The Cytoskeleton—A Complex Interacting Meshwork


Institute of Anatomy and Cell Biology, Martin Luther University Halle-Wittenberg, Grosse Steinstrasse 52, 06108 Halle (Saale), Germany
*
Author to whom correspondence should be addressed.
Cells 2019, 8(4), 362; https://doi.org/10.3390/cells8040362
Submission received: 26 March 2019
/
Revised: 15 April 2019
/
Accepted: 15 April 2019
/
Published: 18 April 2019
Abstract
:The cytoskeleton of animal cells is one of the most complicated and functionally versatile structures, involved in processes such as endocytosis, cell division, intra-cellular transport, motility, force transmission, reaction to external forces, adhesion and preservation, and adaptation of cell shape. These functions are mediated by three classical cytoskeletal filament types, as follows: Actin, microtubules, and intermediate filaments. The named filaments form a network that is highly structured and dynamic, responding to external and internal cues with a quick reorganization that is orchestrated on the time scale of minutes and has to be tightly regulated. Especially in brain tumors, the cytoskeleton plays an important role in spreading and migration of tumor cells. As the cytoskeletal organization and regulation is complex and many-faceted, this review aims to summarize the findings about cytoskeletal filament types, including substructures formed by them, such as lamellipodia, stress fibers, and interactions between intermediate filaments, microtubules and actin. Additionally, crucial regulatory aspects of the cytoskeletal filaments and the formed substructures are discussed and integrated into the concepts of cell motility. Even though little is known about the impact of cytoskeletal alterations on the progress of glioma, a final point discussed will be the impact of established cytoskeletal alterations in the cellular behavior and invasion of glioma.
1. Introduction
A single animal cell has the ability to adapt its shape in response to environmental confinements or chemical cues, to move through tissues (artificial and in vivo, including narrow spaces), and to divide. These processes are all, at least in part, orchestrated by the (dis-)assembly of cytoskeletal proteins. The cytoskeleton is made up of three major types of proteins, as follows: Tubulin, actin, and proteins forming intermediate filaments. These cytoskeletal proteins differ not only in their chemical structure, but also in the type of filaments and structures they form, ranging from fast assembling dendritic actin networks of the lamellipodium, capable of generating forces necessary for cell movement over single microtubule filaments as transport structures, to intermediate filaments capable of promoting or inhibiting cell movement and stabilizing the cell against large stress.
To form a myriad of different cytoskeletal structures, as it is observed in animal cells, the cytoskeletal meshwork does not only need different components with different properties and functions, but also a tight and precise regulation of (dis-)assembly of its components, the respective local regulation of (dis-)assembly-factors, and interactions between the actin, microtubule, and intermediate filament networks.
The ability of cells to migrate is of special interest in glioma spreading, as the success of glioma treatment is crucially coupled to the question of whether a recurrent tumor will arise or not, as resection is successful only if the tumor is completely removed. Consequently, recurrent tumor formation is considered to be the main reason of tumor morbidity [1]. Hence, targeting the migratory machinery in gliomas can be a promising approach for the containment of metastasis. For successful targeting of glioma migration, a broad and detailed knowledge of the cytoskeletal architecture and its alterations is necessary.
Here we provide an overview of different cytoskeletal filaments, including actin, microtubules, and intermediate filaments, their (dis-)assembly, interactions, and function in motility and shape changes of healthy cells. Afterwards, cytoskeletal alterations in glioma and their impact on their migratory behavior are discussed.
2. Actin Regulation and Structure
In cells, actin occurs in two distinct states, as follows: The monomeric G-actin and filamentous F-actin. The modulation of the actin cytoskeleton is regulated by the balance of globular G- and polymeric F-actin and by actin associated proteins [2]. The actin cytoskeleton forms a network consisting of polarized filaments that are mostly associated with force generation necessary for movement, focal adhesion, and shape changes. In the following we describe the building blocks of actin filaments, the assembly and disassembly of filaments, their kinetics, regulation, as well as filament bundles, network structures, and their mechanical properties. A summary of all mentioned actin binding or associated proteins and their function can be found in Table 1.
2.1. Actin Filaments
Actin filaments are, in contrast to intermediate filaments and microtubules, semi-flexible filaments, forming dendritic or cross-linked structures. Semi-flexible means that the persistence length of a single filament is in the order of the filament length, where the persistence length is the length scale on which the correlation between two tangents along the filament drops to 1/e [3]. As a semi-flexible polymer, actin filaments are actively bent by thermal fluctuations, thus generating additional resistance to forces stretching the filament. Actin itself is considered the most dynamic of the three cytoskeletal proteins capable of strong structural changes in the time scale of minutes, thus determining the shape of a cell. A single actin filament consists of actin monomers, called globular actin (G-actin). Under nearly physiological conditions G-actin polymerizes to asymmetric helical structures, filamentous actin (F-actin), with a typical length of 6–7 µm in in vitro studies [4]. The nucleation kinetics is mostly limited by the generation of dimers and trimers [5]. Having reached the trimer state, filament nucleation increases rapidly, but in dependence of the available G-actin pool [6] (Figure 1). The resulting actin filaments have a right handed helical structure. G-actin is polarized, therefore F-actin is polarized as well, with the less dynamic side termed as the (−)-end and the more dynamic (+)-end having a ten times higher polymerization rate than the (−)-end [6]. As actin is an ATPase the (+)- and (−)-ends can also be distinguished by their ATP/ADP status, especially if the growth at the (−)-end is inhibited further. Thus the (+)-end contains higher amounts of ATP bound actin while the (−)-end contains more ADP bound actin.
2.2. Profilin
As F-actin is capable of forming spontaneously above a certain critical concentration of G-actin (≈0.1 µM) a precise cofactor-driven control of actin polymerization is necessary. One such control element is profilin. Profilin is an actin binding protein that regulates actin homeostasis [7,8] by inhibiting the spontaneous formation of actin di- and trimers, but it also catalyzes the transition from ADP- to ATP-actin [7]. Profilin bound G-actin can be used for the construction of actin filaments if nucleation factors like the Arp2/3 (actin-related-proteins 2/3) complex or formins are present [9]. Interestingly, if formins and profilin are present, free actin elongation can be increased by a factor of up to 9 [10,11]. Additionally, profilin was shown to inhibit polymerization at the (−)-end of actin [6].
2.3. Dendritic Actin Networks
Besides the quasi-linear actin filaments there are dendritic actin networks, formed by the Arp2/3 complex [12]. These networks are usually formed at the cell front on a short time scale [13] and, thus, its regulation is of high importance. The generation of a dendritic actin network starts from a so called primer, an existing actin filament at which Arp2/3 binds to its side [9,14]. Arp2/3 is, amongst others, activated by members of the WASP family [6,15]. For the generation of a dense dendritic network not only nucleation factors, but also capping proteins are needed to restrict the elongation of the actin (+)-ends [14,16,17,18]. If capping proteins are present the Arp2/3 complex can generate multiple networks originating from different actin filaments that are able to merge and generate forces near the cell membrane [14,17]. The number of nodes is important for the mechanical properties of the generated network and consequently the elastic modulus scales with the mesh size M by 1/M4. In general the dendritic network behaves visco-elastic, meaning it is mainly elastic on small time scales (<1 min) and viscous on longer time scales (>10 min) [14,17].
2.4. Non-Muscle Myosin
Another important molecule class is the myosin family. Here we will focus exclusively on the non-muscle myosin. Myosin is responsible for the contractility of anti-parallel actin structures using ATP hydrolysis as the energy supply [19,20]. These contractile structures are mainly responsible for the retraction of the cell rear for productive movement, but also for transmitting forces to the surrounding extra-cellular matrix.
Interestingly, myosin II motor activity alone is insufficient to produce contractility. Single myosin II hexamers are unipolar and thus ineffective in generating contractile forces [21], but when assembled into bipolar mini-filaments they are highly processive and capable of generating forces by pulling on anti-parallel actin filaments [22]. Myosin II can be activated via phosphorylation of the regulatory light chain (RLC) or activation of myosin light chain kinases (MLCK). RLC is activated via Rho-associated protein kinase (ROCK) or citron kinases (both activated by RhoA) and MLCK by Ca2+ [23]. After RLC phosphorylation myosin is capable of generating contractile forces [24,25]. Another type of regulation works via the phosphorylation of the myosin heavy chain, utilizing myosin heavy chain kinases (MHCK), casein kinase II (CKII), or protein kinase C (PKC), inhibiting mini-filament assembly or dissociating existing mini-filaments [26,27,28,29]. The switch between those two activation states influences the contractility of the respective acto-myosin network. Consequently, regulation of myosin strongly impacts organization and properties of contractile actin structures, as discussed below.
2.5. Cross-Linked Actin Networks and Actin Bundles
Despite the already mentioned dendritic actin structures, there are actin bundles and networks linked together by cross-linkers. Cross-linkers are molecules that connect single actin filaments either transiently or non-transiently and are either passive (e.g., scruin, fascin, α-actinin, filamin, or fimbrin) or active (myosin). Cross-linked actin bundles and networks largely control shape, mechanical integrity, and contractility of a cell [30,31,32]. Generally, cross-linkers do not influence actin assembly (except for Arp2/3) [32,33,34,35]. Cross-linkers bind actin filaments based on their own size and the position of their binding-sites in different distances, ranging from 10 nm for fimbrin to 160 nm for filamin, and thus determine the density of the resulting actin structure [36,37]. Additionally, the speed of actin polymerization influences the presence of cross-linkers in the resulting network, supposedly by crowding effects, thus excluding larger cross-linkers like α-actinin in quickly polymerizing filaments [33]. If the formed actin structures are subject to a force that acts on a longer time scale than the binding time of the cross-linkers itself, a reorganization of cross-linkers and a subsequent shape change of the bundle occurs [38]. This time scale depends on the type of cross-linker and its binding and unbinding time, which can be in the order of seconds [39,40]. Consequently, cross-linked actin is elastic on short and viscous on long time scales [38] and the presence of cross-linkers generally increases the elastic part of the visco-elastic answer to external stress.
If actin filaments are bundled by cross-linkers the filaments inside the bundle can either be oriented in parallel or anti-parallel, meaning that (+)-ends of neighboring filaments are pointing in the same or the opposite direction. Parallel actin bundles are found amongst others in filopodia [18,41], while anti-parallel bundles are mostly found in stress fibers.
Two mechanisms were proposed to explain the generation of parallel actin bundles. The first mechanism involves the Arp2/3 complex-dependent elongation of filaments in the absence of capping proteins, so that free growing (+)-ends transition into bundles via electro-static interactions between filaments [42]. Here, the geometric constraints and the angle at which filaments make contact to each other determine whether parallel or anti-parallel bundles are formed [42]. Notably, geometric constraints refer to all spatial limitations, such as the available free space, steric effects, etc., that potentially affect the final organization. Parallel bundles are then stabilized via cross-linkers, such as fascin. The second mechanism is formin dependent [43]. Thereby the FH1 (formin homology) domain of formin functions as a ring structure capturing profilin bound actin molecules, while the FH2 domain interacts with the (+)-end of the filament [44,45]. Some members of the formin family also move from the end of the filament into the middle, additionally functioning as cross-linkers to stabilize the generated structure [10,46,47]. As formins do not necessarily bundle filaments, further proteins, such as cross-linkers or Ena/VASP, are also involved in formin dependent bundle formation. Ena/VASP is a protein family associated with anti-capping function and elongation, but has no nucleation activity on its own [48].
In contrast to parallel bundles, anti-parallel bundles are mostly connected with classical cross-linkers and the motor-protein myosin, which has the ability to actively move antiparallel fibers relative to each other. As with parallel bundles, anti-parallel bundles are stabilized by cross-linkers favoring this configuration, like α-actinin or fimbrin [49,50,51]. Through the activation of myosin anti-parallel bundles are pre-stressed, leading to either a contraction or dissociation of the bundle [52,53]. Without further cross-linkers, anti-parallel bundles containing myosin first contract strongly and later disassemble [54].
The mechanical properties of (anti-)parallel bundles depends on the type and density of cross-linkers and, thus, on the compactness of the bundle and whether the bundle allows the sliding of single filaments. For non-cross-linked bundles the persistence length scales with the number of filaments while for cross-linked bundles that allow no filament sliding it scales with the number of filaments squared.
2.6. ADF/Cofilin Induced Actin Disassembly
As most of the actin structures are stable in time, cells need a mechanism to induce actin disassembly to adapt to environmental cues. One such mechanism is governed by the actin binding ADF/cofilin protein family, capable of disassembling and fragmenting actin filaments, but incapable of altering the polymerization rate [55,56]. The efficiency of ADF/cofilin depends on its binding state to actin filaments. Filaments that are fully decorated with ADF/cofilin are stabilized while partially decorated filaments fragment faster [57,58,59]. The induction of fragmentation is likely caused by a reduced persistence length of the filament, that can drop to ≈20% of its initial value through ADF/cofilin binding, locally generating increased mechanical stress [58,59,60]. ADF/cofilin preferentially binds to the (−)-end of actin filaments (ADP bound actin) [61], but binding to ATP bound actin favors its transition from ATP to ADP bound actin and, thus, accelerates filament dissociation [62]. The preference for older ADP bound actin implies that ADF/cofilin disassembly mostly affects inactive compartments of the actin network [55,56,63,64]. As ADF/cofilin cannot bind to the free (+)-end it can only fully saturate actin filaments if the (+)-end is bound by capping proteins and is not capable of further elongation [65]. With the exception of fascin, cross-linkers and tension reduce the efficiency of ADF/cofilin fragmentation [34,66,67,68]. Furthermore, ADF/cofilin is present in high concentrations at the leading edge in dendritic networks, fragmenting links generated by Arp2/3 and the actin filaments itself, generating more free (+)-ends [69].
2.7. Actin Structures Inside the Cell
Looking at a motile cell the net movement is the result of multiple, mostly actin-dependent, processes, as follows: Formation of protrusions in direction of motion, subsequent adhesion to the substrate and loss of adhesion on the rear of the cell, followed by rear-contraction (reviewed in [70,71,72,73]). These processes are governed by sub-cellular structures, like filopodia and the lamellipodium governing cell motion, while stress fibers and the cortex secure mechanical stability and contractility. Further types of protrusions are so called blebs, which are capable of regulating cell movement independently of filopodia and the lamellipodium. The interactions of actin discussed here and its structural and functional integration with microtubules and intermediate filaments are summarized in Figure 2.
2.7.1. The Lamellipodium
The lamellipodium (Figure 3) is a flat structure mainly associated with cell movement, formed by the polymerization of actin at the cell front [13,74], while it is depolymerized at the back of the lamellipodium by ADF/cofilin refilling the G-actin pool [75]. The continuing (de-)polymerization of the whole network creates a treadmilling effect and a retrograde actin flow in the cell [76,77], which is enhanced in some cell types by myosin induced depolymerization at the back of the lamellipodium [78]. Any flow originating from the contraction of the rear via stress fibers generates a flow of opposite direction [77]. The forces generated by actin polymerization in the lamellipodium are up to a few hundred pN/µm [79]. The most important factor for the generation of the lamellipodium is the intrinsically inactive Arp2/3 complex that becomes activated by the Scar/WAVE complex in an activation process by the small Rho GTPase Rac1 [80]. Arp2/3 nucleates a new actin filament at the site of existing filaments [80]. For a three dimensional environment, N-WASP (and not WAVE) was shown to activate Arp2/3 and Rac1 was not found to be strongly localized at the cell front [81,82]. Actin growth is further promoted by the presence of members of the Ena/VASP family accumulating at the lamellipodial tip, promoting further actin elongation and preventing capping [83,84]. Despite the active Arp2/3 complex, a capping protein is needed as well to limit the elongation of single filaments [16,85] so they remain productive and do not form bundles with other uncapped filaments or buckle under the load [86]. For the generation of a stable dendritic network, it is cross-linked by proteins such as cortactin [87]. As the described regulation by Rac1 would result in a constant growth of the lamellipodium, it has to be restricted by a negative feedback loop. One possible mechanism is via the protein arpin, which inhibits Arp2/3 activity in the lamellipodium [88]. It has been postulated that arpin is recruited by Rac1 [88]. Thus, it seems possible that Rac1 activation initiates lamellipodium growth via quick Arp2/3 recruitment and successive actin polymerization and later inhibits its growth via recruitment of arpin. A high turnover rate of arpin or significantly higher concentration might be necessary to inactivate Arp2/3 [89]. A proof for this kind of hypothesis is yet lacking. Despite the actin dynamics, the lamellipodium is also influenced by the cell membrane and its surface tension [90]. A higher membrane surface tension led to a more oriented actin filament polymerization while a lower tension resulted in more protrusions [90], probably related to the finite forces generated by the lamellipodium. Regarding the mechanical properties of the lamellipodium, it has to be noted that myosin was observed to be present at the rear of the lamellipodium, explaining why the lamellipodium is elastic on short and viscous on long time scales [78,91].
Due to Arp2/3 the actin in the lamellipodium is connected to a dendritic structure [92]. Interestingly, an analysis of cell speed relative to the actin orientation in the lamellipodium could demonstrate that faster cells have filaments that are not exactly oriented in the direction of movement and the parallel orientation of filaments is associated with slower movement [93].
2.7.2. Filopodia
Further structures associated with cell motility are filopodia (Figure 3). Filopodia are associated with a sensory function in neurons [94], but do not seem necessary for migration, as the fast moving corneal keratocytes do not possess filopodia in two dimensions and forces generated by filopodia are significantly smaller than those generated by the lamellipodium [95]. In other systems there might be a role for filopodia in migration, e.g., in three dimensional systems [96]. Filopodia form a structure consisting of parallel actin bundles, with their (+)-ends pointing in direction of the cell membrane [97]. This orientation is established via formins (e.g., FMNL2) and Ena/VASP, both being capable of maintaining a prolonged actin polymerization [98]. Some of these formins, like mDia2, can be activated by the small GTPase Cdc42 [99]. Cdc42 is also capable of activating N-WASP and thus Arp2/3, leading to filopodia formation [100]. A common model for filopodia initiation suggests that actin polymerization occurs in the presence of activated Arp2/3 and without capping proteins forming actin bundles [98]. Nevertheless, Arp2/3 does not seem to be necessary for filopodia initiation in adherent cells [101]. A further model of filopodia initiation states that filopodia are initiated by clusters of activated formins near the plasma membrane, nucleating and elongating actin filaments [98]. For both models, subsequent further elongation via formins (e.g., mDia2) and Ena/VASP and stabilization and bundling with cross-linkers, like fascin, generates “mature” filopodia [102]. Besides their role in cell movement, filopodia initiate cell-cell contacts, transmit cell-cell-signals, and respond to the mechanical properties of their surroundings [103]. Interestingly, when filopodia are retracted to the cell the myosins II,V, and VI are not involved in this process [104]. This leads to the idea that only actin (de-)polymerization and changes in the cortex are responsible for filopodia dynamics.
2.7.3. Stress Fibers
Another type of actin related structures are stress fibers (Figure 3) that are neither present in filopodia nor in the lamellipodium. Stress fibers are formed from bundles of anti-parallel actin filaments containing myosin II or parallel filaments [105]. Stress fibers are assembled bundles of 10–30 actin filaments [106], cross-linked by α-actinin in a bipolar fashion, and linked to focal adhesions [105,107]. Focal adhesions are binding sites that connect the cell to the substrate. Contractile stress fibers are one of the main contributors to cell contractility in many animal cells. As the contractility of these stress fibers is regulated by myosin [108], regulation of stress fiber contractility is in many ways similar to the regulation of myosin activity discussed before. In non-motile cells, stress fibers are usually thick and comparably stable, while motile cells typically contain fewer less pronounced fibers with a higher dynamic [109]. Actin and myosin are the two principal constituents of contractile stress fibers, while non-contractile ones do not contain myosin [110]. Despite these components, stress fibers contain actin binding proteins and focal adhesion-associated proteins binding and unbinding in quick succession [110,111,112,113]. The molecules found in stress fibers include cross-linkers, such as α-actinin [114], which does not only function to stabilize the bundle but is also associated with kinases and signaling proteins and, thus, functioning as a signaling mediator [115,116]. Stress fibers can contain further cross-linkers, like fascin, filamin, and paladin, but their precise role despite bundling remains elusive [117,118,119]. One hypothesis states that these proteins function as basis for regulation of cytoskeletal dynamics as, for example, paladin interacts with profilin and VASP [120,121]. Further molecules of e.g., the calponin, tropomyosin, caldesmon family, and others, are found in stress fibers and are all suggested to be part of the cytoskeletal and/or stress fiber regulation [114,122,123,124,125]. Generally speaking, stress fiber formation has been directly associated with an activation of the formin mDia1 and the small Rho GTPase RhoA, activating ROCK [126,127]. The formin favors prolonged actin polymerization of parallel filaments important for dorsal stress fibers [110,128]. In contrast ROCK activates the LIM kinase (LIMK), which inhibits ADF/cofilin induced filament severing [129], and additionally, ROCK activates myosin, favoring stress fiber formation [23,105]. Nevertheless, both the ROCK and formin mechanisms are necessary for the formation of contractile stress fibers [109]. Two other Rho GTPases, Cdc42 and Rac1, act in more indirect ways via the induction of lamellipodial growth via Arp2/3 (Rac1) and filopodia formation via the formin mDia2 (Cdc42) [99,130,131,132]. Collapse of both filament types can function as seeds for either transversal or ventral stress fibers.
Since stress fibers vary in their morphology, molecular signature, and association with focal adhesions, the four following types of stress fibers can be distinguished: The perinuclear actin cap, transverse arcs/stress fibers, and dorsal and ventral stress fibers.
The three classes of contractile stress fibers are the ventral and transverse stress fibers and the perinuclear actin cap, all characterized by the presence of myosin II along the fibers. Even so, the myosin II spacing can change over time, indicating that contractile stress fibers are dynamic structures with non-uniform mechanical properties [133]. Measurements indicate that stress fibers have a stiffness of roughly 12 kPa, constant for strains up to 0.12 [134]. Perturbation of myosin in stress fibers reduces the elastic modulus to 8 kPa, indicating the importance of myosin II in contractile stress fibers [134]. Not surprisingly, the tensile elastic modulus increases from approximately 1.5 MPa to 104 MPa for strains approximating 2 [135]. Ventral stress fibers are oriented parallel to the direction of cell motion and connect adhesion sites of the cell, while transverse fibers are oriented perpendicular to the ventral fibers and are not directly connected to focal adhesions. Even so, transversal stress fibers can contribute to the overall contractility through their connection to dorsal stress fibers [110,136]. The formation of transversal stress fibers is dependent on Arp2/3 and myosin [110] and possess a periodic α-actinin-myosin pattern [137]. Transversal stress fibers form when the dendritic network collapses and is restructured by myosin [138,139]. Notably, simulations on the capability of myosin to generate contractile structures suggest that the presence of myosin and actin is sufficient to generate anti-parallel/contractile bundles, as these were found to be energetically favorable [140]. A further origin of both transversal and ventral fibers is the collapse of filopodia, which functions as a seed for stress fibers [141]. Additionally, ventral stress fibers can be formed from existing dorsal stress fibers and the attached transverse stress fibers, as well as by the fusion of two dorsal stress fibers [110,142]. Ventral stress fibers are also contractile actin-myosin bundles attached to focal adhesions at both ends, thus being directly part of the contractile machinery [143]. Due to their location at the rear of the cell and an orientation that is roughly in the direction of motion, they are part of the rear contraction, and thus associated with cell motility [73,144]. The third type of contractile stress fibers is the so-called perinuclear cap, consisting of stress fibers positioned above the nucleus, regulating the shape of the nucleus. Additionally, they are proposed to serve as a mechanical connection between the nucleus and the rest of the cell [145]. All of these contractile stress fiber types have in common that they are highly dependent on presence and activity of myosin and, thus, on tension. Consequently, myosin inhibition leads to the disassembly of these stress fibers [146].
In contrast to the other stress fiber types, dorsal stress fibers do not contain myosin II [110,111] and are anchored at focal adhesions at their distal ends [110,136]. The lack of myosin directly leads to the lack of contractility of dorsal stress fibers. It is proposed that these fibers consist of fast growing (+)-ends that face the cell periphery and more distant parts consisting of mixed polarity actin filaments [106,109]. Furthermore, paladin and Rac1 are seemingly essential for the formation of dorsal stress fibers. Paladin promotes fiber assembly via VASP recruitment [147,148]. Functionally, these stress fibers seem to be an anchor point for the assembly of the other stress fiber types and a link to focal adhesions [110,111]. It is supposed that dorsal stress fibers are generated via actin polymerization at emerging focal adhesions [110] and stabilized during retraction phases of the lamellipodium via coupling to emerging transverse stress fibers [111,138,149].
2.7.4. Actin Cortex and Blebs
The last cytoplasmic structure described here is the actin cortex (Figure 3), which forms a contractile actin structure at the boarder to the plasma membrane. The cortex is a few hundred nanometer thick layer, consisting of a mixture of filament bundles and cross-linked filaments, with a mesh-size of approximately ≈50–150 nm [150], a thickness of 50–100 nm [151,152], and a distance to the cell membrane of less than 20 nm [151]. The cortex meshwork appears to be mainly isotropic, oriented in parallel to the plasma membrane, but some filaments are also oriented perpendicular to the membrane [153]. Despite actin filaments, the cortex contains a number of cross-linkers (e.g., fascin, actinin, filamin, etc.), myosin, proteins that control actin turnover (like profilin, cofilin), capping proteins, proteins of the ERM (ezrin, radixin, moesin) family, nucleating factors (like Arp2/3, the formin mDia1), and signaling molecules such as RhoGTPases, RhoGEFs (guanine exchange factors), and RhoGAPs (GTPase activating proteins) [154,155]. The two mentioned nucleating factors Arp2/3 and mDia1 were also found to be responsible for the majority of cortical F-actin generation [155,156], while the ERM proteins link the cortex to the membrane and can therewith transmit forces acting on the membrane and determine the cell shape [157,158,159]. Depletion of cofilin-1 or capping proteins in HeLa cells increased cortex thickness but reduced tension, implying a role for actin regulating proteins in cortical tension [160]. Mechanical properties of the cortex determine how the cell deforms in response to external forces. On time scales smaller than the remodulation time of the cortex it behaves elastic [161], with a cell type-dependent elastic modulus in the order of a few hundred to thousands of pascals [162,163]. On long time scales (>1min) the cortex behaves viscous because of the adaption to external forces via actin modulation, dissociation, and (un-)binding of cross-linkers [161]. If myosin is activated the cortex turnover times can be even lower [164,165], perhaps via direct disassembly or enhanced actin breakage [52,78]. Generally speaking, the behavior of the cortex is similar to that of glassy materials [166] and consistent with relaxations of three dimensional in vitro actin networks [167].
One of the main global and local properties of the cortex is its tension, which regulates the cell shape of single cells and tissues [168]. Several studies demonstrated that the cortex tension depends on the myosin activity and actin polymerization, with higher myosin activity and lower actin polymerization leading to an increased cortex tension [169]. A lower cortex tension is further associated with an increased protrusive activity of the cell, thus indirectly regulating cell motility [170]. Interestingly, local drops in cortex tension or cortex-membrane adhesion and local ruptures of the cortex can be the origin of so called blebs (Figure 3), a special, initially actin free, membrane protrusion [171]. Blebs can be initiated by any type of cortex weakening or loss of cortex-membrane adhesion if a given internal hydrostatic pressure threshold is reached [172,173]. Localized myosin contractions, promoting either cortex tearing or increasing local intracellular pressure, are some of the main sources of blebbing [174,175,176], but others are also discussed [177]. Thus, the activation of myosin via the already described activation by ROCK or MLCK are sufficient to induce bleb formation [178,179,180]. The progress of a bleb can be divided in three steps, as follows: Initiation, growth, and retraction. Initially, the growing bleb does not contain actin, but over time, when the bleb expands further, the actin cortex reassembles at the plasma membrane, stalling the bleb growth [157] up to the point of a full restoration where the generated contractile forces retract the bleb [181]. It has to be noted that bleb retraction does not always occur and in some motile cells blebs are stabilized and used as an alternative or additional mode of migration [178,179,180,182]. The expansion of blebs by actomyosin contraction induced pressures lasts 5–30 s, accompanied by a flow of cytosol into the bleb and a concomitant increase in surface area. The surface area is increased by a flow of lipids through the tearing of the membrane from the actin cortex [183]. The maximal bleb size is determined by the initial growth rate and the cortex re-polymerization time [184], both being dependent on cortex tension. The concept of tension inhibiting bleb expansion is further supported by the idea that the needed membrane unfolding is effectively resisting bleb expansion and, thus, slowing down the growth [182,185]. After full maturation the cortex is reconstructed and if the bleb is not stabilized via adhesions it is retracted by the re-established cortex via a myosin induced contraction [178,186].
2.7.5. Nuclear Actin
For completeness it has to be mentioned that actin is not only present in the cytoplasm of eukaryotic cells, but also in the nucleus. As in the cytoplasm, nuclear actin exists in a monomeric and polymeric form [187]. Nuclear G-actin was shown to associate with all three RNA polymerases, participating in transcription initiation and elongation [188]. The exact function of G-actin in the transcription complex remains unclear, but G-actin levels need to be precisely tuned for normal translation [189] and cofilin is required for its elongation [190]. Contrary, stable actin filaments inhibit transcription [191]. Furthermore, actin is implied to affect the nuclear structure. During nuclear expansion at the mitotic exit, chromatin reorganizes depending on the transient formation of polymeric actin, in a seemingly cofilin dependent manner [192]. It is probable that nuclear actin regulates the structure of the nuclear envelope and the nucleus via interactions with the nuclear intermediate filaments lamin [193].
As mentioned, cell nuclei contain polymeric actin that can be generated inside the nucleus via actin nucleation factors [194]. These actin assembly factors include, amongst others, mDia1, Spire1/2, Fmn2, and Arp2/3 [195]. The presence of assembly factors underlines a functional role of polymeric actin and actin binding proteins. Initiation of DNA replication was demonstrated to require formin dependent nuclear actin polymerization [196]. Additionally, the loss of Spire1/2 and Fmn2 resulted in a less efficient clearance of DNA double strand breaks [197]. This is in agreement with two studies demonstrating an association of actin and Arp2/3 with sites of DNA damage and decreased damage repair after reduced Arp2/3 dependent actin nucleation [198,199]. For more information on the role of actin in the nucleus the interested reader is referred to other reviews [188,200].
