani, N.; Irani, B.G.; Davis, K.E.; Zhang, X.; Zou, F.; Gent, L.M.; Hahner, L.D.; et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab. 2011, 14, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Abbott, D.H.; Barnett, D.K.; Bruns, C.M.; Dumesic, D.A. Androgen excess fetal programming of female reproduction: A developmental aetiology for polycystic ovary syndrome? Hum. Reprod. Update. 2005, 11, 357–374. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.T.; Moore, A.M.; Cobern, J.A.; Padmanabhan, V.; Goodman, R.L.; Coolen, L.M.; Lehman, M.N. Prenatal Testosterone Exposure Alters GABAergic Synaptic Inputs to GnRH and KNDy Neurons in a Sheep Model of Polycystic Ovarian Syndrome. Endocrinology 2019. [Google Scholar] [CrossRef]
- Barnes, R.B.; Rosenfield, R.L.; Burstein, S.; Ehrmann, D.A. Pituitary-ovarian responses to nafarelin testing in the polycystic ovary syndrome. N. Engl. J. Med. 1989, 320, 559–565. [Google Scholar] [CrossRef]
- Cheung, A.P.; Chang, R.J. Pituitary responsiveness to gonadotrophin-releasing hormone agonist stimulation: A dose-response comparison of luteinizing hormone/follicle-stimulating hormone secretion in women with polycystic ovary syndrome and normal women. Hum. Reprod. 1995, 10, 1054–1059. [Google Scholar] [CrossRef]
- Catteau-Jonard, S.; Brunel, A.; Dumont, A.; Robin, G.; Pigny, P.; Dewailly, D. Serum FSH level is lower in dysovulating than in ovulating non-PCOS obese women, independently of body mass index. Ann. Endocrinol. 2019. [Google Scholar] [CrossRef]
- Polson, D.W.; Mason, H.D.; Saldahna, M.B.; Franks, S. Ovulation of a single dominant follicle during treatment with low-dose pulsatile follicle stimulating hormone in women with polycystic ovary syndrome. Clin. Endocrinol. 1987, 26, 205–212. [Google Scholar] [CrossRef]
- Hardy, K.; Robinson, F.M.; Paraschos, T.; Wicks, R.; Franks, S.; Winston, R.M. Normal development and metabolic activity of preimplantation embryos in vitro from patients with polycystic ovaries. Hum. Reprod. 1995, 10, 2125–2135. [Google Scholar] [CrossRef]
- Whigham, L.D.; Butz, D.E.; Dashti, H.; Tonelli, M.; Johnson, L.K.; Cook, M.E.; Porter, W.P.; Eghbalnia, H.R.; Markley, J.L.; Lindheim, S.R.; et al. Metabolic Evidence of Diminished Lipid Oxidation in Women With Polycystic Ovary Syndrome. Curr. Metabolomics. 2014, 2, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Broskey, N.T.; Tam, C.S.; Sutton, E.F.; Altazan, A.D.; Burton, J.H.; Ravussin, E.; Redman, L.M. Metabolic inflexibility in women with PCOS is similar to women with type 2 diabetes. Nutr. Metab. 