
De novo Transcriptome Generation and Annotation for Two Korean Endemic Land Snails, Aegista chejuensis and Aegista quelpartensis, Using Illumina Paired-End Sequencing Technology


 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
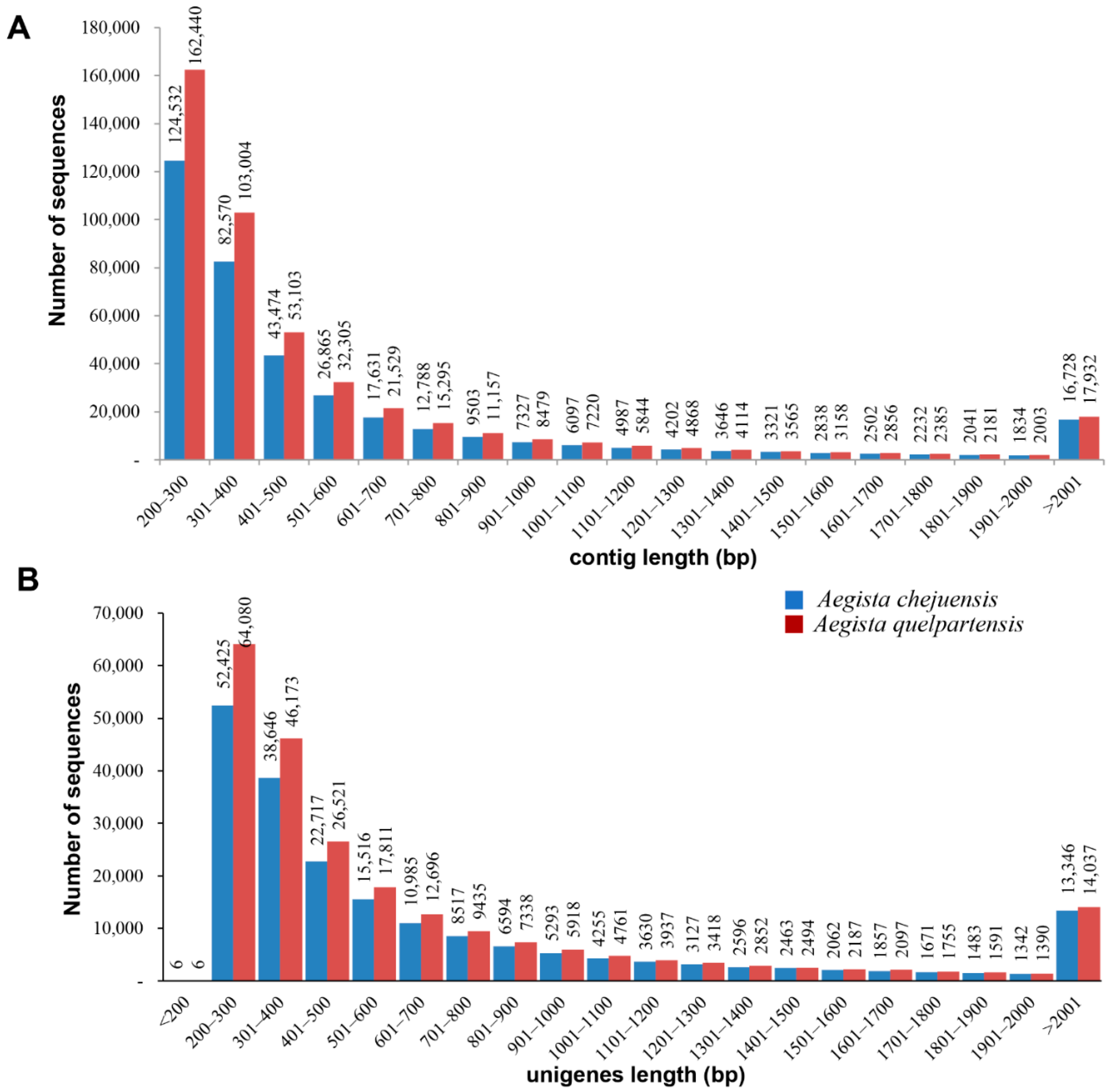
2.1. Illumina Sequence Analysis and Assembly
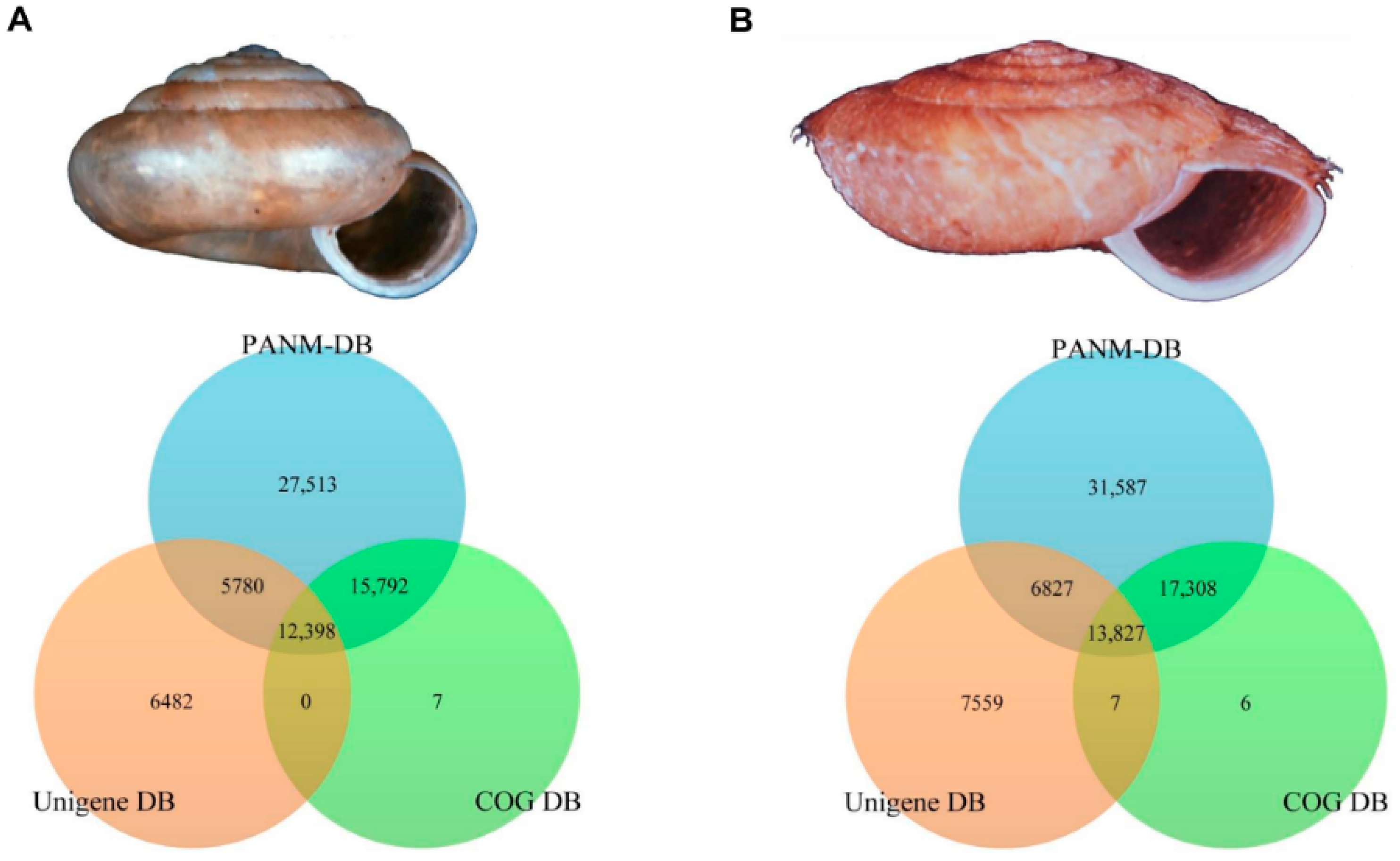
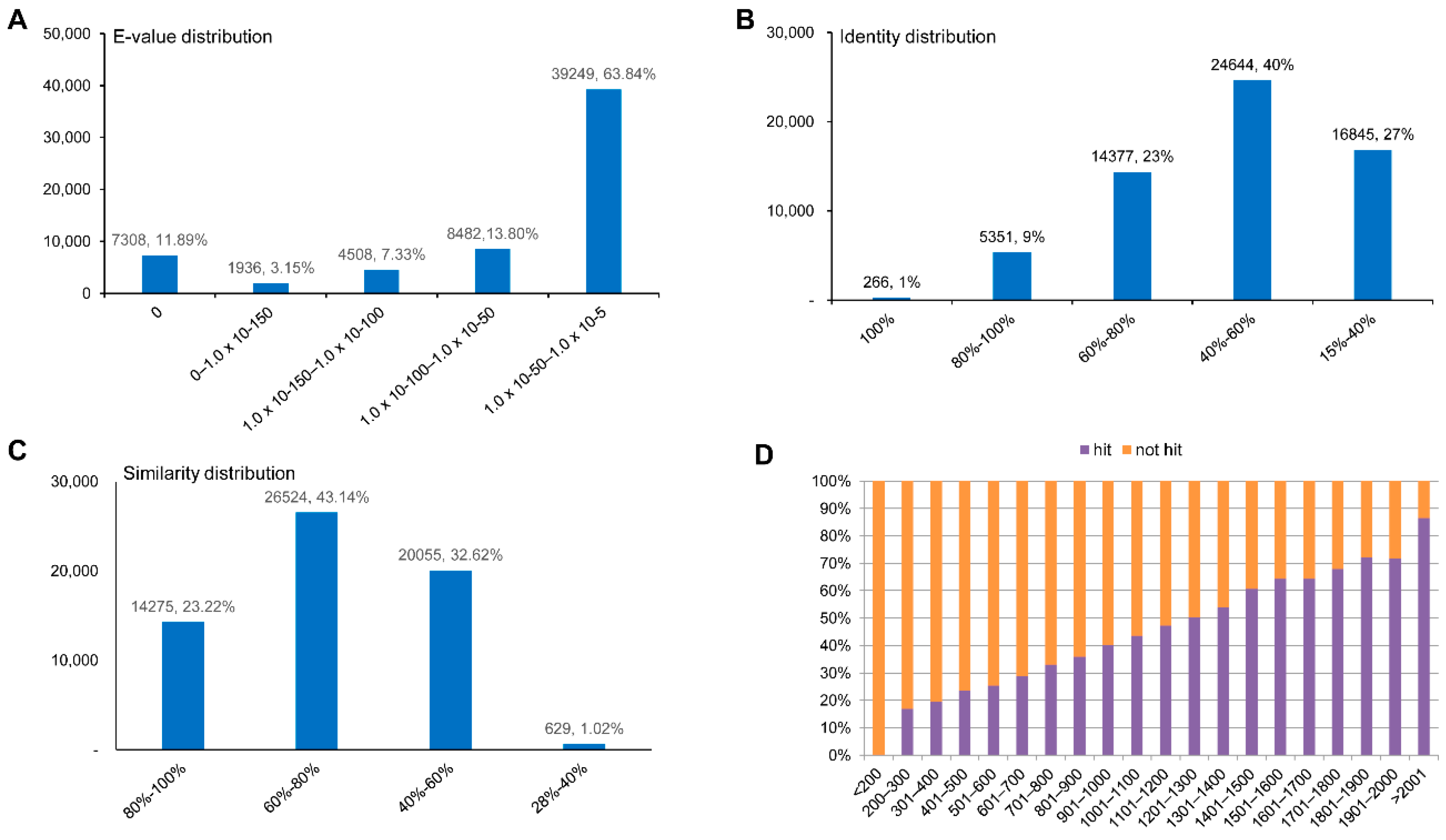
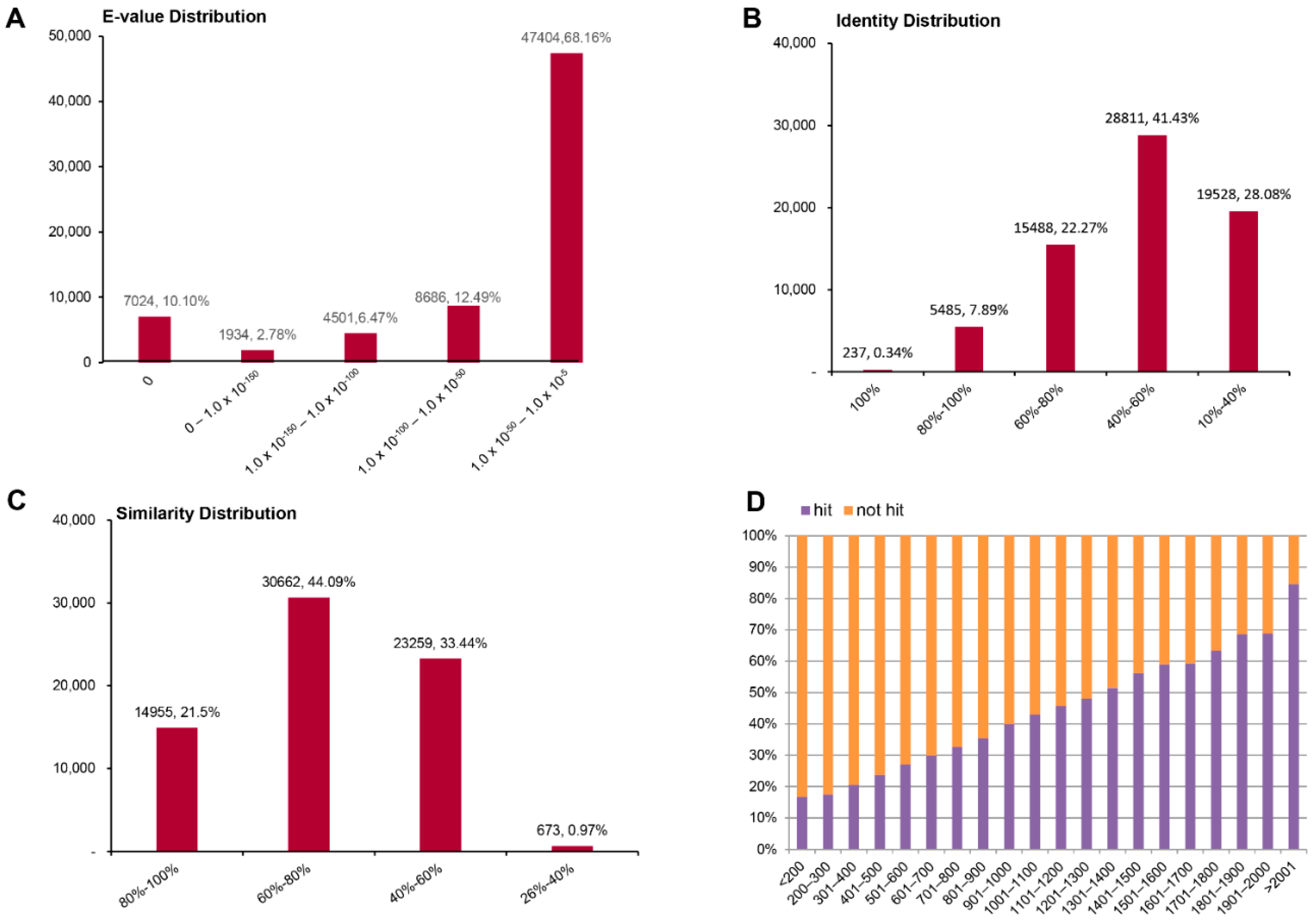
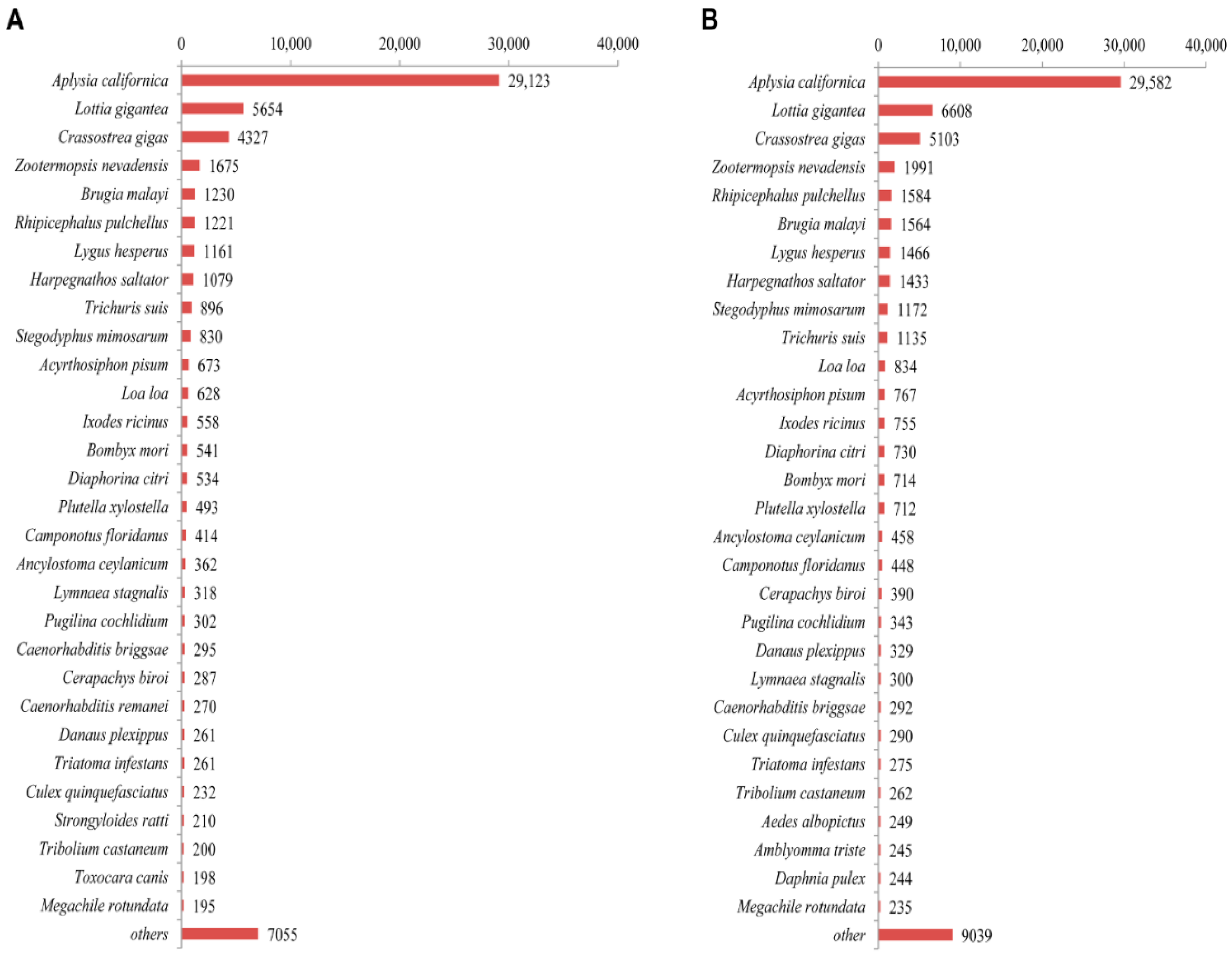
2.2. Sequence Annotation and Homology Characteristics
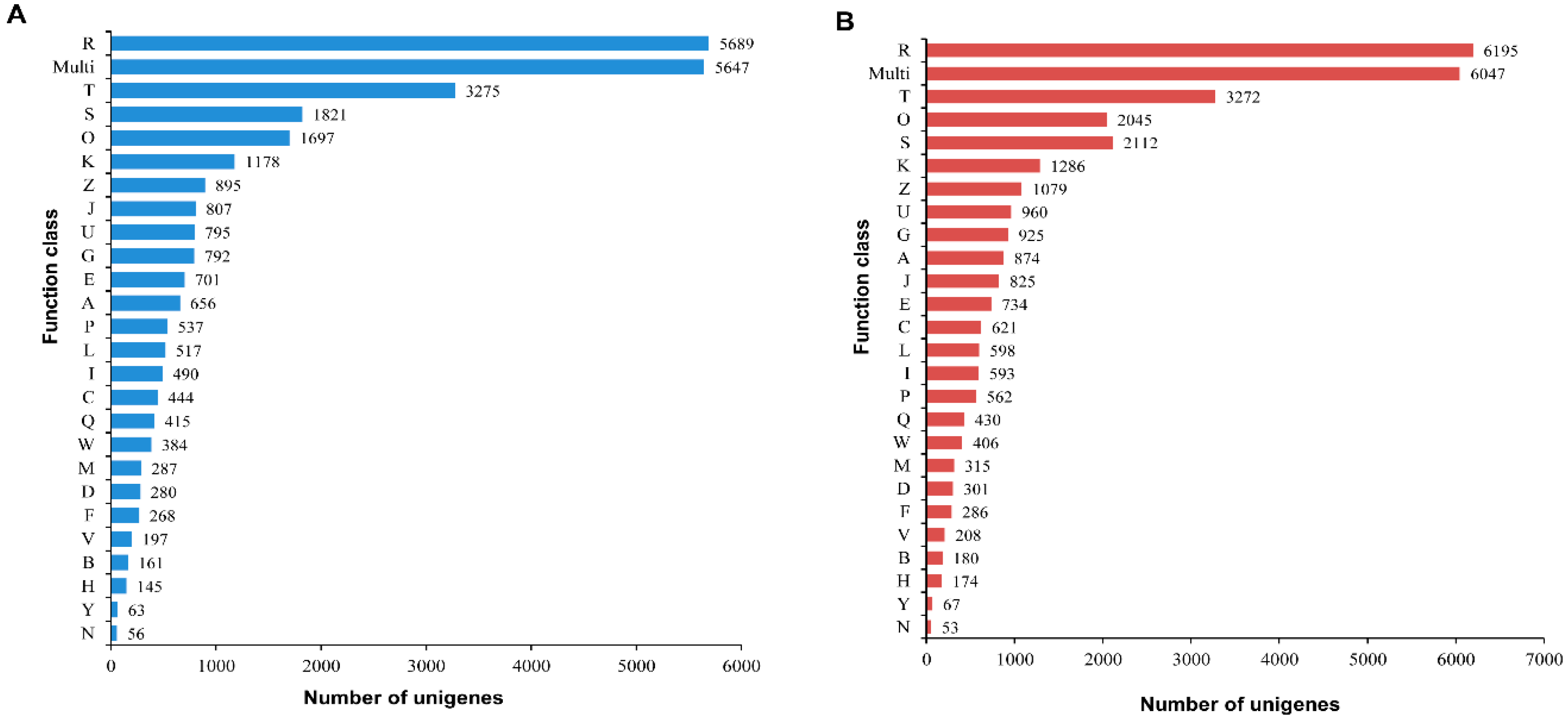
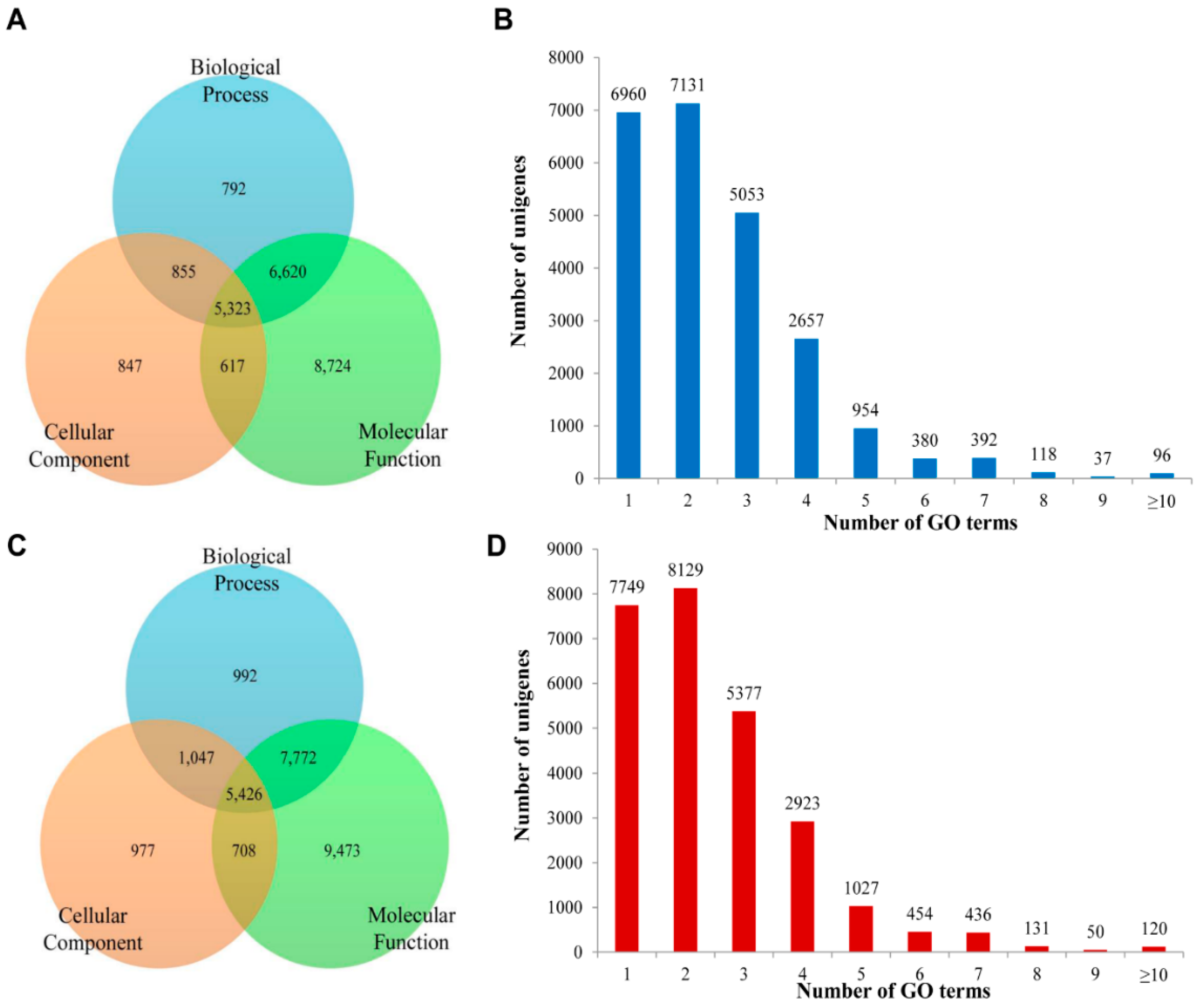
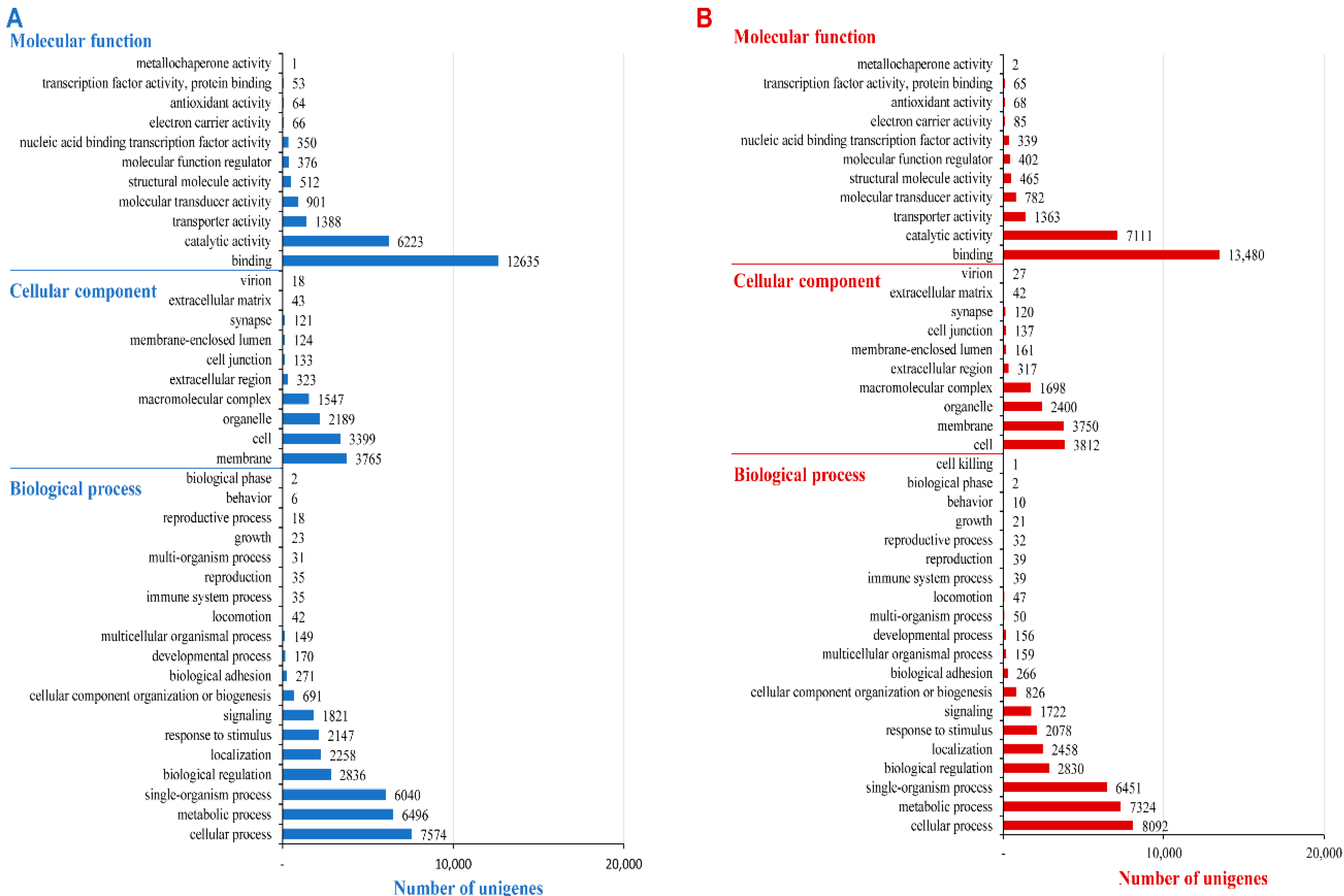
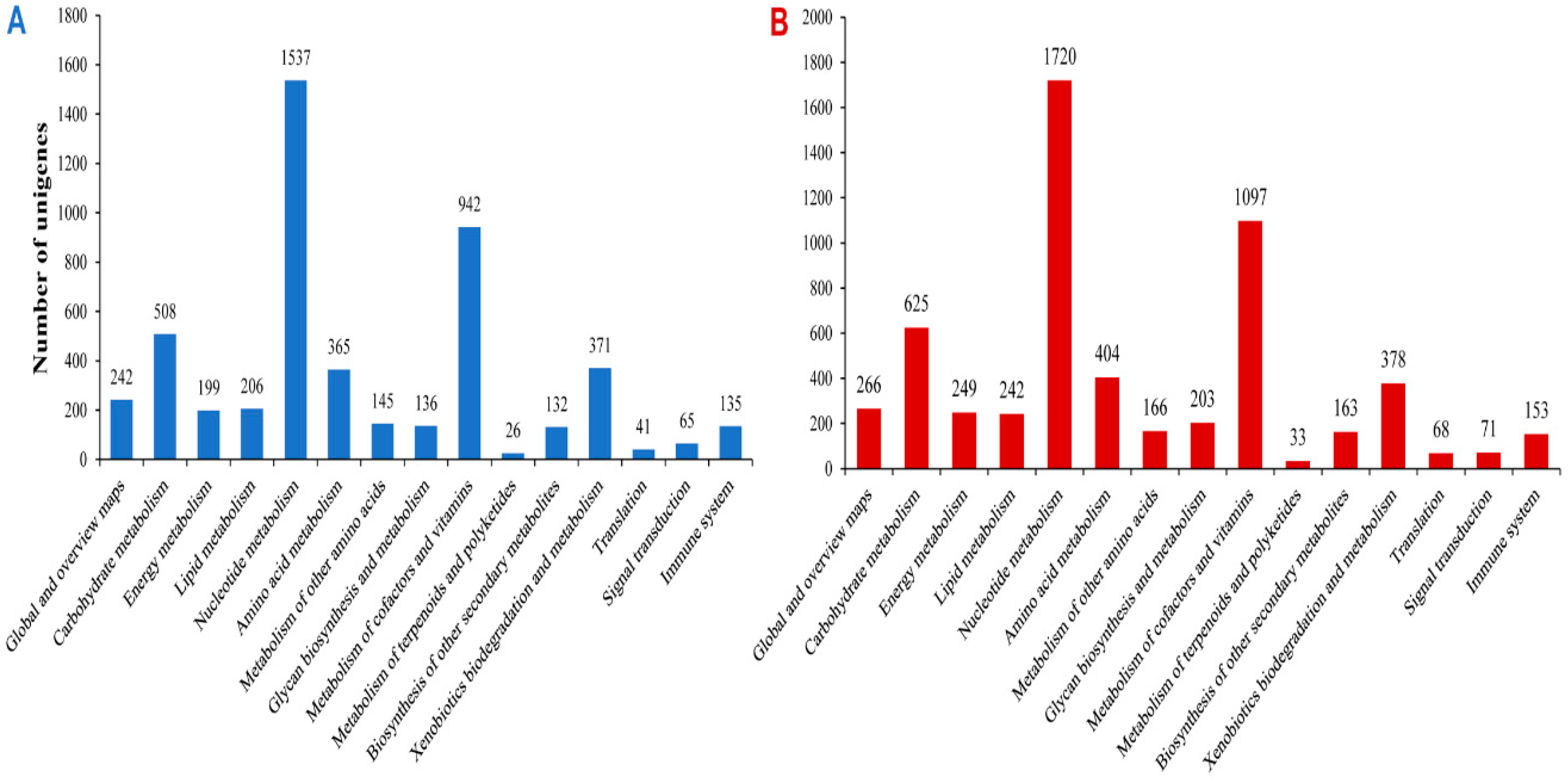
2.3. Functional Prediction Using COG, GO and KEGG
2.4. Protein Domain Identification Using InterProScan Searches
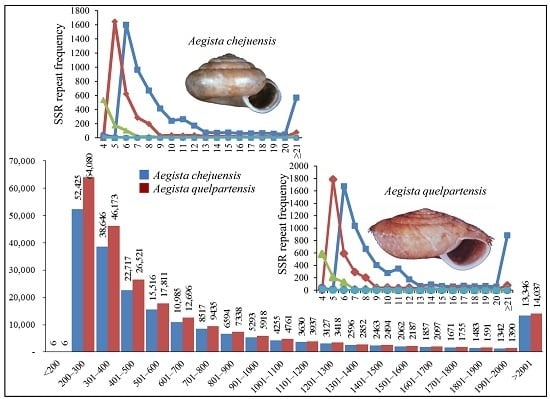
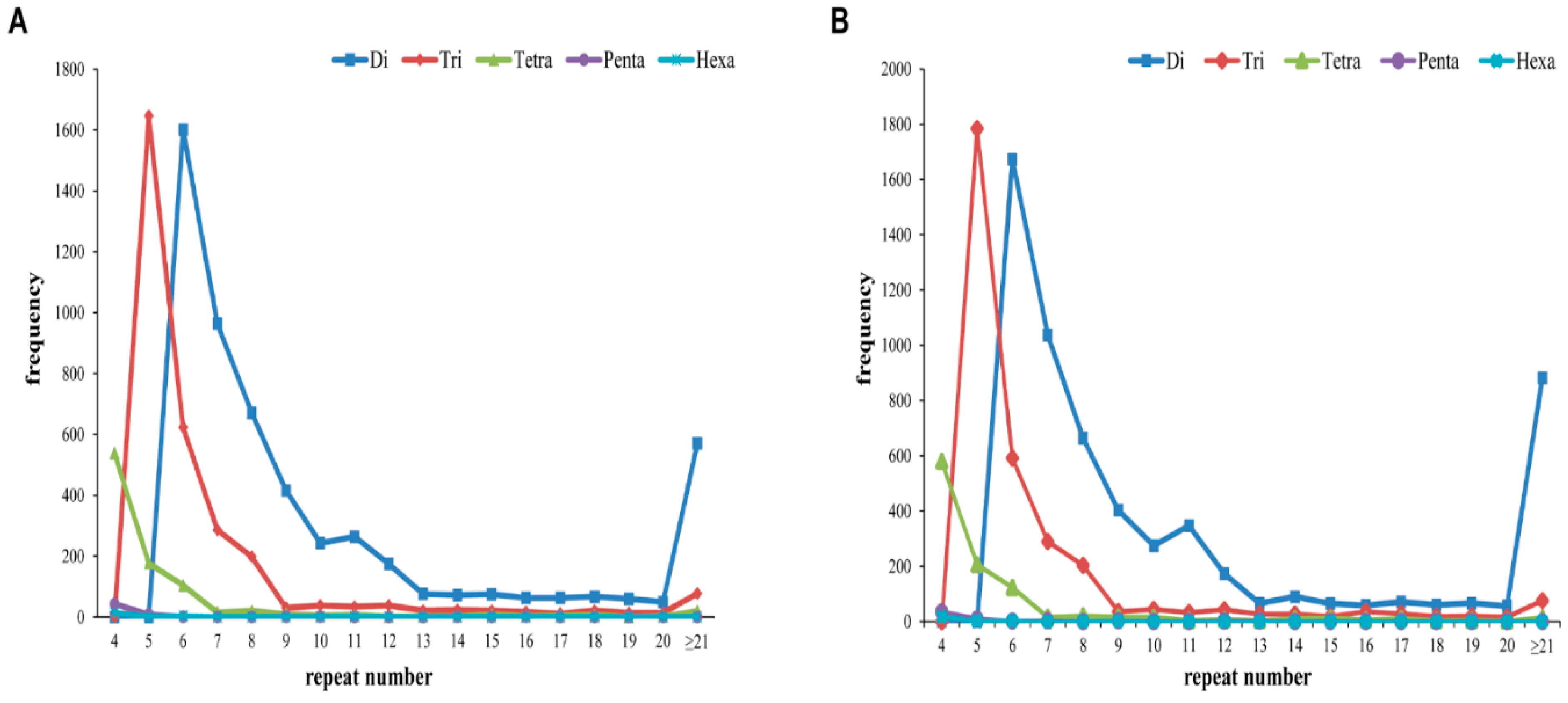
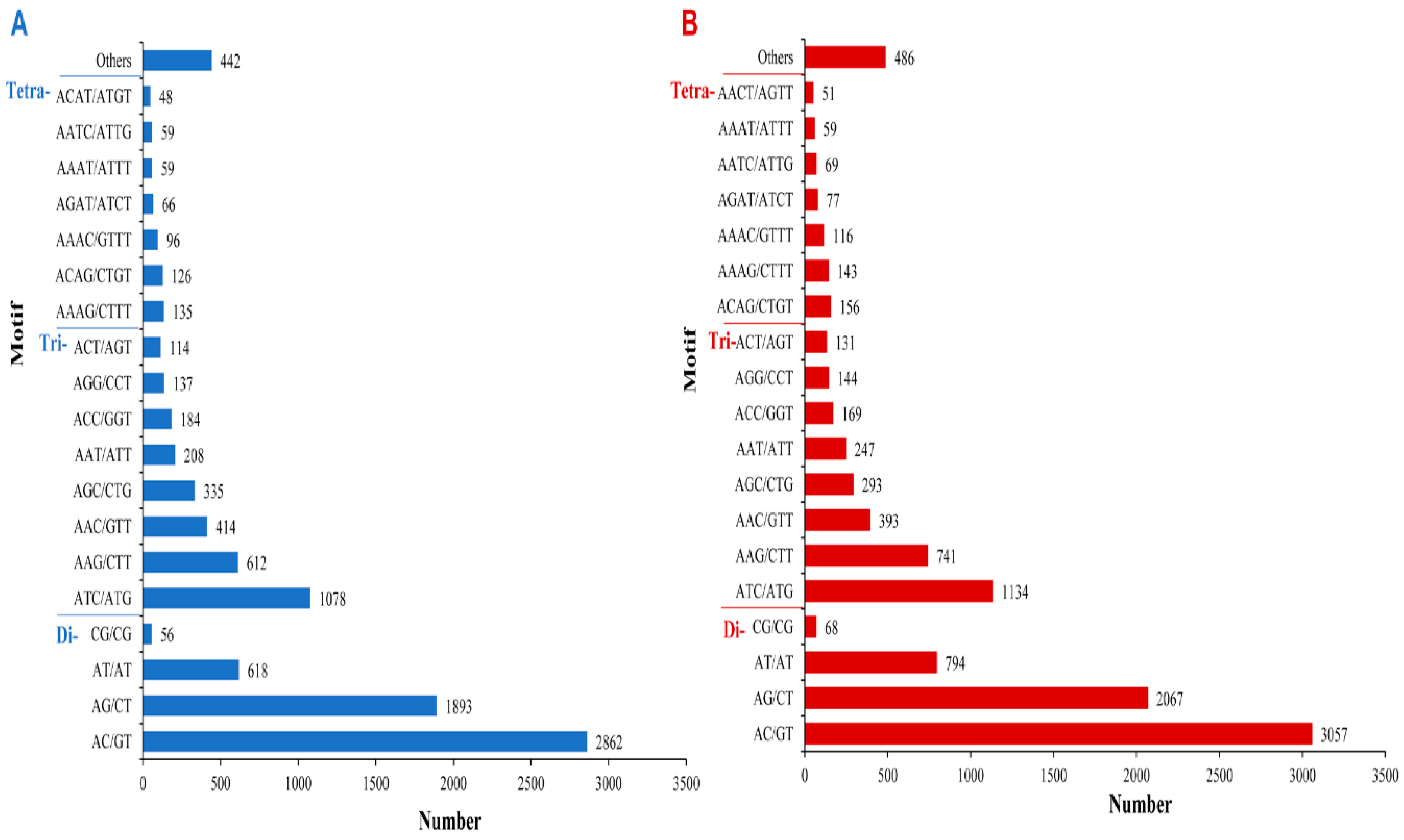
2.5. Simple Sequence Repeat (SSR) Identification
3. Experimental Section
3.1. Sample Preparation, cDNA Synthesis and Illumina Sequencing
3.2. De novo Transcriptome Assembly
3.3. Functional Annotation
3.4. SSR Motifs Detection
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stankowski, S.; Johnson, M.S. Biogeographic discordance of molecular phylogenetic and phenotypic variation in a continental archipelago radiation of land snails. BMC Evol. Biol. 2014, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Kameda, Y.; Kimura, K.; Chiba, S. Divergence in the shell morphology of the land snail genus Aegista. (Pulmonata: Bradybaenidae) under phylogenetic constraints. Biol. J. Linnean. Soc. 2015, 114, 229–241. [Google Scholar] [CrossRef]
- Davison, A.; Chiba, S.; Barton, N.H.; Clarke, B. Speciation and gene flow between snails of opposite chirality. PLoS Biol. 2005, 3, e282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, G.-M. Karyotypes of Korean endemic land snail, Koreanohadra. koreana (Gastropoda: Bradybaenidae). Korean J. Malacol. 2011, 27, 87–90. [Google Scholar] [CrossRef]