3. Microtubules
Microtubules consist of α- and β-tubulin heterodimers forming hollow filaments, usually consisting of 13 protofilaments [201]. The microtubules are seeded by the microtubule organization center (MTOC), generally centrosomal, but in some types of differentiated cell non-radial microtubules are assembled by non-centrosomal MTOCs, for example at the Golgi apparatus [202,203,204]. MTOC, amongst others, contains γ-tubulin as a microtubule nucleator, as well as anchoring and adaptor proteins for attachment of microtubules [205]. Heterodimers interact at the MTOC with the γ-TURC, thus microtubules are nucleated and anchored with their (−)-ends to the MTOC [205]. Microtubules show a behavior called dynamic instability, characterized by a sudden switch from growing to growth arrest and/or quick depolymerization (termed catastrophe), followed by a new growth cycle [201] (Figure 4). A possible explanation for this behavior can be derived from the formation process of single protofilaments. During polymerization, GTP-bound tubulin heterodimers are bound to the (+)-end and normally hydrolyzed shortly after, but sometimes older parts of microtubules still contain GTP-bound tubulin heterodimers [206]. This leads to the following model: If, due to stochastic fluctuations or other perturbations, the (+)-end does not contain the more stable GTP-bound tubulin, it depolymerizes and is stabilized at older GTP-bound tubulin sites [206]. Stochastic fluctuations in the rate of microtubule growth and the stochastic nature of GTP hydrolysis lead to a dynamic GTP cap size [207], with the consequence that faster polymerizing microtubules have a larger GTP cap, resulting in less frequent catastrophe events [208]. The dynamic behavior of microtubules can be regulated by both intrinsic and extrinsic factors (see Table 2 for summary). Microtubule interacting proteins are either microtubule (+)-end-binding proteins (+TIP) or structural microtubule-associated proteins (MAP) interacting with microtubules along their length. These protein classes can have stabilizing or destabilizing effects, changing polymerization dynamics or severing microtubules. Important proteins belonging to the family of the +TIPs are CLASPs (cytoplasmic linker associated protein) and APC (adenomatous polyposis coli) [209,210] that suppress microtubule catastrophe events and promote rescue after catastrophe [211]. Part of the stabilizing effects of CLASPs arises from their capability to modulate interactions between microtubules and the cell cortex [212]. Further important families of +TIPs are the spectraplakins, binding both microtubules and actin [213] and EBs (end binding proteins). EBs are supposed to be a master regulator of +TIP recruitment (e.g., CLASP [214], APC, MACF1 (microtubule-actin crosslinking factor) [215]) and complex assembly [211], generally promoting persistent microtubule growth [216]. EBs are generally associated with an increased polymerization rate and reduced catastrophe number [217,218]. Despite these molecules that mainly (de-)stabilize microtubules, there is a bunch of proteins that sever microtubules, like spastin [219] or katanin [220], or influence depolymerization and polymerization, e.g., stathmin (favors depolymerization) [221] or XMPA215 (increases polymerization rate) [222]. Additionally, there are structural MAPs, like tau protein, MAP2, or DCX (doublecortin), that interact with the filament at its whole length and stabilize it [223,224] by reducing shrinkage speed, promoting filament growth, and reducing catastrophe frequency [223,224]. The effect of structural MAPs can also inhibit the effect of other microtubule associated proteins, as, for example, tau protein can inhibit the katanin induced severing [225]. A further important class of MAPs are the motor proteins kinesin and dynein, both serving as cargo transporters, exploiting the microtubule meshwork [226,227]. In general, kinesin motor proteins transport cargo to the (+)-end, while dynein moves to the (−)-end of microtubules [228,229], transporting diverse cargo types, including membrane components, signaling molecules, such as the small GTPases Rac and Cdc42 [230,231], but also intermediate filaments and their precursors [232,233,234], β-actin coding mRNA, and sub-units of the Arp2/3 complex [235,236]. Motor proteins possess not only a transport function but can stabilize or destabilize microtubules. For example, members of the kinesin-8, kinesin-13 family, or KinI kinesins can induce depolymerization, likely via the destabilization of the GTP cap [237] or the induction of kinks [238]. Despite their transport function and regulatory role in microtubule dynamics, some kinesins organize the microtubule network via the bridging of microtubules, thus favoring the generation of parallel arrays. For example, in neurons kinesin-5 and kinesin-12 are necessary for axonal outgrowth because of their cross-linking ability and the concomitant focus on the extension of microtubule arrays [239,240]. Additionally, kinesin-1 may be involved in this process by sliding filaments alongside each other [241]. A further important aspect regulating microtubule dynamics are the post-translational modifications modifying microtubule properties and affinities of MAPs [242]. Important modifications are, amongst others, tyrosination, glutamylation, and acetylation. Acetylation protects microtubules against repeated mechanical stress via an increased flexibility, but does not protect against depolymerization [243]. Additionally, some severing proteins, such as katanin, preferentially interact with acetylated tubulin [244]. Tyrosination affects the recruitment of microtubule interacting proteins, such as CLIP-170 or kinesin-1, that prefer detyrosinated microtubules, probably facilitating directional transport [245,246]. In contrast, spastin favors cleavage of detyrosinated microtubules [247]. Similar to tyrosination, glutamylation can also affect the interaction with microtubule associated proteins. Map2, tau, and kinesin-1 were reported to preferentially interact with those microtubules with up to three glutamates on their tail [245]. Similarly, both microtubule severing proteins, katanin and spastin, show an increased affinity for glutamylated microtubules [248,249]. Taken together, this data suggests that post translational modifications are important and finely tunable regulators of microtubule dynamics and, consequently, of cell behavior.
For regulation of MAPs, and microtubules in general, the family of Rho GTPases is of major significance. An important example is the stabilizing effect of RhoA, but not Rac1 and Cdc42, on microtubules via the RhoA effector mDia. mDia is capable of interacting with EB1 and APC, leading to a stabilization of microtubules via e.g., Kif4 [250,251]. Furthermore, active mDia induces the alignment of actin and microtubules [251,252]. Interestingly, stathmin action seems to negatively regulate the RhoA/ROCK activity [253], complementing the observation that Cdc42 and Rac1 phosphorylate stathmin via an activation of PAK (p21-activated kinases) [254,255]. In fact, Rac1 activation was demonstrated to decrease catastrophe events and increase microtubule growth time in cells via PAK [255,256]. Furthermore, both Rac1 and Cdc42 activate IQGAP1, interacting with Clip-170, likely providing a stabilization site for microtubule (+)-ends near the cortex [257]. The interactions of microtubules discussed here and their interactions with actin and intermediate filaments are summarized in Figure 2 and Figure 3.
Microtubules typically play a role as tracks for transport, as already mentioned, in spindle positioning during mitosis, migration (discussed below), and in cell shape control [258,259,260]. At the current point, the concept that microtubules control the balance between RhoA and Rac1 and thus influence cell shape and migration is favored over a direct mechanical participation for most cell types [261]. Even so, microtubules are relatively stiff polymers, when compared to actin [262], capable of generating forces of up to 3–4 pN during polymerization [263,264,265]. As a result, microtubules can deform membranes and resist compressional forces in such a way that they act as load bearing fibers in living cells via transversal re-enforcement by other cytoskeletal components [266,267]. Notably, if multiple microtubule filaments grow as a bundle the generated forces increase linearly with the number of microtubules per bundle [268]. Complementary to these observations, the load bearing capacities of microtubules is limited because compressional loads can induce catastrophe events [265,268], in line with the fact that most catastrophes are indeed induced at the cell edge [269,270] and the observed short-wavelength buckles near the boundary of adherent cells [170,271]. A further mechanism of force generation by microtubules is during the shrinkage phase. To actually transmit a force during shrinkage, the (+)-end needs to stay attached to its cargo. When the GTP cap is lost the microtubule protofilaments lose their lateral connection with neighboring protofilaments, bending backwards and forming ring-like shapes [272,273] (Figure 4). If cargo stays attached during this process, a single microtubule can exert forces of up to 30–65 pN [272,273], a magnitude larger than the pushing force [263].
Microtubules also play a key role during the separation of chromosomes during cell division [274]. Depolymerization of microtubules is believed to generate the needed forces to separate the sister chromatids [275]. This aspect will not be discussed in more detail here, but the interested reader may be referred to the following reviews: [274,275,276].
Furthermore, microtubules and actin are linked regarding their functional dynamics and structural organization. On the one hand there is an indirect co-regulation, as microtubules are able to locally regulate and are regulated by RhoGTPases and focal adhesions (see also chapter 5.2), but on the other hand, there are molecules interacting with microtubules and actin. One such molecule is APC, which stabilizes microtubules and nucleates actin filaments, with actin nucleation additionally favored by the formin mDia1 [209,277]. A further formin mDia2 that is capable of nucleating actin can also stabilize microtubules, independent of its nucleation function [278]. There are additional cross-linkers connecting actin and microtubule filaments, such as MACF1 and Arg [279,280]. Consequently, the actin and microtubule cytoskeleton cannot fully be regarded as decoupled systems, as they are not regulated independently and can even be connected physically.
4. Intermediate Filaments
Intermediate filament-forming proteins are a large protein class, encoded by at least 70 genes, organizing filaments with a diameter of 10 nm. Intermediate filaments are grouped in 5 classes according to their structure and sequence homology. Thereby, the first four classes represent cytoplasmic intermediate filaments, while type V are nuclear filaments, so called lamins (lamin A/C, B1, B2). Type I and II are acidic and basic keratins, forming heteropolymers consisting of a mixture of the 54 different type I and II keratins, expressed in dependence of cell type and differentiation status [281]. In contrast, type III intermediate filaments are homopolymers of vimentin, desmin, peripherin, or glial fibrillary acidic protein (GFAP). Vimentin is mainly expressed in fibroblasts, endothelial cells, astrocytes; peripherin in neurons of the peripheral nervous system and desmin in muscle cells and GFAP mainly in astrocytes. Type IV intermediate filaments contain three neurofilament heteropolymers (NF-L/M/H), internexin, synemin, and nestin, mainly expressed in the cells of the nervous system. Nestin and synemin cannot form filaments on their own, but only in conjunction with other intermediate filament proteins [282,283]. Two further intermediate filaments, called filensin and phakinin, cannot be grouped into the mentioned five types. They are expressed in the lens epithelium, forming heteropolymers [284]. All cytoplasmic intermediate filaments have a similar monomer structure, consisting of a central α-helix with a non-helical structure at both of its ends [285]. Two monomers spiral around each other, forming a so-called “coiled-coil” dimer [286] and, subsequently, these dimers form unpolarized tetramers via antiparallel association and 8 tetramers form a cylindrical unit-filament [287]. The unit filaments aggregate further with other unit filaments at the time scale of minutes to form intermediate filaments [288] (Figure 5). After aggregation, the filaments undergo a compaction step during which the filament diameter shrinks to its final size of approximately 10 nm [289,290,291]. For nucleation and polymerization of intermediate filaments co-factors are not needed [292]. Intermediate filaments show a constant, but slow, subunit exchange along the whole filament, occurring at a rate of approximately 1 per 200 tetramers per hour in vitro for vimentin [293].
Inside the cell, cytoplasmic intermediate filaments form a dense meshwork that is mainly located in the perinuclear space, but also reaches the cortex [294]. The form and structure of the network depends on the type of intermediate filament. While keratin forms bundles and fibers that form only weakly connected networks [295], vimentin and desmin form highly connected networks with small mesh size and lamins generate filaments and fibers [296,297,298]. Near the cortex, intermediate filaments interact with focal adhesion sites, desmosomes, and hemidesmosomes, maintaining cell and tissue adhesion [299,300,301,302]. Conversely, desmosomes and focal adhesions function as centers for de novo intermediate filament formation [303]. Via their anchorage with the nuclear and plasma membrane, intermediate filaments form a scaffold for mitochondria, the Golgi apparatus, and further organelles and organize their location [304,305,306]. Due to its network structure and its ability to anchor organelles, intermediate filaments are often considered to be mechanical buffers [284,307,308]. This idea is supported by the single filament properties of intermediate filaments that can withstand deformations of up to 300% of their initial length without rupturing [309]. Looking at the elastic properties of intermediate filaments, they can be considered as flexible polymers with a persistence length of less than 1 µm [309]. Interestingly, intermediate filaments show a strongly increasing elastic modulus with increasing deformation (called strain hardening) [310]. Measurements of single cells and simulations could confirm an important role of intermediate filaments for the overall visco-elastic response of a cell [311,312,313].
Despite its function as a “mechanical buffer” and “organelle anchors” [284,307,308,314], intermediate filaments are highly dynamic components of the cytoskeleton, with multiple functions, including roles in apoptosis, migration, adhesion, and interactions with other cytoskeletal components. For fulfilling these functions, intermediate filaments need to form a defined network capable of (in-)direct interaction with its targets. Organizing factors are, amongst others, post-translational modifications as, for example, phosphorylation and acetylation [315,316], regulating assembly, organization, and function of intermediate filaments [317,318,319]. A further influencer of intermediate filament organization is the plakin family of proteins, connecting microtubules and actin to intermediate filaments [320]. Additionally, plakins connect intermediate filaments in desmosome adhesions and cell-matrix hemidesmosome adhesions to the actin and microtubule cytoskeleton and the nucleus [321,322] and intermediate filaments to one another [323]. Some intermediate filaments can also orient themselves along the actin or microtubule cytoskeleton. Actin and microtubules thereby form the guiding structures where filaments are transported along, either by kinesin and dynein (microtubules) or myosin (actin), resulting in a mutually dependent organization of intermediate filaments and actin and/or microtubules [232,324]. Interestingly, vimentin transport along microtubules can be inhibited by the actin meshwork, demonstrating a complex interaction between the cytoskeletal components [325]. Intermediate filaments are not only associated with other cytoskeletal proteins and (hemi-)desmosomes, but in the case of vimentin, it also binds via plectin or integrin α2β1 to actin and/or focal adhesions and promotes their strengthening [299,326,327,328]. A direct interaction between the vimentin tail domain and actin is also proposed [329]. The interactions of intermediate filaments discussed here and their interactions with microtubules and actin are summarized in Figure 2 and Figure 3 and Table 3.
Intermediate filaments also link the nucleus to the cytoplasmic cytoskeleton via the LINC (linker of nucleoskeleton and cytoskeleton) complex that is present at the nuclear membrane [330], binding to plectin [331] and, therewith, to intermediate filaments. Consequently, a disruption of LINC function leads to a disturbed force transmission [332,333] due to the weakened linkage of the nucleus and cytoskeleton. Similarly, depletion of nestin, vimentin, and GFAP in astrocytes leads to positional and rotational changes of the nucleus [304]. A correlation was found between nuclear rigidity, chromatin organization, and vimentin levels, indicating a crucial role of cytoplasmic intermediate filaments as passive mechanotransducers to the nucleus to control gene expression [334]. Similarly, GFAP mutations and changes in desmin organization alter gene expression [335].
Further types of intermediate filaments are the nuclear lamins. While nuclear lamins near the nuclear periphery form a filament network the organization of lamins in the center of the nucleus is only partly understood [336]. It was proposed that the nucleoplasmic lamins may form filaments, short fibrous structures, foci, or an unstructured “veil” [336]. The presence of seemingly less dense structures of lamins in the nucleoplasm is also in agreement with the observation of their higher mobility [337]. Interestingly, a lamin A knockdown inhibits the expression of actomyosin cytoskeletal related genes, as shown in mesenchymal stem cells [338]. Similarly, increased or reduced expression of lamin A inhibits or favors migration through a porous 3d matrix [339,340]. On a functional level, A-type lamins seem to impact mechanosensing and signaling [341,342] and to contribute to nuclear stiffness [343].
5. Involvement of Cytoskeleton in Cell Motility and Focal Adhesions
One highly important property of a cell is its ability to move, especially conceivable in the context of immune cells chasing pathogens, wound closure, or metastasis of tumor cells. For cells to move efficiently, a few universal steps are necessary, as follows: It needs to form protrusions that attach to its surroundings and, subsequently, a contraction and retraction of the rear is necessary [344]. A characteristic of cell migration is the precise coordination of these events in space and time. If, for example, the maturation of the adhesions at the cell front is not completed, an increase in contraction leads to the rupture of the newly formed adhesions, abandoning productive movement.
To achieve productive movement and the right timing of migration steps, cells have developed two distinct modes of migration, the amoeboid and mesenchymal type. Amoeboid cell migration is characteristic for rounded cells with low adhesion and high Rho-driven contractility, whereas mesenchymal migrating cells show strong adhesion and Rac1-induced protrusions [345], in line with the mutual negative regulation of Rac1 and RhoA [346,347].
5.1. Actin in Motile Processes
Actin filaments are the main contributors to cell migration in terms of force generation at the cell front and contraction at the rear. As stated, a cell can, in principle, use two types of migration, amoeboid and mesenchymal. While mesenchymal motion is mostly achieved via the extension of the lamellipodium or filopodia, the amoeboid migration works via the extension of blebs.
The lamellipodium is one of the main force generating cell structures, generating pushing forces of up to 35 nN in the extreme case of fish keratocytes [348,349]. To generate these forces, actin is polymerized locally at the cell front via Arp2/3 and depolymerized at the back of the lamellipodium by ADF/cofilin [350]. Interestingly, formins such as FMNL2 or FMNL3, seem to participate in lamellipodial extension independent of Arp2/3 complex incorporation and are, in some cell types, major sources of lamellipodial protrusion forces [351]. The protein Arpin inhibits the activity of Arp2/3, leading to pause phases in lamellipodial extensions and less directed motion [352]. The continuous (de-)polymerization of actin creates a treadmilling effect and consequently forces and retrograde flow of actin [76,77]. A flow in the opposite direction of the retrograde flow is generated by stress fiber contraction, transporting actin to the cell front [77]. To hold the lamellipodium in place and prevent retraction via e.g., actin cortex tension, the formation of new cell-ECM contacts is necessary. Generally speaking, during lamellipodium extension nascent adhesions, that mature into focal adhesions or disassemble, form [353]. While Rac1 controls the formation of nascent adhesions [354,355], maturation is controlled by RhoA and myosin II induced contractility [109], making them anchorage sites for stress fibers that generate tension and, thus, also control the composition of foal adhesions [356,357]. The exact actin nucleation mechanism in focal adhesions is only partly understood, but formins, such as FHOD1 or mDia1, are supposed to play a role [358,359]. The formation of nascent adhesions, in contrast, is thought to depend on Arp2/3 activity in the lamellipodium, due to its interaction with vinculin and focal adhesion kinases (FAK) [360,361]. Nevertheless, the lamellipodium is not essential for migration as several cell types, including fibroblasts and melanoblasts, can migrate without Rac or Arp2/3, but significantly slower [362,363,364]. In the absence of the lamellipodium, these cells migrate via filopodia or other, probably formin dependent, pseudo-pods [362,363,364]. For productive movement it is necessary to spatially restrict actin polymerization to one zone. Thus, it is assumed that Rac is only locally active. A mechanism to locally activate Rac1 is believed to function via Cdc42 induced pathways and microtubule capture at the leading edge and the subsequent local RacGEF and vesicle supply (see also next chapter) [365,366,367]. A further possible mechanism involves the Rho/ROCK pathway and actomyosin contractility to inhibit lamellipodium formation in multiple cell regions [368]. This idea is supported by the occurrence of multiple or larger lamellipodia after inhibition of Rho or ROCK activity [368], giving a strong hint to the importance of a finely tuned dynamic equilibrium between contractile and expansive forces. Although RhoA/ROCK is active at the cell front, a too high activity impairs lamellipodium based migration via retraction of the lamellipodium [369].
A second model, describing the generation of protrusions necessary for migration, is so-called blebbing. This mode of migration is observed in e.g., amoebae, tumor cells, neutrophils, or primordial germ cells or more generally in none- or weakly adherent cells, cells moving in a three dimensional matrix, or in confined environments [370]. As previously described, blebs are initially actin free structures that arise by hydrostatic pressure causing a detachment of the actin-cortex from the membrane, thus extending the cell membrane. Hydrostatic pressure is generated mostly by actin cortex contraction. Local RhoA activation inducing myosin activation leads to increased contractility. The so generated hydrostatic pressure causes bleb formation through hydrostatic flow [172,371,372,373]. Similarly to mesenchymal motion, cells using blebbing for migration need to “attach” the newly formed bleb to the surroundings and “detach” the cell rear, according to most models [370]. It seems that adhesion of cells using blebs for migration is very low [374,375], implying that strong cell adhesions may actually impede bleb-based motion. Consequently, both a low adhesion and high cortex contractility favor the amoeboid motion type [370]. One form of attachment of blebbing cells to their surroundings is via “chimneying” [376], which works via forces perpendicular to the direction of motion and, consequently, is independent of specific adhesion molecules. A different model proposed that forces are transmitted via cell-substrate intercalations. If blebs extend and form protrusions at the side of the cell into gaps of the substratum, then contractility of the reestablished cortex can than generate a net force to pull the cell body [377,378]. Despite these models, other forms of bleb based propulsion have been suggested, like a flow friction driven or a “swimming in low Reynolds numbers” model [370]. As bleb initiation and growth is mainly governed by myosin contractility and the actin cortex-membrane linkage, it is not surprising that the level of the actin-membrane cross-linker ezrin is increased at the cells rear and reduced at its front in carcinoma cells using blebbing for migration [181,379,380]. Similarly, increasing the level of other ERM proteins impedes bleb formation and bleb induced migration [172,381,382], while reducing ERM protein levels has the opposite effect [157,172,383]. A further factor that critically limits bleb extension and, thus, migratory properties is the cell membrane, that can usually stretch only about 4% before rupturing [384]. As blebs do not normally contain endosomes [169], it is suggested that bleb expansion is allowed by the local unfolding of the membrane. Furthermore, bleb expansion is faster than lamellipodial growth, can occur in arbitrary directions, and, because they do not contain the cortex, they can naturally adapt to three dimensional environments. Therefore, blebs might be of high importance in complex three dimensional (in vivo) environments, where lamellipodial extension is seriously impeded [345,385,386].
For efficient migration the cell rear needs to contract as well. To actively contract, actin structures use myosin to slide anti-parallel actin fibers along each other, creating contractile forces if the filaments are anchored at e.g., focal adhesions. The best studied contractile structures anchored to the substrate are stress fibers. Typically, stress fibers are directly linked to focal adhesions (except for transversal arcs), connecting the cell via actin fibers to the ECM [105,109]. Interestingly, the formed focal adhesions are stress dependent and inhibition of myosin II generated contractility decreases focal adhesion size [146], while external tension favors focal adhesion maturation [387]. Additionally, the forces acting on focal adhesions can lead to conformational changes of mechanosensitive proteins contained in focal adhesions, like β-integrins or talin [388,389,390], allowing stress fibers to convert mechanical into chemical signals, influencing focal adhesion maturation and turnover [391]. Therefore, dorsal stress fibers help the maturation of focal adhesions via tension at the leading edge and ventral stress fibers at the trailing edge [392]. Thus, stress fibers are highly important for cell adhesion but their function during cell migration remains poorly understood, as they are absent from many fast migrating cells, like leukocytes and Dictyostelium discoideum amoeba, as well as from cells embedded in soft three dimensional matrices [393,394]. Consequently, it was proposed that stress fibers are not necessary for migration. Under certain circumstances, they might have an inhibitory effect on migration because the turnover is comparably slow and contractile forces may impede cell motion [395]. Thus, the significance of stress fibers may be linked to their role in deforming the ECM, stabilizing focal adhesions, and through the generation of tension for rear contraction inside the cell [396]. For rear retraction it seems as if the contractile forces generated by ventral stress fibers are of importance for the disassembly of posterior adhesions and an inhibition of protrusions at the rear [397,398]. As stress fiber contractility in non-motile cells is associated with strengthening of focal adhesions, it needs to be tightly regulated to achieve just the right amount for the detachment of posterior adhesions. Consequently, a too strong RhoA activation inhibits cell migration via increased contractility [395,399] and inhibition of contractility via ROCK inhibition can even increase motility in some cell types under specific conditions [400,401]. The idea of rear retraction via stress fibers is further promoted by an adhesion gradient with lower adhesiveness at the rear [402].
Taken together, actin or, to be more precise, the lamellipodium, filopodia, and blebs are the main causes of force generation for cell motility and contractile structures like stress fibers or the actin cortex are drivers of rear contraction.
5.2. Microtubules in Motile Processes
In contrast to actin, microtubules are mostly not associated with force generation during migration, but rather with cell polarization and focal adhesions. The role of microtubules can, in principle, be divided into three categories, as follows: Participation in cell motility via their own mechanics, via signal transduction, and as a transport structure.
Microtubules are capable of bearing high external pressure and, thus, help to maintain the cells’ shape under physiological conditions [403]. In migrating cells, the microtubule (+)-end points in the direction of the plasma membrane and microtubules reaching the leading edge grow, at least in epithelial cells, more persistent [404], associated with EBs [216]. As discussed before EBs can recruit further +TIPs that promote microtubule stabilization, protrusion formation, and cell migration [212,405,406,407]. Other +TIPs, such as Clip-170 or its binding partner Clasp, act as rescue factors and increase the time of microtubules near the cortex [212,406]. Stabilization of growth can lead to a more persistent force transmission, even though the maximal pushing force decreases quadratically with length, due to buckling. Notably, the stabilization of microtubules does not only promote a more persistent microtubule growth, but also a steadier supply with material needed for migration, as these microtubules persist longer near the leading edge, being ideal tracks for material supply, in agreement with the preference of some kinesin motors for microtubules stabilized by acetylation and detyrosination [408]. Consequently, the polymerization of microtubules can generate a force of a few piconewton, on the same order as the force generated by motor proteins [161,263]. An in vitro study demonstrated that the generated forces can indeed deform membranes [409]. For a direct involvement of microtubules in the force generation process, a significant amount of microtubules actually have to reach the cell front. In most cell types, only very few microtubules reach the lamellipodium and the generated force is insufficient to generate large scale protrusions [410]. In contrast, in neurons and astrocytes, microtubules are capable of generating protrusions. In neurons, microtubules are also found unattached to centrosomes [411,412], forming bundles and generating large enough forces to participate in axon formation [410]. The free bundles (−)-ends are stabilized by members of the CAMSAP/Patronin family [413,414,415]. Notably, in axons these bundles point with their (+)-end away from the cell body [416], thus being capable of creating larger forces than single microtubules [268], sufficient to promote neurite outgrowth [417].
Cell motion associated structures have a high material consumption and, therefore, a steady supply is necessary to allow a continuous movement. For this transport, microtubules and their associated motor proteins are of high importance [226,227] because they can transport membrane components needed for membrane extension, signaling molecules, such as the small GTPases Rac and Cdc42, GEFs, and proteases, but also intermediate filaments and their precursors [230,231,418,419]. Additionally, microtubules transport β-actin coding mRNA and Arp2/3 subunits to the cell front [235,236].
A further indirect way for microtubules to influence cell motility is via the (de-)stabilization of cell-matrix adhesions or regulation of actin (de-)polymerization, as microtubules are associated with focal adhesions [420], their regulation [421], RhoGTPase activity [422], and, consequently, actomyosin contractility [423]. This can easily be seen by experiments with microtubule destabilizing agents like nocodazole, causing cell protrusion defects due to reduced Rac1 induced actin polymerization and increased contractility because of Rho-myosin II signaling [424,425]. This is supported by observations showing that microtubule growth can locally activate Rac1, favoring the generation of new focal adhesion sites [422] and the lamellipodium [425]. In neuronal cells, Rac might be activated by TIAM1 (T-cell lymphoma invasion and metastasis-inducing protein 1) interacting with microtubules via MAP1B [426]. An additional involvement of the Rac activators TRIO (triple functional domain protein) and TIAM2 was suggested in microtubule induced protrusion formation [422,427]. A RhoGEF possibly involved in microtubule dependent regulation of RhoGTPases is H1, which interacts with microtubules, is inactive when bound, and transitions into an active state when microtubules depolymerize [424,428,429,430,431].
Furthermore, microtubules grow in the direction of existing focal adhesions at the cell front where they get entrapped and stabilized [432] and accelerate the maturation of focal adhesion via the transport of integrins [433]. Thereby, stress fibers seem to function as a guidance structure for microtubules, mediated by MACF1 [279,434]. A positive feedback loop is also possible where integrin stimulation could cause a favored delivery of cargo at the site of adhesion [435]. A further possibility is an interaction of FAK or paxilin with APC that clusters at microtubule tips [436,437,438]. An additional signaling mechanism for APC and +TIPs, like Clip170 and CLASPs, is via the Rac and Cdc42 effector IQGAP and the formin mDia, promoting actin nucleation at focal adhesions [277,439,440]. Despite the adhesion favoring effect of microtubules, an opposing destabilizing effect was observed at the cell rear [432,441]. Microtubules actively targeted mature focal adhesions at the rear of motile fibroblasts, accelerating focal adhesion turnover [432,441]. A common model describes the phenomenon via the dynamic instability of microtubule filaments. By growing and targeting focal adhesions, microtubules exert a force on the adhesion site, depolymerize quickly afterwards, and repeat the process [442,443]. This hypothesis is supported by an observation showing a strong correlation between the microtubule poking number and the dissociation of focal adhesions [443]. However, how microtubules find and target focal adhesions is not yet fully understood. It has been observed that they grow along actin bundles towards adhesion sites, potentially cross-linking to actin via e.g., spectraplakins or others [213,444]. This idea is supported by the APC dependent localization of the spectraplakin MACF1 at the cell cortex, close to focal adhesions [445,446]. Additionally, in the absence of MACF1 peripheral microtubules are less well organized and adhesion turnover is inhibited [279]. Still, other mechanisms, involving CLASPs or interactions with integrin-linked kinase (ILK), are also possible [214,421,447,448]. Furthermore, the actin bundling protein fascin interacts with microtubules, promoting focal adhesion turnover via FAK [449]. Other mechanisms of microtubule dependent focal adhesion turnover are via clathrin mediated endocytosis of integrins, NBR1-mediated autophagy, or via vesicles carrying matrix metalloproteinases severing integrin-ECM connections [450,451,452,453].
5.3. Intermediate Filament Involvement in Motile Processes
It is well established that intermediate filaments crucially influence both cell-matrix adhesion and migration. Nevertheless, the precise mechanism of action of intermediate filaments is not fully elucidated. As for microtubules it seems as though intermediate filaments are not a direct part of the force generation mechanism necessary for movement, but rather a signaling platform and mechanical anchor inside the cytoplasm to transduce forces through the whole cell.
For this review we will focus the discussion mainly on vimentin, as vimentin is one of the best investigated intermediate filaments. Notably, we will not discuss keratins, as they are mostly restricted to epithelial cells and keratinocytes and not present in cell of glial origin.
In non-migrating cells intermediate filaments are mostly localized around the nucleus, extending into the periphery [304,454], while they elongate into the lamella, connecting to focal adhesions near the leading edge in migrating cells [455,456]. In the lamellipodium intermediate filaments are found mostly in a non-filamentous state [326,454]. Generally speaking, intermediate filament organization alters the current state of the two other cytoskeletal components [457,458], thus potentially modulating both cell adhesion and migration.
A strong hint for intermediate filaments influencing cell migration comes from the observation that, for example, vimentin can interact with actin and neurofilaments with microtubules [329,459]. Vimentin is indeed necessary for motility of fibroblasts and breast cancer cells [460], epithelial cell wound closure [461], and other migration related phenomena [462]. Vimentin inhibition reduces motility in fibroblasts, astrocytes, and diverse cancer cells [463,464]. Studies evaluating possible mechanisms found evidence for vimentin directly binding to APC. APC regulates vimentin organization in astrocytes to align vimentin along the microtubule network [456]. Post-translational modifications, like detyronsination and acetylation of microtubules, also impact vimentin network organization [294,465]. On the other hand vimentin also affects polarized microtubule organization, amongst others by forming a template for microtubules, guiding microtubule growth, and, thus, favor directed migration [458,466]. The exact mechanism of this interaction is not yet fully understood but may be governed by APC, linking microtubules and vimentin or via vimentin phosphorylation [456,467]. Conversely, the vimentin filament network is dependent on microtubules and its motor proteins. Microtubule disruption leads to vimentin relocalization around the nucleus [234,468]. Furthermore, activation of Cdc42 during scratch-wound assay in astrocytes inhibits dynein mediated rearward transport of GFAP and vimentin containing filaments, promoting intermediate filament network extension in direction of the leading edge [469].