2018, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Shorakae, S.; Jona, E.; de Courten, B.; Lambert, G.W.; Lambert, E.A.; Phillips, S.E.; Clarke, I.J.; Teede, H.J.; Henry, B.A. Brown adipose tissue thermogenesis in polycystic ovary syndrome. Clin. Endocrinol. 2019, 90, 425–432. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Akopians, A.L.; Madrigal, V.K.; Ramirez, E.; Margolis, D.J.; Sarma, M.K.; Thomas, A.M.; Grogan, T.R.; Haykal, R.; Schooler, T.A.; et al. Hyperandrogenism Accompanies Increased Intra-Abdominal Fat Storage in Normal Weight Polycystic Ovary Syndrome Women. J. Clin. Endocrinol. Metab. 2016, 101, 4178–4188. [Google Scholar] [CrossRef]
- Torchen, L.C.; Fogel, N.R.; Brickman, W.J.; Paparodis, R.; Dunaif, A. Persistent apparent pancreatic β-cell defects in premenarchal PCOS relatives. J. Clin. Endocrinol. Metab. 2014, 99, 3855–3862. [Google Scholar] [CrossRef] [Green Version]
- Ek, I.; Arner, P.; Rydén, M.; Holm, C.; Thörne, A.; Hoffstedt, J.; Wahrenberg, H. A unique defect in the regulation of visceral fat cell lipolysis in the polycystic ovary syndrome as an early link to insulin resistance. Diabetes 2002, 51, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Eisner, J.R.; Dumesic, D.A.; Kemnitz, J.W.; Colman, R.J.; Abbott, D.H. Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obes. Res. 2003, 11, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Bruns, C.M.; Baum, S.T.; Colman, R.J.; Dumesic, D.A.; Eisner, J.R.; Jensen, M.D.; Whigham, L.D.; Abbott, D.H. Prenatal androgen excess negatively impacts body fat distribution in a nonhuman primate model of polycystic ovary syndrome. Int. J. Obes. 2007, 31, 1579–1585. [Google Scholar] [CrossRef] [Green Version]
- Eisner, J.R.; Dumesic, D.A.; Kemnitz, J.W.; Abbott, D.H. Timing of prenatal androgen excess determines differential impairment in insulin secretion and action in adult female rhesus monkeys. J. Clin. Endocrinol. Metab. 2000, 85, 1206–1210. [Google Scholar] [CrossRef]
- Keller, E.; Chazenbalk, G.D.; Aguilera, P.; Madrigal, V.; Grogan, T.; Elashoff, D.; Dumesic, D.A.; Abbott, D.H. Impaired preadipocyte differentiation into adipocytes in subcutaneous abdominal adipose of PCOS-like female rhesus monkeys. Endocrinology 2014, 155, 2696–2703. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Cousins, R.S.; Liu, S.; Phelps, B.M.; Promes, J.A. Connecting pancreatic islet lipid metabolism with insulin secretion and the development of type 2 diabetes. Ann. N.Y. Acad. Sci. 2019. [Google Scholar] [CrossRef]