- Hirano, T.; Kameda, Y.; Chiba, S. Phylogeny of the land snails Bradybaena. and Phaeohelix. (Pulmonata: Bradybaenidae) in Japan. J. Mollus. Stud. 2014, 80, 177–183. [Google Scholar] [CrossRef]
- Azuma, M. Colored Illustrations of the Land Snails of Japan; Hoikusha: Osaka, Japan, 1995; p. 359. [Google Scholar]
- Minato, H. Four species of subgenus coelorus pilsbry from Western Japan. Chiribotan 1985, 16, 56–61. [Google Scholar]
- Kurozumi, T.; Ichisawa, K.; Kawakami, Y. Catalogue of the molluscan collection accumulated by the Mr. Hajime Ishizaka-Terrestrial and freshwater gastropods. Bull. Tottori Prefect. Mus. 2011, 48, 119–143. [Google Scholar]
- Noseworthy, R.G.; Lim, N.-R.; Choi, K.-S. A catalogue of the mollusks of Jeju Island, South Korea. Korean J. Malacol. 2007, 23, 65–104. [Google Scholar]
- Min, D.K.; Lee, J.J.; Koh, D.B.; Je, J.G. Mollusks in Korea; Hangul Graphics: Busan, Korea, 2004; p. 566. [Google Scholar]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Hao, Y.; Si, F.; Ren, S.; Hu, G.; Shen, L.; Chen, B. The de novo transcriptome and its analysis in the worldwide vegetable pest, Delia antiqua (Diptera: Anthomyiidae). Genes Genomes Genet. 2014, 4, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Wheat, C.W.; Vogel, H. Transcriptome sequencing goals, assembly, and assessment. Methods Mol. Biol. 2011, 772, 129–144. [Google Scholar] [PubMed]
- Feldmeyer, B.; Wheat, C.W.; Krezdorn, N.; Rotter, B.; Pfenninger, M. Short read Illumina data for the de novo assembly of a non-model snail species transcriptome (Radix balthica, Basommatophora, Pulmonata), and a comparison of assembler performance. BMC Genom. 2011, 12, 317. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hui, J.H.L.; Chan, T.F.; Chu, K.H. De novo transcriptome sequencing of the snail Echinolittorina malaccana: Identification of genes responsive to thermal stress and development of genetic markers for population studies. Mar. Biotechnol. 2014, 16, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhang, Y.; Huang, J.; Xiao, S.; Zhang, Y.; Li, J.; Chen, J.; Yu, Z. Transcriptomics analysis of Crassostrea hongkongensis for the discovery of reproduction-related genes. PLoS ONE 2015, 10, e0134280. [Google Scholar] [CrossRef]
- Senatore, A.; Edirisinghe, N.; Katz, P.S. Deep mRNA sequencing of the Tritonia diomedea brain transcriptome provides access to gene homologues for neuronal excitability, synaptic transmission and peptidergic signaling. PLoS ONE 2015, 10, e0118321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.P.; Xiong, T.; Xu, X.J.; Jiang, M.S.; Dong, H.F. De novo transcriptome analysis of Oncomelania hupensis after molluscicide treatment by Next-Generation Sequencing: Implications for Biology and future snail interventions. PLoS ONE 2015, 10, e0118673. [Google Scholar] [CrossRef] [PubMed]
- Tapia, F.J.; Gallardo-Escarate, C. Spatio-temporal transcriptome analysis in the marine snail Tegula atra along central-northern Chile (28–31°S). Mar. Genom. 2015, 23, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Adamson, K.J.; Wang, T.; Zhao, M.; Bell, F.; Kuballa, A.V.; Storey, K.B.; Cummins, S.F. Molecular insights into land snail neuropeptides through transcriptome and comparative gene analysis. BMC Genom. 2015, 16, 308. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-W.; Wu, W.-L. Genomic resources of two land snail, Aegista diversifamilia and Dolicheulota formosensis, generated by Illumina paired-end sequencing. F1000 Res. 2015, 4, 106. [Google Scholar] [CrossRef]