Another motility associated structure partly regulated by vimentin are focal adhesions. Vimentin is associated with formation, maturation, size, and strength of focal adhesions [300,313,326,327]. Vimentin regulates the Rac1 GEF VAV2 and its localization to focal adhesions to promote their stabilization via Rac1 induced FAK activation [470]. Depletion of vimentin in fibroblasts causes a FAK dependent induction of RhoA and myosin activity to compensate for the loss of tension induced by vimentin depletion [328]. Similarly, vimentin depletion leads to increased stress fiber assembly and myosin activity, via RhoA activation by activating RhoA GEF-H1, but without activating FAK in osteosarcoma cells [471]. Additionally, a triple silencing of vimentin, GFAP, and nestin or of vimentin and GFAP in astrocytes demonstrated that these intermediate filaments help to maintain the polarization of leader cells in collective motion by controlling forces in monolayers [464,472]. Silencing of each individual intermediate filament produced similar but less pronounced results [472]. This effect was attributed to larger and more focal adhesions that were distributed less concentratedly at the cell front [472]. It is supposed that those three intermediate filaments control focal adhesions and traction force in astrocytes via plectin to control vinculin recruitment [472]. In the case of vimentin, an interaction with integrin β3 was also observed [473]. A further hypothesis of how intermediate filaments control focal adhesions and traction forces is via the acto-myosin network, by redirecting forces and restraining actin retrograde flow [474], or by regulation of focal adhesions via microtubules [325,450,466,475]. Another mechanism that might explain the interaction of vimentin with focal adhesions is via the RAF-1/RhoA signaling [476,477], activating ROCK [478] and being able to phosphorylate vimentin, leading to a filament collapse and subsequent release of ROCK at the cells periphery [479]. Thus, the presence of vimentin can influence the RhoA signaling and, consequently, the formation and stability of focal adhesions [480,481].
In line with these findings, transverse arcs interact via plectin with vimentin to promote their retrograde flow and this coupling is necessary for the perinuclear organization of vimentin filaments [482]. Consequently, myosin II driven contractility of transversal arcs was made responsible for retrograde vimentin movement [482]. This is of special interest, as the local activation of Rac1 causes a local disassembly of vimentin at the lamellipodium forming side [454], also preventing polymerization of vimentin via the Rac1 and Cdc42 effector PAK [483]. Conversely, local vimentin depolymerization causes the formation of the lamellipodium at the side of vimentin depletion [454]. Consequently, vimentin co-regulates both contractile actomyosin bundles and protrusive lamellipodial actin, and thus cell polarization. Despite the regulation of actin dynamics in the lamellipodium and lamellum, vimentin seems to form a transport structure for the nucleus in a three dimensional environment (“nuclear piston”) in cooperation with actomyosin, via the creation of a pressure gradient [484].
Taken together, vimentin seems to mainly contribute to focal adhesion maturation and stabilization via the modulation of RhoA by FAK or Rho GEFs by maintenance of directed migration in collective motion and by regulation of the microtubule organization. Additionally, it seems as if vimentin locally suppresses the formation of the lamellipodium, favoring asymmetrical protrusion formation and, thus, generation of a net force.
There have also been studies of other intermediate filaments, but the understanding of their impact on cell migration and adhesion is limited. Here we will only give some non-exhaustive examples. Synemin is associated with migration in astrocytoma via interactions with the focal adhesion associated protein zyxin and regulation of actin dynamics [485,486]. Additionally, synemin was shown to interact with the focal adhesion components talin, vinculin, plectin, and α-actinin [283,487,488,489,490]. Nestin was demonstrated to regulate FAK, integrin α5β1 localization and VASP activity, controlling invasiveness of prostate cancer cells [491].
6. Cytoskeletal Alterations in Glioma
Malignancies belong to the most common causes of death worldwide, with increasing tendency [492,493]. Among them, brain tumors occur at a rate of 5–10 per 100,000 individuals [494]. The most frequent type of brain malignancies are gliomas that can be differentiated in astrocytoma, oligodendroglioma, and oligoastrocytoma. Especially high grade glioma are characterized by a highly infiltrative growth behavior, leading to low patient survival times [495,496,497]. The infiltration of adjacent regions of the brain is one of the most critical parts in patients’ survival, as it makes complete tumor resection almost impossible, consequently leading to frequent recurrences [495,496,498].
As migration and, thus, infiltration of glioma cells is largely governed by reshaping the cytoskeleton, it is no surprise that the composition and organization of the cytoskeleton in glioma cells differs strongly from that of healthy brain cells. In general, the Rho GTPases Rac1, Cdc42, and RhoA play a pivotal role in cell migration and cytoskeletal organization. The same is true for glioma cells, showing an increased expression of the mutually inhibiting GTPases RhoA and Rac1 [499]. Additionally, invading glioma cells exhibit an increased Cdc42 and Rac1 activity, but a decreased activity of RhoA at the cell front, held responsible for the enhanced migration of leader cells [500,501,502,503]. Similarly, cells at the invading front show a markedly increased FAK expression [504]. Thereby, the activity of RhoA seems to cause ambivalent effects. On the one hand, it is strongly expressed at the cell front and necessary for migration and, on the other hand, a marked activation causes an inhibition of glioma migration, via an inhibition of Rac1, and increased contractility, via larger focal adhesions [505,506]. Inhibition of the RhoA target ROCK led to increased migration in glioma cells via an activation of Rac1 [507] and Rac1 inhibition was associated with reduced migration and invasion [507,508,509]. Contrasting these observations, other studies found ROCK inhibition associates with the reduced migration of glioma cells [510,511]. A possible explanation for this discrepancy might be due to biphasic effects of ROCK activity. While low ROCK activity inhibits motion via low contractility and adhesion, a too high activity may impede migration due to contraction of protrusions and large adhesion sites, in a similar manner as proposed for the relation between adhesion and migration speed [512]. The mutual inhibition of RhoA and Rac1 is further complicated by Cdc42 being capable of activating Rac1 in glioma [513]. Interestingly, the formin mDia1, which is activated by RhoA, plays a major role in glioma cell polarization. For mDia1 expression, an association was demonstrated with the microtubule dependent localization of APC and Cdc42 at the cell front, regulating cell polarization [514]. The impact of RhoA and Rac1 on glioma migration is in good agreement with the observation of an increased FAK activation, negatively regulating RhoA and activating Rac1 in glioma, being associated with staging and a poorer prognosis [504,515]. In contrast, downregulation of FAK is associated with reduced migration [516,517]. The important role of RhoA points to myosin as a further important player in glioma migration. Indeed, myosin was found to be crucial for glioma cell migration in vivo, likely for rear retraction and nucleus deformation [518,519,520]. The lamellipodium is another structure associated with migration of glioma cells. Even though its main organizing protein Arp2/3 is associated with glioma staging, its exact role in glioma migration is not yet clear [521]. While Arp2/3 inhibition was reported to reduce lamellipodial size and migration in a scratch wound assay, another study recognized no such relationship on linear functionalized tracks [521,522]. Interestingly, Arp2/3 might also be involved in the maintenance of the glioma stem cell character [523]. When cortactin, an important cross-linker in the dendritic actin network with overexpression in glioma, is inhibited the size of the lamellipodium and scratch wound closure is reduced [524].
Silencing of the actin bundling protein fascin was demonstrated to reduce migratory capabilities of glioma cells, their division rate, and to increase the sensitivity to cytotoxic lymphocytes [525,526]. The drop in migration was attributed to a loss of filopodia after fascin-1 silencing [525,526].
Glioma cells are also capable of generating blebs for cell protrusion [527,528,529,530]. The switch from mesenchymal to amoeboid motion occurs under conditions of low adhesion, via e.g., integrin β1 blockade, p130Cas silencing, or high contractility via RhoA activation [527,528,529,530]. This is of special interest as migration capabilities of these cells was largely unchanged in 3D, pointing to an escape mechanism for interventions targeting “classical” migration associated molecules, such as e.g., integrin β1 or, potentially, others associated with mesenchymal migration [527,528,529,530]. Still, the exact role of amoeboid motion in glioma invasion remains largely unknown.
Glioma cells may express the intermediate filaments vimentin, GFAP, nestin, synemin, and α-internexin [531,532]. Studies evaluating the organization and expression of intermediate filaments in glioblastoma found the following three distinct subpopulations with: (1) High vimentin, GFAP, and synemin expression or (2) low vimentin, GFAP, and synemin expression or (3) high nestin but low vimentin, GFAP, and synemin expression [533]. Whether this expression pattern is associated with the origin of these tumors or has a distinct impact on migratory behavior is not yet elucidated. These patterns might alternatively be related with the molecular sub-classes of glioma (proneural, proliferative, mesenchymal). As opposed to healthy glia cells, glioma generally possess increased amounts of the intermediate filaments vimentin, nestin, and GFAP [534,535,536]. While the amount of vimentin and the glioma stem cell marker nestin showed a negative correlation with patient prognosis and/or glioma staging [537,538,539,540], an association of GFAP expression with staging is controversial. A recent meta-study reported about a missing correlation between the amount of GFAP and the grade of gliomas [536]. Interestingly, GFAP serum levels were proposed as a marker to differentiate between primary brain tumors and brain metastasis [541]. The α-internexin levels correlated negatively with staging, but only in oligodendroglioma [532]. These findings correlate well with the infiltrative or migratory phenotype obtained by glioma cells, as vimentin and nestin are associated with more motile characteristics (see before), while a forced expression of GFAP was shown to inhibit glioma motility in vitro [542,543]. Vimentin may additionally have a role in radiation induced migration of glioma cells. After non-lethal irradiation, the expression of vimentin was increased and associated with faster migration [544]. Furthermore, a nestin down-regulation is associated with an increased adhesion to collagen, fibronectin, and laminin, leading to an inhibition of migration of glioma cells, while overexpression had the reverse effect [545]. This might be of further interest, as nestin was especially found in cells at the invasive front of glioma [546]. The underlying molecular mechanisms of these nestin induced effects remained unclear so far. It remains tempting to speculate that, in glioma, a similar mechanism as discussed before might be responsible for the increased migratory capacity, via an activation of FAK and regulation of integrin localization [491]. The intermediate filament synemin seems to be an additional part of the motile machinery of astrocytoma cells. Synemin is co-localized with the actin cross-linker α-actinin at the cell front and down-regulation of synemin impaired astrocytoma motility, via reduced F-actin and α-actinin amounts [486,547]. Nevertheless, precise mechanisms of synemin actions in glioma are missing.
Intermediate filaments are not only differentially expressed in glioma, but β-III, β-IV, and γ-tubulin are also overexpressed in glioma [548,549,550]. Special attention should be given to β-III tubulin, which is not expressed in glia cells under physiologic conditions, but is up-regulated in high-grade glioma [551]. β-III tubulin might be responsible for the resistance of glioblastoma to the microtubule stabilizing agent taxol, as observed in carcinoma cells [552,553], but this concept is still under debate [551]. Notably, taxol was suggested to trigger differentiation in some glioma cells and in one study associated with an increase in GFAP expression [554,555]. The higher expression of γ-tubulin might be related to centrosome abnormalities, altered microtubule dynamics, and consequently, adhesion dynamics and cell polarization [556]. Furthermore, the microtubule severing protein spastin and the destabilizing protein stathmin are both associated with increased motility and, in case of spastin, also with reduced proliferation [557,558]. Spastin expression is increased in glioma and mainly located at the cell front and the mitotic spindle, implying an indirect role in both motility and cell division by destabilization of microtubules, inducing spindle formation defects during division and facilitating microtubule turnover at the front to adapt to changes of the microenvironment. For the microtubule associated protein DCX different, contradictory results were found. Few studies reported that DCX is preferentially expressed at the invasive front of glioma [559,560], indicating a pro-invasive role. Other authors demonstrated a very low DCX expression in glioma and that a forced expression causes apoptosis and inhibits invasion [561,562]. Accordingly, the role of DCX in glioma needs to be critically evaluated. In contrast, the role of EB1 seems to be unambiguous, the reduced accumulation of EB1 at microtubule tips is associated with higher microtubule instability, a less migratory and motile phenotype of glioma cells, caused by a lower amount of microtubules reaching the leading edge [563]. The microtubule stabilizing factor APC, despite regulating differentiation and cell cycle arrest [564,565], might also be part of the migratory machinery of glioma cells, as it was recently shown that vimentin, GFAP, and nestin organization, along microtubules in the glioblastoma cell line U138-MG, is critically dependent on APC [456]. APC may, additionally, increase the vimentin polymerization rate and, thus, influence migration [456]. The motor protein dynein is not differentially expressed in glioma cells in general, but migrating glioma cells express increased amounts of the protein [566]. Dynein is, in part, responsible for microtubule dependent intracellular transport and it can be speculated that dynein is necessary to maintain cell polarization by transporting inhibitory signals away from the cell front [567]. Similarly, kinesin-5 and microtubule-actin linker protein MACF1 are involved in increased migration and up-regulated similar to KiF2C and KiF14 in glioma cells [568,569,570,571]. The exact mechanism of MACF1 action is not elucidated yet, but a contribution of Wnt-signaling was proposed [571]. Further microtubule and actin-associated proteins are differentially expressed in glioma. A summary can be found in Table 4.
For productive migration, glioma cells generate an ECM differing from the normal micro-milieu of the central nervous system. This includes an overexpression of tenascin-C at the invasive front [590], increasing motility of glioma cells via interactions with β1 or αvβ3 integrins [591], probably via an activation of FAK and inhibition of RhoA [592,593,594]. The idea of such a mechanism is supported by the observed drop in glioma migration after inhibition of β1- or αv-integrin and increased motility after β1 overexpression [595,596,597,598]. In line with the association of integrins with migration is the elevated α-actinin expression in glioma, which is associated with a worse prognosis and connects focal adhesions to the cytoskeleton [577]. Despite tenascin-C other ECM components were also associated with a more migratory phenotype of glioma, such as laminin, fibronectin, and collagen [522,599,600], but myelin biomembranes are also efficient substrates for migration [601]. For thin linear laminin coatings trying to imitate properties of the basal lamina, a favored invasion route of glioma, it could be demonstrated that polarization of collective migration of glioma cells was microtubule dependent [522]. This migration mode could largely be inhibited by formin inhibition, but not by inhibition of Arp2/3 that led to an increased migration speed. This is in line with the general importance of formins for glioma migration, demonstrating that mDia antagonism/depletion and agonism/overexpression impaired migration and invasion [514,572]. The observed effect was, thereby, likely mediated via increased F-actin assembly and the stabilization of microtubules, leading to defects in protrusion formation, cell-cycle arrest, and apoptosis [572,602]. Hyaluronic acid/hyaluronan (HA), the main ECM component of the healthy brain, as well as their main receptors CD44 and RHAMM (receptor of hyaluronan-mediated motility) are also upregulated in glioma [603,604,605,606], but their exact role is not yet determined. In general, increased HA expression is associated with an increase in motility and migration of glioma [607,608], probably via a CD44 or RHAMM induced activation of Rho GTPases, such as Rac1 or Cdc42 [609,610,611]. Even so, the role of CD44 remains ambivalent in glioma cell migration. On the one hand low and high levels of CD44 are associated with a less migratory phenotype and on the other hand intermediate expression levels facilitate migration [612]. This biphasic effect is probably related to the substrate adhesiveness.
7. Conclusions
The actin and microtubule cytoskeleton, their interactions, and relation to cell migration are comparably well understood for healthy cells, but the impact of their interplay with intermediate filaments just started to gain attention. Consequently, the role of intermediate filaments during migration is less well understood and many questions, especially regarding the role of GFAP, vimentin, synemin, and nestin in glia cells, remain open. For malignant cells the situations worsen, due to altered expression and localization profiles of cytoskeleton associated proteins, leading to changes in the global organization of the cytoskeleton, the signaling, and turnover of structures. Additional attention should be paid to the heterogeneity of glioma, showing significant differences in expression profile and behavior between “tumor-core” and “invading” tumor cells, but also between glioma of different patients. In case of glioma, the impact of cytoskeletal variations is only sparsely investigated and will need a significantly increased amount of data to understand the migration of glioma in the central nervous system. This includes the effect of the observed overexpressions, like spastin, vimentin, nestin, actin cross-linkers, etc., on migration and the underlying mechanisms, but also how migration associated signaling is altered in glioma and whether this can be used for specifically targeting glioma migration. Furthermore, the function of stress fibers in glioma migration and whether blebs play a significant role during brain infiltration by glioma are also not yet addressed adequately. Current approaches targeting the cytoskeleton in glioma mainly focus on microtubules, but mostly not to alter migratory properties but to induce cell cycle arrest or apoptosis. Nevertheless, these approaches remained futile because of glioma heterogeneity, therapeutic side effects, or resistance mechanisms.
Author Contributions
T.H. writing—original draft, writing—review & editing; F.D. writing—review & editing.
Funding
T.H. was supported by the funding program Open Access Publishing by the German Research Foundation (DFG).
Acknowledgments
The authors thank Prof. Wolfgang Ballhausen for critical reading of the manuscript. We thank Dr. Urszula Grabiec for helpful discussions. We acknowledge the financial support within the funding program Open Access Publishing by the German Research Foundation (DFG).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef]
- Rotty, J.D.; Bear, J.E. Competition and collaboration between different actin assembly pathways allows for homeostatic control of the actin cytoskeleton. Bioarchitecture 2014, 5, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Janmey, P.A. Polymer physics of the cytoskeleton. Curr. Opin. Solid State Mater. Sci. 2011, 15, 177–182. [Google Scholar] [CrossRef]
- Sept, D.; Xu, J.; Pollard, T.D.; McCammon, J.A. Annealing accounts for the length of actin filaments formed by spontaneous polymerization. Biophys. J. 1999, 77, 2911–2919. [Google Scholar] [CrossRef]
- Sept, D.; Elcock, A.H.; McCammon, J.A. Computer simulations of actin polymerization can explain the barbed-pointed end asymmetry. J. Mol. Biol. 1999, 294, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Blanchoin, L.; Mullins, R.D. Molecular Mechanisms Controlling Actin Filament Dynamics in Nonmuscle Cells. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 545–576. [Google Scholar] [CrossRef]
- Kang, F.; Purich, D.L.; Frederick, S.; Southwick, F.S. Profilin Promotes Barbed-end Actin Filament Assembly without Lowering the Critical Concentration. J. Biol. Chem. 1999, 274, 36963–36972. [Google Scholar] [CrossRef] [PubMed]
- Machesky, L.M.; Mullins, R.D.; Higgs, H.N.; Kaiser, D.A.; Blanchoin, L.; May, R.C.; Hall, M.E.; Pollard, T.D. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc. Natl. Acad. Sci. USA 1999, 96, 3739–3744. [Google Scholar] [CrossRef] [PubMed]
- Kovar, D.R.; Harris, E.S.; Mahaffy, R.; Higgs, H.N.; Pollard, T.D. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell 2006, 124, 423–435. [Google Scholar] [CrossRef]
- Romero, S.; Le Clainche, C.; Didry, D.; Egile, C.; Pantaloni, D.; Carlier, M.F. Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell 2004, 119, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Swaney, K.F.; Li, R. Function and regulation of the Arp2/3 complex during cell migration in diverse environments. Curr. Opin. Cell Biol. 2016, 42, 63–72. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Borisy, G.G. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 1999, 145, 1009–1026. [Google Scholar] [CrossRef]
- Achard, V.; Martiel, J.L.; Michelot, A.; Guérin, C.; Reymann, A.C.; Blanchoin, L.; Boujemaa-Paterski, R. A “Primer”-Based Mechanism Underlies Branched Actin Filament Network Formation and Motility. Curr. Biol. 2010, 20, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Molinie, N.; Gautreau, A. The Arp2/3 Regulatory System and Its Deregulation in Cancer. Physiol. Rev. 2018, 98, 215–238. [Google Scholar] [CrossRef] [PubMed]
- Akin, O.; Mullins, R.D. Capping Protein Increases the Rate of Actin-Based Motility by Promoting Filament Nucleation by the Arp2/3 Complex. Cell 2008, 133, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Kawska, A.; Carvalho, K.; Manzi, J.; Boujemaa-Paterski, R.; Blanchoin, L.; Martiel, J.-L.; Sykes, C. How actin network dynamics control the onset of actin-based motility. Proc. Natl. Acad. Sci. USA 2012, 109, 14440–14445. [Google Scholar] [CrossRef] [PubMed]
- Vignjevic, D.; Yarar, D.; Welch, M.D.; Peloquin, J.; Svitkina, T.; Borisy, G.G. Formation of filopodia-like bundles in vitro from a dendritic network. J. Cell Biol. 2003, 160, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Bendix, P.M.; Koenderink, G.H.; Cuvelier, D.; Dogic, Z.; Koeleman, B.N.; Brieher, W.M.; Field, C.M.; Mahadevan, L.; Weitz, D.A. A quantitative analysis of contractility in active cytoskeletal protein networks. Biophys. J. 2008, 94, 3126–3136. [Google Scholar] [CrossRef] [PubMed]
- Koenderink, G.H.; Dogic, Z.; Nakamura, F.; Bendix, P.M.; MacKintosh, F.C.; Hartwig, J.H.; Stossel, T.P.; Weitz, D.A. An active biopolymer network controlled by molecular motors. Proc. Natl. Acad. Sci. USA 2009, 106, 15192–15197. [Google Scholar] [CrossRef]
- Finer, J.T.; Simmons, R.M.; Spudich, J.A. Single myosin molecule mechanics: Piconewton forces and nanometre steps. Nature 1994, 368, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R.K.; Pardee, J.D. Assembly mechanism of Dictyostelium myosin II: Regulation by K+, Mg2+, and actin filaments. Biochemistry 1996, 35, 15504–15514. [Google Scholar] [CrossRef]
- Matsumura, F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends Cell Biol. 2005, 15, 371–377. [Google Scholar] [CrossRef]
- Jung, H.S.; Komatsu, S.; Ikebe, M.; Craig, R. Head–Head and Head–Tail Interaction: A General Mechanism for Switching Off Myosin II Activity in Cells. Mol. Biol. Cell 2008, 19, 3234–3242. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Smith, R.; Kendrick-Jones, J. Light-chain phosphorylation controls the conformation of vertebrate non-muscle and smooth muscle myosin molecules. Nature 1983, 302. [Google Scholar] [CrossRef]
- Yumura, S.; Yoshida, M.; Betapudi, V.; Licate, L.S.; Iwadate, Y.; Nagasaki, A.; Uyeda, T.Q.P.; Egelhoff, T.T. Multiple Myosin II Heavy Chain Kinases: Roles in Filament Assembly Control and Proper Cytokinesis in Dictyostelium. Mol. Biol. Cell 2005, 16, 4256–4266. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Kotula, L.; Hwang, Y.W. Two distinct mechanisms for regulation of nonmuscle myosin assembly via the heavy chain: Phosphorylation for MIIB and Mts 1 binding for MIIA. Biochemistry 2000, 39, 11441–11451. [Google Scholar] [CrossRef] [PubMed]