- True, C.; Abbott, D.H.; Roberts, C.T., Jr.; Varlamov, O. Sex Differences in Androgen Regulation of Metabolism in Nonhuman Primates. Adv. Exp. Med. Biol. 2017, 1043, 559–574. [Google Scholar]
- Noroozzadeh, M.; Ramezani Tehrani, F.; Bahri Khomami, M.; Azizi, F. A Comparison of Sexual Function in Women with Polycystic Ovary Syndrome (PCOS) Whose Mothers Had PCOS During Their Pregnancy Period with Those Without PCOS. Arch. Sex. Behav. 2017, 46, 2033–2042. [Google Scholar] [CrossRef]
- Stener-Victorin, E.; Manti, M.; Fornes, R.; Risal, S.; Lu, H.; Benrick, A. Origins and Impact of Psychological Traits in Polycystic Ovary Syndrome. Med. Sci. 2019, 7, 86. [Google Scholar] [CrossRef] [Green Version]
- Goto, M.; Piper Hanley, K.; Marcos, J.; Wood, P.J.; Wright, S.; Postle, A.D.; Cameron, I.T.; Mason, J.I.; Wilson, D.I.; Hanley, N.A. In humans, early cortisol biosynthesis provides a mechanism to safeguard female sexual development. J. Clin. Invest. 2006, 116, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Tomaszycki, M.L.; Davis, J.E.; Gouzoules, H.; Wallen, K. Sex differences in infant rhesus macaque separation-rejection vocalizations and effects of prenatal androgens. Horm. Behav. 2001, 39, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Tomaszycki, M.L.; Gouzoules, H.; Wallen, K. Sex differences in juvenile rhesus macaque (Macaca mulatta) agonistic screams: Life history differences and effects of prenatal androgens. Dev. Psychobiol. 2005, 47, 318–327. [Google Scholar] [CrossRef]
- Wallen, K. Hormonal influences on sexually differentiated behavior in nonhuman primates. Front. Neuroendocrinol. 2005, 26, 7–26. [Google Scholar] [CrossRef]
- Pomerantz, S.M.; Roy, M.M.; Thornton, J.E.; Goy, R.W. Expression of adult female patterns of sexual behavior by male, female, and pseudohermaphroditic female rhesus monkeys. Biol. Reprod. 1985, 33, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, S.M.; Goy, R.W.; Roy, M.M. Expression of male-typical behavior in adult female pseudohermaphroditic rhesus: Comparisons with normal males and neonatally gonadectomized males and females. Horm. Behav. 1986, 20, 483–500. [Google Scholar] [CrossRef]
