Synaptic determinants of Rett syndrome
- 1 Istituto di Neuroscienze CNR, Pisa, Italy
- 2 Dipartimento di Anatomia, Farmacologia e Medicina Legale, Università di Torino and National Institute of Neuroscience-Italy, Torino, Italy
- 3 Dipartimento di Psicologia, Università di Firenze, Firenze, Italy
There is mounting evidence showing that the structural and molecular organization of synaptic connections is affected both in human patients and in animal models of neurological and psychiatric diseases. As a consequence of these experimental observations, it has been introduced the concept of synapsopathies, a notion describing brain disorders of synaptic function and plasticity. A close correlation between neurological diseases and synaptic abnormalities is especially relevant for those syndromes including also mental retardation in their symptomatology, such as Rett syndrome (RS). RS (MIM312750) is an X-linked dominant neurological disorder that is caused in the majority of cases by mutations in methyl-CpG-binding protein 2 (MeCP2). This review will focus on the current knowledge of the synaptic alterations produced by mutations of the gene MeCP2 in mouse models of RS and will highlight prospects experimental therapies currently in use. Different experimental approaches have revealed that RS could be the consequence of an impairment in the homeostasis of synaptic transmission in specific brain regions. Indeed, several forms of experience-induced neuronal plasticity are impaired in the absence of MeCP2. Based on the results presented in this review, it is reasonable to propose that understanding how the brain is affected by diseases such as RS is at reach. This effort will bring us closer to identify the neurobiological bases of human cognition.
Introduction
Loss-of-function mutations in the X-linked methyl-CpG binding protein 2 (Mecp2) gene, a transcriptional regulator that acts through epigenetic mechanisms on chromatin structure, cause the 95% of Rett’s syndrome (RS; MIM312750) cases, a severe neurological disorder that affects about 1:10000 girls worldwide (Amir et al., 1999). Importantly, alterations in MeCP2 expression are associated to several other neurological conditions such as Angelman-like syndrome, motor deficits, learning disabilities, seizures, bipolar disease, juvenile-onset schizophrenia, autism-like features, psychosis, etc (Chahrour and Zoghbi, 2007).
Interestingly, the discovery that target mutation of MeCP2 in the central nervous system may produce a phenotype similar to the whole body mutation have indicated that impairment of MeCP2 function in the brain is crucial for the pathogenesis of the disease (Chen et al., 2001; Guy et al., 2001). Despite the severe neurological abnormalities, the brain of RS patients and of animal models does not show obvious anatomical alterations apart from a decreased weight and volume of about 12–34% (Chahrour and Zoghbi, 2007). Such changes are not generalized but more pronounced in specific brain areas (Reiss et al., 1993; Subramaniam et al., 1997; Kishi and Macklis, 2004; Armstrong, 2005; Stearns et al., 2007). Importantly, RS brain does not show obvious signs of neurodegeneration, atrophy, gliosis, demyelination, or neuronal migration defects (Jellinger et al., 1988; Reiss et al., 1993) suggesting that neurological symptoms may primarily stem from subtle defects of subcellular compartments such as dendrites, axons, or synaptic structures.
Synaptic Alterations in Rett Syndrome
In agreement with this hypothesis, direct evidences supporting the involvement of MeCP2 in the regulation of synaptic connectivity are now available. First of all, postmortem analyses of the brain of RS patients revealed structural abnormalities of synapses such as decreased number of dendritic spines in discrete cortical areas (Belichenko et al., 1994). A more recent quantitative analysis showed that both secondary and tertiary apical dendrites of pyramidal neurons in the CA1 area of the hippocampus of RS patients displayed less dendritic spines than unaffected individuals, with no significant correlation between age and spine density (Chapleau et al., 2009). Interestingly, autoradiographic analyses in the cortex and in the basal ganglia of patients revealed that the density of NMDA, AMPA, and GABA receptors was significantly altered (Johnston et al., 2005), a result indicating that RS is associated with abnormalities in the expression of molecules that are crucial for both excitatory and inhibitory synaptic transmission.
Similarly to RS patients, a number of recent studies pointed out morphological, functional and molecular alterations of synapses in neurons of different mice models of the disease (see Table 1). Hippocampal neurons cultured from MeCP2-KO mouse brains showed a decreased frequency of spontaneous excitatory synaptic transmission (EPSCs, Nelson et al., 2006). Moreover, autaptic hippocampal cultures from MeCP2-KO mice displayed a reduction in both frequency and amplitude of action-potential evoked EPSCs, whereas neurons from MeCP2Tg1 mice, in which wild-type (WT) human MeCP2 is approximately two-fold higher than in normal animals (Collins et al., 2004), showed an increase of these parameters (Chao et al., 2007). In line with these functional changes, the number of immunopositive puncta for the excitatory synapse molecules PSD-95 and vesicular glutamate transporter-1 (VGLUT1) was found to be decreased in MeCP2-KO cultures brains while it was increased in cultures deriving from MeCP2Tg1 mice. Indeed, recent analyses showed that overexpression of both WT and mutated forms (MeCP2R106W and MeCP2T158M) of MeCP2 proteins in organotypic hippocampal slices caused a reduction of dendritic spine density in CA1 pyramidal neurons 48 h after viral infection (Zhou et al., 2006; Chapleau et al., 2009). However, the overexpression of the WT form of the protein led only to a transient alteration in dendritic spine number that returned normal after 96 h. Accordingly, a previous study had shown that overexpression of MeCP2 in hippocampal slices did not affect dendritic spine density 5 days after transfection but it altered spine morphology leading to an increased proportion of longer and thinner dendritic spines (Zhou et al., 2006). Moreover, knockdown of endogenous MeCP2 with a specific small hairpin interference RNA induced a decrease in spine density appearing after 96 h of expression, and produced a specific loss of mature-shaped dendritic spines (Chapleau et al., 2009). In contrast with this study, previous data had indicated that 5-days-long down-regulation of MePC2 expression did not affect dendritic spine density but instead decreased neuronal dendritic complexity (Zhou et al., 2006). Altogether, these studies indicate that MeCP2 expression may contribute to the normal density and morphology of dendritic spines in CA1 pyramidal neurons of the hippocampus.