- Murakami, N.; Chauhan, V.P.S.; Elzinga, M. Two nonmuscle myosin II heavy chain isoforms expressed in rabbit brains: Filament forming properties, the effects of phosphorylation by protein kinase C and casein kinase II, and location of the phosphorylation sites. Biochemistry 1998, 37, 1989–2003. [Google Scholar] [CrossRef]
- Rosenberg, M.; Ravid, S. Protein Kinase C Regulates Myosin IIB Phosphorylation, Cellular Localization, and Filament Assembly. Mol. Biol. Cell 2006, 17, 1364–1374. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Schaller, V.; Bausch, A.R. Collective dynamics of active cytoskeletal networks. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Wagner, B.; Tharmann, R.; Haase, I.; Fischer, M.; Bausch, A.R. Cytoskeletal polymer networks: The molecular structure of cross-linkers determines macroscopic properties. Proc. Natl. Acad. Sci. USA 2006, 103, 13974–13978. [Google Scholar] [CrossRef] [PubMed]
- Falzone, T.T.; Lenz, M.; Kovar, D.R.; Gardel, M.L. Assembly Kinetics Determine the Architecture of α-actinin Crosslinked F-actin Networks. Nat. Commun. 2013, 3. [Google Scholar] [CrossRef]
- Huang, S. Arabidopsis VILLIN1 Generates Actin Filament Cables That Are Resistant to Depolymerization. Plant Cell Online 2005, 17, 486–501. [Google Scholar] [CrossRef]
- Kasza, K.E.; Broedersz, C.P.; Koenderink, G.H.; Lin, Y.C.; Messner, W.; Millman, E.A.; Nakamura, F.; Stossel, T.P.; MacKintosh, F.C.; Weitz, D.A. Actin filament length tunes elasticity of flexibly cross-linked actin networks. Biophys. J. 2010, 99, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.G.; Shi, W.; Ramagopal, U.; Tseng, Y.; Wirtz, D.; Kovar, D.R.; Staiger, C.J.; Almo, S.C. Structure of the actin crosslinking core of fimbrin. Structure 2004, 12, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Stossel, T.P.; Condeelis, J.; Cooley, L.; Hartwig, J.H.; Noegel, A.; Schleicher, M.; Shapiro, S.S. Filamins as integrattors of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2001, 2, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Schwarz, W.H.; Käs, J.A.; Stossel, T.P.; Janmey, P.A.; Pollard, T.D. Mechanical properties of actin filament networks depend on preparation, polymerization conditions, and storage of actin monomers. Biophys. J. 1998, 74, 2731–2740. [Google Scholar] [CrossRef]
- Bartles, J.R. Parallel actin bundles and their multiple actin-bundling proteins. Curr. Opin. Cell Biol. 2000, 12, 72–78. [Google Scholar] [CrossRef]
- Ferrer, J.M.; Lee, H.; Chen, J.; Pelz, B.; Nakamura, F.; Kamm, R.D.; Lang, M.J. Measuring molecular rupture forces between single actin filaments and actin-binding proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 9221–9226. [Google Scholar] [CrossRef]
- Revenu, C.; Athman, R.; Robine, S.; Louvard, D. The co-workers of actin filaments: From cell structures to signals. Nat. Rev. Mol. Cell Biol. 2004, 5, 635–646. [Google Scholar] [CrossRef]
- Reymann, A.C.; Martiel, J.L.; Cambier, T.; Blanchoin, L.; Boujemaa-Paterski, R.; Théry, M. Nucleation geometry governs ordered actin networks structures. Nat. Mater. 2010, 9, 827–832. [Google Scholar] [CrossRef]
- Chesarone, M.A.; Dupage, A.G.; Goode, B.L. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 2010, 11, 62–74. [Google Scholar] [CrossRef]
- Chen, Q.; Nag, S.; Pollard, T.D. Formins filter modified actin subunits during processive elongation. J. Struct. Biol. 2012, 177, 32–39. [Google Scholar] [CrossRef]
- Goode, B.L.; Eck, M.J. Mechanism and Function of Formins in the Control of Actin Assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef]
- Michelot, A.; Derivery, E.; Paterski-Boujemaa, R.; Guérin, C.; Huang, S.; Parcy, F.; Staiger, C.J.; Blanchoin, L. A Novel Mechanism for the Formation of Actin-Filament Bundles by a Nonprocessive Formin. Curr. Biol. 2006, 16, 1924–1930. [Google Scholar] [CrossRef]
- Esue, O.; Harris, E.S.; Higgs, H.N.; Wirtz, D. The Filamentous Actin Cross-Linking/Bundling Activity of Mammalian Formins. J. Mol. Biol. 2008, 384, 324–334. [Google Scholar] [CrossRef]
- Edwards, M.; Zwolak, A.; Schafer, D.A.; Sept, D.; Dominguez, R.; Cooper, J.A. Capping protein regulators fine-tune actin assembly dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 677–689. [Google Scholar] [CrossRef]
- Laporte, D.; Ojkic, N.; Vavylonis, D.; Wu, J.-Q. a-Actinin and fimbrin cooperate with myosin II to organize actomyosin bundles during contractile-ring assembly. Mol. Biol. Cell 2012, 23, 3094–3110. [Google Scholar] [CrossRef]
- Köhler, S.; Bausch, A.R. Contraction mechanisms in composite active actin networks. PLoS ONE 2012, 7, 1–8. [Google Scholar] [CrossRef]
- Murrell, M.; Thoresen, T.; Gardel, M. Reconstitution of contractile actomyosin arrays. Methods Enzymol. 2014, 540, 265–282. [Google Scholar] [CrossRef] [PubMed]
- Haviv, L.; Gillo, D.; Backouche, F.; Bernheim-Groswasser, A. A Cytoskeletal Demolition Worker: Myosin II Acts as an Actin Depolymerization Agent. J. Mol. Biol. 2008, 375, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.K.; Petrasek, Z.; Heinemann, F.; Schwille, P. Myosin motors fragment and compact membrane-bound actin filaments. Elife 2013, 2013, 1–18. [Google Scholar] [CrossRef]
- Reymann, A.C.; Boujemaa-Paterski, R.; Martiel, J.L.; Guérin, C.; Cao, W.; Chin, H.F.; De La Cruz, E.M.; Théry, M.; Blanchoin, L. Actin network architecture can determine myosin motor activity. Science 2012, 336, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Ingerman, E.; Hsiao, J.Y.; Mullins, R.D. Arp2/3 complex ATP hydrolysis promotes lamellipodial actin network disassembly but is dispensable for assembly. J. Cell Biol. 2013, 200, 619–633. [Google Scholar] [CrossRef]
- Reymann, A.-C.; Suarez, C.; Guerin, C.; Martiel, J.-L.; Staiger, C.J.; Blanchoin, L.; Boujemaa-Paterski, R. Turnover of branched actin filament networks by stochastic fragmentation with ADF/cofilin. Mol. Biol. Cell 2011, 22, 2541–2550. [Google Scholar] [CrossRef]
- Andrianantoandro, E.; Pollard, T.D. Mechanism of Actin Filament Turnover by Severing and Nucleation at Different Concentrations of ADF/Cofilin. Mol. Cell 2006, 24, 13–23. [Google Scholar] [CrossRef]
- Ngo, K.X.; Kodera, N.; Katayama, E.; Ando, T.; Uyeda, T.Q. Cofilin-induced unidirectional cooperative conformational changes in actin filaments revealed by high-speed atomic force microscopy. Elife 2015, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Elam, W.A.; Kang, H.; De La Cruz, E.M. Biophysics of actin filament severing by cofilin. FEBS Lett. 2013, 587, 1215–1219. [Google Scholar] [CrossRef]
- Mccullough, B.R.; Blanchoin, L.; Martiel, J.; La, E.M. De Cofilin Increases the Bending Flexibility of Actin Filaments: Implications for Severing and Cell Mechanics. J. Mol. Biol. 2009, 381, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Goodarzi, J.P.; De La Cruz, E.M. Energetics and Kinetics of Cooperative Cofilin-Actin Filament Interactions. J. Mol. Biol. 2006, 361, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Pollard, T.D. Mechanism of interaction of Acanthamoeba actophorin (ADF/Cofilin) with actin filaments. J. Biol. Chem. 1999, 274, 15538–15546. [Google Scholar] [CrossRef] [PubMed]
- Kueh, H.Y.; Brieher, W.M.; Mitchison, T.J. Quantitative analysis of actin turnover in listeria comet tails: Evidence for catastrophic filament turnover. Biophys. J. 2010, 99, 2153–2162. [Google Scholar] [CrossRef]
- Hao, Y.K.; Charras, G.T.; Mitchison, T.J.; Brieher, W.M. Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J. Cell Biol. 2008, 182, 341–353. [Google Scholar] [CrossRef]
- Suarez, C.; Roland, J.; Boujemaa-Paterski, R.; Kang, H.; McCullough, B.R.; Reymann, A.C.; Guérin, C.; Martiel, J.L.; De La Cruz, E.M.; Blanchoin, L. Cofilin tunes the nucleotide state of actin filaments and severs at bare and decorated segment boundaries. Curr. Biol. 2011, 21, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Breitsprecher, D.; Koestler, S.A.; Chizhov, I.; Nemethova, M.; Mueller, J.; Goode, B.L.; Small, J.V.; Rottner, K.; Faix, J. Cofilin cooperates with fascin to disassemble filopodial actin filaments. J. Cell Sci. 2011, 124, 3305–3318. [Google Scholar] [CrossRef]
- Schmoller, K.M.; Semmrich, C.; Bausch, A.R. Slow down of actin depolymerization by cross-linking molecules. J. Struct. Biol. 2011, 173, 350–357. [Google Scholar] [CrossRef]
- Hayakawa, K.; Tatsumi, H.; Sokabe, M. Actin filaments function as a tension sensor by tension-dependent binding of cofilin to the filament. J. Cell Biol. 2011, 195, 721–727. [Google Scholar] [CrossRef]
- Chan, C.; Beltzner, C.C.; Pollard, T.D. Cofilin Dissociates Arp2/3 Complex and Branches from Actin Filaments. Curr. Biol. 2009, 19, 537–545. [Google Scholar] [CrossRef]
- Abercrombie, M.; Heaysman, J.E.M.; Pegrum, S.M. Locomotion of fibroblasts in culture. V. Surface marking with concanavalin A. Exp. Cell Res. 1972, 73, 536–539. [Google Scholar] [CrossRef]
- Abercrombie, M.; Heaysman, J.E.M.; Pegrum, S.M. The locomotion of fibroblasts in culture. IV. Electron microscopy of the leading lamella. Exp. Cell Res. 1971, 67, 359–367. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A physically integrated molecular process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef]
- Mitchison, T.J.; Cramer, L.P. Actin-based cell motility and cell locomotion. Cell 1996, 84, 371–379. [Google Scholar] [CrossRef]
- Xu, K.; Babcock, H.P.; Zhuang, X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat. Methods 2012, 9, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Ydenberg, C.A.; Padrick, S.B.; Sweeney, M.O.; Gandhi, M.; Sokolova, O.; Goode, B.L. GMF severs actin-Arp2/3 complex branch junctions by a cofilin- like mechanism. Curr. Biol. 2013, 23, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Vallotton, P.; Gupton, S.L.; Waterman-Storer, C.M.; Danuser, G. Simultaneous mapping of filamentous actin flow and turnover in migrating cells by quantitative fluorescent speckle microscopy. Proc. Natl. Acad. Sci. USA 2004, 101, 9660–9665. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.A.; Tsuchida, M.A.; Allen, G.M.; Barnhart, E.L.; Applegate, K.T.; Yam, P.T.; Ji, L.; Keren, K.; Danuser, G.; Theriot, J.A. Myosin II contributes to cell-scale actin network treadmilling via network disassembly. Nature 2010, 465, 373–377. [Google Scholar] [CrossRef]
- Prass, M.; Jacobson, K.; Mogilner, A.; Radmacher, M. Direct measurement of the lamellipodial protrusive force in a migrating cell. J. Cell Biol. 2006, 174, 767–772. [Google Scholar] [CrossRef]
- Krause, M.; Gautreau, A. Steering cell migration: Lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 2014, 15, 577–590. [Google Scholar] [CrossRef]
- Tang, H.; Li, A.; Bi, J.; Veltman, D.M.; Zech, T.; Spence, H.J.; Yu, X.; Timpson, P.; Insall, R.H.; Frame, M.C.; et al. Loss of scar/WAVE complex promotes N-WASP- and FAK-dependent invasion. Curr. Biol. 2013, 23, 107–117. [Google Scholar] [CrossRef]
- Petrie, R.J.; Gavara, N.; Chadwick, R.S.; Yamada, K.M. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J. Cell Biol. 2012, 197, 439–455. [Google Scholar] [CrossRef]
- Breitsprecher, D.; Kiesewetter, A.K.; Linkner, J.; Vinzenz, M.; Stradal, T.E.B.; Small, J.V.; Curth, U.; Dickinson, R.B.; Faix, J. Molecular mechanism of Ena/VASP-mediated actin-filament elongation. EMBO J. 2011, 30, 456–467. [Google Scholar] [CrossRef]
- Hansen, S.D.; Mullins, R.D. VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J. Cell Biol. 2010, 191, 571–584. [Google Scholar] [CrossRef]
- Mejillano, M.R.; Kojima, S.I.; Applewhite, D.A.; Gertler, F.B.; Svitkina, T.M.; Borisy, G.G. Lamellipodial versus filopodial mode of the actin nanomachinery: Pivotal role of the filament barbed end. Cell 2004, 118, 363–373. [Google Scholar] [CrossRef]
- Small, J.V.; Herzog, M.; Anderson, K. Actin filament organization in the fish keratocyte lamellipodium. J. Cell Biol. 1995, 129, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Helgeson, L.A.; Nolen, B.J. Mechanism of synergistic activation of Arp2/3 complex by cortactin and N-WASP. Elife 2013, 2013, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Dang, I.; Gorelik, R.; Sousa-Blin, C.; Derivery, E.; Guérin, C.; Linkner, J.; Nemethova, M.; Dumortier, J.G.; Giger, F.A.; Chipysheva, T.A.; et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature 2013, 503, 281–284. [Google Scholar] [CrossRef]
- Veltman, D. Actin dynamics: Cell migration takes a new turn with arpin. Curr. Biol. 2014, 24, R31–R33. [Google Scholar] [CrossRef]
- Houk, A.R.; Jilkine, A.; Mejean, C.O.; Boltyanskiy, R.; Dufresne, E.R.; Angenent, S.B.; Altschuler, S.J.; Wu, L.F.; Weiner, O.D. Membrane tension maintains cell polarity by confining signals to the leading edge during neutrophil migration. Cell 2012, 148, 175–188. [Google Scholar] [CrossRef]
- Ofer, N.; Mogilner, A.; Keren, K. Actin disassembly clock determines shape and speed of lamellipodial fragments. PNAS 2011, 108, 20394–20399. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Verkhovsky, A.B.; McQuade, K.M.; Borisy, G.G. Analysis of the actin-myosin II system in fish epidermal keratocytes: Mechanism of cell body translocation. J. Cell Biol. 1997, 139, 397–415. [Google Scholar] [CrossRef]
- Weichsel, J.; Urban, E.; Small, J.V.; Schwarz, U.S. Reconstructing the orientation distribution of actin filaments in the lamellipodium of migrating keratocytes from electron microscopy tomography data. Cytom. Part A 2012, 81 A, 496–507. [Google Scholar] [CrossRef]
- Faix, J.; Rottner, K. The making of filopodia. Curr. Opin. Cell Biol. 2006, 18, 18–25. [Google Scholar] [CrossRef]
- Cojoc, D.; Difato, F.; Ferrari, E.; Shahapure, R.B.; Laishram, J.; Righi, M.; Di Fabrizio, E.M.; Torre, V. Properties of the force exerted by filopodia and lamellipodia and the involvement of cytoskeletal components. PLoS ONE 2007, 2. [Google Scholar] [CrossRef]
- Jacquemet, G.; Hamidi, H.; Ivaska, J. Filopodia in cell adhesion, 3D migration and cancer cell invasion. Curr. Opin. Cell Biol. 2015, 36, 23–31. [Google Scholar] [CrossRef]
- Vignjevic, D.; Peloquin, J.; Borisy, G.G. In vitro assembly of filopodia-like bundles. Methods Enzymol. 2006, 406, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Svitkina, T. Filopodia initiation: Focus on the Arp2/3 complex and formins. Cell Adhes. Migr. 2011, 5, 402–408. [Google Scholar] [CrossRef]
- Block, J.; Breitsprecher, D.; Kühn, S.; Winterhoff, M.; Kage, F.; Geffers, R.; Duwe, P.; Rohn, J.L.; Baum, B.; Brakebusch, C.; et al. FMNL2 drives actin-based protrusion and migration downstream of Cdc42. Curr. Biol. 2012, 22, 1005–1012. [Google Scholar] [CrossRef]
- Miki, H.; Sasaki, T.; Takai, Y.; Takenawa, T. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature 1998, 391, 93–96. [Google Scholar] [CrossRef]
- Young, L.E.; Heimsath, E.G.; Higgs, H.N. Cell type-dependent mechanisms for formin-mediated assembly of filopodia. Mol. Biol. Cell 2015, 26, 4646–4659. [Google Scholar] [CrossRef]
- Khurana, S.; George, S.P. The role of actin bundling proteins in the assembly of filopodia in epithelial cells. Cell Adh. Migr. 2011, 5, 409–420. [Google Scholar] [CrossRef]
- Chan, C.E.; Odde, D.J. Traction dynamics of filopodia on compliant substrates. Science 2008, 322, 1687–1691. [Google Scholar] [CrossRef]
- Kress, H.; Stelzer, E.H.K.; Holzer, D.; Buss, F.; Griffiths, G.; Rohrbach, A. Filopodia act as phagocytic tentacles and pull with discrete steps and a load-dependent velocity. Proc. Natl. Acad. Sci. USA 2007, 104, 11633–11638. [Google Scholar] [CrossRef]
- Naumanen, P.; Lappalainen, P.; Hotulainen, P. Mechanisms of actin stress fibre assembly. J. Microsc. 2008, 231, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Cramer, L.P.; Siebert, M.; Mitchison, T.J. Identification of novel graded polarity actin filament bundles in locomoting heart fibroblasts: Implications for the generation of motile force. J. Cell Biol. 1997, 136, 1287–1305. [Google Scholar] [CrossRef] [PubMed]
- Sjöblom, B.; Salmazo, A.; Djinović-Carugo, K. α-Actinin structure and regulation. Cell. Mol. Life Sci. 2008, 65, 2688–2701. [Google Scholar] [CrossRef] [PubMed]
- Koenderink, G.H.; Paluch, E.K. Architecture shapes contractility in actomyosin networks. Curr. Opin. Cell Biol. 2018, 50, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, S.; Mellor, H. Actin stress fibres. J. Cell Sci. 2007, 120, 3491–3499. [Google Scholar] [CrossRef]
- Hotulainen, P.; Lappalainen, P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol. 2006, 173, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Tojkander, S.; Gateva, G.; Schevzov, G.; Hotulainen, P.; Naumanen, P.; Martin, C.; Gunning, P.W.; Lappalainen, P. A molecular pathway for myosin II recruitment to stress fibers. Curr. Biol. 2011, 21, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Endlich, N.; Schordan, E.; Cohen, C.D.; Kretzler, M.; Lewko, B.; Welsch, T.; Kriz, W.; Otey, C.A.; Endlich, K. Palladin is a dynamic actin-associated protein in podocytes. Kidney Int. 2009, 75, 214–226. [Google Scholar] [CrossRef]
- Schmidt, K.; Nichols, B.J. Functional interdependence between septin and actin cytoskeleton. BMC Cell Biol. 2004, 5, 1–13. [Google Scholar] [CrossRef]
- Lazarides, E.; Burridge, K. α-Actinin: Immunofluorescent localization of a muscle structural protein in nonmuscle cells. Cell 1975, 6, 289–298. [Google Scholar] [CrossRef]
- Vallenius, T.; Makela, T.P. Clik1: A novel kinase targeted to actin stress fibers by the CLP-36 PDZ-LIM protein. J. Cell Sci. 2002, 115, 2067–2073. [Google Scholar]
- Vallenius, T.; Luukko, K.; Mäkelä, T.P. CLP-36 PDZ-LIM Protein Associates with Nonmuscle alpha -Actinin-1 and alpha -Actinin-4. J. Biol. Chem. 2000, 275, 11100–11105. [Google Scholar] [CrossRef]
- Wang, K.; Ash, J.F.; Singer, S.J. Filamin, a New High-Molecular-Weight Protein Found in Smooth Muscle and Nonmuscle Cells. Purification and Properties of Chicken Gizzard Filamin. Proc. Natl. Acad. Sci. USA 1975, 72, 4483–4486. [Google Scholar] [CrossRef]
- Dixon, R.D.S.; Arneman, D.K.; Rachlin, A.S.; Sundaresan, N.R.; Costello, M.J.; Campbell, S.L.; Otey, C.A. Palladin is an actin cross-linking protein that uses immunoglobulin-like domains to bind filamentous actin. J. Biol. Chem. 2008, 283, 6222–6231. [Google Scholar] [CrossRef]
- Elkhatib, N.; Neu, M.B.; Zensen, C.; Schmoller, K.M.; Louvard, D.; Bausch, A.R.; Betz, T.; Vignjevic, D.M. Fascin plays a role in stress fiber organization and focal adhesion disassembly. Curr. Biol. 2014, 24, 1492–1499. [Google Scholar] [CrossRef]
- Boukhelifa, M.; Moza, M.; Johansson, T.; Rachlin, A.; Parast, M.; Huttelmaier, S.; Roy, P.; Jockusch, B.M.; Carpen, O.; Karlsson, R.; et al. The proline-rich protein palladin is a binding partner for profilin. FEBS J. 2006, 273, 26–33. [Google Scholar] [CrossRef]
- Boukhelifa, M.; Parast, M.M.; Bear, J.E.; Gertler, F.B.; Otey, C.A. Palladin Is a Novel Binding Partner for Ena/VASP Family Members. Cell Motil. Cytoskeleton 2004, 58, 17–29. [Google Scholar] [CrossRef]
- Strasser, P.; Gimona, M.; Moessler, H.; Herzog, M.; Small, J.V. Mammalian calponin: Identification and expression of genetic variants. FEBS J. 1993, 330, 13–18. [Google Scholar] [CrossRef]
- Weber, K.; Groeschel-Stewart, U. Antibody to myosin: The specific visualization of myosin-containing filaments in nonmuscle cells. Proc. Natl. Acad. Sci. USA 1974, 71, 4561–4564. [Google Scholar] [CrossRef]
- Yamashiro-Matsumura, S.; Matsumura, F. Characterization of 83-kilodalton nonmuscle caldesmon from cultured rat cells: Stimulation of actin binding of nonmuscle tropomyosin and periodic localization along microfilaments like tropomyosin. J. Cell Biol. 1988, 106, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Castellino, F.; Ono, S.; Matsumura, F.; Luini, A. Essential role of caldesmon in the actin filament reorganization induced by glucocorticoids. J. Cell Biol. 1995, 131, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.; Chen, X.Q.; Manser, E.; Lim, L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 1996, 16, 5313–5327. [Google Scholar] [CrossRef]
- Watanabe, N.; Madaule, P.; Reid, T.; Ishizaki, T.; Watanabe, G.; Kakizuka, A.; Saito, Y.; Nakao, K.; Jockusch, B.M.; Narumiya, S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997, 16, 3044–3056. [Google Scholar] [CrossRef]
- Tominaga, T.; Sahai, E.; Chardin, P.; McCormick, F.; Courtneidge, S.A.; Alberts, A.S. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell 2000, 5, 13–25. [Google Scholar] [CrossRef]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef]
- Chen, Z.; Borek, D.; Padrick, S.B.; Gomez, T.S.; Metlagel, Z.; Ismail, A.M.; Umetani, J.; Billadeau, D.D.; Otwinowski, Z.; Rosen, M.K. Structure and control of the actin regulatory WAVE complex. Nature 2010, 468, 533–538. [Google Scholar] [CrossRef]
- Lebensohn, A.M.; Kirschner, M.W. Activation of the WAVE Complex by Coincident Signals Controls Actin Assembly. Mol. Cell 2009, 36, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.J.; Rajfur, Z.; Maddox, A.S.; Freel, C.D.; Chen, Y.; Edlund, M.; Otey, C.; Burridge, K. Simultaneous Stretching and Contraction of Stress Fibers In Vivo. Mol. Biol. Cell 2004, 15, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Oswald, S.J.; Ngu, H.; Yin, F.C.P. Mechanical properties of actin stress fibers in living cells. Biophys. J. 2008, 95, 6060–6071. [Google Scholar] [CrossRef]
- Deguchi, S.; Ohashi, T.; Sato, M. Tensile properties of single stress fibers isolated from cultured vascular smooth muscle cells. J. Biomech. 2006, 39, 2603–2610. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.W.; Vaughan, A.N.; Cramer, L.P. Retrograde Flow and Myosin II Activity within the Leading Cell Edge Deliver F-Actin to the Lamella to Seed the Formation of Graded Polarity Actomyosin II Filament Bundles. Mol. Biol. Cell 2008, 19, 5006–5018. [Google Scholar] [CrossRef]
- Langanger, G.; Moeremans, M.; Daneels, G.; Sobieszek, A.; De Brabander, M.; De Mey, J. The molecular organisation of myosin in stress fibers of cultured cells. J. Cell Biol. 1986, 102, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Burnette, D.T.; Manley, S.; Sengupta, P.; Sougrat, R.; Davidson, M.W.; Kachar, B.; Lippincott-Schwartz, J. A role for actin arcs in the leading-edge advance of migrating cells. Nat. Cell Biol. 2011, 13, 371–382. [Google Scholar] [CrossRef]
- Hu, S.; Dasbiswas, K.; Guo, Z.; Tee, Y.H.; Thiagarajan, V.; Hersen, P.; Chew, T.L.; Safran, S.A.; Zaidel-Bar, R.; Bershadsky, A.D. Long-range self-organization of cytoskeletal myosin II filament stacks. Nat. Cell Biol. 2017, 19, 133–141. [Google Scholar] [CrossRef]
- Dasanayake, N.L.; Michalski, P.J.; Carlsson, A.E. General mechanism of actomyosin contractility. Phys. Rev. Lett. 2011, 107, 1–4. [Google Scholar] [CrossRef]
- Nemethova, M.; Auinger, S.; Small, J.V. Building the actin cytoskeleton: Filopodia contribute to the construction of contractile bundles in the lamella. J. Cell Biol. 2008, 180, 1233–1244. [Google Scholar] [CrossRef]
- Tojkander, S.; Gateva, G.; Husain, A.; Krishnan, R.; Lappalainen, P. Generation of contractile actomyosin bundles depends on mechanosensitive actin filament assembly and disassembly. Elife 2015, 4. [Google Scholar] [CrossRef]
- Small, J.V.; Rottner, K.; Kaverina, I.; Anderson, K.I. Assembling an actin cytoskeleton for cell attachment and movement. Biochim. Biophys. Acta Mol. Cell Res. 1998, 1404, 271–281. [Google Scholar] [CrossRef]
- Chen, W.-T. Mechanism of the trailing edge during fibroblast movement. J.Cell Biol. 1981, 90, 187–200. [Google Scholar] [CrossRef]
- Khatau, S.B.; Hale, C.M.; Stewart-Hutchinson, P.J.; Patel, M.S.; Stewart, C.L.; Searson, P.C.; Hodzic, D.; Wirtz, D. A perinuclear actin cap regulates nuclear shape. Proc. Natl. Acad. Sci. USA 2009, 106, 19017–19022. [Google Scholar] [CrossRef]
- Burridge, K.; Guilluy, C. Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 2016, 343, 14–20. [Google Scholar] [CrossRef]
- Gateva, G.; Tojkander, S.; Koho, S.; Carpen, O.; Lappalainen, P. Palladin promotes assembly of non-contractile dorsal stress fibers through VASP recruitment. J. Cell Sci. 2014, 127, 1887–1898. [Google Scholar] [CrossRef]
- Kovac, B.; Teo, J.L.; Mäkelä, T.P.; Vallenius, T. Assembly of non-contractile dorsal stress fibers requires α-actinin-1 and Rac1 in migrating and spreading cells. J. Cell Sci. 2013, 126, 263–273. [Google Scholar] [CrossRef]
- Shemesh, T.; Verkhovsky, A.B.; Svitkina, T.M.; Bershadsky, A.D.; Kozlov, M.M. Role of focal adhesions and mechanical stresses in the formation and progression of the lamellum interface. Biophys. J. 2009, 97, 1254–1264. [Google Scholar] [CrossRef]
- Eghiaian, F.; Rigato, A.; Scheuring, S. Structural, mechanical, and dynamical variability of the actin cortex in living cells. Biophys. J. 2015, 108, 1330–1340. [Google Scholar] [CrossRef]
- Clausen, M.P.; Colin-York, H.; Schneider, F.; Eggeling, C.; Fritzsche, M. Dissecting the actin cortex density and membrane-cortex distance in living cells by super-resolution microscopy. J. Phys. D Appl. Phys. 2017, 50. [Google Scholar] [CrossRef]
- Hanakam, F.; Albrecht, R.; Eckerskorn, C.; Matzner, M.; Gerisch, G. Myristoylated and non-myristoylated forms of the pH sensor protein hisactophilin II: Intracellular shuttling to plasma membrane and nucleus monitored in real time by a fusion with green fluorescent protein. EMBO J. 1996, 15, 2935–2943. [Google Scholar] [CrossRef]
- Morone, N.; Usukura, J.; Kusumi, A. Three-dimensional reconstruction of the membrane skelton at the plama membrane interface by electron tomography. J. Cell Biol. 2006, 174, 851–862. [Google Scholar] [CrossRef]
- Biro, M.; Romeo, Y.; Kroschwald, S.; Bovellan, M.; Boden, A.; Tcherkezian, J.; Roux, P.P.; Charras, G.; Paluch, E.K. Cell cortex composition and homeostasis resolved by integrating proteomics and quantitative imaging. Cytoskeleton 2013, 70, 741–754. [Google Scholar] [CrossRef]
- Bovellan, M.; Romeo, Y.; Biro, M.; Boden, A.; Chugh, P.; Yonis, A.; Vaghela, M.; Fritzsche, M.; Moulding, D.; Thorogate, R.; et al. Cellular control of cortical actin nucleation. Curr. Biol. 2014, 24, 1628–1635. [Google Scholar] [CrossRef]
- Roh-johnson, M.; Shemer, G.; Higgins, C.D.; Mcclellan, J.H.; Werts, A.D.; Tulu, U.S.; Gao, L.; Betzig, E.; Kiehart, D.P.; Goldstein, B. Triggering a cell shape change by exploiting preexisting actomyosin contractions. Science 2012, 335, 1232–1235. [Google Scholar] [CrossRef]
- Charras, G.T.; Hu, C.K.; Coughlin, M.; Mitchison, T.J. Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 2006, 175, 477–490. [Google Scholar] [CrossRef]