- Meng, X.I.; Liu, M.; Jiang, K.-Y.; Wang, B.-J.; Tian, X.; Sun, S.-J.; Luo, Z.-Y.; Qiu, C.-W.; Wang, L. De novo characterization of Japanese scallop Mizuhopecten yessoensis transcriptome and analysis of its gene expression following cadmium exposure. PLoS ONE 2013, 8, e64485. [Google Scholar] [CrossRef] [PubMed]
- Prentis, P.J.; Pavasovic, A. The Anadara trapezia transcriptome: A resource for molluscan physiological genomics. Mar. Genom. 2014, 18, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, Y.; Xu, G.; Xie, L.; Hu, X.; Bao, Z.; Zhang, R. Characterization of the Zhikong scallop (Chlamys farreri) mantle transcriptome and identification of biomineralization-related genes. Mar. Biotechnol. 2013, 15, 706–715. [Google Scholar] [PubMed]
- Garg, R.; Patel, R.K.; Jhanwar, S.; Priya, P.; Bhattacharjee, A.; Yadav, G.; Bhatia, S.; Chattopadhyay, D.; Tyagi, A.K.; Jain, M. Gene discovery and tissue-specific transcriptome analysis in Chickpea with massively parallel pyrosequencing and web resource development. Plant. Physiol. 2011, 156, 1661–1678. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, S.T.; Emrich, S.J. Assessing de novo transcriptome assembly metrices for consistency and utility. BMC Genom. 2013, 14, 465. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Xia, C.; Zhao, G.; Jia, J.; Kong, X. Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genom. 2012, 13, 392. [Google Scholar] [CrossRef] [PubMed]
- Gerdol, M.; de Moro, G.; Manfrin, C.; Milandri, A.; Riccardi, E.; Beran, A.; Venier, P.; Pallavicini, A. RNA sequencing and de novo assembly of the digestive gland transcriptome in Mytilus galloprovincialis fed with toxinogenic and non-toxic strains of Alexandrium minutum. BMC Res. Notes 2014, 7, 722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Lei, Q.; Tian, Q.; Xie, S.; Du, X.; Li, J.; Wang, L.; Xiong, Y. De novo assembly, gene annotation, and simple sequence repeat marker development using Illumina paired-end transcriptome sequences in the pearl oyster Pinctada maxima. Biosci. Biotechnol. Biochem. 