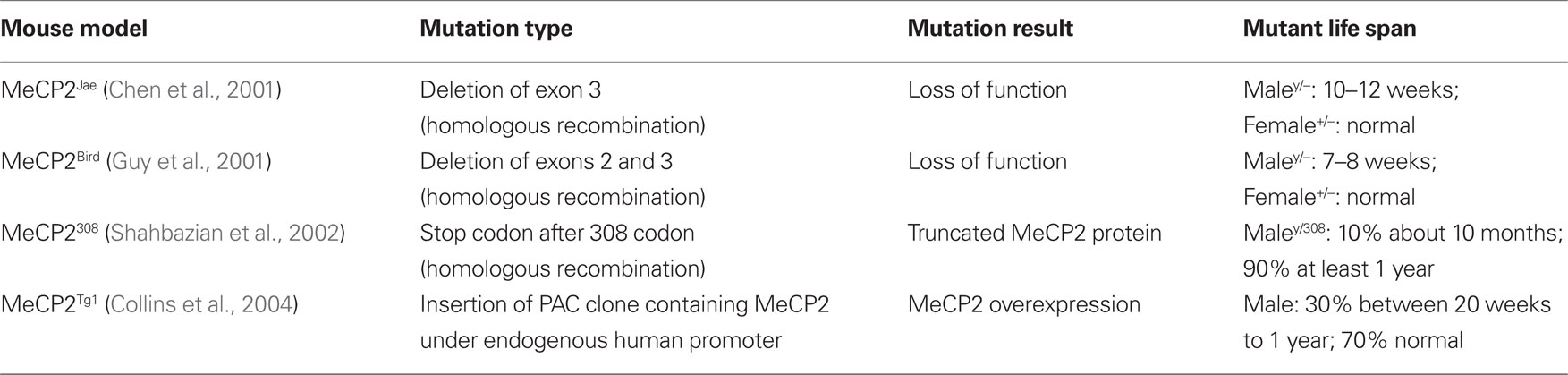
Table 1. Types of MeCP2 mouse mutants tested for defects in synaptic connectivity and plasticity.
Importantly, synaptic alterations produced by the manipulation of MeCP2 expression have been described in the brain of different mouse models of RS (see Table 2), further indicating that these animals are suitable to investigate the neurobiological bases of the disease. Indeed, newly generated neurons in the hippocampal dentate gyrus of 8 weeks old MeCP2Jae-KO male mice showed altered density and distribution of dendritic spines (Smrt et al., 2007). Moreover, recent studies reported that in the hippocampus and the primary motor (M1) cortex of both young and adult MeCP2Bird-KO and MeCP2Jae-KO male mice there is a decrease of dendritic spine density (Fukuda et al., 2005; Belichenko et al., 2009b; Tropea et al., 2009). Interestingly, neural circuits of MeCP2Bird-KO mice seem to be more severely affected as they also showed abnormalities in presynaptic structures reported as defects of axonal fasciculation in the M1 cortex. Finally, it was found that synaptic defects may appear early during development as a consequence of altered levels of MeCP2 expression. Indeed, VGLUT1 immunolabeling intensity is reduced in 2 weeks old MeCP2Bird-KO mice and increased in MeCP2Tg1 mutants. In contrast, these immunolabeling changes appeared to be milder at 5 weeks of age (Chao et al., 2007). Also, 6–7 months old MeCP2Bird heterozygous female mice displayed a decrease of dendritic spine density in layer V pyramidal neurons of the M1 cortex (Belichenko et al., 2009a). As observed in RS girls, patients, female MeCP2+/− mice show mosaic expression of mutant and wild-type Mecp2 alleles caused by the random inactivation of one X-linked Mecp2 allele (Chahrour and Zoghbi, 2007). The density of dendritic spines in neurons expressing MeCP2 seemed to be more severely affected than in MeCP2-negative ones, thus indicating both a cell autonomous and a non-cell autonomous effect of MeCP2 deletion in the organization of synaptic structures (Belichenko et al., 2009a).
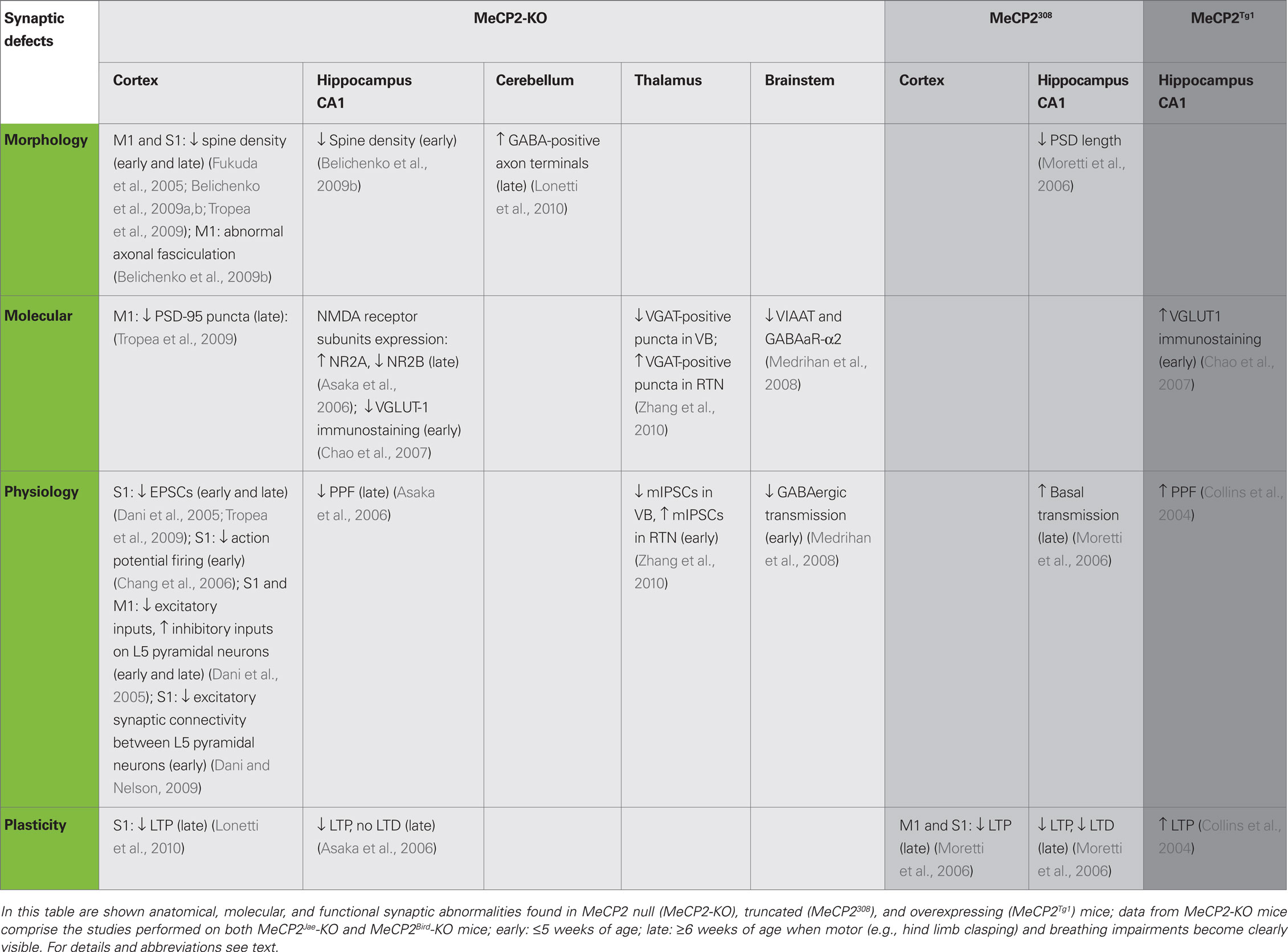
Table 2. Synaptic alterations in MeCP2 mice models.