- True, C.A.; Takahashi, D.L.; Burns, S.E.; Mishler, E.C.; Bond, K.R.; Wilcox, M.C.; Calhoun, A.R.; Bader, L.A.; Dean, T.A.; Ryan, N.D.; et al. Chronic combined hyperandrogenemia and western-style diet in young female rhesus macaques causes greater metabolic impairments compared to either treatment alone. Hum. Reprod. 2017, 32, 1880–1891. [Google Scholar] [CrossRef]
- Berenbaum, S.A.; Meyer-Bahlburg, H.F. Gender development and sexuality in disorders of sex development. Horm. Metab. Res. 2015, 47, 361–366. [Google Scholar] [CrossRef]
- Hines, M.; Ahmed, S.F.; Hughes, I.A. Psychological outcomes and gender-related development in complete androgen insensitivity syndrome. Arch. Sex. Behav. 2003, 32, 93–101. [Google Scholar] [CrossRef]
- Hamann, S.; Stevens, J.; Vick, J.H.; Bryk, K.; Quigley, C.A.; Berenbaum, S.A.; Wallen, K. Brain responses to sexual images in 46, XY women with complete androgen insensitivity syndrome are female-typical. Horm. Behav. 2014, 66, 724–730. [Google Scholar] [CrossRef]
- Köhler, B.; Kleinemeier, E.; Lux, A.; Hiort, O.; Grüters, A.; Thyen, U.; DSD Network Working Group. Satisfaction with genital surgery and sexual life of adults with XY disorders of sex development: Results from the German clinical evaluation study. J. Clin. Endocrinol. Metab. 2012, 97, 577–588. [Google Scholar]
- Kight, K.E.; McCarthy, M.M. Sex differences and estrogen regulation of BDNF gene expression, but not propeptide content, in the developing hippocampus. J. Neurosci. Res. 2017, 95, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Pavey, T.G.; Brown, W.J. Sitting time and depression in young women over 12-years: The effect of physical activity. J. Sci. Med. Sport. 2019. [Google Scholar] [CrossRef]
- Manti, M.; Fornes, R.; Qi, X.; Folmerz, E.; Lindén Hirschberg, A.; de Castro Barbosa, T.; Maliqueo, M.; Benrick, A.; Stener-Victorin, E. Maternal androgen excess and obesity induce sexually dimorphic anxiety-like behavior in the offspring. FASEB J. 2018, 32, 4158–4171. [Google Scholar] [CrossRef] [Green Version]
- Legro, R.S.; Dodson, W.C.; Kunselman, A.R.; Stetter, C.M.; Kris-Etherton, P.M.; Williams, N.I.; Gnatuk, C.L.; Estes, S.J.; Allison, K.C.; Sarwer, D.B.; et al. Benefit of Delayed Fertility Therapy With Preconception Weight Loss Over Immediate Therapy in Obese Women With PCOS. J. Clin. Endocrinol. Metab. 2016, 101, 2658–2666. [Google Scholar] [CrossRef]
- Hanem, L.G.E.; Salvesen, Ø.; Juliusson, P.B.; Carlsen, S.M.; Nossum, M.C.F.; Vaage, M.Ø.; Ødegård, R.; Vanky, E. Intrauterine metformin exposure and offspring cardiometabolic risk factors (PedMet study): A 5–10 year follow-up of the PregMet randomized controlled trial. Lancet Child Adolesc. Health 2019, 3, 166–174. [Google Scholar] [CrossRef]