2014, 78, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Prentis, P.J.; Gilding, E.K.; Pavasovic, A. Assembly and annotation of a non-model gastropod (Nerita melanotragus) transcriptome: A comparison of De novo assemblers. BMC Res. Notes 2014, 7, 488. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Liu, P.; Gao, B.; Wang, Y.; Wang, Z.; Chen, P.; Li, J. Transcriptome analysis of the Portunus trituberculatus: De novo assembly, growth-related gene identification and marker discovery. PLoS ONE 2014, 9, e94055. [Google Scholar] [CrossRef] [PubMed]
- Feldmesser, E.; Rosenwasser, S.; Vardi, A.; Ben-Dor, S. Improving transcriptome construction in non-model organisms: Integrating manual and automated gene definition in Emiliania huxleyi. BMC Genom. 2014, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, B.B.; Hwang, H.-J.; Kang, S.W.; Park, S.Y.; Wang, T.H.; Park, E.B.; Chung, J.M.; Song, D.K.; Kim, C.; Kin, S.; et al. Transcriptome characterization for non-model endangered lycaenids, Protantigius superans and Spindasis takanosis, using Illumina HiSeq 2500 sequencing. Int. J. Mol. Sci. 2015, 16, 29948–29970. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Park, S.Y.; Patnaik, B.B.; Hwang, H.J.; Kim, C.; Kim, S.; Lee, J.S.; Han, Y.S.; Lee, Y.S. Construction of PANM Database (Protostome DB) for rapid annotation of NGS data in Mollusks. Korean J. Malacol. 2015, 31, 243–247. [Google Scholar] [CrossRef]
- Knudsen, B.; Kohn, A.B.; Nahir, B.; McFadden, C.S.; Moroz, L.L. Complete DNA sequence of the mitochondrial genome of the sea-slug, Aplysia californica: Conservation of the gene order in Euthyneura. Mol. Phylogenet. Evol. 2006, 38, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Heyland, A.; Vue, Z.; Voolstra, C.R.; Medina, M.; Moroz, L.L. Developmental transcriptome of Aplysia californica. J. Exp. Zool. B