Several electrophysiological properties of synaptic connections were also found abnormal in MeCP2 mutants (see Table 2). Whole-cell patch clamp recordings of layer V pyramidal neurons in the primary somatosensory (S1) cortex revealed a reduction of both spontaneous EPSCs and spontaneous action potential firing in MeCP2-KO male mice (Dani et al., 2005; Chang et al., 2006; Tropea et al., 2009). Intriguingly, the drive of excitatory inputs on layer V cortical neuron was reduced in MeCP2-KO animals whereas the total inhibitory input was enhanced, indicating that the absence of MeCP2 produces a shift of the homeostatic balance between excitation and inhibition in favor of inhibition (Dani et al., 2005). In addition, using quadruple whole-cell recordings on cortical slices obtained from 4 weeks old MeCP2-KO mice, it has been recently reported that excitatory synaptic connectivity between layer V thick-tufted pyramidal neurons is reduced (Dani and Nelson, 2009). In agreement with these functional impairments, immunofluorescence signal of the synaptic scaffolding protein PSD-95 is reduced in layer V of the M1 cortex of MeCP2Jae mutants compared with WT mice (Tropea et al., 2009). Significative alterations of neurosecretory processes have also been reported in association with MeCP2 deletion (Wang et al., 2006). This study indicated that cultured nodose ganglia neurons from P0 and P35 KO-mice show an increased secretion of BDNF in basal conditions compared with WT neurons. Moreover, these authors used an adrenal medulla slice preparation to show that the size of the ready releasable granule pool is increased in MeCP2-KO chromaffin cells. These data suggest that alterations of the level of both catecholamine and neurotrophin release may have an aversive effect on the correct formation of synaptic circuits in MeCP2 mutant mice during development (Wang et al., 2006).
So far, the analysis of the organization of synaptic circuits in mouse models of RS suggested that MeCP2 may have an important role in the formation/maturation of neuronal GABAergic connectivity (Table 2). In one study, the authors analyzed inhibitory circuits in the ventro-lateral medulla, an area involved in the generation of the respiratory rhythm, of P7 old MeCP2Bird-KO male mice (Medrihan et al., 2008). This brain area showed a decrease of GABAergic, but not glycinergic, inhibitory neurotransmission at P7. Moreover, they found that the expression levels of both mRNA and protein of the vesicular-aminobutyric acid transporter (VIAAT) as well as of GABAA-α2 receptor subunit is reduced in MeCP2-KO mice suggesting that very early molecular and cellular impairments of inhibitory circuits may occur in the brain stem of mutant animals. Moreover, it has been recently shown that GABAergic transmission is selectively altered in two different thalamic nuclei. While quantal GABAergic events (mIPSCs) are decreased in neurons of the ventral basal complex (VB) in P14-16 MeCP2-KO mice, mIPSCs are increased in neurons of the reticular thalamic nucleus (RTN) at this age (Zhang et al., 2010), a difference that was detected also in P21-23 mutant mice. However, changes in the functional properties of inhibitory synapses were found in VB but not in RTN neurons of P6 MeCP2-KO mice. Consistently, the VB of MeCP2-KO mice show fewer VGAT-positive immunopuncta than WT littermates whereas VGAT immunolabeling is increased in the RTN of mutants (Zhang et al., 2010). Very recently, we tackled the study of the structural organization of GABAergic inhibitory circuits in both the cerebral and the cerebellar cortex of MeCP2Jae-KO male mice (Lonetti et al., 2010). Using electron microscopy and immunogold labeling, we showed that at P54 both stellate cell- and basket cell-Purkinje cell GABA-positive synapses are more abundant in the cerebellum of mutants than in WT animals. In contrast, there was no difference between genotypes in the number of GABA-positive synaptic in layer III of the S1 cerebral cortex. Taken together, these data suggest that MeCP2 may subserve unique functions in diverse inhibitory neurons subpopulations throughout the brain.
In summary these studies strongly indicate that RS may alter different aspects of neuronal connectivity by affecting the structure, number, distribution and functional properties of synapses. The remainder of this review focuses mainly on the available data regarding the effects of different MeCP2 mutations on the expression of synaptic plasticity.
Synaptic Plasticity in MeCP2 Mutants
Synaptic plasticity is particularly high during specific temporal windows of development and it is crucial for correct circuit formation. Importantly, these developmental periods coincide with the period of emergence of RS symptoms, therefore impairments of synaptic plasticity could play an important role in the genesis of RS symptoms.
The first demonstration that alteration of MeCP2 expression may influence synaptic plasticity came from ex vivo electrophysiological recordings on brain slices. Asaka et al. (2006) showed that in the hippocampus of symptomatic MeCP2-KO mice basal synaptic transmission was normal whereas long-term synaptic plasticity was impaired. Long-term potentiation (LTP) of the Schaffer’s collateral-CA1 pathway (Sc-CA1) in the CA1 area of the hippocampus represents an activity-dependent increase of the strength of synaptic transmission that is thought to underlie many forms of explicit memory formation. Acute hippocampal slices from MeCP2-KO male mice (age >6 weeks) showed a clear reduction in the magnitude of LTP induced by high-frequency stimulation, an impairment that becomes visible minutes after the tetanic stimulation. However, LTP evoked by theta-burst stimulation, which reproduces a physiological pattern of neuronal stimulation, outlined synaptic defects that were present only in the maintenance but not in the induction phase of potentiation. Thus, these experiments indicate that the neuronal machinery underlying LTP is not entirely impaired by the loss of MeCP2 but it could affect the stimulus threshold for the induction/maintenance phases of synaptic potentiation (Asaka et al., 2006). Interestingly, LTP induction and maintenance in hippocampal slices from 3 to 5 weeks old KO mice appeared normal suggesting that synaptic plasticity in this area might be preserved at early postnatal stages. Similarly, Asaka et al. (2006) demonstrated that long-term depression (LTD) at hippocampal Sc-CA1 synapses, another form of learning-related synaptic plasticity, is abolished in KO male mice of age >6 weeks while no alterations were observed in 3–5 weeks old mutants in comparison with age-matched WT mice. Also short-term plasticity is affected in the hippocampus of MeCP2 mutants. Indeed, the study of presynaptic functions tested by paired-pulse facilitation (PPF) analysis revealed alterations in >6 weeks old animals, but not in younger MeCP2-KO male mice (Asaka et al., 2006). These electrophysiological data came with the intriguing observation that the molecular composition of the NMDA type of glutamate receptor (NMDAR) is abnormal in symptomatic KO mice (Table 2). Mutant animals displayed an increased expression of NR2B subunit and a decreased expression of NR2A subunit, without changes in the total amount of NMDARs (Asaka et al., 2006). These data indicate that the loss of MeCP2 may affect the normal shift from NR2B to NR2A subunit expression occurring during postnatal development, a molecular change that is important for specifying the biophysical and pharmacological properties of excitatory synapses in the adult brain. Finally, given that NMDARs are crucially involved in synaptic plasticity, these authors suggested that such defect in NMDARs expression at synapses might underlie the deficits in long-term hippocampal plasticity showed by symptomatic MeCP2-KO mice.