- Herman, R.A.; Wallen, K. Cognitive performance in rhesus monkeys varies by sex and prenatal androgen exposure. Harm. Behav. 2007, 51, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Holt, R.I.; Lambert, K.D. The use of oral hypoglycaemic agents in pregnancy. Diabet Med. 2014, 31, 282–291. [Google Scholar] [CrossRef]
- Glueck, C.J.; Pranikoff, J.; Aregawi, D.; Wang, P. Prevention of gestational diabetes by metformin plus diet in patients with polycystic ovary syndrome. Fertil. Steril. 2008, 89, 625–634. [Google Scholar] [CrossRef]
- Hjorth-Hansen, A.; Salvesen, O.; Engen Hanem, L.G.; Eggebø, T.; Salvesen, K.Å.; Vanky, E.; Ødegård, R. Fetal growth and birth anthropometrics in metformin-exposed offspring born to mothers with PCOS. J. Clin. Endocrinol. Metab. 2018, 103, 740–747. [Google Scholar] [CrossRef]
- Hanem, L.G.E.; Stridsklev, S.; Juliusson, P.B.; Salvesen, Ø.; Roelants, M.; Carlsen, S.M.; Ødegård, R.; Vanky, E. Metformin use in PCOS pregnancies increases the risk of offspring overweight at 4 years of age: Follow-up of two RCTs. J. Clin. Endocrinol. Metab. 2018, 103, 1612–1621. [Google Scholar]
- de Zegher, F.; Lopez-Bermejo, A.; Ibanez, L. Adipose tissue expandability and the early origins of PCOS. Trends Endocrinol. Metab. 2009, 20, 418–423. [Google Scholar] [CrossRef]
- Abbott, D.H.; Levine, J.E.; Dumesic, D.A. Translational insight into Polycystic Ovary Syndrome (PCOS) from female monkeys with PCOS-like traits. Curr. Pharm. Des. 2016, 22, 5625–5633. [Google Scholar] [CrossRef] [Green Version]



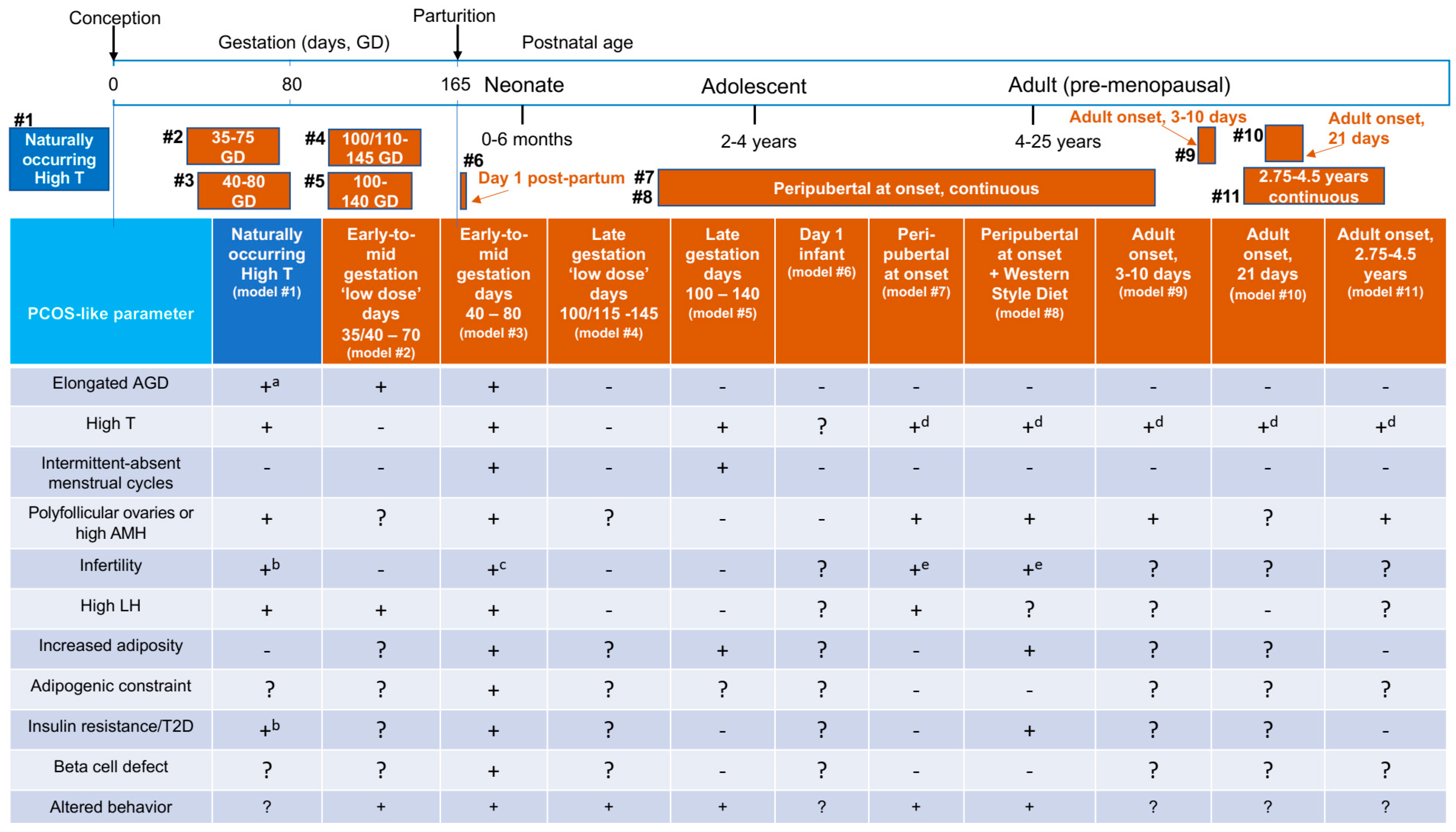{kind=link}
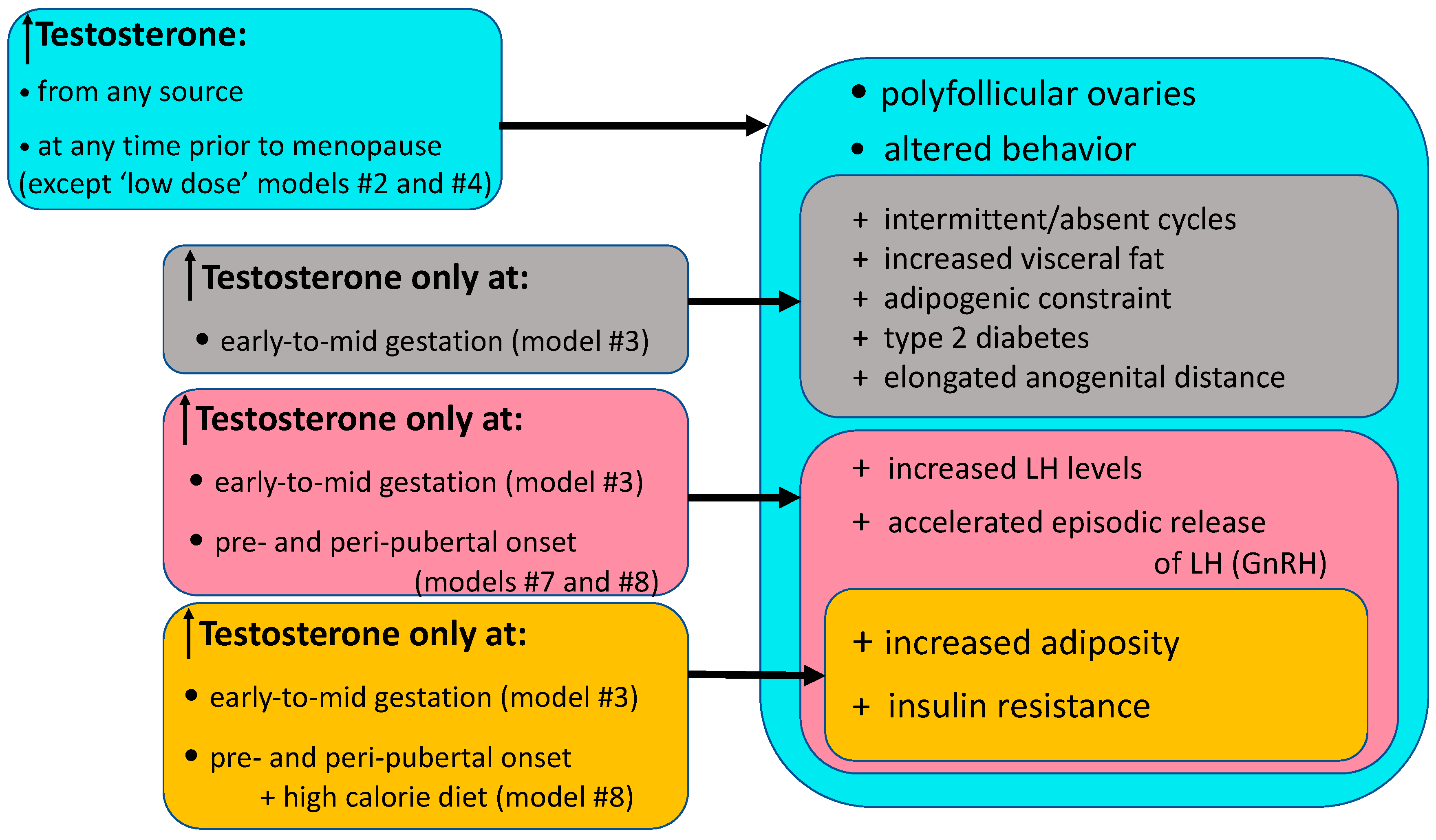{kind=link}
| PCOS and PCOS-Like Phenotypes a | ||||
|---|---|---|---|---|
| (% of PCOS Women and PCOS-Like Monkeys) | ||||
| Classic Phenotypes | Non-Classic Phenotypes | |||
| Female Population | Type A | Type B | Type C | Type D |
| PCOS women (clinical referrals) | 49 | 13 | 14 | 17 |
| -----------62%----------- | ||||
| Female macaques | ||||
| Early-to-mid gestation T-exposed | 38 | 25 | 12 | 25 |
| -----------63%----------- | ||||
| Late gestation T-exposed | 20 | 60 | 20 | 0 |
| -----------80%----------- | ||||
| PCOS women (from local, unselected populations) | 25 | 19 | 35 | 20 |
| -----------55%----------- | ||||
| Female macaques | ||||
| Naturally hyperandrogenic (High T) | 25 | 8 | 42 | 25 |