Mol. Dev. Ecol. 2011, 0, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Hou, G.; Song, H.; Xu, P.; Luo, D.; Gu, D.; Xu, M.; Luo, J.; Zhang, J.; Hu, Y. Transcriptome analysis between invasive Pomacea canaliculata and indigenous Cipangopaludina cahayensis reveals genomic divergence and diagnostic microsatellite/SSR markers. BMC Genet. 2015, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Wood, V.; Dolinski, K.; Draghici, S. Use and misuse of the gene ontology annotations. Nat. Rev. Genet. 2008, 9, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Brayer, K.J.; Segal, D.J. Keep your fingers off my DNA: Protein-protein interactions mediated by C2H2 zinc finger domains. Cell. Biochem. Biophys. 2008, 50, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, Z.; Xu, X.; Zhang, H.; Li, C. Genome-wide analysis of C2H2 Zinc-finger family transcriptome factors and their responses to abiotic stresses in Poplar (Populus trichocarpa). PLoS ONE 2015, 10, e0134753. [Google Scholar]
- Albertin, C.B.; Simakov, O.; Mitros, T.; Wang, Z.Y.; Pungor, J.R.; Edsinger-Gonzales, E.; Brenner, S.; Ragsdale, C.W.; Rokhsar, D.S. The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 2015, 524, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Roberts, R. WD-repeat proteins: Structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 2001, 58, 2085–2097. [Google Scholar] [CrossRef] [PubMed]
- Annadurai, R.S.; Jayakumar, V.; Mugasimangalam, R.C.; Katta, M.A.V.S.K.; Anand, S.; Gopinathan, S.; Sarma, S.P.; Fernandes, S.J.; Mullapudi, N.; Murugesan, S.; et al. Next generation sequencing and de novo transcriptome analysis of Costus pictus D. Don, a non-model plant with potent anti-diabetic properties. BMC Genom. 2012, 13, 663. [Google Scholar] [CrossRef] [PubMed]