Likewise, male mice expressing a truncated form of MeCP2 obtained by positioning a premature stop sequence after 308 codon (MeCP2308) display impairments in synaptic plasticity (Table 2). These mutants show a reduction of both LTP and LTD at Sc-CA1 synapses in the hippocampus at 18–22 weeks, a defect in synaptic function that was accompanied by a reduction of the length of postsynaptic densities at asymmetric contacts (Moretti et al., 2006). Moreover, MeCP2308 mice showed increased basal synaptic transmission and decreased PPF at Sc-CA1 pathway suggesting that the release probability of synaptic vesicles is enhanced in this model (Moretti et al., 2006). In contrast, MeCP2Tg1 mice showed normal basal synaptic transmission but displayed an enhancement of both PPF and LTP measured in acute hippocampal slices, indicating that the effects of MeCP2 overexpression on synaptic plasticity in the hippocampus are opposite to those produced by the deletion of MeCP2 (Collins et al., 2004). More recently we tested synaptic plasticity in the S1 cortex of 8 weeks old MeCP2-KO mice. Field potential recordings from layer II–III in response to white matter-layer VI theta-burst stimulation revealed that in MeCP2Jae-KO male animals the response amplitude returned to baseline levels within 45 min, showing that the expression of LTP is severely impaired in the cerebral cortex of these mutants (Lonetti et al., 2010). These data are in agreement with another study indicating that cortical LTP defects manifest only in older mutants and, importantly, after the appearance of deficits in excitatory synaptic transmission in younger, 2- to 3-week-old, MeCP2-null mice (Dani and Nelson, 2009). Interestingly, truncated Mecp2308 male mice showed a similar reduction of LTP tested in layer II–III of the M1 and S1 cortices (Moretti et al., 2006).
MeCP2 Target Genes in Neuronal Cells
Despite significant advances in our understanding of the abnormalities in synaptic connectivity produced by MeCP2 mutation, the precise molecular causes are still far from being understood. MeCP2 protein shows a methyl-CpG binding domain (MBD), through that it can bind methylated-CpG islands on genomic DNA, and a transcription repressor domain (TRD), that allows the interaction with co-repressors Sin3 and histone deacetylases (HDACs) 1 and 2. When assembled in such molecular complex, MeCP2 is able to induce epigenetic modifications of chromatin structure leading to the silencing of downstream genes. Although MeCP2 has been widely considered a general inhibitor of transcription for years, a recent study has indicated that MeCP2 may also act as a gene activator in the brain through its association with CREB1, a major transcription factor that is crucially involved in activity-dependent modifications of synapses. MeCP2 gene activation function has been revealed by comparing results of genomic analyses conducted in the hypothalamus and in the cerebellum of WT mice and MeCP2 mutants (Chahrour et al., 2008; Ben-Shachar et al., 2009). Importantly, Chahrour et al. (2008) showed that MeCP2 may bind directly to the promoter region of Creb1, a strong indication that CREB1 might represent one of the activated MeCP2 target.
An important discovery in RS research was the demonstration that MeCP2 regulates the expression of brain-derived neurotrophic factor (bdnf) in neurons (Chen et al., 2003; Martinowich et al., 2003). BDNF is a neurotrophin crucially involved both in the formation of synaptic circuits during brain development (Huang et al., 1999) and in adult synaptic plasticity (Zakharenko et al., 2003; Monteggia et al., 2004). The loss of MeCP2 in mice leads to the misregulation of BDNF expression, an alteration with important consequences on the disease’s symptoms. Indeed, symptomatic MeCP2-KO male mice show decreased levels of BDNF in different brain areas (Chang et al., 2006; Chahrour et al., 2008; Lonetti et al., 2010). By crossing MeCP2 heterozygous female mutants with transgenic mice overexpressing bdnf resulted in MeCP2 mutant male mice with ameliorated symptoms, at both cellular and behavioral level (Chang et al., 2006). Thus far, whether the action of MeCP2 on bdnf gene expression is direct or whether is an indirect effect remains a matter of discussion. The original in vitro experiments have indicated that MeCP2 acts as a repressor of bdnf by binding to its promoter III (Chen et al., 2003; Martinowich et al., 2003). Moreover, these studies showed that neuronal activity leads to the release of MeCP2 from bdnf promoter region thus allowing its transcription. In contrast, a more recent study suggested that in the hypothalamus of mutants overexpressing MeCP2 bdnf transcripts are upregulated while in Mecp2-null animals bdnf is downregulated, a finding that is in support of the role of MeCP2 as a transcriptional activator (Chahrour et al., 2008). Therefore, whether the level reduction of BDNF shown by MeCP2-KO mice is a direct consequence of the loss of MeCP2 or whether it is an indirect effect of the decreased cortical activity shown by these animals is still not completely clear.