- McClatchey, A.I.; Fehon, R.G. Merlin and the ERM proteins - regulators of receptor distribution and signaling at the cell cortex. Trends Cell Biol. 2009, 19, 198–206. [Google Scholar] [CrossRef]
- Carlier, M.; Laurent, V.; Santolini, J.; Melki, R.; Didry, D.; Xia, G.; Hong, Y.; Chua, N.; Pantaloni, D. Actin depolymerizing factor (ADF/Cofilin) enhances the rate of filament turnover: Implication in actin-based motility. J. Cell Biol. 1997, 136, 1307–1322. [Google Scholar] [CrossRef]
- Chugh, P.; Clark, A.G.; Smith, M.B.; Cassani, D.A.D.; Dierkes, K.; Ragab, A.; Roux, P.P.; Charras, G.; Salbreux, G.; Paluch, E.K. Actin cortex architecture regulates cell surface tension. Nat. Cell Biol. 2017, 19, 689–697. [Google Scholar] [CrossRef]
- Howard, J. Mechanics of Motor Proteins and the Cytoskeleton; Oxford University Press: Oxford, UK, 2005; ISBN 0-87893-334-4. [Google Scholar]
- Ananthakrishnan, R.; Guck, J.; Wottawah, F.; Schinkinger, S.; Lincoln, B.; Romeyke, M.; Moon, T.; Käs, J. Quantifying the contribution of actin networks to the elastic strength of fibroblasts. J. Theor. Biol. 2006, 242, 502–516. [Google Scholar] [CrossRef]
- Tee, S.Y.; Fu, J.; Chen, C.S.; Janmey, P.A. Cell shape and substrate rigidity both regulate cell stiffness. Biophys. J. 2011, 100, L25–L27. [Google Scholar] [CrossRef]
- Guha, M.; Zhou, M.; Wang, Y.L. Cortical actin turnover during cytokinesis requires myosin II. Curr. Biol. 2005, 15, 732–736. [Google Scholar] [CrossRef]
- Murthy, K.; Wadsworth, P. Myosin-II-dependent localization and dynamics of F-actin during cytokinesis. Curr. Biol. 2005, 15, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Bursac, P.; Lenormand, G.; Fabry, B.; Oliver, M.; Weitz, D.A.; Viasnoff, V.; Butler, J.P.; Fredberg, J.J. Cytoskeletal remodelling and slow dynamics in the living cell. Nat. Mater. 2005, 4, 557–561. [Google Scholar] [CrossRef]
- Gardel, M.L.; Nakamura, F.; Hartwig, J.H.; Crocker, J.C.; Stossel, T.P.; Weitz, D.A. Prestressed F-actin networks cross-linked by hinged filamins replicate mechanical properties of cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1762–1767. [Google Scholar] [CrossRef]
- Lecuit, T.; Lenne, P.F. Cell surface mechanics and the control of cell shape, tissue patterns and morphogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 633–644. [Google Scholar] [CrossRef]
- Charras, G.T.; Coughlin, M.; Mitchison, T.J.; Mahadevan, L. Life and times of a cellular bleb. Biophys. J. 2008, 94, 1836–1853. [Google Scholar] [CrossRef]
- Kasza, K.E.; Rowat, A.C.; Liu, J.; Angelini, T.E.; Brangwynne, C.P.; Koenderink, G.H.; Weitz, D.A. The cell as a material. Curr. Opin. Cell Biol. 2007, 19, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Paluch, E.K.; Raz, E. The role and regulation of blebs in cell migration. Curr. Opin. Cell Biol. 2013, 25, 582–590. [Google Scholar] [CrossRef]
- Goudarzi, M.; Banisch, T.U.; Mobin, M.B.; Maghelli, N.; Tarbashevich, K.; Strate, I.; van den Berg, J.; Blaser, H.; Bandemer, S.; Paluch, E.; et al. Identification and Regulation of a Molecular Module for Bleb-Based Cell Motility. Dev. Cell 2012, 23, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Tinevez, J.-Y.; Schulze, U.; Salbreux, G.; Roensch, J.; Joanny, J.-F.; Paluch, E. Role of cortical tension in bleb growth. Proc. Natl. Acad. Sci. USA 2009, 106, 18581–18586. [Google Scholar] [CrossRef] [PubMed]
- Paluch, E.; Piel, M.; Prost, J.; Bornens, M.; Sykes, C. Cortical actomyosin breakage triggers shape oscillations in cells and cell fragments. Biophys. J. 2005, 89, 724–733. [Google Scholar] [CrossRef]
- Charras, G.T.; Yarrow, J.C.; Horton, M.A.; Mahadevan, L.; Mitchison, T.J. Non-equilibration of hydrostatic pressure in blebbing cells. Nature 2005, 435, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Paluch, E.; Van Der Gucht, J.; Sykes, C. Cracking up: Symmetry breaking in cellular systems. J. Cell Biol. 2006, 175, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Loitto, V.M.; Karlsson, T.; Magnusson, K.E. Water flux in cell motility: Expanding the mechanisms of membrane protrusion. Cell Motil. Cytoskeleton 2009, 66, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Reichman-Fried, M.; Castanon, I.; Dumstrei, K.; Marlow, F.L.; Kawakami, K.; Solnica-Krezel, L.; Heisenberg, C.P.; Raz, E. Migration of Zebrafish Primordial Germ Cells: A Role for Myosin Contraction and Cytoplasmic Flow. Dev. Cell 2006, 11, 613–627. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. Differing modes for tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Tournaviti, S.; Hannemann, S.; Terjung, S.; Kitzing, T.M.; Stegmayer, C.; Ritzerfeld, J.; Walther, P.; Grosse, R.; Nickel, W.; Fackler, O.T. SH4-domain-induced plasma membrane dynamization promotes bleb-associated cell motility. J. Cell Sci. 2007, 120, 3820–3829. [Google Scholar] [CrossRef] [PubMed]
- Rossy, J.; Gutjahr, M.C.; Blaser, N.; Schlicht, D.; Niggli, V. Ezrin/moesin in motile Walker 256 carcinosarcoma cells: Signal-dependent relocalization and role in migration. Exp. Cell Res. 2007, 313, 1106–1120. [Google Scholar] [CrossRef]
- Brugues, J.; Maugis, B.; Casademunt, J.; Nassoy, P.; Amblard, F.; Sens, P. Dynamical organization of the cytoskeletal cortex probed by micropipette aspiration. Proc. Natl. Acad. Sci. USA 2010, 107, 15415–15420. [Google Scholar] [CrossRef] [PubMed]
- Charras, G.T. A short history of blebbing. J. Microsc. 2008, 231, 466–478. [Google Scholar] [CrossRef]
- Cunningham, C.C. Actin polymerization and intracellular solvent flow in cell surfase blebbing. J. Cell Biol. 1995, 129, 1589–1599. [Google Scholar] [CrossRef]
- Strychalski, W.; Guy, R.D. A computational model of bleb formation. Math. Med. Biol. 2013, 30, 115–130. [Google Scholar] [CrossRef]
- Keller, H.; Rentsch, P.; Hagmann, J. Differences in cortical actin structure and dynamics document that different types of blebs are formed by distinct mechanisms. Exp. Cell Res. 2002, 277, 161–172. [Google Scholar] [CrossRef]
- Kristó, I.; Bajusz, I.; Bajusz, C.; Borkúti, P.; Vilmos, P. Actin, actin-binding proteins, and actin-related proteins in the nucleus. Histochem. Cell Biol. 2016, 145, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, C.; Borkúti, P.; Kristó, I.; Kovács, Z.; Abonyi, C.; Vilmos, P. Nuclear actin: Ancient clue to evolution in eukaryotes? Histochem. Cell Biol. 2018, 150, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Le, H.Q.; Ghatak, S.; Yeung, C.Y.C.; Tellkamp, F.; Günschmann, C.; Dieterich, C.; Yeroslaviz, A.; Habermann, B.; Pombo, A.; Niessen, C.M.; et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 2016, 18, 864–875. [Google Scholar] [CrossRef]
- Obrdlik, A.; Percipalle, P. The F-actin severing protein cofilin-1 is required for RNA polymerase II transcription elongation. Nucleus 2011, 2, 72–79. [Google Scholar] [CrossRef]
- Serebryannyy, L.A.; Parilla, M.; Annibale, P.; Cruz, C.M.; Laster, K.; Gratton, E.; Kudryashov, D.; Kosak, S.T.; Gottardi, C.J.; de Lanerolle, P. Persistent nuclear actin filaments inhibit transcription by RNA polymerase II. J. Cell Sci. 2016, 129, 3412–3425. [Google Scholar] [CrossRef] [PubMed]
- Baarlink, C.; Plessner, M.; Sherrard, A.; Morita, K.; Misu, S.; Virant, D.; Kleinschnitz, E.M.; Harniman, R.; Alibhai, D.; Baumeister, S.; et al. A transient pool of nuclear F-actin at mitotic exit controls chromatin organization. Nat. Cell Biol. 2017, 19, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef]
- Baarlink, C.; Wang, H.; Grosse, R. Nuclear Actin Network Assembly by Formins Regulates the SRF Coactivator MAL. Science 2013, 2, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Plessner, M.; Grosse, R. Dynamizing nuclear actin filaments. Curr. Opin. Cell Biol. 2019, 56, 1–6. [Google Scholar] [CrossRef]
- Parisis, N.; Krasinska, L.; Harker, B.; Urbach, S.; Rossignol, M.; Camasses, A.; Dewar, J.; Morin, N.; Fisher, D. Initiation of DNA replication requires actin dynamics and formin activity. EMBO J. 2017, e201796585. [Google Scholar] [CrossRef] [PubMed]
- Belin, B.J.; Lee, T.; Mullins, R.D. DNA damage induces nuclear actin filament assembly by formin-2 and spire-1/2 that promotes efficient DNA repair. Elife 2015, 4, 1–21. [Google Scholar] [CrossRef]
- Caridi, C.P.; D’agostino, C.; Ryu, T.; Zapotoczny, G.; Delabaere, L.; Li, X.; Khodaverdian, V.Y.; Amaral, N.; Lin, E.; Rau, A.R.; et al. Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature 2018, 559, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Schrank, B.R.; Aparicio, T.; Li, Y.; Chang, W.; Chait, B.T.; Gundersen, G.G.; Gottesman, M.E.; Gautier, J. Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature 2018, 559, 61–66. [Google Scholar] [CrossRef]
- Kelpsch, D.J.; Tootle, T.L. Nuclear Actin: From Discovery to Function. Anat. Rec. 2018, 2013, 1999–2013. [Google Scholar] [CrossRef] [PubMed]
- Ohi, R.; Zanic, M. Ahead of the Curve: New Insights into Microtubule Dynamics. F1000Research 2016, 5, 314. [Google Scholar] [CrossRef] [PubMed]
- Doxsey, S. Re-evaluating centrosome function. Nat. Rev. Mol. Cell Biol. 2001, 2, 688–698. [Google Scholar] [CrossRef]
- Vinogradova, T.; Miller, P.M.; Kaverina, I. Microtubule network asymmetry in motile cells: Role of Golgi-derived array. Cell Cycle 2009, 8, 2168–2174. [Google Scholar] [CrossRef]
- Sanchez, A.D.; Feldman, J.L. Microtubule-organizing centers: From the centrosome to non-centrosomal sites. Curr. Opin. Cell Biol. 2017, 44, 93–101. [Google Scholar] [CrossRef]
- Wu, J.; Akhmanova, A. Microtubule-Organizing Centers. Annu. Rev. Cell Dev. Biol. 2017, 33, 51–75. [Google Scholar] [CrossRef]
- Dimitrov, A.; Quesnoit, M.; Moutel, S.; Cantaloube, I.; Poüs, C.; Perez, F. Detection of GTP-tubulin conformation in vivo reveals a role for GTP remnants in microtubule rescues. Science 2008, 322, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.; Hyman, A.A. Growth, fluctuation and switching at microtubule plus ends. Nat. Rev. Mol. Cell Biol. 2009, 10, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Duellberg, C.; Cade, N.I.; Holmes, D.; Surrey, T. The size of the EB cap determines instantaneous microtubule stability. Elife 2016, 5, e13470. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Wittmann, T.; Näthke, I.S.; Waterman-Storer, C.M. Adenomatous polyposis coli on microtubule plus ends in cell extensions can promote microtubule net growth with or without EB1. Mol. Biol. Cell 2006, 17, 2331–2345. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Zhou, X.Z.; Lu, K.P. Critical role for the EB1 and APC interaction in the regulation of microtubule polymerization. Curr. Biol. 2001, 11, 1062–1067. [Google Scholar] [CrossRef]
- Akhmanova, A.; Steinmetz, M.O. Control of microtubule organization and dynamics: Two ends in the limelight. Nat. Rev. Mol. Cell Biol. 2015, 16, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Mimori-Kiyosue, Y.; Grigoriev, I.; Lansbergen, G.; Sasaki, H.; Matsui, C.; Severin, F.; Galjart, N.; Grosveld, F.; Vorobjev, I.; Tsukita, S.; et al. CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J. Cell Biol. 2005, 168, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Suozzi, K.C.; Wu, X.; Fuchs, E. Spectraplakins: Master orchestrators of cytoskeletal dynamics. J. Cell Biol. 2012, 197, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Drabek, K.; van Ham, M.; Stepanova, T.; Draegestein, K.; van Horssen, R.; Sayas, C.L.; Akhmanova, A.; ten Hagen, T.; Smits, R.; Fodde, R.; et al. Role of CLASP2 in Microtubule Stabilization and the Regulation of Persistent Motility. Curr. Biol. 2006, 16, 2259–2264. [Google Scholar] [CrossRef]
- Leung, C.L.; Sun, D.; Zheng, M.; Knowles, D.R.; Liem, R.K. Microtubule actin cross-linking factor (MACF): A hybrid of dystonin and dystrophin that can interact with the actin and microtubule cytoskeletons. J. Cell Biol. 1999, 147, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.; De Groot, C.O.; Grigoriev, I.; Gouveia, S.M.; Munteanu, E.L.; Schober, J.M.; Honnappa, S.; Buey, R.M.; Hoogenraad, C.C.; Dogterom, M.; et al. Mammalian end binding proteins control persistent microtubule growth. J. Cell Biol. 2009, 184, 691–706. [Google Scholar] [CrossRef]
- Maurer, S.P.; Cade, N.I.; Bohner, G.; Gustafsson, N.; Boutant, E.; Surrey, T. EB1 Accelerates Two Conformational Transitions Important for Microtubule Maturation and Dynamics. Curr. Biol. 2014, 24, 372–384. [Google Scholar] [CrossRef]
- Dong, Z.; Zhou, L.; Han, N.; Zhang, M.; Lyu, X. Wnt/β-catenin pathway involvement in ionizing radiation-induced invasion of U87 glioblastoma cells. Strahlentherapie und Onkol. 2015, 191, 672–680. [Google Scholar] [CrossRef]
- Evans, K.J.; Gomes, E.R.; Reisenweber, S.M.; Gundersen, G.G.; Lauring, B.P. Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J. Cell Biol. 2005, 168, 599–606. [Google Scholar] [CrossRef]
- Sharp, D.J.; Ross, J.L. Microtubule-severing enzymes at the cutting edge. J. Cell Sci. 2012, 125, 2561–2569. [Google Scholar] [CrossRef]
- Gupta, K.K.; Li, C.; Duan, A.; Alberico, E.O.; Kim, O.V.; Alber, M.S.; Goodson, H. V Mechanism for the catastrophe-promoting activity of the microtubule destabilizer Op18/stathmin. Proc. Natl. Acad. Sci. USA 2013, 110, 20449–20454. [Google Scholar] [CrossRef]
- Brouhard, G.J.; Stear, J.H.; Noetzel, T.L.; Al-Bassam, J.; Kinoshita, K.; Harrison, S.C.; Howard, J.; Hyman, A.A. XMAP215 Is a Processive Microtubule Polymerase. Cell 2008, 132, 79–88. [Google Scholar] [CrossRef]
- Moores, C.A.; Perderiset, M.; Kappeler, C.; Kain, S.; Drummond, D.; Perkins, S.J.; Chelly, J.; Cross, R.; Houdusse, A.; Francis, F. Distinct roles of doublecortin modulating the microtubule cytoskeleton. EMBO J. 2006, 25, 4448–4457. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2004, 6, 204. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Yu, W.; Andreadis, A.; Luo, M.; Baas, P.W. Tau Protects Microtubules in the Axon from Severing by Katanin. J. Neurosci. 2006, 26, 3120–3129. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.M.; Folkmann, A.W.; Maia, A.R.R.; Efimova, N.; Efimov, A.; Kaverina, I. Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat. Cell Biol. 2009, 11, 1069–1080. [Google Scholar] [CrossRef]
- Yadav, S.; Puri, S.; Linstedt, A. A Primary Role for Golgi Positioning in Directed Secretion, Cell Polarity, and Wound Healing. Mol. Biol. Cell 2009, 20, 1728–1736. [Google Scholar] [CrossRef]
- Verhey, K.J.; Kaul, N.; Soppina, V. Kinesin Assembly and Movement in Cells. Annu. Rev. Biophys. 2011, 40, 267–288. [Google Scholar] [CrossRef]
- Cianfrocco, M.A.; DeSantis, M.E.; Leschziner, A.E.; Reck-Peterson, S.L. Mechanism and regulation of cytoplasmic dynein. Annu. Rev. Cell Dev. Biol. 2015, 31, 83–108. [Google Scholar] [CrossRef]
- Osmani, N.; Peglion, F.; Chavrier, P.; Etienne-Manneville, S. Cdc42 localization and cell polarity depend on membrane traffic. J. Cell Biol. 2010, 191, 1261–1269. [Google Scholar] [CrossRef]
- Palamidessi, A.; Frittoli, E.; Garré, M.; Faretta, M.; Mione, M.; Testa, I.; Diaspro, A.; Lanzetti, L.; Scita, G.; Di Fiore, P.P. Endocytic Trafficking of Rac Is Required for the Spatial Restriction of Signaling in Cell Migration. Cell 2008, 134, 135–147. [Google Scholar] [CrossRef]
- Hookway, C.; Ding, L.; Davidson, M.W.; Rappoport, J.Z.; Danuser, G.; Gelfand, V.I. Microtubule-dependent transport and dynamics of vimentin intermediate filaments. Mol. Biol. Cell 2015, 26, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.; Tian, P.; Adam, S.A.; Kittisopikul, M.; Jaqaman, K.; Goldman, R.D.; Gelfand, V.I. Kinesin-dependent transport of keratin filaments: A unified mechanism for intermediate filament transport. FASEB J. 2018, fj.201800604R. [Google Scholar] [CrossRef] [PubMed]
- Helfand, B.T.; Mikami, A.; Vallee, R.B.; Goldman, R.D. A requirement for cytoplasmic dynein and dynactin in intermediate filament network assembly and organization. J. Cell Biol. 2002, 157, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Mingle, L.A. Localization of all seven messenger RNAs for the actin-polymerization nucleator Arp2/3 complex in the protrusions of fibroblasts. J. Cell Sci. 2005, 118, 2425–2433. [Google Scholar] [CrossRef] [PubMed]
- Oleynikov, Y.; Singer, R.H. RNA localization: Different zipcodes, same postman? Trends Cell Biol. 1998, 8, 381–383. [Google Scholar] [CrossRef]
- Su, X.; Ohi, R.; Pellman, D. Move in for the kill: Motile microtubule regulators. Trends Cell Biol. 2012, 22, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Moores, C.A.; Yu, M.; Guo, J.; Beraud, C.; Sakowicz, R.; Milligan, R.A. A Mechanism for Microtubule Depolymerization by KinI Kinesins. Mol. Cell 2002, 9, 903–909. [Google Scholar] [CrossRef]
- Liu, M.; Nadar, V.C.; Kozielski, F.; Kozlowska, M.; Yu, W.; Baas, P.W. Kinesin-12, a mitotic microtubule-associated motor protein, impacts axonal growth, navigation, and branching. J. Neurosci. 2010, 30, 14896–14906. [Google Scholar] [CrossRef]
- Myers, K.A.; Baas, P.W. Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J. Cell Biol. 2007, 178, 1081–1091. [Google Scholar] [CrossRef]
- Jolly, A.L.; Kim, H.; Srinivasan, D.; Lakonishok, M.; Larson, A.G.; Gelfand, V.I. Kinesin-1 heavy chain mediates microtubule sliding to drive changes in cell shape. Proc. Natl. Acad. Sci. USA 2010, 107, 12151–12156. [Google Scholar] [CrossRef]
- Song, Y.; Brady, S.T. Posttranslational Modifications of Tubulin: Pathways to Functional Diversity of Microtubules. Trends Cell Biol. 2015, 25, 125–136. [Google Scholar] [CrossRef]
- Portran, D.; Schaedel, L.; Xu, Z.; Théry, M.; Nachury, M.V. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat. Cell Biol. 2017, 19, 391–398. [Google Scholar] [CrossRef]
- Sudo, H.; Baas, P.W. Acetylation of microtubules influences their sensitivity to severing by katanin in neurons and fibroblasts. J. Neurosci. 2010, 30, 7215–7226. [Google Scholar] [CrossRef]
- Roll-Mecak, A. Intrinsically disordered tubulin tails: Complex tuners of microtubule functions? Semin. Cell Dev. Biol. 2015, 37, 11–19. [Google Scholar] [CrossRef]
- Konishi, Y.; Setou, M. Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat. Neurosci. 2009, 12, 559–567. [Google Scholar] [CrossRef]
- Roll-Mecak, A.; Vale, R.D. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 2008, 451, 363–367. [Google Scholar] [CrossRef]
- Lacroix, B.; Van Dijk, J.; Gold, N.D.; Guizetti, J.; Aldrian-Herrada, G.; Rogowski, K.; Gerlich, D.W.; Janke, C. Tubulin polyglutamylation stimulates spastin-mediated microtubule severing. J. Cell Biol. 2010, 189, 945–954. [Google Scholar] [CrossRef]
- Valenstein, M.L.; Roll-Mecak, A. Graded Control of Microtubule Severing by Tubulin Glutamylation. Cell 2016, 164, 911–921. [Google Scholar] [CrossRef]
- Morris, E.J.; Nader, G.P.F.; Ramalingam, N.; Bartolini, F.; Gundersen, G.G. Kif4 interacts with EB1 and stabilizes microtubules downstream of Rho-mDia in migrating fibroblasts. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Wen, Y.; Eng, C.H.; Schmoranzer, J.; Cabrera-Poch, N.; Morris, E.J.S.; Chen, M.; Wallar, B.J.; Alberts, A.S.; Gundersen, G.G. EB1 and APC bind to mDia to EB1 and APC bind to mDia to stabilize stabilize microtubules downstream of Rho and promote cell migration. Nat. Cell Biol. 2004, 6, 820–830. [Google Scholar] [CrossRef]
- Ishizaki, T.; Morishima, Y.; Okamoto, M.; Furuyashiki, T.; Kato, T.; Narumiya, S. Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1. Nat. Cell Biol. 2001, 3, 8–14. [Google Scholar] [CrossRef]
- Fife, C.M.; Sagnella, S.M.; Teo, W.S.; Po’Uha, S.T.; Byrne, F.L.; Yeap, Y.Y.C.; Ng, D.C.H.; Davis, T.P.; McCarroll, J.A.; Kavallaris, M. Stathmin mediates neuroblastoma metastasis in a tubulin-independent manner via RhoA/ROCK signaling and enhanced transendothelial migration. Oncogene 2017, 36, 501–511. [Google Scholar] [CrossRef]
- Daub, H.; Gevaert, K.; Vandekerckhove, J.; Sobel, A.; Hall, A. Rac/Cdc42 and p65PAK regulate the microtubule-destabilizing protein stathmin through phosphorylation at serine 16. J. Biol. Chem. 2001, 276, 1677–1680. [Google Scholar] [CrossRef]
- Wittmann, T.; Bokoch, G.M.; Waterman-Storer, C.M. Regulation of microtubule destabilizing activity of Op18/stathmin downstream of Rac1. J. Biol. Chem. 2004, 279, 6196–6203. [Google Scholar] [CrossRef]
- Wittmann, T.; Bokoch, G.M.; Waterman-Storer, C.M. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J. Cell Biol. 2003, 161, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Watanabe, T.; Noritake, J.; Nakagawa, M.; Yamaga, M.; Kuroda, S.; Matsuura, Y.; Iwamatsu, A.; Perez, F.; Kaibuchi, K. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 2002, 109, 873–885. [Google Scholar] [CrossRef]
- Brown, R.A.; Talas, G.; Porter, R.A.; McGrouther, D.A.; Eastwood, M. Balanced mechanical forces and microtubule contribution to fibroblast contraction. J. Cell. Physiol. 1996, 169, 439–447. [Google Scholar] [CrossRef]
- Rudolph, R.; Woodward, M. Spatial orientation of microtubules in contractile fibroblasts in vivo. Anat. Rec. 1978, 191, 169–181. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Hay, E.D. Analysis of the role of microfilaments and microtubules in acquisition of bipolarity and elongation of fibroblasts in hydrated collagen gels. J. Cell Biol. 1984, 99, 536–549. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Microtubules in Cell Migration. Annu. Rev. Cell Dev. Biol. 2013, 29, 471–499. [Google Scholar] [CrossRef]
- Hawkins, T.; Mirigian, M.; Selcuk Yasar, M.; Ross, J.L. Mechanics of microtubules. J. Biomech. 2010, 43, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Dogterom, M.; Yurke, B. Measurement of the force-velocity relation for growing microtubules. Science 1997, 278, 856–860. [Google Scholar] [CrossRef]
- Kolomeisky, A.B.; Fisher, M.E. Force-velocity relation for growing microtubules. Biophys. J. 2001, 80, 149–154. [Google Scholar] [CrossRef]
- Janson, M.E.; de Dood, M.E.; Dogterom, M. Dynamic instability of microtubules is regulated by force. J. Cell Biol. 2003, 161, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Robison, P.; Caporizzo, M.A.; Ahmadzadeh, H.; Bogush, A.I.; Chen, C.Y.; Margulies, K.B.; Shenoy, V.B.; Prosser, B.L. Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 2016, 352, aaf0659. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; MacKintosh, F.C.; Kumar, S.; Geisse, N.A.; Talbot, J.; Mahadevan, L.; Parker, K.K.; Ingber, D.E.; Weitz, D.A. Microtubules can bear enhanced compressive loads in living cells because of lateral reinforcement. J. Cell Biol. 2006, 173, 733–741. [Google Scholar] [CrossRef]
- Laan, L.; Husson, J.; Munteanu, E.L.; Kerssemakers, J.W.J.; Dogterom, M. Force-generation and dynamic instability of microtubule bundles. Proc. Natl. Acad. Sci. USA 2008, 105, 8920–8925. [Google Scholar] [CrossRef]
- Komarova, Y.A.; Vorobjev, I.A.; Borisy, G.G. Life cycle of MTs: Persistent growth in the cell interior, asymmetric transition frequencies and effects of the cell boundary. J. Cell Sci. 2002, 115, 3527–3539. [Google Scholar]
- Drummond, D.R.; Cross, R.A. Dynamics of interphase microtubules in Schizosaccharomyces pombe. Curr. Biol. 2000, 10, 766–775. [Google Scholar] [CrossRef]
- Bicek, A.D.; Tüzel, E.; Demtchouk, A.; Uppalapati, M.; Hancock, W.O.; Kroll, D.M.; Odde, D.J. Anterograde microtubule transport drives microtubule bending in LLC-PK1 epithelial cells. Mol. Biol. Cell 2009, 20, 2943–2953. [Google Scholar] [CrossRef]
- Desai, A.; Mitchison, T.J. Microtubule Polymerization Dynamics. Annu. Rev. Cell Dev. Biol. 1997, 13, 83–117. [Google Scholar] [CrossRef]
- Grishchuk, E.L.; Molodtsov, M.I.; Ataullakhanov, F.I.; McIntosh, J.R. Force production by disassembling microtubules. Nature 2005, 438, 384–388. [Google Scholar] [CrossRef]
- Petry, S. Mechanisms of Mitotic Spindle Assembly. Annu. Rev. Biochem. 2016, 85, 659–683. [Google Scholar] [CrossRef]
- Duro, E.; Marston, A.L. From equator to pole: Splitting chromosomes in mitosis and meiosis. Genes Dev. 2015, 29, 109–122. [Google Scholar] [CrossRef]
- Gadde, S.; Heald, R. Mechanisms and Molecules of the Mitotic Spindle. Curr. Biol. 2004, 14, R797–R805. [Google Scholar] [CrossRef]
- Okada, K.; Bartolini, F.; Deaconescu, A.M.; Moseley, J.B.; Dogic, Z.; Grigorieff, N.; Gundersen, G.G.; Goode, B.L. Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J. Cell Biol. 2010, 189, 1087–1096. [Google Scholar] [CrossRef]
- Bartolini, F.; Moseley, J.B.; Schmoranzer, J.; Cassimeris, L.; Goode, B.L.; Gundersen, G.G. The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J. Cell Biol. 2008, 181, 523–536. [Google Scholar] [CrossRef]
- Wu, X.; Kodama, A.; Fuchs, E. ACF7 Regulates Cytoskeletal-Focal Adhesion Dynamics and Migration and Has ATPase Activity. Cell 2008, 135, 137–148. [Google Scholar] [CrossRef]
- Miller, A.L.; Wang, Y.; Mooseker, M.S.; Koleske, A.J. The Abl-related gene (Arg) requires its F-actin–microtubule cross-linking activity to regulate lamellipodial dynamics during fibroblast adhesion. J. Cell Biol. 2004, 165, 407–420. [Google Scholar] [CrossRef]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef]
- Eliasson, C.; Sahlgren, C.; Berthold, C.H.; Stakeberg, J.; Celis, J.E.; Betsholtz, C.; Eriksson, J.E.; Pekny, M. Intermediate filament protein partnership in astrocytes. J. Biol. Chem. 1999, 274, 23996–24006. [Google Scholar] [CrossRef]
- Bellin, R.M.; Sernett, S.W.; Becker, B.; Ip, W.; Huiatt, T.W.; Robson, R.M. Molecular characteristics and interactions of the intermediate filament protein synemin. Interactions with alpha-actinin may anchor synemin-containing heterofilaments. J. Biol. Chem. 1999, 274, 29493–29499. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Molecular Structure, Assembly Mechanism, and Integration Into Functionally Distinct Intracellular Scaffolds. Annu. Rev. Biochem. 2004, 73, 749–789. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E. Intermediate Filaments: Structure, Dynamics, Function, and Disease. Annu. Rev. Biochem. 1994, 63, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.A.; Strelkov, S.V.; Burkhard, P.; Aebi, U.; Parry, D.A.D. Sequence comparisons of intermediate filament chains: Evidence of a unique functional/structural role for coiled-coil segment 1A and linker L1. J. Struct. Biol. 2002, 137, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.V.; Kreplak, L.; Wedig, T.; Mucke, N.; Svergun, D.I.; Herrmann, H.; Aebi, U.; Strelkov, S.V. Monitoring intermediate filament assembly by small-angle x-ray scattering reveals the molecular architecture of assembly intermediates. Proc. Natl. Acad. Sci. USA 2006, 103, 16206–16211. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Häner, M.; Brettel, M.; Ku, N.O.; Aebi, U. Characterization of distinct early assembly units of different intermediate filament proteins. J. Mol. Biol. 1999, 286, 1403–1420. [Google Scholar] [CrossRef]