| -----------67%----------- | ||||
| Phenotype | Sample Size (n) | h2 | Reference |
|---|---|---|---|
| Duration of freezing behavior (inhibition) | 285 | 0.38 | [85] |
| Anxious temperament | 238 | 0.36 | [82] |
| Glucose metabolism in hippocampus | 238 | Right: 0.65 Left: 0.76 | [82] |
| Infant exploratory behavior | 428 | 0.25 | [85] |
| Infant reaction to novel threat | 428 | 0.24 | [85] |
| PCOS-Like Model | Naturally Occurring or Experimentally Induced | Testosterone (T) Regimen | Circulating T Levels Achieved |
|---|---|---|---|
| High T [13] | Naturally occurring (model #1) | None | ≥0.31 ng/mL (1.1 nmol/L) |
| Gestation days [111], 35/40–75 ‘Low dose’ | Experimentally induced (model #2) | T enanthate 20 mg/week, IM to dam, Early-to-mid gestation 35–40 consecutive days | ? |
| Gestation days [100,112], 40–80 | Experimentally induced (model #3) | T propionate 10–15 mg/day, SC to dam, Early-to-mid gestation 15–40 consecutive days | ~0.30 ng/mL (1.1 nmol/L) |
| Gestation days [111], 100/110–145 ‘Low dose’ | Experimentally induced (model #4) | T enanthate 20 mg/week, IM to dam, Late gestation 35–45 consecutive days | ? |
| Gestation days [112], 110–140 | Experimentally induced (model #5) | T propionate 10 mg/day, SC to dam, Late gestation 25–30 consecutive days | ? |
| Postpartum day 1 [113] | Experimentally induced (model #6) | T 35 mg/kg, SC, Neonate 1 day | ? |
| Onset at 1–2.5 years of age [35,114], (±Western Style diet) | Experimentally induced (models #7 and #8) | T Silastic capsules, SC, Pre/Peri pubertal, Continuous ~4 years | ~1.35 ng/mL (4.7 nmol/L) |
| Adult onset [115,116] | Experimentally induced (model #9) | T or Dihydrotestosterone (DHT), SC, Adult 4 mg/kg T, Continuous, 3 days, 20 µg/kg T or 145 µg/kg DHT, Continuous, 5 days, 400 µg/kg T, Continuous 10 days | 4 mg/kg T: ~31 ng/mL (108 nmol/L) 20 µg/kg T: ~4 ng/mL (14 nmol/L) 145 µg/kg DHT: ~6 ng/mL (21 nmol/L) 400 µg/kg T: ~13 ng/mL (45 nmol/L) |
| Adult onset [117] | Experimentally induced (model #10) | GnRH antagonist + exogenous LH + FSH (to control LH/FSH, ovarian hormones), Adult T or DHT, Silastic capsules, SC, 10 T capsules or 150 µg/kg DHT, DHT Continuous 21 days | 30–40 ng/mL T (150 nmol/L) ~30 ng/mL DHT (70 nmol/L) |
| Adult onset [118,119,120] | Experimentally induced (model #11) | T or Androstenedione Adult, 10-25 mg, Silastic capsules, SC, Continuous ~1–4.5 years | 0.8–1.2 ng/mL (2.8–4.2 nmol/L) |
| Model Traits | Model # Exhibiting Traits (# from Figure 1) |