Other relevant MeCP2 targets are the imprinted genes ube3a (ubiquitin protein ligase E3A), gabrb3 (γ-aminobutyric-acid receptor β3) and dlx5 (distal-less homeobox 5) whose expression is altered both in RS patients and in MeCP2-KO mice (Horike et al., 2005; Makedonski et al., 2005; Samaco et al., 2005). Interestingly, ube3a is a gene that is found deregulated in other autism spectrum disorders, while dlx5 is involved in forebrain development and neurogenesis (Perera et al., 2004) as well as in the differentiation of GABAergic interneurons (Stühmer et al., 2002). Recently, gene expression analyses conducted on selected brain regions have pointed out interesting results, supporting the idea that MeCP2 may produce unique effects in specific neuronal populations. The study of the olfactory system showed that at least 13 different proteins with various functions (e.g., chromatin remodeling, energy metabolism, cell signaling, and neuroprotection) are abnormally expressed in MeCP2-KO male mice (Matarazzo and Ronnett, 2004). Microarray analyses in the hypothalamus revealed a massive transcriptional deregulation (2582 genes) in both MeCP2Tg1 and MeCP2-KO mice. These two mouse models largely shared the same misregulated genes strongly suggesting that these genes may represent direct targets of MeCP2 (Chahrour et al., 2008). To validate this hypothesis, these authors used chromatin immune-precipitation (Chip) and showed that MeCP2 can bind to the promoter region and activate the expression of genes encoding somatostatin (Sst), opioid receptor kappa 1 (Oprk1), guanidinoacetate methyltransferase (Gamt), G protein-regulated inducer of neurite outgrowth 1 (Gprin1). Intriguingly, GAMT deficiency has been found in patients showing severe mental retardation, absent or limited speech development, seizures, and hypotonia (Sykut-Cegielska et al., 2004; Caldeira Araújo et al., 2005). Moreover, other genes were found to be repressed by MeCP2, such as myocyte enhancer factor 2C (Mef2c) and ataxin 2 binding protein 1 (A2bp1). A2BP1 regulates the splicing of neuronal genes and its disruption seems to be involved in mental retardation, epilepsy and autism susceptibility (Chahrour et al., 2008). Finally, proteomic analysis of the cerebellum by the same authors showed that several molecules involved in the transmission of nerve impulse, dendrite development and neuroblast proliferation are upregulated in mice overexpressing MeCP2 (Ben-Shachar et al., 2009). Thus, despite the initial difficulties to find direct targets of MeCP2, the data obtained more recently using different RS animal models provide strong evidences that MeCP2 is involved in the regulation of genes crucial for the proper formation of neuronal circuits and synaptic plasticity. Importantly, for some of the genes target of MeCP2 there are evidences that their expression is misregulated in other neurological diseases associated with mental retardation further indicating that they are fundamental for normal brain functions.
Activity-Dependent Pathways Underlying MeCP2 Functions
Importantly, it has been recently disclosed a role of MeCP2 in the modulation of activity-dependent gene regulation in neurons (Chen et al., 2003) indicating that MeCP2 can be directly engaged in the molecular processes underlying dynamic changes of synapses in the brain. Membrane depolarization induced by KCl application on cortical neuronal cultures leads to the phosphorylation of MeCP2 that correlates with increased rates of transcription of bdnf (Chen et al., 2003) (Figure 1A). Indeed, Ca2+ influx in neurons produces covalent modifications on specific amino acid residues of the MeCP2 protein – e.g., S80 and S421 – (Chen et al., 2003; Zhou et al., 2006; Chao and Zoghbi, 2009) that modulate its binding affinity to the DNA in response to neuronal activity. It has been shown using mass spectrometry that human MeCP2 contains a phosphorylation site at Serine 80 (S80) that is also present both in mouse and rat proteins (Tao et al., 2009). Moreover, the analysis of brain extracts from epileptic mice revealed that multiple amino acid residues (S80, S399, T148/S149, S164, S229, S421, S424) in MeCP2 protein can be phosphorylated in mice after kainic acid-based seizure induction while only two sites, S80 and S399, were found phosphorylated in control mouse brains.
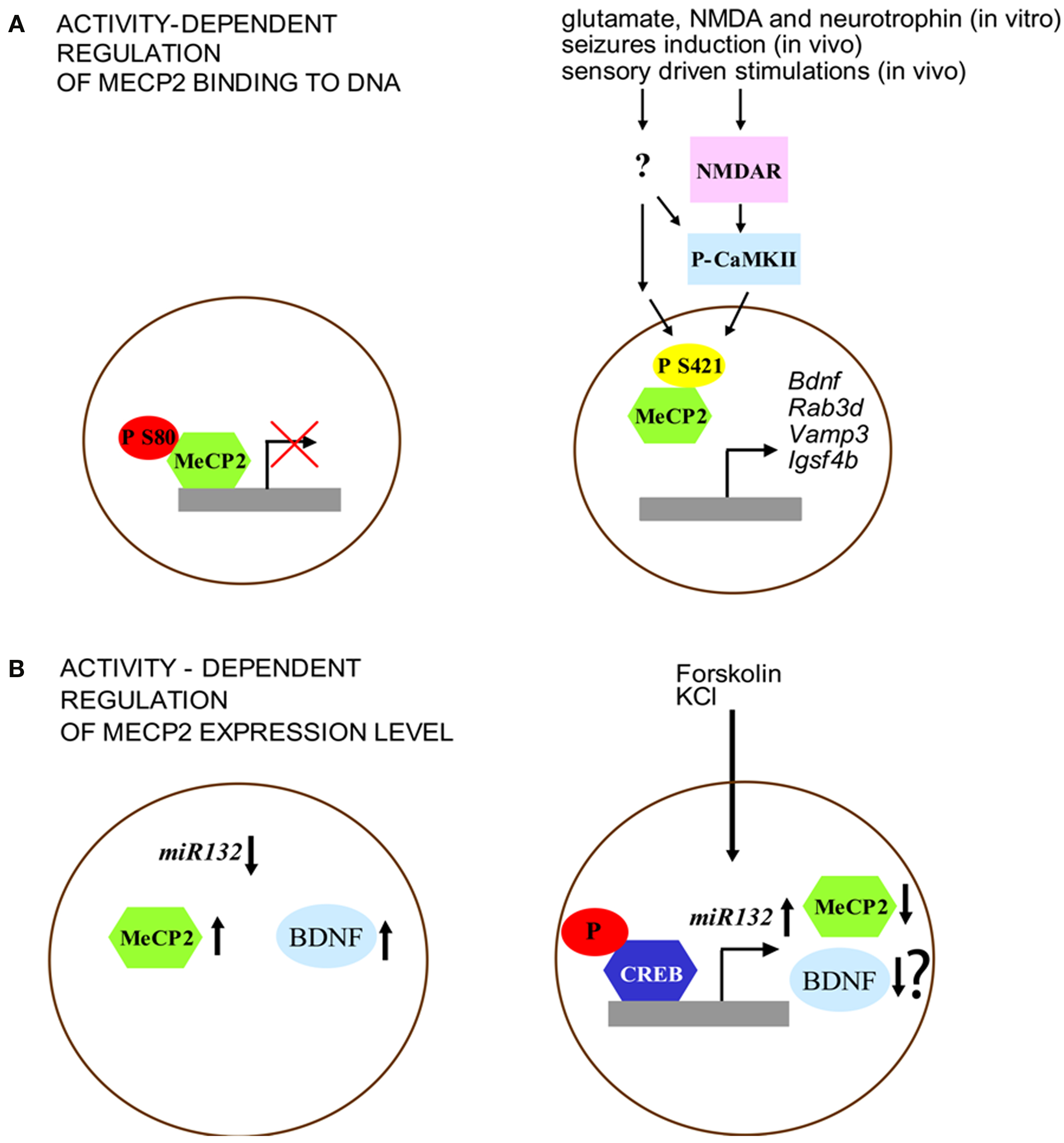
Figure 1. Activity-dependent regulation of MeCP2 expression and function. (A) MeCP2 binding to the promoter region is regulated by its phosphorylation. Left: under basal conditions, MeCP2 is phosphorylated at S80 site. Right: neuronal activity (e.g., through NMDA glutamate receptors and CaMKII activation) is associated with decreased phospho-S80 and increased levels of phospho-S421 MeCP2. (B) Expression of MeCP2 is negatively regulated by miR132 at the translational level. Left: MeCP2 overexpression, as well as blockade of miR132 repression increases BDNF III transcript levels. Right: Forskolin and KCl induce CREB phosphorylation that binds to the miR132 promoter. Increased levels of miR132 reduce MeCP2 expression. Although this model may induce to hypothesize a reduction of BDNF expression, it was not shown by the original paper of Klein et al. (2007) whether the interplay between miR132 and MeCP2, after forskolin and KCl treatment, may affect this neurotrophin level.