- Mücke, N.; Wedig, T.; Bürer, A.; Marekov, L.N.; Steinert, P.M.; Langowski, J.; Aebi, U.; Herrmann, H. Molecular and biophysical characterization of assembly-starter units of human vimentin. J. Mol. Biol. 2004, 340, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulou, S.; Möller, D.; Sachs, N.; Herrmann, H.; Aebi, U. Near-UV Circular Dichroism Reveals Structural Transitions of Vimentin Subunits during Intermediate Filament Assembly. J. Mol. Biol. 2009, 386, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Steinert, P.M.; Marekov, L.N.; Parry, D.A. Diversity of intermediate filament structure. J. Biol. Chem. 1993, 268, 24916–24925. [Google Scholar] [PubMed]
- Herrmann, H.; Kreplak, L.; Aebi, U. Isolation, Characterization, and In Vitro Assembly of Intermediate Filaments. Methods Cell Biol. 2004, 78, 3–24. [Google Scholar] [PubMed]
- Nöding, B.; Herrmann, H.; Köster, S. Direct Observation of Subunit Exchange along Mature Vimentin Intermediate Filaments. Biophys. J. 2014, 107, 2923–2931. [Google Scholar] [CrossRef]
- Chang, L.; Goldman, R.D. Intermediate filaments mediate cytoskeletal crosstalk. Mol. Cell Biol. 2004, 5, 601–613. [Google Scholar] [CrossRef]
- Kayser, J.; Haslbeck, M.; Dempfle, L.; Krause, M.; Grashoff, C.; Buchner, J.; Herrmann, H.; Bausch, A.R. The Small Heat Shock Protein Hsp27 Affects Assembly Dynamics and Structure of Keratin Intermediate Filament Networks. Biophys. J. 2013, 105, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Heitlinger, E.; Peter, M.; Lustig, A.; Villiger, W.; Nigg, E.A.; Aebi, U. The role of the head and tail domain in lamin structure and assembly: Analysis of bacterially expressed chicken Lamin A and truncated B2 lamins. J. Struct. Biol. 1992, 108, 74–91. [Google Scholar] [CrossRef]
- Heitlinger, E.; Peter, M.; Häner, M.; Lustig, A.; Aebi, U.; Nigg, E.A. Expression of chicken lamin B2 in Escherichia coli: Characterization of its structure, assembly, and molecular interactions. J. Cell Biol. 1991, 113, 485–495. [Google Scholar] [CrossRef]
- Aebi, U.; Cohn, J.; Buhle, L.; Gerace, L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature 1986, 323, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Kreis, S.; Schönfeld, H.J.; Melchior, C.; Steiner, B.; Kieffer, N. The intermediate filament protein vimentin binds specifically to a recombinant integrin α2/β1 cytoplasmic tail complex and co-localizes with native α2/β1 in endothelial cell focal adhesions. Exp. Cell Res. 2005, 305, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, D. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J. Cell Sci. 2003, 116, 4977–4984. [Google Scholar] [CrossRef]
- Seltmann, K.; Cheng, F.; Wiche, G.; Eriksson, J.E.; Magin, T.M. Keratins stabilize hemidesmosomes through regulation of β4-integrin turnover. J. Invest. Dermatol. 2015, 135, 1609–1620. [Google Scholar] [CrossRef]
- Loschke, F.; Homberg, M.; Magin, T.M. Keratin isotypes control desmosome stability and dynamics through PKCα. J. Invest. Dermatol. 2016, 136, 202–213. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Cytoplasmic Intermediate Filaments in Cell Biology. Annu. Rev. Cell Dev. Biol. 2018, 34, 1–28. [Google Scholar] [CrossRef]
- Dupin, I.; Sakamoto, Y.; Etienne-Manneville, S. Cytoplasmic intermediate filaments mediate actin-driven positioning of the nucleus. J. Cell Sci. 2011, 124, 865–872. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Venkova, L.S.; Chernoivanenko, I.S.; Minin, A.A. Vimentin is involved in regulation of mitochondrial motility and membrane potential by Rac1. Biol. Open 2015, 4, 1290–1297. [Google Scholar] [CrossRef]
- Nekrasova, O.E.; Mendez, M.G.; Chernoivanenko, I.S.; Tyurin-Kuzmin, P.A.; Kuczmarski, E.R.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Vimentin intermediate filaments modulate the motility of mitochondria. Mol. Biol. Cell 2011, 22, 2282–2289. [Google Scholar] [CrossRef]
- Wang, N.; Stamenović, D. Contribution of intermediate filaments to cell stiffness, stiffening, and growth. Am. J. Physiol. Cell Physiol. 2000, 279, C188–C194. [Google Scholar] [CrossRef]
- Godsel, L.M.; Hobbs, R.P.; Green, K.J. Intermediate filament assembly: Dynamics to disease. Trends Cell Biol. 2008, 18, 28–37. [Google Scholar] [CrossRef]
- Sanghvi-Shah, R.; Weber, G.F. Intermediate Filaments at the Junction of Mechanotransduction, Migration, and Development. Front. Cell Dev. Biol. 2017, 5, 1–19. [Google Scholar] [CrossRef]
- Storm, C.; Pastore, J.J.; Mackintosh, F.C.; Lubensky, T.C.; Janmey, P.A. Nonlinear elasticity in biological gels. Nature 2005, 435, 191–194. [Google Scholar] [CrossRef]
- Guo, M.; Ehrlicher, A.J.; Mahammad, S.; Fabich, H.; Jensen, M.H.; Moore, J.R.; Fredberg, J.J.; Goldman, R.D.; Weitz, D.A. The Role of Vimentin Intermediate Filaments in Cortical and Cytoplasmic Mechanics. Biophys. J. 2013, 105, 1562–1568. [Google Scholar] [CrossRef]
- Mendez, M.G.; Restle, D.; Janmey, P.A. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys. J. 2014, 107, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C. Cytoskeleton structure and dynamic behaviour: Quick excursus from basic molecular mechanisms to some implications in cancer chemotherapy. Eur. Rev. Med. Pharmacol. Sci. 2009, 13, 13–21. [Google Scholar] [PubMed]
- Izawa, I.; Inagaki, M. Regulatory mechanisms and functions of intermediate filaments: A study using site- and phosphorylation state-specific antibodies. Cancer Sci. 2006, 97, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Hyder, C.L.; Pallari, H.-M.; Kochin, V.; Eriksson, J.E. Providing cellular signposts - Post-translational modifications of intermediate filaments. FEBS Lett. 2008, 582, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Hobbs, R.P.; Coulombe, P.A. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr. Opin. Cell Biol. 2013, 25, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Rogel, M.R.; Jaitovich, A.; Ridge, K.M. The Role of the Ubiquitin Proteasome Pathway in Keratin Intermediate Filament Protein Degradation. Proc. Am. Thorac. Soc. 2010, 7, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Coulombe, P.A.; McLean, W.H.I. Intermediate Filament Proteins and Their Associated Diseases. N. Engl. J. Med. 2004, 351, 2087–2100. [Google Scholar] [CrossRef]
- Leung, C.L.; Liem, R.K.H.; Parry, D.A.D.; Green, K.J. The plakin family. J. Cell Sci. 2001, 114, 3409–3410. [Google Scholar] [PubMed]
- Leung, C.L.; Green, K.J.; Liem, R.K.H. Plakins: A family of versatile cytolinker proteins. Trends Cell Biol. 2002, 12, 37–45. [Google Scholar] [CrossRef]
- Desai, B.V.; Harmon, R.M.; Green, K.J. Desmosomes at a glance. J. Cell Sci. 2009, 122, 4401–4407. [Google Scholar] [CrossRef]
- Wiche, G.; Osmanagic-Myers, S.; Castañón, M.J. Networking and anchoring through plectin: A key to IF functionality and mechanotransduction. Curr. Opin. Cell Biol. 2015, 32, 21–29. [Google Scholar] [CrossRef]
- Kölsch, A.; Windoffer, R.; Leube, R.E. Actin-dependent dynamics of keratin filament precursors. Cell Motil. Cytoskeleton 2009, 66, 976–985. [Google Scholar] [CrossRef]
- Robert, A.; Herrmann, H.; Davidson, M.W.; Gelfand, V.I. Microtubule-dependent transport of vimentin filament precursors is regulated by actin and by the concerted action of Rho- and p21-activated kinases. FASEB J. 2014, 28, 2879–2890. [Google Scholar] [CrossRef]
- Burgstaller, G.; Gregor, M.; Winter, L.; Wiche, G. Keeping the vimentin network under control: Cell-matrix adhesion-associated plectin 1f affects cell shape and polarity of fibroblasts. Mol. Biol. Cell 2010, 21, 3362–3375. [Google Scholar] [CrossRef]
- Lynch, C.D.; Lazar, A.M.; Iskratsch, T.; Zhang, X.; Sheetz, M.P. Endoplasmic spreading requires coalescence of vimentin intermediate filaments at force-bearing adhesions. Mol. Biol. Cell 2013, 24, 21–30. [Google Scholar] [CrossRef]
- Gregor, M.; Osmanagic-Myers, S.; Burgstaller, G.; Wolfram, M.; Fischer, I.; Walko, G.; Resch, G.P.; Jörgl, A.; Herrmann, H.; Wiche, G. Mechanosensing through focal adhesion-anchored intermediate filaments. FASEB J. 2014, 28, 715–729. [Google Scholar] [CrossRef]
- Esue, O.; Carson, A.A.; Tseng, Y.; Wirtz, D. A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J. Biol. Chem. 2006, 281, 30393–30399. [Google Scholar] [CrossRef]
- Mellad, J.A.; Warren, D.T.; Shanahan, C.M. Nesprins LINC the nucleus and cytoskeleton. Curr. Opin. Cell Biol. 2011, 23, 47–54. [Google Scholar] [CrossRef]
- Wilhelmsen, K.; Litjens, S.H.M.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; van den Bout, I.; Raymond, K.; Sonnenberg, A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol. 2005, 171, 799–810. [Google Scholar] [CrossRef]
- Lombardi, M.L.; Jaalouk, D.E.; Shanahan, C.M.; Burke, B.; Roux, K.J.; Lammerding, J. The Interaction between Nesprins and Sun Proteins at the Nuclear Envelope Is Critical for Force Transmission between the Nucleus and Cytoskeleton. J. Biol. Chem. 2011, 286, 26743–26753. [Google Scholar] [CrossRef]
- Morgan, J.T.; Pfeiffer, E.R.; Thirkill, T.L.; Kumar, P.; Peng, G.; Fridolfsson, H.N.; Douglas, G.C.; Starr, D.A.; Barakat, A.I. Nesprin-3 regulates endothelial cell morphology, perinuclear cytoskeletal architecture, and flow-induced polarization. Mol. Biol. Cell 2011, 22, 4324–4334. [Google Scholar] [CrossRef]
- Keeling, M.C.; Flores, L.R.; Dodhy, A.H.; Murray, E.R.; Gavara, N. Actomyosin and vimentin cytoskeletal networks regulate nuclear shape, mechanics and chromatin organization. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Toivola, D.M.; Tao, G.Z.; Habtezion, A.; Liao, J.; Omary, M.B. Cellular integrity plus: Organelle-related and protein-targeting functions of intermediate filaments. Trends Cell Biol. 2005, 15, 608–617. [Google Scholar] [CrossRef]
- Naetar, N.; Ferraioli, S.; Foisner, R. Lamins in the nuclear interior—Life outside the lamina. J. Cell Sci. 2017, 130, 2087–2096. [Google Scholar] [CrossRef]
- Turgay, Y.; Eibauer, M.; Goldman, A.E.; Shimi, T.; Khayat, M.; Ben-Harush, K.; Dubrovsky-Gaupp, A.; Sapra, K.T.; Goldman, R.D.; Medalia, O. The molecular architecture of lamins in somatic cells. Nature 2017, 543, 261–264. [Google Scholar] [CrossRef]
- Buxboim, A.; Swift, J.; Irianto, J.; Spinler, K.R.; Dingal, P.C.D.P.; Athirasala, A.; Kao, Y.C.; Cho, S.; Harada, T.; Shin, J.-W.; et al. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 2015, 24, 1909–1917. [Google Scholar] [CrossRef]
- Davidson, P.M.; Denais, C.; Bakshi, M.C.; Lammerding, J. Nuclear deformability constitutes a rate-limiting step during cell migration in 3-D environments. Cell. Mol. Bioeng. 2014, 7, 293–306. [Google Scholar] [CrossRef]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.D.P.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef]
- Cho, S.; Irianto, J.; Discher, D.E. Mechanosensing by the nucleus: From pathways to scaling relationships. J. Cell Biol. 2017, 1–11. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Dechat, T.; Foisner, R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015, 29, 225–237. [Google Scholar] [CrossRef]
- Stephens, A.D.; Banigan, E.J.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol. Biol. Cell 2017, 28, 1984–1996. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Webb, D.J.; Horwitz, A.R. Cell migration at a glance. J. Cell Sci. 2005, 118, 4917–4919. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Plasticity of cell migration: A multiscale tuning model. J. Cell Biol. 2010, 188, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef]
- Nguyen, L.K.; Kholodenko, B.N.; von Kriegsheim, A. Rac1 and RhoA: Networks, loops and bistability. Small GTPases 2016, 0, 1–6. [Google Scholar] [CrossRef]
- Brunner, C.A.; Ehrlicher, A.; Kohlstrunk, B.; Knebel, D.; Käs, J.A.; Goegler, M. Cell migration through small gaps. Eur. Biophys. J. 2006, 35, 713–719. [Google Scholar] [CrossRef]
- Heinemann, F.; Doschke, H.; Radmacher, M. Keratocyte lamellipodial protrusion is characterized by a concave force-velocity relation. Biophys. J. 2011, 100, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef]
- Kage, F.; Winterhoff, M.; Dimchev, V.; Mueller, J.; Thalheim, T.; Freise, A.; Brühmann, S.; Kollasser, J.; Block, J.; Dimchev, G.; et al. FMNL formins boost lamellipodial force generation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, R.; Gautreau, A. The Arp2/3 inhibitory protein arpin induces cell turning by pausing cell migration. Cytoskeleton 2015, 72, 362–371. [Google Scholar] [CrossRef]
- Choi, C.K.; Vicente-Manzanaras, M.; Zareno, J.; Whitmore, L.A.; Mogilner, A.; Horwitz, A.R. Actin and α-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 2008, 9, 1039–1050. [Google Scholar] [CrossRef]
- Ridley, A.J. Life at the Leading Edge. Cell 2011, 145, 1012–1022. [Google Scholar] [CrossRef]
- Pasapera, A.M.; Plotnikov, S.V.; Fischer, R.S.; Case, L.B.; Egelhoff, T.T.; Waterman, C.M. Rac1-Dependent Phosphorylation and Focal Adhesion Recruitment of Myosin IIA Regulates Migration and Mechanosensing. Curr. Biol. 2015, 25, 175–186. [Google Scholar] [CrossRef]
- Kuo, J.-C.; Han, X.; Hsiao, C.-T.; Yates III, J.R.; Waterman, C.M. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011, 13, 383–393. [Google Scholar] [CrossRef]
- Schiller, H.B.; Friedel, C.C.; Boulegue, C.; Fässler, R. Quantitative proteomics of the integrin adhesome show a myosin II-dependent recruitment of LIM domain proteins. EMBO Rep. 2011, 12, 259–266. [Google Scholar] [CrossRef]
- Peng, J.; Wallar, B.J.; Flanders, A.; Swiatek, P.J.; Alberts, A.S. Disruption of the Diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr. Biol. 2003, 13, 534–545. [Google Scholar] [CrossRef]
- Iskratsch, T.; Yu, C.H.; Mathur, A.; Liu, S.; Stévenin, V.; Dwyer, J.; Hone, J.; Ehler, E.; Sheetz, M. FHOD1 is needed for directed forces and adhesion maturation during cell spreading and migration. Dev. Cell 2013, 27, 545–559. [Google Scholar] [CrossRef]
- DeMali, K.A.; Barlow, C.A.; Burridge, K. Recruitment of the Arp2/3 complex to vinculin: Coupling membrane protrusion to matrix adhesion. J. Cell Biol. 2002, 159, 881–891. [Google Scholar] [CrossRef]
- Serrels, B.; Serrels, A.; Brunton, V.G.; Holt, M.; McLean, G.W.; Gray, C.H.; Jones, G.E.; Frame, M.C. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 2007, 9, 1046–1056. [Google Scholar] [CrossRef]
- Suraneni, P.; Rubinstein, B.; Unruh, J.R.; Durnin, M.; Hanein, D.; Li, R. The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol. 2012, 197, 239–251. [Google Scholar] [CrossRef]
- Wu, C.; Asokan, S.B.; Berginski, M.E.; Haynes, E.M.; Sharpless, N.E.; Griffith, J.D.; Gomez, S.M.; Bear, J.E. Arp2/3 Is Critical for Lamellipodia and Response to Extracellular Matrix Cues but Is Dispensable for Chemotaxis. Cell 2012, 148, 973–987. [Google Scholar] [CrossRef]
- Li, A.; Ma, Y.; Yu, X.; Mort, R.L.; Lindsay, C.R.; Stevenson, D.; Strathdee, D.; Insall, R.H.; Chernoff, J.; Snapper, S.B.; et al. Rac1 Drives Melanoblast Organization during Mouse Development by Orchestrating Pseudopod- Driven Motility and Cell-Cycle Progression. Dev. Cell 2011, 21, 722–734. [Google Scholar] [CrossRef]
- O’Neill, P.R.; Kalyanaraman, V.; Gautman, N. Subcellular optogenetic activation of Cdc42 controls local and distal signaling to drive immune cell migration. Mol. Biol. Cell 2016, 27, 1442–1450. [Google Scholar] [CrossRef]
- Yang, H.W.; Collins, S.R.; Meyer, T. Locally excitable Cdc42 signals steer cells during chemotaxis. Nat. Cell Biol. 2016, 18, 191–201. [Google Scholar] [CrossRef]
- Harris, K.P.; Tepass, U. Cdc42 and Vesicle Trafficking in Polarized Cells. Traffic 2010, 11, 1272–1279. [Google Scholar] [CrossRef]
- Vega, F.M.; Fruhwirth, G.; Ng, T.; Ridley, A.J. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J. Cell Biol. 2011, 193, 655–665. [Google Scholar] [CrossRef]
- Petrie, R.J.; Yamada, K.M. At the leading edge of three-dimensional cell migration. J. Cell Sci. 2012, 125, 5917–5926. [Google Scholar] [CrossRef]
- Paluch, E.K.; Aspalter, I.M.; Sixt, M. Focal Adhesion–Independent Cell Migration. Annu. Rev. Cell Dev. Biol. 2016, 32, 469–490. [Google Scholar] [CrossRef]
- Maugis, B.; Brugues, J.; Nassoy, P.; Guillen, N.; Sens, P.; Amblard, F. Dynamic instability of the intracellular pressure drives bleb-based motility. J. Cell Sci. 2010, 123, 3884–3892. [Google Scholar] [CrossRef]
- Bergert, M.; Chandradoss, S.D.; Desai, R.A.; Paluch, E. Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc. Natl. Acad. Sci. USA 2012, 109, 14434–14439. [Google Scholar] [CrossRef]
- Weiser, D.C.; Row, R.H.; Kimelman, D. Rho-regulated Myosin phosphatase establishes the level of protrusive activity required for cell movements during zebrafish gastrulation. Development 2009, 136, 2375–2384. [Google Scholar] [CrossRef] [PubMed]
- Sroka, J.; von Gunten, M.; Dunn, G.A.; Keller, H.U. Phenotype modulation in non-adherent and adherent sublines of Walker carcinosarcoma cells: The role of cell-substratum contacts and microtubules in controlling cell shape, locomotion and cytoskeletal structure. Int. J. Biochem. Cell Biol. 2002, 34, 882–899. [Google Scholar] [CrossRef]
- Blaser, H.; Eisenbeiss, S.; Neumann, M.; Reichman-Fried, M.; Thisse, B.; Thisse, C.; Raz, E. Transition from non-motile behaviour to directed migration during early PGC development in zebrafish. J. Cell Sci. 2005, 118, 4027–4038. [Google Scholar] [CrossRef]
- Malawista, S.E.; de Boisfleury Chevance, A.; Boxer, L.A. Random locomotion and chemotaxis of human blood polymorphonuclear leukocytes from a patient with Leukocyte Adhesion Deficiency-1: Normal displacement in close quarters via chimneying. Cell Motil. Cytoskeleton 2000, 46, 183–189. [Google Scholar] [CrossRef]
- Tozluoǧlu, M.; Tournier, A.L.; Jenkins, R.P.; Hooper, S.; Bates, P.A.; Sahai, E. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat. Cell Biol. 2013, 15, 751–762. [Google Scholar] [CrossRef]
- Cattin, A.L.; Burden, J.J.; Van Emmenis, L.; MacKenzie, F.E.; Hoving, J.J.A.; Garcia Calavia, N.; Guo, Y.; McLaughlin, M.; Rosenberg, L.H.; Quereda, V.; et al. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 2015, 162, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, S.; Chen, E.J.H.; Clarke, F.; Lyck, R.; Affentranger, S.; Burkhardt, J.K.; Niggli, V. Ezrin/Radixin/Moesin Proteins and Flotillins Cooperate to Promote Uropod Formation in T Cells. Front. Immunol. 2013, 4, 84. [Google Scholar] [CrossRef]
- Niggli, V.; Rossy, J. Ezrin/radixin/moesin: Versatile controllers of signaling molecules and of the cortical cytoskeleton. Int. J. Biochem. Cell Biol. 2008, 40, 344–349. [Google Scholar] [CrossRef]
- Lorentzen, A.; Bamber, J.; Sadok, A.; Elson-Schwab, I.; Marshall, C.J. An ezrin-rich, rigid uropod-like structure directs movement of amoeboid blebbing cells. J. Cell Sci. 2011, 124, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Yanase, Y.; Hide, I.; Mihara, S.; Shirai, Y.; Saito, N.; Nakata, Y.; Hide, M.; Sakai, N. A critical role of conventional protein kinase C in morphological changes of rodent mast cells. Immunol. Cell Biol. 2011, 89, 149–159. [Google Scholar] [CrossRef]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.A.; Skalak, R. Mechanics and Thermodynamics of Biomembranes: Part 1. CRC Crit. Rev. Bioeng. 1979, 3, 181–330. [Google Scholar] [PubMed]
- Pinner, S.; Sahai, E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat. Cell Biol. 2008, 10, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Trinkaus, J.P. Ingression during Early Gastrulation of Fundulus. Dev. Biol. 1996, 177, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Oakes, P.W.; Beckham, Y.; Stricker, J.; Gardel, M.L. Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 2012, 196, 363–374. [Google Scholar] [CrossRef]
- Puklin-Faucher, E.; Gao, M.; Schulten, K.; Vogel, V. How the headpiece hinge angle is opened: New insights into the dynamics of integrin activation. J. Cell Biol. 2006, 175, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.R.; Vogel, K.-P.; Steinhoff, H.-J.; Zoegler, W.H.; Patel, B.; Emsley, J.; Critchley, D.R.; Roberts, G.C.K.; Barsukov, I.L. Structural and Dynamic Characterization of a Vinculin Binding Site in the Talin Rod. Biochemistry 2006. [Google Scholar] [CrossRef]
- del Rio, A.; Perez-Jimenez, R.; Liu, R.; Roca-Cusachs, P.; Fernandez, J.M.; Sheetz, M.P. Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science 2009, 323, 638–641. [Google Scholar] [CrossRef]
- Johnson, C.P.; Tang, H.-Y.; Carag, C.; Speicher, D.W.; Discher, D.E. Forced unfolding of proteins within cells. Science 2007, 317, 663–666. [Google Scholar] [CrossRef]
- Vallenius, T. Actin stress fibre subtypes in mesenchymal-migrating cells. Open Biol. 2013, 3, 130001. [Google Scholar] [CrossRef]
- Valerius, N.H.; Stendahl, O.; Hartwig, J.H.; Stossel, T.P. Distribution of actin-binding protein and myosin in polymorphonuclear leukocytes during locomotion and phagocytosis. Cell 1981, 24, 195–202. [Google Scholar] [CrossRef]
- Rubino, S.; Fighetti, M.; Unger, E.; Cappuccinelli, P. Location of actin, myosin, and microtubular structures during directed locomotion of Dictyostelium amebae. J. Cell Biol. 1984, 98, 382–390. [Google Scholar] [CrossRef]
- Cheng, S.; Castillo, V.; Welty, M.; Eliaz, I.; Sliva, D. Honokiol inhibits migration of renal cell carcinoma through activation of RhoA/ROCK/MLC signaling pathway. Int. J. Oncol. 2016, 49, 1525–1530. [Google Scholar] [CrossRef]
- Castella, L.F.; Buscemi, L.; Godbout, C.; Meister, J.-J.; Hinz, B. A new lock-step mechanism of matrix remodelling based on subcellular contractile events. J. Cell Sci. 2010, 123, 1751–1760. [Google Scholar] [CrossRef]
- Kirfel, G.; Rigort, A.; Borm, B.; Herzog, V. Cell migration: Mechanisms of rear detachment and the formation of migration tracks. Eur. J. Cell Biol. 2004, 83, 717–724. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Koach, M.A.; Whitmore, L.; Lamers, M.L.; Horwitz, A.F. Segregation and activation of myosin IIB creates a rear in migrating cells. J. Cell Biol. 2008, 183, 543–554. [Google Scholar] [CrossRef]
- Cuddihy, A.R.; Bristow, R.G. The p53 protein family and radiation sensitivity: Yes or no? Cancer Metastasis Rev. 2004, 23, 237–257. [Google Scholar] [CrossRef]
- Totsukawa, G.; Wu, Y.; Sasaki, Y.; Hartshorne, D.J.; Yamakita, Y.; Yamashiro, S.; Matsumura, F. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 2004, 164, 427–439. [Google Scholar] [CrossRef]
- Aifuwa, I.; Giri, A.; Longe, N.; Lee, S.H.; An, S.S.; Wirtz, D. Senescent stromal cells induce cancer cell migration via inhibition of RhoA/ROCK/myosin-based cell contractility. Oncotarget 2015, 6, 30516–30531. [Google Scholar] [CrossRef]
- Schmidt, C.E.; Horwitz, A.F.; Lauffenburger, D.A.; Sheetz, M.P. Integrin-cytoskeletal interactions in migrating fibroblasts are dynamic, asymmetric, and regulated. J. Cell Biol. 1993, 123, 977–991. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; MacKintosh, F.C.; Weitz, D.A. Force fluctuations and polymerization dynamics of intracellular microtubules. Proc. Natl. Acad. Sci. USA 2007, 104, 16128–16133. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, P. Regional regulation of microtubule dynamics in polarized, motile cells. Cell Motil. Cytoskeleton 1999, 42, 48–59. [Google Scholar] [CrossRef]
- Schober, J.M.; Cain, J.M.; Komarova, Y.A.; Borisy, G.G. Migration and actin protrusion in melanoma cells are regulated by EB1 protein. Cancer Lett. 2009, 284, 30–36. [Google Scholar] [CrossRef]
- Al-Bassam, J.; Kim, H.; Brouhard, G.; van Oijen, A.; Harrison, S.C.; Chang, F. CLASP Promotes Microtubule Rescue by Recruiting Tubulin Dimers to the Microtubule. Dev. Cell 2010, 19, 245–258. [Google Scholar] [CrossRef]
- Steinmetz, M.O.; Jahnke, W.; Towbin, H.; Garcia-Echeverria, C.; Voshol, H.; Muller, D.; van Oostrum, J. Phosphorylation disrupts the central helix in Op18/stathmin and suppresses binding to tubulin. EMBO Rep. 2001, 2, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; McEwen, D.P.; Martens, J.R.; Meyhofer, E.; Verhey, K.J. Single Molecule Imaging Reveals Differences in Microtubule Track Selection Between Kinesin Motors. PLoS Biol. 2009, 7, e1000216. [Google Scholar] [CrossRef] [PubMed]
- Fygenson, D.K.; Marko, J.F.; Libchaber, A. Mechanics of microtubule-based membrane extension. Phys. Rev. Lett. 1997, 79, 4497–4500. [Google Scholar] [CrossRef]
- Mogilner, A.; Oster, G. Polymer motors: Pushing out the front and pulling up the back. Curr. Biol. 2003, 13, 721–733. [Google Scholar] [CrossRef]
- Yu, W.; Centonze, V.E.; Ahmad, F.J.; Baas, P.W. Microtubule nucleation and release from the neuronal centrosome. J. Cell Biol. 1993, 122, 349–359. [Google Scholar] [CrossRef]
- Roll-Mecak, A.; McNally, F.J. Microtubule-severing enzymes. Curr. Opin. Cell Biol. 2010, 22, 96–103. [Google Scholar] [CrossRef]
- Marcette, J.D.; Chen, J.J.; Nonet, M.L. The Caenorhabditis elegans microtubule minus-end binding homolog PTRN-1 stabilizes synapses and neurites. Elife 2014, 3, e01637. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.E.; Spilker, K.A.; Cueva, J.G.; Perrino, J.; Goodman, M.B.; Shen, K. PTRN-1, a microtubule minus end-binding CAMSAP homolog, promotes microtubule function in Caenorhabditis elegans neurons. Elife 2014, 3, e01498. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.W.; Schatzle, P.; Tortosa, E.; Pages, S.; Holtmaat, A.; Kapitein, L.C.; Hoogenraad, C.C. Dendrites In Vitro and In Vivo Contain Microtubules of Opposite Polarity and Axon Formation Correlates with Uniform Plus-End-Out Microtubule Orientation. J. Neurosci. 2016, 36, 1071–1085. [Google Scholar] [CrossRef]
- Baas, P.W.; Deitch, J.S.; Black, M.M.; Banker, G.A. Polarity orientation of microtubules in hippocampal neurons: Uniformity in the axon and nonuniformity in the dendrite. Proc. Natl. Acad. Sci. USA 1988, 85, 8335–8339. [Google Scholar] [CrossRef]
- Bradke, F.; Dotti, C.G. The role of local actin instability in axon formation. Science 1999, 283, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, C.; Faix, J.; Himmel, M.; Bentzien, F.; Linder, S. KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix degradation in primary macrophages. Blood 2010, 116, 1559–1569. [Google Scholar] [CrossRef]