- Potapov, V.; Sobolev, V.; Edelman, M.; Kister, A.; Gelfand, I. Protein-protein recognition: Juxtaposition of domain and interface cores in immunoglobulins and other sandwich-like proteins. J. Mol. Biol. 2004, 10, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Berisio, R.; Ciccarelli, L.; Squeglia, F.; de Simone, A.; Vitagliano, L. Structural and dynamic properties of incomplete Immunoglobulin-like fold domains. Prot. Pep. Lett. 2012, 19, 1045–1053. [Google Scholar] [CrossRef]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity (Edinb.) 2007, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, L.; Sanchez, R.; Gomez, D.; Fuenzalida, G.; Gallardo-Escarate, C.; Tanguy, A. Transcriptome analysis in Concholepas concholepas (Gastropoda, Muricidae): Mining and characterization of new genomic and molecular markers. Mar. Genom. 2011, 4, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jia, H.; Yin, Y.; Wu, G.; Xia, H.; Wang, X.; Fu, C.; Li, M.; Wu, J. Transcriptome analysis of leaf tissue of Raphanus sativus by RNA sequencing. PLoS ONE 2013, 8, e80350. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Lu, C.; Yuan, C.Q.; Cui, B.B.; Qiu, Q.D.; Sun, P.; Hu, R.Y.; Wu, D.C.; Sun, Y.H.; Li, Y. Characterization of ESTs from black locust for gene discovery and marker development. Genet. Mol. Res. 2015, 14, 12684–12691. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Database page. Available online: http://bioinfo.sch.ac.kr/submission/ (accessed on 1 January 2016).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices Clustering Tool (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Protostome DB (PANM-DB). Available online: http://malacol.or.kr/blast/aminoacid.html (accessed on 1 January 2016).
- Consea, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; Wang, J. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- MISA-MicroSAtellite Identification Tool. Available online: http://pgrc.ipk-gatersleben.de/misa/ (accessed on 14 September 2015).