The different pattern of MeCP2 phosphorylation suggests that the function of this gene may be finely tuned through dynamic covalent modifications induced by external stimuli. Further support for this idea comes from the results of experiments showing that phosphorylation of MeCP2 at S80 and S421 is modulated in different directions by neuronal activity, probably reflecting diverse functional modifications. Brain extracts of mice that have undergone seizures showed a decrease of phospho-S80 MeCP2 levels while phospho-S421 MeCP2 was increased in these animals (Tao et al., 2009). While activity-dependent phosphorylation of S421 reduced the binding of MeCP2 to methylated DNA (Chen et al., 2003; Zhou et al., 2006), the phosphorylation of S80 seems to have an opposite effect (Figure 1A). Indeed MeCP2-S80A mutation decreases the binding level of MeCP2 at promoter region of target genes (i.e. Rab3d, Vamp3 and Igsf4b) indicating that phosphorylation at this site may be important for the modulation of transcription (Tao et al., 2009). On the other hand, it has been shown that MeCP2 can be phosphorylated at S421 selectively in the nervous system. This covalent modification is induced by synaptic activity both in vitro, through applications of glutamate, NMDA or neurotrophins on cultured hippocampal neurons, and in vivo in the parietal cortex by seizures induction and in the suprachiasmatic nucleus through sensory driven stimulations (Zhou et al., 2006). Moreover, MeCP2 phosphorylation on S421 can be mediated by the activation of the calcium-calmodulin kinase II (CaMKII), a protein kinase that is important for the molecular steps underlying the maintenance of later phases of synaptic plasticity as well as cognitive functions (Zhou et al., 2006). Finally, the mutation of MeCP2 at S421 in hippocampal organotypic slices blocks Ca2+-dependent bdnf transcription (Zhou et al., 2006).
More recently, it has been shown that MeCP2 expression can be regulated (Figure 1B) at the translational level by miR132, a microRNA (miRNA) highly expressed in the brain, through the binding of the miRNA recognition element in the MeCP2 3′-UTR (Klein et al., 2007). Indeed, treatments with both forskolin and KCl induce the phosphorylation of CREB and miR132 expression, a molecular modification that results in the decrease of MeCP2 level. Moreover, while the introduction of miR132 into primary cortical neurons lowers the level of expression of MeCP2 protein, the block of miR132 causes a very different effect by increasing the expression of BDNF (Figure 2B). In sum, these data lead to the conclusion that one of the main effect of the disruption of MeCP2 protein in the brain is a critical perturbation of activity-dependent gene transcription, a pathological process that may underlie the distinctive alterations of synaptic function and plasticity that are associated with RS.
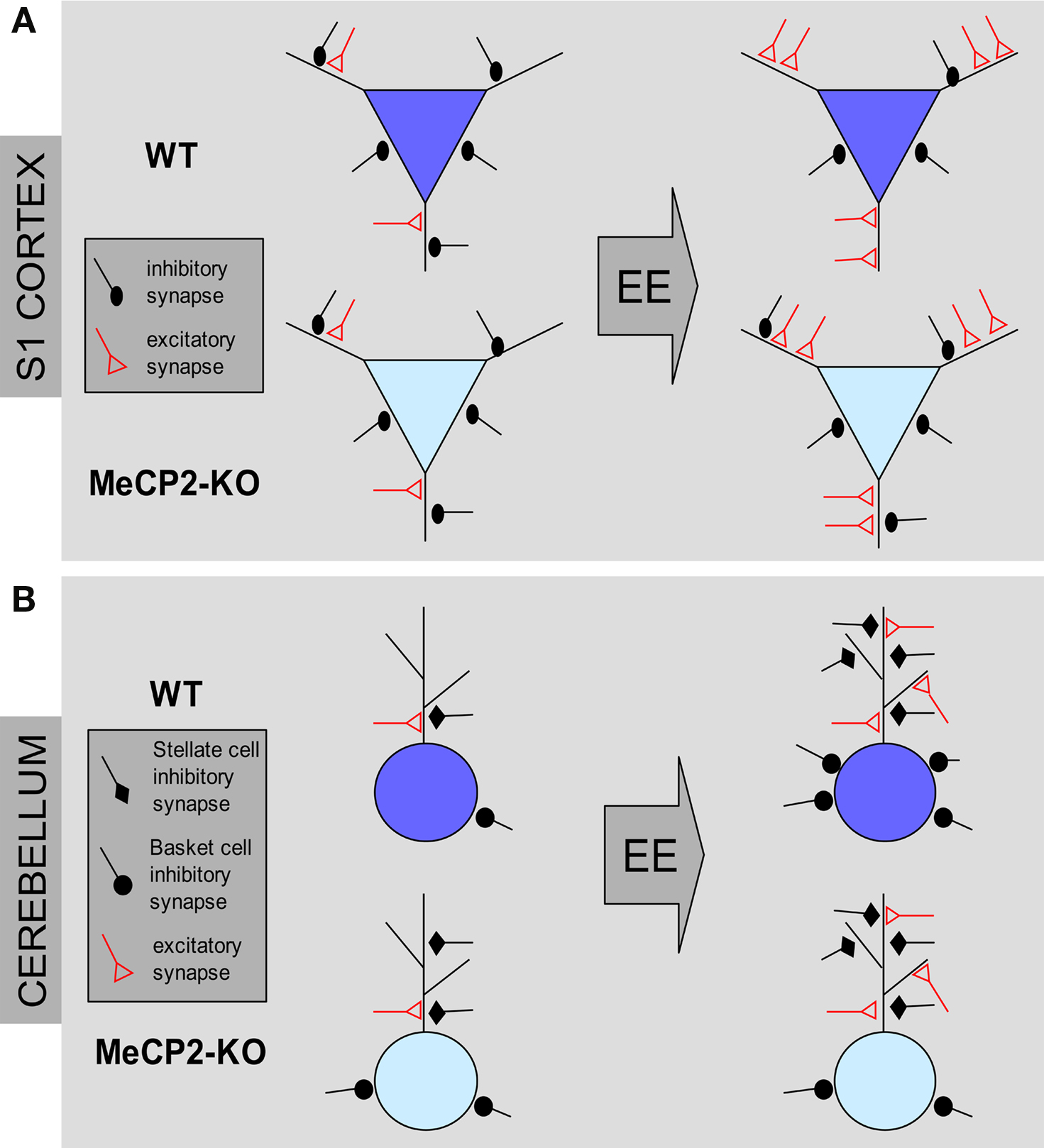
Figure 2. Structural effects of environmental enrichment on synaptic circuits of MeCP2 mutant mice. (A) MeCP2-KO and WT mice reared in standard conditions (ST) show similar excitatory and inhibitory synaptic density in the S1 cortex. Environmental enrichment (EE) leads to an increase of excitatory synapses and a loss of inhibitory synapses in WT male animals (blue pyramidal neurons). In KO male mice, EE produces an increase of excitatory contacts but no changes of inhibitory synapse density (cyan pyramidal neurons). (B) ST-MeCP2-KO mice show an increased number of inhibitory synapses on the soma and dendrites of Purkinje cells (PC) in the cerebellum compared to ST–WT. EE induce an increase of excitatory contacts in both genotypes. However, while EE increases the density of inhibitory synapses on both dendrites and soma in WT animals, it is able to increase only the density of inhibitory synapses on dendrites MeCP2-KO mice with no effect on inhibitory synapse density on the PC soma.