- Steffen, A.; Le Dez, G.; Poincloux, R.; Recchi, C.; Nassoy, P.; Rottner, K.; Galli, T.; Chavrier, P. MT1-MMP-Dependent Invasion Is Regulated by TI-VAMP/VAMP7. Curr. Biol. 2008, 18, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Bouchet, B.P.; Gough, R.E.; Ammon, Y.C.; van de Willige, D.; Post, H.; Jacquemet, G.; Maarten Altelaar, A.F.; Heck, A.J.R.; Goult, B.T.; Akhmanova, A. Talin-KANK1 interaction controls the recruitment of cortical microtubule stabilizing complexes to focal adhesions. Elife 2016, 5, 1–23. [Google Scholar] [CrossRef]
- Stehbens, S.; Wittmann, T. Targeting and transport: How microtubules control focal adhesion dynamics. J. Cell Biol. 2012, 198, 481–489. [Google Scholar] [CrossRef]
- Rooney, C.; White, G.; Nazgiewicz, A.; Woodcock, S.A.; Anderson, K.I.; Ballestrem, C.; Malliri, A. The Rac activator STEF (Tiam2) regulates cell migration by microtubule-mediated focal adhesion disassembly. EMBO Rep. 2010, 11, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, O.C.; Schaefer, A.W.; Mandato, C.A.; Forscher, P.; Bement, W.M.; Waterman-Storer, C.M. Conserved microtubule–actin interactions in cell movement and morphogenesis. Nat. Cell Biol. 2003, 5, 599–609. [Google Scholar] [CrossRef]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002, 4, 294–301. [Google Scholar] [CrossRef]
- Waterman-Storer, C.M.; Worthylake, R.A.; Liu, B.P.; Burridge, K.; Salmon, E.D. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat. Cell Biol. 1999, 1, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Montenegro-Venegas, C.; Tortosa, E.; Rosso, S.; Peretti, D.; Bollati, F.; Bisbal, M.; Jausoro, I.; Avila, J.; Cáceres, A.; Gonzalez-Billault, C. MAP1B regulates axonal development by modulating Rho-GTPase Rac1 activity. Mol. Biol. Cell 2010, 21, 3518–3528. [Google Scholar] [CrossRef]
- Van Haren, J.; Boudeau, J.; Schmidt, S.; Basu, S.; Liu, Z.; Lammers, D.; Demmers, J.; Benhari, J.; Grosveld, F.; Debant, A.; et al. Dynamic microtubules catalyze formation of navigator-TRIO complexes to regulate neurite extension. Curr. Biol. 2014, 24, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Nalbant, P.; Chang, Y.-C.; Birkenfeld, J.; Chang, Z.-F.; Bokoch, G.M. Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge. Mol. Biol. Cell 2009, 20, 4070–4082. [Google Scholar] [CrossRef]
- Callow, M.G.; Zozulya, S.; Gishizky, M.L.; Jallal, B.; Smeal, T. PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 2005, 118, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Li, R.; Zheng, Y.; Busch, H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 1998, 273, 34954–34960. [Google Scholar] [CrossRef]
- Zenke, F.T.; Krendel, M.; DerMardirossian, C.; King, C.C.; Bohl, B.P.; Bokoch, G.M. p21-activated Kinase 1 Phosphorylates and Regulates 14-3-3 Binding to GEF-H1, a Microtubule-localized Rho Exchange Factor. J. Biol. Chem. 2004, 279, 18392–18400. [Google Scholar] [CrossRef]
- Kaverina, I.; Rottner, K.; Small, J.V. Targeting, capture, and stabilization of microtubules at early focal adhesions. J. Cell Biol. 1998, 142, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Noss, E.H.; Hsu, V.W.; Brenner, M.B. Integrins traffic rapidly via circular dorsal ruffles and macropinocytosis during stimulated cell migration. J. Cell Biol. 2011, 193, 61–70. [Google Scholar] [CrossRef] [PubMed]
- López, M.P.; Huber, F.; Grigoriev, I.; Steinmetz, M.O.; Akhmanova, A.; Koenderink, G.H.; Dogterom, M. Actin–microtubule coordination at growing microtubule ends. Nat. Commun. 2014, 5, 4778. [Google Scholar] [CrossRef]
- Wickström, S.A.; Fässler, R. Regulation of membrane traffic by integrin signaling. Trends Cell Biol. 2011, 21, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S. APC in cell migration. Adv. Exp. Med. Biol. 2009, 656, 30–40. [Google Scholar] [PubMed]
- Matsumoto, S.; Fumoto, K.; Okamoto, T.; Kaibuchi, K.; Kikuchi, A. Binding of APC and dishevelled mediates Wnt5a-regulated focal adhesion dynamics in migrating cells. EMBO J. 2010, 29, 1192–1204. [Google Scholar] [CrossRef]
- Juanes, M.A.; Bouguenina, H.; Eskin, J.A.; Jaiswal, R.; Badache, A.; Goode, B.L. Adenomatous polyposis coli nucleates actin assembly to drive cell migration and microtubuleinduced focal adhesion turnover. J. Cell Biol. 2017, 216, 2859–2875. [Google Scholar] [CrossRef]
- Breitsprecher, D.; Jaiswal, R.; Bombardier, J.P.; Gould, C.J.; Gelles, J.; Goode, B.L. Rocket Launcher Mechanism of Collaborative Actin Assembly Defined by Single-Molecule Imaging. Science 2012, 336, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Brandt, D.T.; Grosse, R. Get to grips: Steering local actin dynamics with IQGAPs. EMBO Rep. 2007, 8, 1019–1023. [Google Scholar] [CrossRef]
- Krylyshkina, O.; Anderson, K.I.; Kaverina, I.; Upmann, I.; Manstein, D.J.; Small, J.V.; Toomre, D.K. Nanometer targeting of microtubules to focal adhesions. J. Cell Biol. 2003, 161, 853–859. [Google Scholar] [CrossRef]
- Kaverina, I.; Krylyshkina, O.; Small, J.V. Microtubule targeting of substrate contacts promotes their relaxation and dissociation. J. Cell Biol. 1999, 146, 1033–1043. [Google Scholar] [CrossRef]
- Rid, R.; Schiefermeier, N.; Grigoriev, I.; Small, J.V.; Kaverina, I. The last but not the least: The origin and significance of trailing adhesions in fibroblastic cells. Cell Motil. Cytoskeleton 2005, 61, 161–171. [Google Scholar] [CrossRef]
- Applewhite, D.A.; Grode, K.D.; Keller, D.; Zadeh, A.D.; Zadeh, A.; Slep, K.C.; Rogers, S.L. The spectraplakin Short stop is an actin-microtubule cross-linker that contributes to organization of the microtubule network. Mol. Biol. Cell 2010, 21, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Shen, Q.-T.; Oristian, D.S.; Lu, C.P.; Zheng, Q.; Wang, H.-W.; Fuchs, E. Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3β. Cell 2011, 144, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Zaoui, K.; Benseddik, K.; Daou, P.; Salaun, D.; Badache, A. ErbB2 receptor controls microtubule capture by recruiting ACF7 to the plasma membrane of migrating cells. Proc. Natl. Acad. Sci. USA 2010, 107, 18517–18522. [Google Scholar] [CrossRef] [PubMed]
- Higashida, C.; Miyoshi, T.; Fujita, A.; Oceguera-Yanez, F.; Monypenny, J.; Andou, Y.; Narumiya, S.; Watanabe, N. Actin Polymerization-Driven Molecular Movement of mDia1 in Living Cells. Science 2004, 303, 2007–2010. [Google Scholar] [CrossRef] [PubMed]
- Wickström, S.A.; Lange, A.; Hess, M.W.; Polleux, J.; Spatz, J.P.; Krüger, M.; Pfaller, K.; Lambacher, A.; Bloch, W.; Mann, M.; et al. Integrin-Linked Kinase Controls Microtubule Dynamics Required for Plasma Membrane Targeting of Caveolae. Dev. Cell 2010, 19, 574–588. [Google Scholar] [CrossRef] [PubMed]
- Villari, G.; Jayo, A.; Zanet, J.; Fitch, B.; Serrels, B.; Frame, M.; Stramer, B.; Goult, B.; Parsons, M. A direct interaction between fascin and microtubules contributes to adhesion dynamics and cell migration. J. Cell Sci. 2015, 128, 4601–4604. [Google Scholar] [CrossRef]
- Ezratty, E.J.; Partridge, M.A.; Gundersen, G.G. Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat. Cell Biol. 2005, 7, 581–590. [Google Scholar] [CrossRef]
- Ezratty, E.J.; Bertaux, C.; Marcantonio, E.E.; Gundersen, G.G. Clathrin mediates integrin endocytosis for focal adhesion disassembly in migrating cells. J. Cell Biol. 2009, 187, 733–747. [Google Scholar] [CrossRef]
- Stehbens, S.J.; Paszek, M.; Pemble, H.; Ettinger, A.; Gierke, S.; Wittmann, T. CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat. Cell Biol. 2014, 16, 558–570. [Google Scholar] [CrossRef]
- Kenific, C.M.; Stehbens, S.J.; Goldsmith, J.; Leidal, A.M.; Faure, N.; Ye, J.; Wittmann, T.; Debnath, J. NBR 1 enables autophagy-dependent focal adhesion turnover. J. Cell Biol. 2016, 212, 577–590. [Google Scholar] [CrossRef]
- Helfand, B.T.; Mendez, M.G.; Murthy, S.N.P.; Shumaker, D.K.; Grin, B.; Mahammad, S.; Aebi, U.; Wedig, T.; Wu, Y.I.; Hahn, K.M.; et al. Vimentin organization modulates the formation of lamellipodia. Mol. Biol. Cell 2011, 22, 1274–1289. [Google Scholar] [CrossRef]
- Menko, A.S.; Bleaken, B.M.; Libowitz, A.A.; Zhang, L.; Stepp, M.A.; Walker, J.L. A central role for vimentin in regulating repair function during healing of the lens epithelium. Mol. Biol. Cell 2014, 25, 776–790. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Boëda, B.; Etienne-Manneville, S. APC binds intermediate filaments and is required for their reorganization during cell migration. J. Cell Biol. 2013, 200, 249–258. [Google Scholar] [CrossRef]
- Huber, F.; Boire, A.; López, M.P.; Koenderink, G.H. Cytoskeletal crosstalk: When three different personalities team up. Curr. Opin. Cell Biol. 2015, 32, 39–47. [Google Scholar] [CrossRef]
- Shabbir, S.H.; Cleland, M.M.; Goldman, R.D.; Mrksich, M. Geometric control of vimentin intermediate filaments. Biomaterials 2014, 35, 1359–1366. [Google Scholar] [CrossRef]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.-I.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef]
- Rogel, M.R.; Soni, P.N.; Troken, J.R.; Sitikov, A.; Trejo, H.E.; Ridge, K.M. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 2011, 25, 3873–3883. [Google Scholar] [CrossRef]
- Chung, B.M.; Rotty, J.D.; Coulombe, P.A. Networking galore: Intermediate filaments and cell migration. Curr. Opin. Cell Biol. 2013, 25, 600–612. [Google Scholar] [CrossRef]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lepekhin, E.A.; Eliasson, C.; Berthold, C.H.; Berezin, V.; Bock, E.; Pekny, M. Intermediate filaments regulate astrocyte motility. J. Neurochem. 2001, 79, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Rathje, L.-S.Z.; Nordgren, N.; Pettersson, T.; Ronnlund, D.; Widengren, J.; Aspenstrom, P.; Gad, A.K.B. Oncogenes induce a vimentin filament collapse mediated by HDAC6 that is linked to cell stiffness. Proc. Natl. Acad. Sci. USA 2014, 111, 1515–1520. [Google Scholar] [CrossRef]
- Gan, Z.; Ding, L.; Burckhardt, C.J.; Lowery, J.; Zaritsky, A.; Sitterley, K.; Mota, A.; Costigliola, N.; Starker, C.G.; Voytas, D.F.; et al. Vimentin Intermediate Filaments Template Microtubule Networks to Enhance Persistence in Cell Polarity and Directed Migration. Cell Syst. 2016, 3, 252–263. [Google Scholar] [CrossRef]
- Duan, G.; Walther, D. The Roles of Post-translational Modifications in the Context of Protein Interaction Networks. PLoS Comput. Biol. 2015, 11, 1–23. [Google Scholar] [CrossRef]
- Nieuwenhuizen, R.P.J.; Nahidiazar, L.; Manders, E.M.M.; Jalink, K.; Stallinga, S.; Rieger, B. Co-orientation: Quantifying simultaneous co-localization and orientational alignment of filaments in light microscopy. PLoS ONE 2015, 10, 1–21. [Google Scholar] [CrossRef]
- Leduc, C.; Manneville, S.E. Regulation of microtubule-associated intermediate filament network polarization. J. Cell Biol 2017, 216, 1689–1703. [Google Scholar] [CrossRef] [PubMed]
- Havel, L.; Kline, E.; Salgueiro, A.; Marcus, A. Vimentin regulates lung cancer cell adhesion through a VAV2–Rac1 pathway to control focal adhesion kinase activity. Oncogene 2014, 34, 1–12. [Google Scholar] [CrossRef]
- Jiu, Y.; Peränen, J.; Schaible, N.; Cheng, F.; Eriksson, J.E.; Krishnan, R.; Lappalainen, P. Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and RhoA. J. Cell Sci. 2017, 130, 892–902. [Google Scholar] [CrossRef]
- De Pascalis, C.; Perez-Gonzalez, C.; Seetharaman, S.; Boeda, B.; Vianay, B.; Burute, M.; Leduc, C.; Borghi, N.; Trepat, X.; Etienne-Manneville, S. Intermediate filaments control collective migration by restricting traction forces and sustaining cell-cell contacts. bioRxiv 2018. [Google Scholar] [CrossRef]
- Kim, J.; Yang, C.; Kim, E.J.; Jang, J.; Kim, S.-J.; Kang, S.M.; Kim, M.G.; Jung, H.; Park, D.; Kim, C. Vimentin filaments regulate integrin–ligand interactions by binding to the cytoplasmic tail of integrin β3. J. Cell Sci. 2016, 129, 2030–2042. [Google Scholar] [CrossRef]
- Costigliola, N.; Ding, L.; Burckhardt, C.J.; Han, S.J.; Gutierrez, E.; Mota, A.; Groisman, A.; Mitchison, T.J.; Danuser, G. Vimentin fibers orient traction stress. Proc. Natl. Acad. Sci. USA 2017, 114, 5195–5200. [Google Scholar] [CrossRef]
- Akhmanova, A.; Stehbens, S.J.; Yap, A.S. Touch, Grasp, Deliver and Control: Functional Cross-Talk Between Microtubules and Cell Adhesions. Traffic 2009, 10, 268–274. [Google Scholar] [CrossRef]
- Janosch, P.; Kieser, A.; Eulitz, M.; Lovric, J.; Sauer, G.; Reichert, M.; Gounari, F.; Büscher, D.; Baccarini, M.; Mischak, H.; et al. The Raf-1 kinase associates with vimentin kinases and regulates the structure of vimentin filaments. FASEB J. 2000, 14, 2008–2021. [Google Scholar] [CrossRef]
- Ehrenreiter, K.; Piazzolla, D.; Velamoor, V.; Sobczak, I.; Small, J.V.; Takeda, J.; Leung, T.; Baccarini, M. Raf-1 regulates Rho signaling and cell migration. J. Cell Biol. 2005, 168, 955–964. [Google Scholar] [CrossRef]
- Amano, M.; Fukata, Y.; Kaibuchi, K. Regulation and Functions of Rho-Associated Kinase. Exp. Cell Res. 2000, 261, 44–51. [Google Scholar] [CrossRef]
- Sin, W.C.; Chen, X.Q.; Leung, T.; Lim, L. RhoA-binding kinase alpha translocation is facilitated by the collapse of the vimentin intermediate filament network. Mol. Cell. Biol. 1998, 18, 6325–6339. [Google Scholar] [CrossRef]
- Ren, X.D.; Kiosses, W.B.; Sieg, D.J.; Otey, C.A.; Schlaepfer, D.D.; Schwartz, M.A. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 2000, 113, 3673–3678. [Google Scholar]
- Ren, X.D.; Kiosses, W.B.; Schwartz, M.A. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999, 18, 578–585. [Google Scholar] [CrossRef]
- Jiu, Y.; Lehtimäki, J.; Tojkander, S.; Cheng, F.; Jäälinoja, H.; Liu, X.; Varjosalo, M.; Eriksson, J.E.; Lappalainen, P. Bidirectional Interplay between Vimentin Intermediate Filaments and Contractile Actin Stress Fibers. Cell Rep. 2015, 11, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Tanabe, K.; Manser, E.; Lim, L.; Yasui, Y.; Inagaki, M. Phosphorylation and reorganization of vimentin by p21-activated kinase (PAK). Genes Cells 2002, 7, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Petrie, R.J.; Koo, H.; Yamada, K.M. Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 2014, 345, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Huiatt, T.W.; Paulin, D.; Li, Z.; Robson, R.M. Synemin interacts with the LIM domain protein zyxin and is essential for cell adhesion and migration. Exp. Cell Res. 2010, 316, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Jing, R.; Pitre, A.; Williams, B.J.; Skalli, O. Intermediate filament protein synemin contributes to the migratory properties of astrocytoma cells by influencing the dynamics of the actin cytoskeleton. FASEB J. 2008, 22, 3196–3206. [Google Scholar] [CrossRef] [PubMed]
- Uyama, N.; Zhao, L.; Van Rossen, E.; Hirako, Y.; Reynaert, H.; Adams, D.H.; Xue, Z.; Li, Z.; Robson, R.; Pekny, M.; et al. Hepatic stellate cells express synemin, a protein bridging intermediate filaments to focal adhesions. Gut 2006, 55, 1276–1289. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Critchley, D.R.; Paulin, D.; Li, Z.; Robson, R.M. Identification of a repeated domain within mammalian α-synemin that interacts directly with talin. Exp. Cell Res. 2008, 314, 1839–1849. [Google Scholar] [CrossRef]
- Sun, N.; Critchley, D.R.; Paulin, D.; Li, Z.; Robson, R.M. Human alpha-synemin interacts directly with vinculin and metavinculin. Biochem. J. 2008, 409, 657–667. [Google Scholar] [CrossRef]
- Hijikata, T.; Nakamura, A.; Isokawa, K.; Imamura, M.; Yuasa, K.; Ishikawa, R.; Kohama, K.; Takeda, S.; Yorifuji, H. Plectin 1 links intermediate filaments to costameric sarcolemma through -synemin, -dystrobrevin and actin. J. Cell Sci. 2008, 121, 2062–2074. [Google Scholar] [CrossRef]
- Hyder, C.L.; Lazaro, G.; Pylvänäinen, J.W.; Roberts, M.W.G.; Qvarnström, S.M.; Eriksson, J.E. Nestin regulates prostate cancer cell invasion by influencing the localisation and functions of FAK and integrins. J. Cell Sci. 2014, 127, 2161–2173. [Google Scholar] [CrossRef]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, 2011–2030. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: Globocan 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Legler, J.M.; Ries, L.A.; Smith, M.A.; Warren, J.L.; Heineman, E.F.; Kaplan, R.S.; Linet, M.S. Cancer surveillance series [corrected]: Brain and other central nervous system cancers: Recent trends in incidence and mortality. J. Natl. Cancer Inst. 1999, 91, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.G.; McCarthy, B.J.; Freels, S.; Kupelian, V.; Bondy, M.L. The conditional probability of survival of patients with primary malignant brain tumors: Surveillance, epidemiology, and end results (SEER) data. Cancer 1999, 85, 485–491. [Google Scholar] [CrossRef]
- Scherer, H.J. The forms of growth in gliomas and their practical significance. Brain 1940, 63, 1–35. [Google Scholar] [CrossRef]
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Radiographic patterns of relapse in glioblastoma. J. Neurooncol. 2011, 101, 319–323. [Google Scholar] [CrossRef]
- Iwadate, Y.; Sakaida, T.; Saegusa, T.; Hiwasa, T.; Takiguchi, M.; Fujimoto, S.; Yamaura, A. Proteome-based identification of molecular markers predicting chemosensitivity to each category of anticancer agents in human gliomas. Int. J. Oncol. 2005, 26, 993–998. [Google Scholar] [CrossRef]
- Hirata, E.; Yukinaga, H.; Kamioka, Y.; Arakawa, Y.; Miyamoto, S.; Okada, T.; Sahai, E.; Matsuda, M. In vivo fluorescence resonance energy transfer imaging reveals differential activation of Rho-family GTPases in glioblastoma cell invasion. J. Cell Sci. 2012, 125, 858–868. [Google Scholar] [CrossRef]
- Salhia, B.; Tran, N.L.; Chan, A.; Wolf, A.; Nakada, M.; Rutka, F.; Ennis, M.; McDonough, W.S.; Berens, M.E.; Symons, M.; et al. The Guanine Nucleotide Exchange Factors Trio, Ect2, and Vav3 Mediate the Invasive Behavior of Glioblastoma. Am. J. Pathol. 2008, 173, 1828–1838. [Google Scholar] [CrossRef]
- Malchinkhuu, E.; Sato, K.; Maehama, T.; Mogi, C.; Tomura, H.; Ishiuchi, S.; Yoshimoto, Y.; Kurose, H.; Okajima, F. S1P2 receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem. Biophys. Res. Commun. 2008, 366, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.L.; McDonough, W.S.; Savitch, B.A.; Fortin, S.P.; Winkles, J.A.; Symons, M.; Nakada, M.; Cunliffe, H.E.; Hostetter, G.; Hoelzinger, D.B.; et al. Increased Fibroblast Growth Factor-Inducible 14 Expression Levels Promote Glioma Cell Invasion via Rac1 and Nuclear Factor-κB and Correlate with Poor Patient Outcome. Cancer Res. 2006, 66, 9535–9542. [Google Scholar] [CrossRef]
- Zagzag, D.; Friedlander, D.R.; Margolis, B.; Grumet, M.; Semenza, G.L.; Zhong, H.; Simons, J.W.; Holash, J.; Wiegand, S.J.; Yancopoulos, G.D. Molecular Events Implicated in Brain Tumor Angiogenesis and Invasion. Pediatr. Neurosurg. 2000, 33, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.D.; Hanna, S.; Saykali, B.A.; El-Sitt, S.; Nasrallah, A.; Marston, D.; El-Sabban, M.; Hahn, K.M.; Symons, M.; El-Sibai, M. The regulation of RhoA at focal adhesions by StarD13 is important for astrocytoma cell motility. Exp. Cell Res. 2014, 321, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Seasholtz, T.M.; Radeff-Huang, J.; Sagi, S.A.; Matteo, R.; Weems, J.M.; Cohen, A.S.; Feramisco, J.R.; Brown, J.H. Rho-mediated cytoskeletal rearrangement in response to LPA is functionally antagonized by Rac1 and PIP2. J. Neurochem. 2004, 91, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Salhia, B.; Rutten, F.; Nakada, M.; Beaudry, C.; Berens, M.; Kwan, A.; Rutka, J.T. Inhibition of Rho-kinase affects astrocytoma morphology, motility, and invasion through activation of Rac1. Cancer Res. 2005, 65, 8792–8800. [Google Scholar] [CrossRef]
- Chan, A.Y.; Coniglio, S.J.; Chuang, Y.; Michaelson, D.; Knaus, U.G.; Philips, M.R.; Symons, M. Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene 2005, 24, 7821–7829. [Google Scholar] [CrossRef]
- Chuang, Y.; Tran, N.L.; Rusk, N.; Nakada, M.; Berens, M.E.; Symons, M. Role of Synaptojanin 2 in Glioma Cell Migration and Invasion. Cancer Res. 2004, 64, 8271–8275. [Google Scholar] [CrossRef]
- Yin, M.; Lu, Q.; Liu, X.; Wang, T.; Liu, Y.; Chen, L. Silencing Drp1 inhibits glioma cells proliferation and invasion by RHOA/ROCK1 pathway. Biochem. Biophys. Res. Commun. 2016, 478, 663–668. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, K.; Pillai, P.P. PDGFR inhibition mediated intracellular signalling in C6 glioma growth and migration: Role of ERK and ROCK pathway. Cytotechnology 2018, 70, 465–477. [Google Scholar] [CrossRef]
- Kim, D.-H.; Wirtz, D. Predicting how cells spread and migrate. Cell Adh. Migr. 2013, 7, 293–296. [Google Scholar] [CrossRef]
- Fortin, S.P.; Ennis, M.J.; Schumacher, C.A.; Zylstra-Diegel, C.R.; Williams, B.O.; Ross, J.T.D.; Winkles, J.A.; Loftus, J.C.; Symons, M.H.; Tran, N.L. Cdc42 and the Guanine Nucleotide Exchange Factors Ect2 and Trio Mediate Fn14-Induced Migration and Invasion of Glioblastoma Cells. Mol. Cancer Res. 2012, 10, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Yamana, N.; Arakawa, Y.; Nishino, T.; Kurokawa, K.; Tanji, M.; Itoh, R.E.; Monypenny, J.; Ishizaki, T.; Bito, H.; Nozaki, K.; et al. The Rho-mDia1 Pathway Regulates Cell Polarity and Focal Adhesion Turnover in Migrating Cells through Mobilizing Apc and c-Src. Mol. Cell. Biol. 2006, 26, 6844–6858. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Sun, X.; You, Y.; Liu, N.; Fu, Z. Expression of focal adhesion kinase and phosphorylated focal adhesion kinase in human gliomas is associated with unfavorable overall survival. Transl. Res. 2010, 156, 45–52. [Google Scholar] [CrossRef]
- Jones, G.; Machado, J.; Merlo, A. Loss of focal adhesion kinase (FAK) inhibits epidermal growth factor receptor-dependent migration and induces aggregation of nh(2)-terminal FAK in the nuclei of apoptotic glioblastoma cells. Cancer Res. 2001, 61, 4978–4981. [Google Scholar] [PubMed]
- Obara, S.; Nakata, M.; Takeshima, H.; Kuratsu, J.; Maruyama, I.; Kitajima, I. Inhibition of migration of human glioblastoma cells by cerivastatin in association with focal adhesion kinase (FAK). Cancer Lett. 2002, 185, 153–161. [Google Scholar] [CrossRef]
- Beadle, C.; Assanah, M.; Monzo, P.; Vallee, R.; Rosenfeld, S.; Canoll, P. The Role of Myosin II in Glioma Invasion of the Brain Christopher. Mol. Biol. Cell 2008, 19, 3357–3368. [Google Scholar] [CrossRef] [PubMed]
- Ivkovic, S.; Beadle, C.; Noticewala, S.; Massey, S.C.; Swanson, K.R.; Toro, L.N.; Bresnick, A.R.; Canoll, P.; Rosenfeld, S.S. Direct inhibition of myosin II effectively blocks glioma invasion in the presence of multiple motogens. Mol. Biol. Cell 2012, 23, 533–542. [Google Scholar] [CrossRef]
- Lee, W.; Lim, S.; Kim, Y. The role of myosin II in glioma invasion: A mathematical model. PLoS ONE 2017, 12, 1939–4586. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, X.; Chen, C.; Liu, B.; Ren, B.; Wang, L.; Zhao, K.; Yu, S.; Ming, H. Expression of the Arp2/3 complex in human gliomas and its role in the migration and invasion of glioma cells. Oncol. Rep. 2013, 30, 2127–2136. [Google Scholar] [CrossRef]
- Monzo, P.; Chong, Y.K.; Guetta-Terrier, C.; Krishnasamy, A.; Sathe, S.R.; Yim, E.K.F.; Ng, W.H.; Ang, B.T.; Tang, C.; Ladoux, B.; et al. Mechanical confinement triggers glioma linear migration dependent on formin FHOD3. Mol. Biol. Cell 2016, 27, 1246–1261. [Google Scholar] [CrossRef]
- Zhang, C.; Hai, L.; Zhu, M.; Yu, S.P.; Li, T.; Lin, Y.; Liu, B.; Zhou, X.C.; Chen, L.; Zhao, P.F.; et al. Actin cytoskeleton regulator Arp2/3 complex is required for DLL1 activating Notch1 signaling to maintain the stem cell phenotype of glioma initiating cells. Oncotarget 2017, 8, 33353–33364. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, K.; Ren, B.; Zhu, M.; Zhang, C.; Zhao, P.; Zhou, H.; Chen, L.; Yu, S.; Yang, X. Expression of cortactin in human gliomas and its effect on migration and invasion of glioma cells. Oncol. Rep. 2015, 34, 1815–1824. [Google Scholar] [CrossRef]
- Hwang, J.H.; Smith, C.A.; Salhia, B.; Rutka, J.T. The Role of Fascin in the Migration and Invasiveness of Malignant Glioma Cells. Neoplasia 2008, 10, 149–159. [Google Scholar] [CrossRef]
- Hoa, N.T.; Ge, L.; Erickson, K.L.; Kruse, C.A.; Cornforth, A.N.; Kuznetsov, Y.; McPherson, A.; Martini, F.; Jadus, M.R. Fascin-1 knock-down of human glioma cells reduces their microvilli/filopodia while improving their susceptibility to lymphocyte-mediated cytotoxicity. Am. J. Transl. Res. 2015, 7, 271–284. [Google Scholar]
- Eke, I.; Storch, K.; Kästner, I.; Vehlow, A.; Faethe, C.; Mueller-Klieser, W.; Taucher-Scholz, G.; Temme, A.; Schackert, G.; Cordes, N. Three-dimensional invasion of human glioblastoma cells remains unchanged by X-ray and carbon ion irradiation in vitro. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84. [Google Scholar] [CrossRef]
- Frankel, P.; Pellet-Many, C.; Lehtolainen, P.; D’Abaco, G.M.; Tickner, M.L.; Cheng, L.; Zachary, I.C. Chondroitin sulphate-modified neuropilin 1 is expressed in human tumour cells and modulates 3D invasion in the U87MG human glioblastoma cell line through a p130Cas-mediated pathway. EMBO Rep. 2008, 9, 983–989. [Google Scholar] [CrossRef]
- Oppel, F.; Müller, N.; Schackert, G.; Hendruschk, S.; Martin, D.; Geiger, K.D.; Temme, A. SOX2-RNAi attenuates S-phase entry and induces RhoA-dependent switch to protease-independent amoeboid migration in human glioma cells. Mol. Cancer 2011, 10, 1–16. [Google Scholar] [CrossRef]
- Weeks, A.; Okolowsky, N.; Golbourn, B.; Ivanchuk, S.; Smith, C.; Rutka, J.T. ECT2 and RASAL2 mediate mesenchymal-amoeboid transition in human astrocytoma cells. Am. J. Pathol. 2012, 181, 662–674. [Google Scholar] [CrossRef]
- Quick, Q.; Paul, M.; Skalli, O. Roles and potential clinical applications of intermediate filament proteins in brain tumors. Semin. Pediatr. Neurol. 2015, 22, 40–48. [Google Scholar] [CrossRef]