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.-C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer 3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008, 9, 253. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics | A. chejuensis | A. quelpartensis |
|---|---|---|
| Raw Reads | ||
| Number of sequences | 256,655,870 | 239,242,058 |
| Number of total nucleotides | 32,338,639,620 | 30,144,499,308 |
| Clean Reads | ||
| Number of sequences | 253,220,985 | 235,525,993 |
| Number of total nucleotides | 31,397,895,789 | 29,188,212,057 |
| Mean length (bp) | 124 | 123.9 |
| High-quality reads (%) | 98.66 (sequences); 97.09 (bases) | 98.45 (sequences); 96.83 (bases) |
| Number of reads discarded (%) | 1.34 (sequences); 2.91 (bases) | 1.55 (sequences); 3.17 (bases) |
| Assembled contigs | ||
| Number of contigs | 375,118 | 463,438 |
| Number of total nucleotides | 229,108,084 | 269,776,350 |
| Mean length (bp) | 610.8 | 582.1 |
| N50 length (bp) | 788 | 719 |
| GC% of contig | 42.02 | 41.53 |
| Largest contig (bp) | 34,543 | 26,467 |
| Number of contigs ≥500 bp | 124,882 | 145,244 |
| Assembled unigenes | ||
| Number of unigenes | 198,531 | 230,497 |
| Number of total nucleotides | 145,998,300 | 162,627,732 |
| Mean length (bp) | 735.4 | 705.6 |
| N50 length (bp) | 1073 | 1001 |
| GC% of unigene | 41.98 | 41.40 |
| Length ranges (bp) | 105–34,543 | 100–29,273 |
| Databases | All Annotated Unigenes | ≤300 bp | 300–1000 bp | ≥1000 bp | ||||
|---|---|---|---|---|---|---|---|---|
| A. chejuensis | A. quelpartensis | A. chejuensis | A. quelpartensis | A. chejuensis | A. quelpartensis | A. chejuensis | A. quelpartensis | |
| PANM | 61,483 | 69,549 | 8882 | 11,191 | 27,318 | 32,401 | 25,283 | 25,957 |
| Unigene | 24,660 | 28,220 | 3857 | 4564 | 10,543 | 12,971 | 10,260 | 10,685 |
| COG | 28,197 | 31,148 | 2443 | 3085 | 8859 | 11,297 | 16,895 | 16,766 |
| GO | 23,778 | 26,396 | 2747 | 3467 | 8670 | 10,734 | 12,361 | 12,195 |
| KEGG | 2246 | 2537 | 190 | 230 | 634 | 877 | 1422 | 1430 |
| ALL | 68,484 | 77,745 | 10,515 | 13,037 | 31,552 | 37,410 | 26,417 | 27,298 |
| Domain | Description | Number of Unigenes |
|---|---|---|
| IPR015880 | Zinc finger, C2H2-like domain | 1655 |
| IPR000276 | G protein-coupled receptor, rhodopsin-like family | 338 |
| IPR012337 | Ribonuclease H-like domain | 322 |
| IPR002110 | Ankyrin repeat | 307 |
| IPR000477 | Reverse transcriptase domain | 299 |
| IPR013087 | Zinc finger C2H2-type/integrase DNA-binding domain | 290 |
| IPR027417 | P-loop containing nucleoside triphosphate hydrolase domain | 254 |
| IPR002290 | Serine/threonine/dual specificity protein kinase, catalytic domain | 238 |
| IPR003591 | Leucine-rich repeat, typical subtype repeat | 174 |
| IPR002126 | Cadherin domain | 171 |
| IPR000504 | RNA recognition motif domain | 169 |
| IPR001680 | WD40 repeat | 164 |
| IPR000742 | EGF-like domain | 162 |
| IPR002048 | EF-hand domain | 154 |
| IPR005135 | Endonuclease/exonuclease/phosphatase domain | 138 |
| IPR013783 | Immunoglobulin-like fold domain | 136 |
| IPR011701 | Major facilitator superfamily | 130 |
| IPR002035 | von Willebrand factor, type A domain | 124 |
| IPR012336 | Thioredoxin-like fold domain | 124 |
| IPR001304 | C-type lectin domain | 118 |
| IPR001478 | PDZ domain | 114 |
| IPR019734 | Tetratricopeptide repeat | 114 |
| IPR001841 | Zinc finger, RING-type domain | 111 |
| IPR001245 | Serine-threonine/tyrosine-protein kinase catalytic domain | 104 |
| IPR001849 | Pleckstrin homology domain | 103 |
| IPR002172 | Low-density lipoprotein (LDL) receptor class A repeat | 103 |
| IPR003593 | AAA+ ATPase domain | 103 |
| IPR001452 | SH3 domain | 100 |
| IPR003599 | Immunoglobulin subtype domain | 100 |
| IPR000008 | C2 domain | 96 |
| IPR011989 | Armadillo-like helical domain | 92 |
| IPR007087 | Zinc finger, C2H2 domain | 90 |
| IPR001888 | Transposase, type 1 family | 86 |
| IPR000859 | CUB domain | 83 |
| IPR001881 | EGF-like calcium-binding domain | 82 |
| IPR003961 | Fibronectin type III domain | 80 |
| IPR019427 | 7TM GPCR, serpentine receptor class w (Srw) family | 79 |
| IPR020846 | Major facilitator superfamily domain | 77 |
| IPR002557 | Chitin binding domain | 75 |
| Domain | Description | Number of Unigenes |
|---|---|---|
| IPR015880 | Zinc finger, C2H2-like domain | 1658 |
| IPR000477 | Reverse transcriptase domain | 378 |
| IPR012337 | Ribonuclease H-like domain | 360 |
| IPR027417 | P-loop containing nucleoside triphosphate hydrolase domain | 322 |
| IPR002110 | Ankyrin repeat | 322 |
| IPR000276 | G protein-coupled receptor, rhodopsin-like family | 280 |
| IPR013087 | Zinc finger C2H2-type/integrase DNA-binding domain | 249 |
| IPR000504 | RNA recognition motif domain | 223 |
| IPR002290 | Serine/threonine/dual specificity protein kinase, catalytic domain | 217 |
| IPR001680 | WD40 repeat | 198 |
| IPR002048 | EF-hand domain | 179 |
| IPR005135 | Endonuclease/exonuclease/phosphatase domain | 176 |
| IPR013783 | Immunoglobulin-like fold domain | 175 |
| IPR003591 | Leucine-rich repeat, typical subtype repeat | 165 |
| IPR002126 | Cadherin domain | 153 |
| IPR000742 | EGF-like domain | 146 |
| IPR011701 | Major facilitator superfamily | 145 |
| IPR012336 | Thioredoxin-like fold domain | 140 |
| IPR002035 | von Willebrand factor, type A domain | 126 |
| IPR011989 | Armadillo-like helical domain | 116 |
| IPR001841 | Zinc finger, RING-type domain | 109 |
| IPR001888 | Transposase, type 1 family | 105 |
| IPR019734 | Tetratricopeptide repeat | 104 |
| IPR001304 | C-type lectin domain | 104 |
| IPR001245 | Serine-threonine/tyrosine-protein kinase catalytic domain | 102 |
| IPR016040 | NAD(P)-binding domain | 101 |
| IPR001478 | PDZ domain | 101 |
| IPR002347 | Glucose/ribitol dehydrogenase family | 100 |
| IPR000008 | C2 domain | 98 |
| IPR003593 | AAA+ ATPase domain | 97 |
| IPR020846 | Major facilitator superfamily domain | 96 |
| IPR001849 | Pleckstrin homology domain | 93 |
| IPR029058 | Alpha/Beta hydrolase fold domain | 92 |
| IPR001452 | SH3 domain | 92 |
| IPR003599 | Immunoglobulin subtype domain | 90 |
| IPR015943 | WD40/YVTN repeat-like-containing domain | 89 |
| IPR007087 | Zinc finger, C2H2 domain | 89 |
| IPR002172 | Low-density lipoprotein (LDL) receptor class A repeat | 86 |
| IPR000859 | CUB domain | 86 |
| IPR000719 | Protein kinase domain | 81 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.W.; Patnaik, B.B.; Hwang, H.-J.; Park, S.Y.; Wang, T.H.; Park, E.B.; Chung, J.M.; Song, D.K.; Patnaik, H.H.; Lee, J.B.; et al. De novo Transcriptome Generation and Annotation for Two Korean Endemic Land Snails, Aegista chejuensis and Aegista quelpartensis, Using Illumina Paired-End Sequencing Technology. Int. J. Mol. Sci. 2016, 17, 379. https://doi.org/10.3390/ijms17030379
Kang SW, Patnaik BB, Hwang H-J, Park SY, Wang TH, Park EB, Chung JM, Song DK, Patnaik HH, Lee JB, et al. De novo Transcriptome Generation and Annotation for Two Korean Endemic Land Snails, Aegista chejuensis and Aegista quelpartensis, Using Illumina Paired-End Sequencing Technology. International Journal of Molecular Sciences. 2016; 17(3):379. https://doi.org/10.3390/ijms17030379
Chicago/Turabian StyleKang, Se Won, Bharat Bhusan Patnaik, Hee-Ju Hwang, So Young Park, Tae Hun Wang, Eun Bi Park, Jong Min Chung, Dae Kwon Song, Hongray Howrelia Patnaik, Jae Bong Lee, and et al. 2016. "De novo Transcriptome Generation and Annotation for Two Korean Endemic Land Snails, Aegista chejuensis and Aegista quelpartensis, Using Illumina Paired-End Sequencing Technology" International Journal of Molecular Sciences 17, no. 3: 379. https://doi.org/10.3390/ijms17030379