Reversing Synaptic Defects in Rett Syndrome Models
Studies aiming at rescuing RS phenotype in animal models showed that nerve cells lacking MeCP2 can respond, at least partially, to genetic, pharmacological and behavioral treatments (see Table 3) further stimulating the research of efficient therapies (for a recent in-depth review of this topic see Cobb et al., 2010). Indeed, symptoms amelioration shown by RS mouse models was associated with structural and functional modifications of synapses. By crossing MeCP2Jae-KO mice with transgenic mice overexpressing BDNF protein it was recently obtained a novel KO male mouse line (CAGGsBDNF;Mecp2KO) in which the expression level of BDNF was increased and the appearance of RS symptoms was significantly delayed (Chang et al., 2006). These mice showed in comparison with MeCP2Jae-KO male mice a significantly longer lifespan, improvements of locomotor functions and activity, as well as increased brain weight. Interestingly, increased BDNF levels were able to restore normal firing rate of layer V pyramidal neurons in the S1 cortex indicating that this genetic treatment is effective against synaptic alterations in RS. Finally by achieving the proper expression of MeCP2 in null-mutant mice, behavioral, cellular, and synaptic symptoms were reversed even in adult animals (Giacometti et al., 2007; Guy et al., 2007), implying that neurons are not permanently damaged by the absence of MeCP2. A remarkable set of experiments performed in the laboratory of Dr. Adrian Bird demonstrated that the disease in mice can be completely rescued, once the symptoms are fully expressed through the reintroduction of MeCP2 expression in cells (Guy et al., 2007). This work has taken advantage of a newly generated line of transgenic mice in which the endogenous MeCP2 is silenced through the insertion of a Stop cassette flanked by LoxP sequences (MeCP2LoxP-Stop). The induction of Cre expression obtained by tamoxifen (TM) injection in adult transgenic mice produced the deletion of the LoxP-Stop-LoxP region and allowed the expression of MeCP2. Acute daily TM-injection in MeCP2LoxP-Stop male mice at 3–4 weeks of age and the consequently reactivation of MeCP2 expression prevented the development of symptoms and mutant mice survived without differences from WT mice. Moreover, the reactivation of MeCP2 expression by TM-injection in MeCP2LoxP-Stop heterozygous female that showed typical neurological defects was able to revert the behavioral symptoms and rescued the abnormalities of LTP expression induced by theta-burst stimulation in the hippocampus. These data demonstrated that the absence of normal levels of MeCP2 during development does not irreversibly damage both neuronal function and the molecular machinery underlying synaptic circuit properties, and most importantly, that most of RS symptoms may be reversible (Guy et al., 2007).

Table 3. Synaptic effects of pharmacological and behavioral treatments in MeCP2 mutant mice.
Although genetic interventions have proven very promising in resolving, or at least ameliorating, neuropathological signs of RS in animal models, the application of such therapies in humans are not foreseen to be accessible in a near future. Thus, a primary goal of RS research is to focus toward the development of affordable non-genetic therapies aimed to resolve malfunctions in neural circuits. Intriguingly, a recent pharmacological treatment in MeCP2-KO mice with insulin-like growth factor 1 (IGF-1) produced very promising results both at the cellular and the behavioral level (Tropea et al., 2009). It has been previously demonstrated that IGF-1, a pleiotrophic growth factor that is able to cross the hematoencephalic barrier, has beneficial effect on cognition in aging animals (Markowska et al., 1998; Trejo et al., 2004). Moreover transgenic mice with IGF-1 deficiency show defects in hippocampal LTP and reduction of glutamatergic boutons (Trejo et al., 2007). Importantly, it was shown that prolonged systemic administration of IGF-1 is able to rescue these synaptic alterations (Trejo et al., 2007). Tropea et al. (2009) showed that the early (2 weeks after birth) intraperitoneal injection of IGF-1 improved survival, locomotory activity, breathing irregularity, and cardiac arrhythmias in MeCP2Jae-KO male mice Tropea et al. (2009). Together with these behavioral and systemic improvements, IGF-1 was able to partially restore brain weight deficits in MeCP2 mutants and was also effective on synapses. Indeed, the motor cortex of KO-mice treated with IGF-1 showed an augmented level of expression of the postsynaptic protein PSD-95 compared with untreated MeCP2-KO animals. This result was paralleled by an increase in dendritic spine density and in spontaneous EPSCs frequency of layer V pyramidal neurons of the M1 cortex. Moreover, the administration of IGF-1 was effective in rescuing synaptic defects in heterozygous female (Tropea et al., 2009). These authors found that the neural circuits of the visual cortex of adult MeCP2Jae heterozygous females showed typical synaptic features of immature animals when tested for ocular dominance plasticity induced by monocular deprivation. Importantly, this synaptic phenotype was normalized in mutants that received IGF-1 treatment (Tropea et al., 2009), a result further supporting the idea that activity-dependent synaptic abnormalities shown by MeCP2 mutants may be reversed by an appropriate treatment.