|---|---|
| Equivalent to diagnostic criteria | |
| Rotterdam criteria | 1,3,5,7–9,11 |
| Elevated adult T levels | 1,3,5,7–11 |
| Intermittent or absent menstrual cycles | 3,5 |
| Polycystic ovaries or elevated adult AMH (≥10 ng/mL) | 1,3,7–9,11 |
| Reproductive and endocrine traits | |
| Delayed menarche | 3,5 |
| Ovarian hyperandrogenism | 3 |
| Adrenal hyperandrogenism | 3 |
| Diminished oocyte or embryo quality | 3,7,8 |
| Diminished embryo quality | 3,7,8 |
| Accelerated episodic LH release (hypothalamic GnRH) | 3,7 |
| LH hypergonadotropism | 1–3,7,8 |
| Uterine endometrial abnormalities | 1,7,8 |
| Metabolic traits | |
| Newborn hypoglycemia | 3 |
| Infant accelerated weight gain | 3 |
| Infant insulin hypersensitivity | 3 |
| Infant pancreatic beta cell over-compensation | 3 |
| Adult hyperlipidemia | 3 |
| Adult increased adiposity | 3,8 |
| Adult adipogenic constraint | 3 |
| Adult lipolytic constraint | 7,8 |
| Adult hyperinsulinemia | 1,3,8 |
| Adult insulin resistance | 1,3,8 |
| Adult pancreatic beta cell decompensation | 3 |
| Adult increased type 2 diabetes | 3 |
| Behavioral and neural traits | |
| Altered infant/juvenile behavior | 2,3,5,7,8 |
| Altered infant vocalizations | 2 |
| Increased adult sedentary behavior | 7,8 |
| Altered adult sexual behavior | 3,5 |
| Anatomical traits | |
| Elongated anogenital distance | 2,3 |
| Gestational traits | |
| Gestational hyperglycemia | 3 |
| Fetal hypolipidemia | 3 |
| Compromised placental structure and function | 7,8 |
| Altered fetal growth | 3,8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbott, D.H.; Rogers, J.; Dumesic, D.A.; Levine, J.E. Naturally Occurring and Experimentally Induced Rhesus Macaque Models for Polycystic Ovary Syndrome: Translational Gateways to Clinical Application. Med. Sci. 2019, 7, 107. https://doi.org/10.3390/medsci7120107
Abbott DH, Rogers J, Dumesic DA, Levine JE. Naturally Occurring and Experimentally Induced Rhesus Macaque Models for Polycystic Ovary Syndrome: Translational Gateways to Clinical Application. Medical Sciences. 2019; 7(12):107. https://doi.org/10.3390/medsci7120107
Chicago/Turabian StyleAbbott, David H., Jeffrey Rogers, Daniel A. Dumesic, and Jon E. Levine. 2019. "Naturally Occurring and Experimentally Induced Rhesus Macaque Models for Polycystic Ovary Syndrome: Translational Gateways to Clinical Application" Medical Sciences 7, no. 12: 107. https://doi.org/10.3390/medsci7120107