- Ducray, F.; Mokhtari, K.; Crinire, E.; Idbaih, A.; Marie, Y.; Dehais, C.; Paris, S.; Carpentier, C.; Dieme, M.J.; Adam, C.; et al. Diagnostic and prognostic value of alpha internexin expression in a series of 409 gliomas. Eur. J. Cancer 2011, 47, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Skalli, O.; Wilhelmsson, U.; Örndahl, C.; Fekete, B.; Malmgren, K.; Rydenhag, B.; Pekny, M. Astrocytoma grade IV (glioblastoma multiforme) displays 3 subtypes with unique expression profiles of intermediate filament proteins. Hum. Pathol. 2013, 44, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Paetau, A. Glial fibrillary acidic protein, vimentin and fibronectin in primary cultures of human glioma and fetal brain. Acta Neuropathol. 1988, 75, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Yung, W.A.; Luna, M.; Borit, A. Vimentin and Glial Fibrillary Acidic Protein in Human Brain Tumors. J. Neuro-Oncol. 1985, 38, 35–38. [Google Scholar] [CrossRef]
- van Bodegraven, E.J.; van Asperen, J.V.; Robe, P.A.J.; Hol, E.M. Importance of GFAP isoform-specific analyses in astrocytoma. Glia 2019, 1–17. [Google Scholar] [CrossRef]
- Lin, L.; Wang, G.; Ming, J.; Meng, X.; Han, B.; Sun, B.; Cai, J.; Jiang, C. Analysis of expression and prognostic significance of vimentin and the response to temozolomide in glioma patients. Tumor Biol. 2016, 37, 15333–15339. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, L.; Dong, X.; Liu, L.; Huo, L.; Chen, H. High Expression of Vimentin is Associated with Progression and a Poor Outcome in Glioblastoma. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Mahesparan, R.; Read, T.-A.; Lund-Johansen, M.; Skaftnesmo, K.O.; Bjerkvig, R.; Engebraaten, O. Expression of extracellular matrix components in a highly infiltrative in vivo glioma model. Acta Neuropathol. 2003, 105, 49–57. [Google Scholar] [CrossRef]
- Lv, D.; Lu, L.; Hu, Z.; Fei, Z.; Liu, M.; Wei, L.; Xu, J. Nestin Expression Is Associated with Poor Clinicopathological Features and Prognosis in Glioma Patients: An Association Study and Meta-analysis. Mol. Neurobiol. 2017, 54, 727–735. [Google Scholar] [CrossRef]
- Baumgarten, P.; Quick-Weller, J.; Gessler, F.; Wagner, M.; Tichy, J.; Forster, M.T.; Foerch, C.; Seifert, V.; Mittelbronn, M.; Senft, C. Pre- and early postoperative GFAP serum levels in glioma and brain metastases. J. Neurooncol. 2018, 139, 541–546. [Google Scholar] [CrossRef]
- Elobeid, A.; Bongcam-Rudloff, E.; Westermark, B.; Nistér, M. Effects of inducible glial fibrillary acidic protein on glioma cell motility and proliferation. J. Neurosci. Res. 2000, 60, 245–256. [Google Scholar] [CrossRef]
- Toda, M.; Miura, M.; Asou, H.; Toya, S.; Uyemura, K. Cell growth suppression of astrocytoma C6 cells by glial fibrillary acidic protein cDNA transfection. J. Neurochem. 1994, 63, 1975–1978. [Google Scholar] [CrossRef]
- Kouam, P.N.; Rezniczek, G.A.; Kochanneck, A.; Priesch-Grzeszkowiak, B.; Hero, T.; Adamietz, I.A.; Bühler, H. Robo1 and vimentin regulate radiation-induced motility of human glioblastoma cells. PLoS ONE 2018, 13, 1–18. [Google Scholar] [CrossRef]
- Ishiwata, T.; Teduka, K.; Yamamoto, T.; Kawahara, K.; Matsuda, Y.; Naito, Z. Neuroepithelial stem cell marker nestin regulates the migration, invasion and growth of human gliomas. Oncol. Rep. 2011, 26, 91–99. [Google Scholar] [CrossRef]
- Kitai, R.; Horita, R.; Sato, K.; Yoshida, K.; Arishima, H.; Higashino, Y.; Hashimoto, N.; Takeuchi, H.; Kubota, T.; Kikuta, K.-I. Nestin expression in astrocytic tumors delineates tumor infiltration. Brain Tumor Pathol. 2010, 27, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Jing, R.; Pizzolato, G.; Robson, R.M.; Gabbiani, G.; Skalli, O. Intermediate filament protein synemin is present in human reactive and malignant astrocytes and associates with ruffled membranes in astrocytoma cells. Glia 2005, 50, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Bobek, M.P.; Misek, D.E.; Kuick, R.; Blaivas, M.; Kurnit, D.M.; Taylor, J.; Hanash, S.M. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res. 2001, 61, 6885–6891. [Google Scholar] [PubMed]
- Katsetos, C.D.; Legido, A.; Perentes, E.; Mörk, S.J. Class III β-Tubulin Isotype: A Key Cytoskeletal Protein at the Crossroads of Developmental Neurobiology and Tumor Neuropathology. J. Child Neurol. 2003, 18, 851–866. [Google Scholar] [CrossRef] [PubMed]
- Katsetos, C.D.; Reddy, G.; Dráberová, E.; Šmejkalová, B.; Del Valle, L.; Ashraf, Q.; Tadevosyan, A.; Yelin, K.; Maraziotis, T.; Mishra, O.P.; et al. Altered Cellular Distribution and Subcellular Sorting of γ-Tubulin in Diffuse Astrocytic Gliomas and Human Glioblastoma Cell Lines. J. Neuropathol. Exp. Neurol. 2006, 65, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Katsetos, C.D.; Reginato, M.J.; Baas, P.W.; D’Agostino, L.; Legido, A.; Tuszyński, J.A.; Dráberová, E.; Dráber, P. Emerging microtubule targets in glioma therapy. Semin. Pediatr. Neurol. 2015, 22, 49–72. [Google Scholar] [CrossRef]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [PubMed]
- Kavallaris, M.; Kuo, D.Y.; Burkhart, C.A.; Regl, D.L.; Norris, M.D.; Haber, M.; Horwitz, S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J. Clin. Investig. 1997, 100, 1282–1293. [Google Scholar] [CrossRef]
- Karmakar, S.; Banik, N.L.; Ray, S.K. Combination of all-trans retinoic acid and paclitaxel-induced differentiation and apoptosis in human glioblastoma U87MG xenografts in nude mice. Cancer 2008, 112, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Kan, D.; Lo, T.H.; Lu, K.S.; Chien, C.-L. Induction of neural differentiation in rat C6 glioma cells with taxol. Brain Behav. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Godinho, S.A.; Pellman, D. Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef]
- Dráberová, E.; Vinopal, S.; Morfini, G.; Liu, P.S.; Sládková, V.; Sulimenko, T.; Burns, M.R.; Solowska, J.; Kulandaivel, K.; De Chadarévian, J.P.; et al. Microtubule-severing ATPase spastin in glioblastoma: Increased expression in human glioblastoma cell lines and inverse roles in cell motility and proliferation. J. Neuropathol. Exp. Neurol. 2011, 70, 811–826. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Mu, L.; Han, X.; Liu, X.; Fu, S. SiRNA targeting stathmin inhibits invasion and enhances chemotherapy sensitivity of stem cells derived from Glioma cell lines. Acta Biochim. Biophys. Sin. (Shanghai) 2014, 46, 1034–1040. [Google Scholar] [CrossRef]
- Masui, K.; Mawatari, S.; Suzuki, S.O.; Iwaki, T. Evaluation of sensitivity and specificity of doublecortin immunostatining for the detection of infiltrating glioma cells. Brain Tumor Pathol. 2008, 25, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Daou, M.-C.; Smith, T.W.; Litofsky, N.S.; Hsieh, C.C.; Ross, A.H. Doublecortin is preferentially expressed in invasive human brain tumors. Acta Neuropathol. 2005, 110, 472–480. [Google Scholar] [CrossRef]
- Santra, M.; Santra, S.; Roberts, C.; Zhang, R.L.; Chopp, M. Doublecortin induces mitotic microtubule catastrophe and inhibits glioma cell invasion. J. Neurochem. 2009, 108, 231–245. [Google Scholar] [CrossRef]
- Santra, M.; Zhang, X.; Santra, S.; Jiang, F.; Chopp, M. Ectopic Doublecortin Gene Expression Suppresses the Malignant Phenotype in Glioblastoma Cells. Cancer Res. 2006, 66, 11726–11735. [Google Scholar] [CrossRef] [PubMed]
- Pagano, A.; Honoré, S.; Mohan, R.; Berges, R.; Akhmanova, A.; Braguer, D. Epothilone B inhibits migration of glioblastoma cells by inducing microtubule catastrophes and affecting EB1 accumulation at microtubule plus ends. Biochem. Pharmacol. 2012, 84, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Näthke, I.S. THE ADENOMATOUS POLYPOSIS COLI PROTEIN: The Achilles Heel of the Gut Epithelium. Annu. Rev. Cell Dev. Biol. 2004, 20, 337–366. [Google Scholar] [CrossRef] [PubMed]
- Segditsas, S.; Tomlinson, I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006, 25, 7531–7537. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.O.; McKenney, R.J.; Mawatari, S.Y.; Mizuguchi, M.; Mikami, A.; Iwaki, T.; Goldman, J.E.; Canoll, P.; Vallee, R.B. Expression patterns of LIS1, dynein and their interaction partners dynactin, NudE, NudEL and NudC in human gliomas suggest roles in invasion and proliferation. Acta Neuropathol. 2007, 113, 591–599. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, W.-H.; Wang, Y.-L. Microtubules stabilize cell polarity by localizing rear signals. Proc. Natl. Acad. Sci. USA 2014, 111, 16383–16388. [Google Scholar] [CrossRef]
- Bie, L.; Zhao, G.; Wang, Y.P.; Zhang, B. Kinesin family member 2C (KIF2C/MCAK) is a novel marker for prognosis in human gliomas. Clin. Neurol. Neurosurg. 2012, 114, 356–360. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, L.; Li, D.; Deng, J.; Zhao, Z.; He, S.; Zhang, Y.; Tu, Y. Kinesin family member 14 is a candidate prognostic marker for outcome of glioma patients. Cancer Epidemiol. 2013, 37, 79–84. [Google Scholar] [CrossRef]
- Venere, M.; Horbinski, C.; Crish, J.F.; Jin, X.; Vasanji, A.; Major, J.; Burrows, A.C.; Chang, C.; Prokop, J.; Wu, Q.; et al. The mitotic kinesin KIF11 is a driver of invasion, proliferation, and self-renewal in glioblastoma. Sci. Transl. Med. 2015, 7, 304ra143. [Google Scholar] [CrossRef]
- Afghani, N.; Mehta, T.; Wang, J.; Tang, N.; Skalli, O.; Quick, Q.A. Microtubule actin cross-linking factor 1, a novel target in glioblastoma. Int. J. Oncol. 2017, 50, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Arden, J.D.; Lavik, K.I.; Rubinic, K.A.; Chiaia, N.; Khuder, S.A.; Howard, M.J.; Nestor-Kalinoski, A.L.; Alberts, A.S.; Eisenmann, K.M. Small-molecule agonists of mammalian Diaphanous-related (mDia) formins reveal an effective glioblastoma anti-invasion strategy. Mol. Biol. Cell 2015, 26, 3704–3718. [Google Scholar] [CrossRef]
- Hiratsuka, M.; Inoue, T.; Toda, T.; Kimura, N.; Shirayoshi, Y.; Kamitani, H.; Watanabe, T.; Ohama, E.; Tahimic, C.G.T.; Kurimasa, A.; et al. Proteomics-based identification of differentially expressed genes in human gliomas: Down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 2003, 309, 558–566. [Google Scholar] [CrossRef]
- Hoelzinger, D.B.; Mariani, L.; Weis, J.; Woyke, T.; Berens, T.J.; McDonough, W.; Sloan, A.; Coons, S.W.; Berens, M.E. Gene Expression Profile of Glioblastoma Multiforme Invasive Phenotype Points to New Therapeutic Targets. Neoplasia 2005, 7, 7–16. [Google Scholar] [CrossRef]
- Geiger, K.D.; Stoldt, P.; Schlote, W.; Derouiche, A. Ezrin Immunoreactivity Is Associated with Increasing Malignancy of Astrocytic Tumors but Is Absent in Oligodendrogliomas. Am. J. Pathol. 2000, 157, 1785–1793. [Google Scholar] [CrossRef]
- Tynninen, O.; Carpen, O.; Jaaskelainen, J.; Paavonen, T.; Paetau, A. Ezrin expression in tissue microarray of primary and recurrent gliomas. Neuropathol. Appl. Neurobiol. 2004, 30, 472–477. [Google Scholar] [CrossRef]
- Belot, N.; Rorive, S.; Doyen, I.; Lefranc, F.; Bruyneel, E.; Dedecker, R.; Micik, S.; Brotchi, J.; Decaestecker, C.; Salmon, I.; et al. Molecular characterization of cell substratum attachments in human glial tumors relates to prognostic features. Glia 2001, 36, 375–390. [Google Scholar]
- Adams, J.C. Roles of fascin in cell adhesion and motility. Curr. Opin. Cell Biol. 2004, 16, 590–596. [Google Scholar] [CrossRef]
- Peraud, A.; Mondal, S.; Hawkins, C.; Mastronardi, M.; Bailey, K.; Rutka, J.T. Expression of fascin, an actin-bundling protein, in astrocytomas of varying grades. Brain Tumor Pathol. 2003, 20, 53–58. [Google Scholar] [CrossRef]
- Ng, H.K.; Ko, H.C.W.; Tse, C.C.H. Immunohistochemical and Ultrastructural Studies of Oligodendrogliomas Revealed Features of Neuronal Differentiation. Int. J. Surg. Pathol. 1994, 2, 47–55. [Google Scholar] [CrossRef]
- Wharton, S.B.; Chan, K.K.; Whittle, I.R. Microtubule-associated protein 2 (MAP-2) is expressed in low and high grade diffuse astrocytomas. J. Clin. Neurosci. 2002, 9, 165–169. [Google Scholar] [CrossRef]
- Zhang, Y.; Ni, S.; Huang, B.; Wang, L.; Zhang, X.; Li, X.; Wang, H.; Liu, S.; Hao, A.; Li, X. Overexpression of SCLIP promotes growth and motility in glioblastoma cells. Cancer Biol. Ther. 2015, 16, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Jin, X.; Jung, J.E.; Beck, S.; Kim, H. Cell surface Nestin is a biomarker for glioma stem cells. Biochem. Biophys. Res. Commun. 2013, 433, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Strojnik, T.; Røsland, G.V.; Sakariassen, P.O.; Kavalar, R.; Lah, T. Neural stem cell markers, nestin and musashi proteins, in the progression of human glioma: Correlation of nestin with prognosis of patient survival. Surg. Neurol. 2007, 68, 133–143. [Google Scholar] [CrossRef]
- Dehghani, F.; Schachenmayr, W.; Laun, A.; Korf, H.W. Prognostic implication of histopathological, immunohistochemical and clinical features of oligodendrogliomas: A study of 89 cases. Acta Neuropathol. 1998, 95, 493–504. [Google Scholar] [CrossRef]
- Forget, M.-A.; Desrosiers, R.R.; Del, M.; Moumdjian, R.; Shedid, D.; Berthelet, F.; Béliveau, R. The expression of rho proteins decreases with human brain tumor progression: Potential tumor markers. Clin. Exp. Metastasis 2002, 19, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, A.; Didier, S.; Fortin, S.; Chuang, Y.; White, T.; Berens, M.E.; Rushing, E.; Eschbacher, J.; Tran, N.L.; Chan, A.; et al. The small GTPase RhoG mediates glioblastoma cell invasion. Mol. Cancer 2012, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Rutka, J.T.; Muller, M.; Hubbard, S.L.; Forsdike, J.; Dirks, P.B.; Jung, S.; Tsugu, A.; Ivanchuk, S.; Costello, P.; Mondal, S.; et al. Astrocytoma Adhesion to Extracellular Matrix: Functional Significance of Integrin and Focal Adhesion Kinase Expression. J. Neuropathol. Exp. Neurol. 1999, 58, 198–209. [Google Scholar] [CrossRef]
- Natarajan, M.; Hecker, T.P.; Gladson, C.L. FAK signaling in anaplastic astrocytoma and glioblastoma tumors. Cancer J. 2003, 9, 126–133. [Google Scholar] [CrossRef]
- Mariani, L.; Beaudry, C.; McDonough, W.S.; Hoelzinger, D.B.; Demuth, T.; Ross, K.R.; Berens, T.; Coons, S.W.; Watts, G.; Trent, J.M.; et al. Glioma cell motility is associated with reduced transcription of proapoptotic and proliferation genes: A cDNA microarray analysis. J. Neurooncol. 2001, 53, 161–176. [Google Scholar] [CrossRef]
- Giese, A.; Loo, A.; Norman, S.A.; Treasurywala, S.; Berens, M.E. Contrasting migratory response of astrocytoma cells to tenascin mediated by different integrins. J. Cell Sci. 1996, 109, 2161–2168. [Google Scholar]
- Plopper, G.E.; McNamee, H.P.; Dike, L.E.; Bojanowski, K.; Ingber, D.E. Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol. Biol. Cell 1995, 6, 1349–1365. [Google Scholar] [CrossRef]
- Wenk, M.B.; Midwood, K.S.; Schwarzbauer, J.E. Tenascin-C suppresses Rho activation. J. Cell Biol. 2000, 150, 913–920. [Google Scholar] [CrossRef]
- Hirata, E.; Arakawa, Y.; Shirahata, M.; Yamaguchi, M.; Kishi, Y.; Okada, T.; Takahashi, J.A.; Matsuda, M.; Hashimoto, N. Endogenous tenascin-C enhances glioblastoma invasion with reactive change of surrounding brain tissue. Cancer Sci. 2009, 100, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; Wild-Bode, C.; Aulwurm, S.; Dichgans, J.; Weller, M. Transforming Growth Factors β1 (TGF-β1) and TGF-β2 Promote Glioma Cell Migration via Up-Regulation of αVβ3 Integrin Expression. Biochem. Biophys. Res. Commun. 2000, 268, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Tonn, J.C.; Wunderlich, S.; Kerkau, S.; Klein, C.E.; Roosen, K. Invasive behaviour of human gliomas is mediated by interindividually different integrin patterns. Anticancer Res. 1998, 18, 2599–2605. [Google Scholar] [PubMed]
- Rooprai, H.K.; Vanmeter, T.; Panou, C.; Schnüll, S.; Trillo-Pazos, G.; Davies, D.; Pilkington, G.J. The role of integrin receptors in aspects of glioma invasion in vitro. Int. J. Dev. Neurosci. 1999, 17, 613–623. [Google Scholar] [CrossRef]
- Gritsenko, P.G.; Friedl, P. Adaptive adhesion systems mediate glioma cell invasion in complex environments. J. Cell Sci. 2018, 131, jcs216382. [Google Scholar] [CrossRef]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462. [Google Scholar] [CrossRef]
- Demuth, T.; Berens, M.E. Molecular mechanisms of glioma cell migration and invasion. J. Neurooncol. 2004, 70, 217–228. [Google Scholar] [CrossRef]
- Giese, A.; Kluwe, L.; Laube, B.; Meissner, H.; Berens, M.E.; Westphal, M. Migration of human glioma cells on myelin. Neurosurgery 1996, 38, 755–764. [Google Scholar] [CrossRef]
- Lash, L.L.; Wallar, B.J.; Turner, J.D.; Vroegop, S.M.; Kilkuskie, R.E.; Kitchen-Goosen, S.M.; Xu, H.E.; Alberts, A.S. Small-Molecule Intramimics of Formin Autoinhibition: A New Strategy to Target the Cytoskeletal Remodeling Machinery in Cancer Cells. Cancer Res. 2013, 73, 6793–6803. [Google Scholar] [CrossRef]
- Wiranowska, M.; Ladd, S.; Smith, S.R.; Gottschall, P.E. CD44 adhesion molecule and neuro-glial proteoglycan NG2 as invasive markers of glioma. Brain Cell Biol. 2006, 35, 159–172. [Google Scholar] [CrossRef]
- Akiyama, Y.; Jung, S.; Salhia, B.; Lee, S.; Hubbard, S.; Taylor, M.; Mainprize, T.; Akaishi, K.; van Furth, W.; Rutka, J.T. Hyaluronate Receptors Mediating Glioma Cell Migration and Proliferation. J. Neurooncol. 2001, 53, 115–127. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef]
- Merzak, A.; Koocheckpour, S.; Pilkington, G.J. CD44 mediates human glioma cell adhesion and invasion in vitro. Cancer Res. 1994, 54, 3988–3992. [Google Scholar]
- Park, J.B.; Kwak, H.-J.; Lee, S.-H. Role of hyaluronan in glioma invasion. Cell Adh. Migr. 2008, 2, 202–207. [Google Scholar] [CrossRef]
- Karousou, E.; Misra, S.; Ghatak, S.; Dobra, K.; Götte, M.; Vigetti, D.; Passi, A.; Karamanos, N.K.; Skandalis, S.S. Roles and targeting of the HAS/hyaluronan/CD44 molecular system in cancer. Matrix Biol. 2017, 59, 3–22. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W. Hyaluronan-mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. Semin. Cancer Biol. 2008, 18, 251–259. [Google Scholar] [CrossRef]
- Park, D.; Kim, Y.; Kim, H.; Kim, K.; Lee, Y.-S.; Choe, J.; Hahn, J.-H.; Lee, H.; Jeon, J.; Choi, C.; et al. Hyaluronic acid promotes angiogenesis by inducing RHAMM-TGFβ receptor interaction via CD44-PKCδ. Mol. Cells 2012, 33, 563–574. [Google Scholar] [CrossRef]
- Murai, T.; Miyazaki, Y.; Nishinakamura, H.; Sugahara, K.N.; Miyauchi, T.; Sako, Y.; Yanagida, T.; Miyasaka, M. Engagement of CD44 Promotes Rac Activation and CD44 Cleavage during Tumor Cell Migration. J. Biol. Chem. 2004, 279, 4541–4550. [Google Scholar] [CrossRef]
- Klank, R.L.; Decker Grunke, S.A.; Bangasser, B.L.; Forster, C.L.; Price, M.A.; Odde, T.J.; SantaCruz, K.S.; Rosenfeld, S.S.; Canoll, P.; Turley, E.A.; et al. Biphasic Dependence of Glioma Survival and Cell Migration on CD44 Expression Level. Cell Rep. 2017, 18, 23–31. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Scheme of actin filament formation. First G-actin binds to ATP. Afterwards, it forms stable di- or trimers and, finally, filaments elongate by addition of monomers. Hydrolysis of ATP to ADP leads to a distinction between the fast growing (+)-end and the slower growing or dissociating (−)-end.
Figure 1.
Scheme of actin filament formation. First G-actin binds to ATP. Afterwards, it forms stable di- or trimers and, finally, filaments elongate by addition of monomers. Hydrolysis of ATP to ADP leads to a distinction between the fast growing (+)-end and the slower growing or dissociating (−)-end.

Figure 2.
Illustration of actin, microtubule, and intermediate filament signaling, with focus on migration associated structures and signaling cascades.
Figure 2.
Illustration of actin, microtubule, and intermediate filament signaling, with focus on migration associated structures and signaling cascades.

Figure 3.
Organizational structures of actin, microtubules, and intermediate filaments inside of a cell and their physical interactions. Notably, all three cytoskeletal proteins interact directly with each other.
Figure 3.
Organizational structures of actin, microtubules, and intermediate filaments inside of a cell and their physical interactions. Notably, all three cytoskeletal proteins interact directly with each other.

Figure 4.
Scheme of microtubule formation and dynamic instability. Microtubules consist of α- and β-heterodimers, forming a hollow tube elongating by the addition of heterodimers, forming a GTP-cap at the (+)-end of the microtubule, protecting microtubules from shrinkage. If the (+)-end loses its GTP-cap it induces microtubule shrinkage.
Figure 4.
Scheme of microtubule formation and dynamic instability. Microtubules consist of α- and β-heterodimers, forming a hollow tube elongating by the addition of heterodimers, forming a GTP-cap at the (+)-end of the microtubule, protecting microtubules from shrinkage. If the (+)-end loses its GTP-cap it induces microtubule shrinkage.

Figure 5.
Illustration of intermediate filament assembly. Intermediate filaments arise from the monomers spiraling around each other to form dimers. Two dimers aggregate to a tetramer and eight tetramers to a unit length filament. Unit filaments form the final filament via end-to-end aggregation. Notably, this process is independent of ATP or GTP.
Figure 5.
Illustration of intermediate filament assembly. Intermediate filaments arise from the monomers spiraling around each other to form dimers. Two dimers aggregate to a tetramer and eight tetramers to a unit length filament. Unit filaments form the final filament via end-to-end aggregation. Notably, this process is independent of ATP or GTP.


{kind=link}
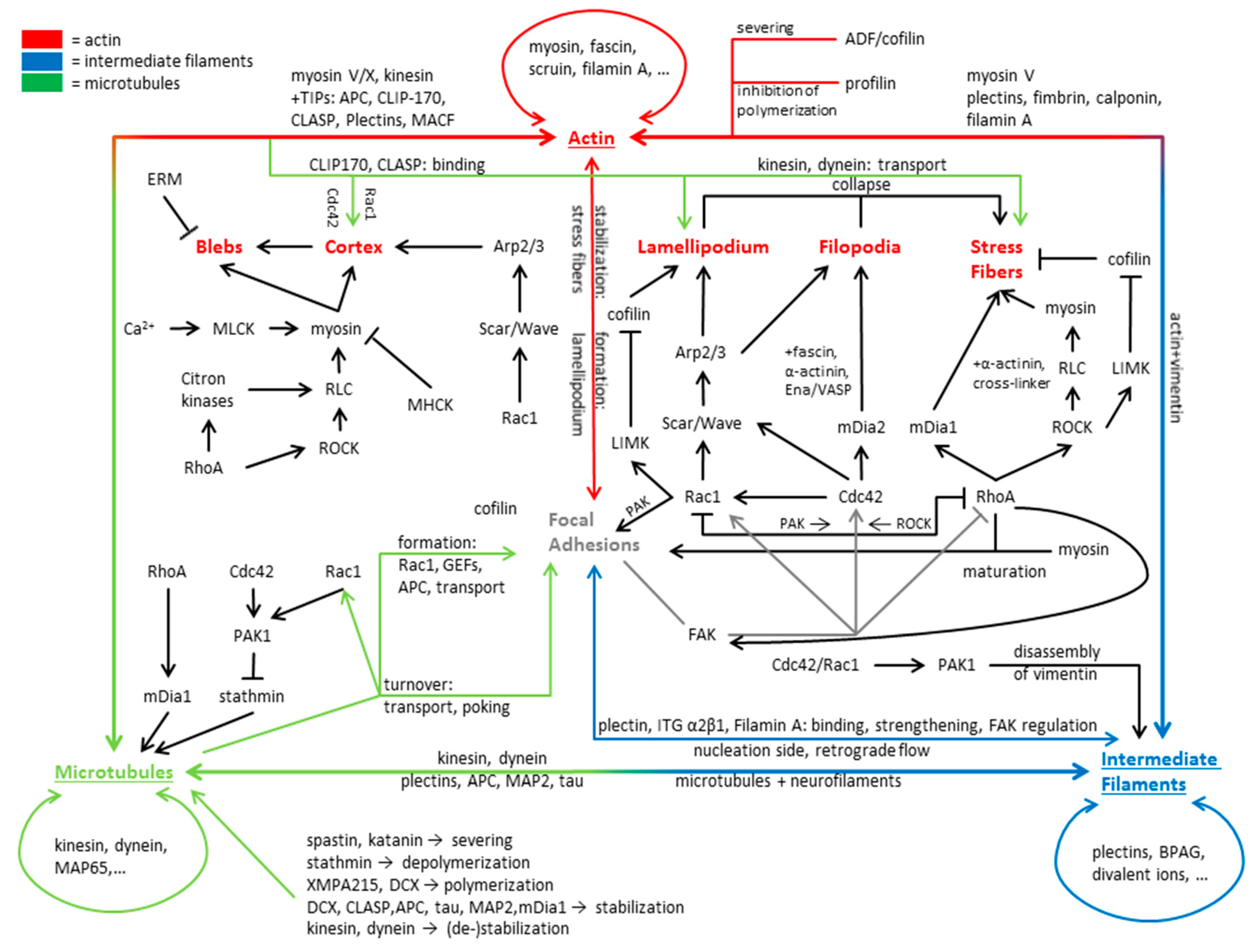{kind=link}
{kind=link}
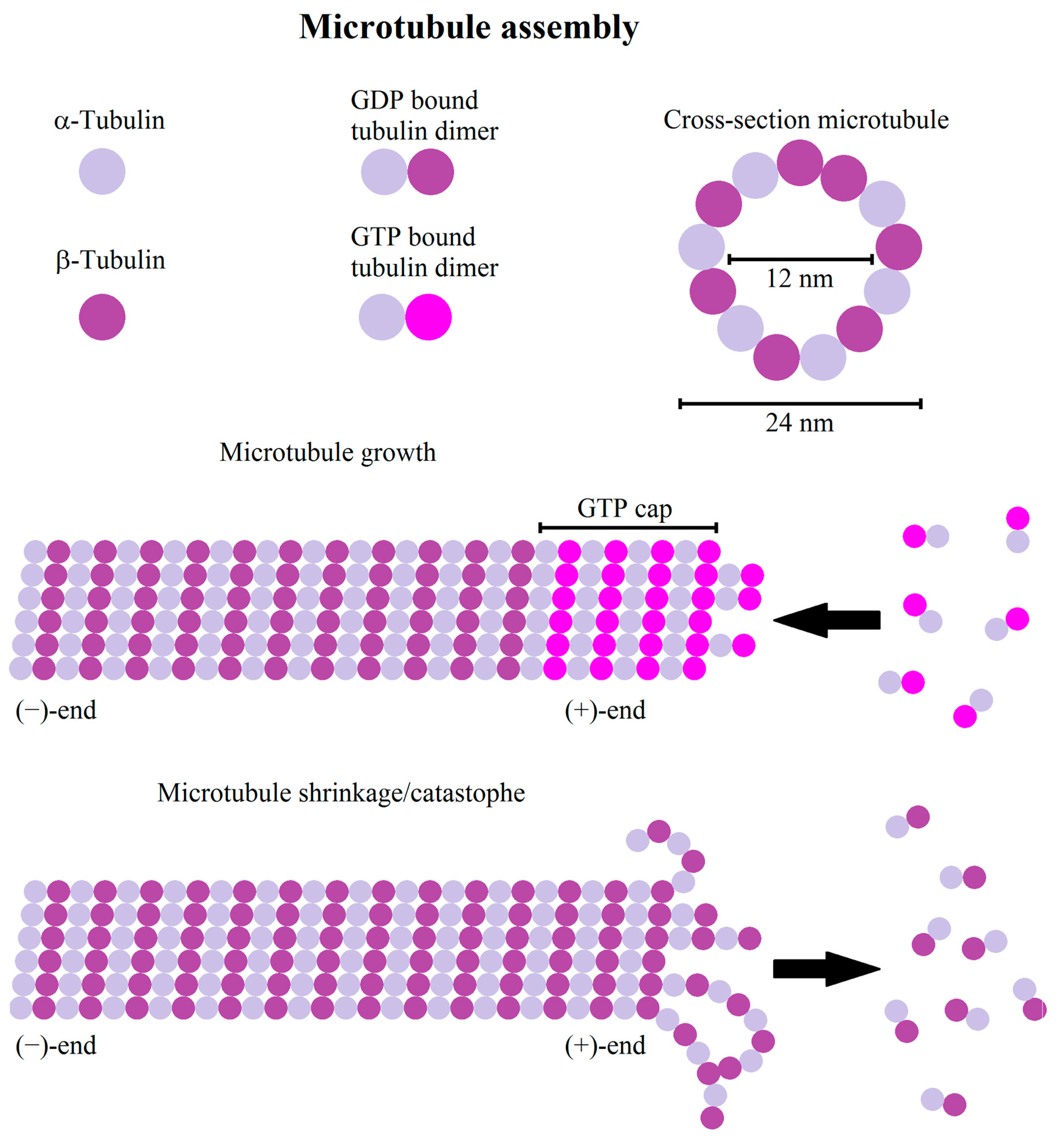{kind=link}
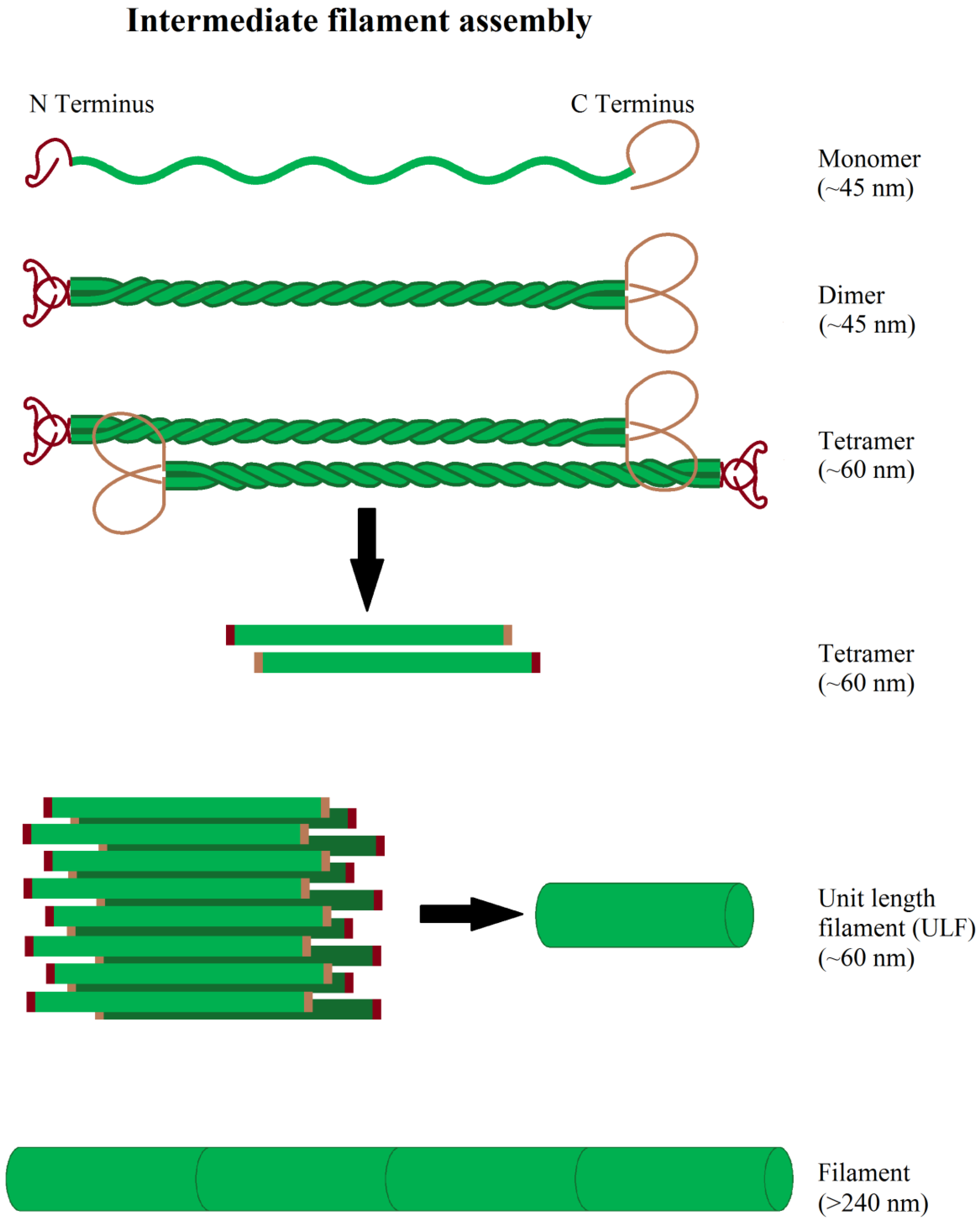{kind=link}
Table 1.
Summary of mentioned actin associated proteins and their direct or indirect functions.
| Actin Associated Proteins | Function |
|---|---|
| Arp2/3 | Polymerization factor |
| Ena/VASP | Polymerization factor, anti-capping function |
| FMNL2 | Polymerization factor |
| mDia1 | Polymerization factor |
| mDia2 | Polymerization factor |
| Profilin | Inhibits actin polymerization |
| ADF/Cofilin | Actin severing |
| Arpin | Inhibits Arp2/3 |
| Myosin II | Cell/actin contractility, cross linker |
| RLC | Activates myosin II |
| MLCK | Activates myosin II |
| MHCK | Inhibits myosin II activity |
| PKC | Inhibits myosin II activity |
| CKII | Inhibits myosin II activity |
| Scruin | Cross linker |
| Fascin | Cross linker |
| α-actinin | Cross linker |
| Filamin | Cross linker |
| Fimbrin | Cross linker |
| Paladin | Cross linker |
| Ezrin | Membrane-cortex linker |
| Radixin | Membrane-cortex linker |
| Moesin | Membrane-cortex linker |
| Cdc42 | Signaling molecule, activates mDia2, WAVE, N-WASP |
| Rac1 | Signaling molecule, activates WASP/WAVE, arpin |
| RhoA | Signaling molecule, activates ROCK, mDia1, LIMK |
| ROCK | Signaling molecule, activates myosin II |
| WASP/WAVE | Signaling molecule, activates Arp2/3 |
| N-WASP | Signaling molecule, activates Arp2/3 |
| LIMK | Signaling molecule, inhibits ADF/cofilin |
Table 2.
Summary of mentioned microtubule associated proteins and their direct or indirect functions.
Table 2.
Summary of mentioned microtubule associated proteins and their direct or indirect functions.
| Microtubule Associated Proteins | Function |
|---|---|
| Stathmin | Depolymerization |
| XMPA215 | Polymerization factor |
| EB | Polymerization, Stabilization, Recruitment of proteins |
| DCX | Polymerization factor, Stabilization |
| CLASP | Stabilization |
| APC | Stabilization |
| mDia1 | Stabilization |
| mDia2 | Stabilization |
| Tau | Stabilization |
| MAP2 | Stabilization |
| Spastin | Microtubule severing |
| Katanin | Microtubule severing |
| Kinesin | Cargo transport |
| Dynein | Cargo transport |
| MACF1 | Actin-Microtubule interactions |
| Cdc42 | Signaling molecule, activates PAK |
| Rac1 | Signaling molecule, activates PAK |
| RhoA | Signaling molecule, mDia1 |
| PAK | Signaling molecule, inhibits stathmin |
Table 3.
Summary of mentioned intermediate filament associated proteins and their function.
| Intermediate Filament Associated Proteins | Function |
|---|---|
| LINC | Nucleus—intermediate filament linkage |
| Plakins | Linkage to adhesion sites |
| Plectin | Intermediate filament—integrin linkage |
| Kinesin * | Filament transport |
| Dynein * | Filament transport |
| Myosin * | Filament transport |
* Involved in the transport of filaments as their cargo.
Table 4.
Summary of motility associated proteins differentially expressed in glioma, compared to healthy glia cells.
Table 4.
Summary of motility associated proteins differentially expressed in glioma, compared to healthy glia cells.
| Function | Expression/Activity | Sources | |
|---|---|---|---|
| Actin associated proteins | |||
| Arp2/3 | polymerization | high | [521] |
| mDia2 (formin family) | polymerization | high | [572] |
| Profilin | polymerization | low | [573] |
| Moesin/Ezrin | membrane to actin cortex linkage | high | [574,575,576] |
| Cortactin | actin cross-linker | high | [524] |
| Filamin | actin cross-linker | high | [548] |
| α-actinin | actin cross-linker | high | [577] |
| Fascin | actin cross-linker | high | [578,579] |
| Microtubule associated proteins | |||
| MAP2 | stabilization | high | [580,581] |
| Sclip (Stathmin family) | destabilization | high | [582] |
| Spastin | destabilization | high | [557] |
| MACF1 | microtubule-actin linkage | high | [571] |
| Dynein | cargo transport | unchanged | [566] |
| Kinesin-5, KiF2C, KiF14 | cargo transport | high | [568,569,570] |
| Β-III, β-IV, γ tubulin | microtubule formation and anchorage | high | [548,549,550] |
| Intermediate filaments | |||
| Vimentin/Nestin | cytoskeletal meshwork | high | [538,539,545,546,583,584,585] |
| GFAP | cytoskeletal meshwork | high | [536] |
| α-Internexin | cytoskeletal meshwork | high | [532] |
| Signaling molecules | |||
| RhoA | contractility | high/low | [499,586] |
| RhoB | contractility | low | [586] |
| RhoG | contractility, protrusion formation | high | [587] |
| Rac1 | protrusion formation | high | [499] |
| FAK | protrusion formation, adhesion turnover | high | [504,588,589] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hohmann, T.; Dehghani, F. The Cytoskeleton—A Complex Interacting Meshwork. Cells 2019, 8, 362. https://doi.org/10.3390/cells8040362
AMA Style
Hohmann T, Dehghani F. The Cytoskeleton—A Complex Interacting Meshwork. Cells. 2019; 8(4):362. https://doi.org/10.3390/cells8040362
Chicago/Turabian StyleHohmann, Tim, and Faramarz Dehghani. 2019. "The Cytoskeleton—A Complex Interacting Meshwork" Cells 8, no. 4: 362. https://doi.org/10.3390/cells8040362
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.