Recently we asked whether environmental enrichment (EE), a behavioral treatment that is known to induce structural remodeling in the brain, was able to produce beneficial effects on the phenotypic features expressed by null MeCP2 mice (Lonetti et al., 2010). The correct formation of neural circuits requires experience-dependent stabilization and remodeling of synaptic connections. When reared in EE animals are placed in large groups in cages containing toys and running wheels enhancing and facilitating social interactions with other mice, as well as sensory-motor and cognitive stimulations. It has been previously shown that this housing condition exerts several beneficial effects on many mouse models of neurological disorders like Huntington, Parkinson, and Alzheimer disease at a behavioral, cellular, and molecular level (Nithianantharajah and Hannan, 2006). Moreover, this treatment is able to enhance synapse formation and plasticity and to increase the production of BDNF, a neurotrophin that is strongly reduced in MeCP2-KO mice. Intriguingly, we have shown that rearing of MeCP2Jae-KO mice in enriched conditions from P10 rescued cortical LTP deficits shown by mutants reared in standard conditions (Lonetti et al., 2010). Moreover, using unbiased stereology applied to electron microscopy, we found that both in the III layer of the S1 cortex and in the molecular layer of the cerebellum EE is able to induce the growth of new excitatory synapses both in WT and KO male mice, indicating that nerve cells lacking MeCP2 may show structural remodeling of excitatory synapses in response to environmental stimulation (Figure 2). Finally, combined with the beneficial effects at the synaptic level, we found that early intervention also increased the levels of BDNF expression in the cortex of MeCP2 mutants and ameliorated several behavioral features of RS (Lonetti et al., 2010).
Activity-Dependent Defects of Inhibitory Synapses in MeCP2-Null Brain
The employment of early EE allowed us also to disclose defects at GABAergic synapses produced by MeCP2 null mutation that were not clearly evident in mutant mice reared in ST conditions (Figure 2). MeCP2 mutants in ST conditions display WT-like numbers of GABA-positive inhibitory synapses in the S1 area of the cortex. Whereas S1 cortices of WT animals showed a robust reduction in the density of GABAergic axo-dendritic contacts in EE conditions, these contacts were unchanged in MeCP2Jae-KO mice after EE (Lonetti et al., 2010). Intriguingly, no change was detected in the number of axo-somatic inhibitory synapses between WT and KO animals in both ST and EE rearing conditions.
Thus, in WT animals EE results in an increased ratio between excitatory and inhibitory synapses due to a new growth of excitatory synapses and a parallel retraction of inhibitory synapses (see also Nithianantharajah and Hannan, 2006; Alvarez and Sabatini, 2007; Sale et al., 2007). This effect was present also in the cortex of EE MeCP2-KO mice, but differently from normal animals it derived exclusively from an increase of excitatory synapses with no changes in inhibitory synapse number. These results thus lead to the interesting conclusion that structural synaptic plasticity in the S1 cortex of 8 weeks old MeCP2-KO mice is normal in excitatory circuits whereas is altered in inhibitory circuits. Furthermore, we found that inhibitory circuits in the cerebellum responded differently to EE (Lonetti et al., 2010). Indeed, both in WT and in MeCP2-KO animals stellate cells-Purkinje cells GABAergic synapse density was strongly increased under EE conditions. However, in the same region we found that perisomatic synapse number between basket cells and Purkinje cells is increased in EE WT mice but not in EE KO mice, thus indicating that MeCP2 deletion may produce cell-specific impairments in structural plasticity of GABAergic synapses in the cerebellum. Altogether, these data strongly suggest that, in addition to alterations in excitatory connectivity, cortical interneurons could be important substrates of the phenotypic alterations of MeCP2 mutants and that the integrity of GABAergic circuits is affected in RS.
Conclusions
The wide range of the phenotypes shown both by patients with MeCP2 mutations and murine models carrying alterations in MeCP2 expression and function indicated that behavioral abnormalities stem from MeCP2 dysfunction taking place in specific neuronal subpopulations. Indeed, genetic manipulations that produced the removal of MeCP2 functions in selected brain areas (e.g., hypothalamus) or neuronal types (e.g., serotonergic, dopaminergic, and noradrenergic) caused specific behavioral, cellular, and molecular changes (Fyffe et al., 2008; Samaco et al., 2009). Similarly, several evidences have indicated that loss of MeCP2 may affect synaptic connectivity with variable degrees of intensity in specific brain areas and neural circuits. For example, the loss of MeCP2 seems to affect structural plasticity of inhibitory synapses induced by behavioral stimulation only within selected populations of interneurons both in the cerebral and the cerebellar cortex. In contrast, the capabilities to respond with structural changes to external stimulation are preserved at excitatory synapses in MeCP2-KO mice (Lonetti et al., 2010). Therefore, it is clear that a detailed brain map of the anatomical and physiological alterations in synaptic plasticity is needed to understand how neuronal circuits are ultimately compromised in RS. Moreover, testing the temporal progression of the neural dysfunctions produced by MeCP2 mutation will contribute to an earlier identification of RS thus providing useful indications to model efficient protocols for future therapies. Although an apparently normal early development had initially been considered as one of the criteria for RS, several retrospective studies indicated the disorder to manifest during the first year of life (e.g., Kerr and Witt Engerström, 2001). While alterations have been reported in dendritic spine morphology of MeCP2 mutant mice at early ages ( Belichenko et al., 2009b; our unpublished observations), little is known about the structure and function of dendritic spines and synapses in very young mutants. Knowledge about the exact mechanisms of synapse formation, stabilization, or pruning that are affected by MeCP2 mutation at early stages of the disease could reveal the primary deficits underlying the phenotype of MeCP2 mutants. Intriguingly, we found that early EE improves several phenotypes of female MeCP2+/− and male MeCP2Jae-KO mice(Lonetti et al., 2010). By contrast, late onset EE has more subtle effects (Kondo et al., 2008; Nag et al., 2009) suggesting that early EE could be particularly valuable to normalize the initial pathological signs occurring during early postnatal development. Thus, it is reasonable to envision that early intervention after early diagnosis might achieve the best results in RS patients. Finally, the results showing that the treatments used so far produced only partial recovery of the symptoms indicate that there are unknown neuronal defects that are particularly resilient. Thus, future studies focused on the characteristics of population-specific function of MeCP2 in neurons will prevent possible confounding dilution effects due to the extreme neuronal heterogeneity existing in the brain, and will provide new knowledge helping to identify the molecular bases of the clinical problems.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Sources of support: Fondazione Telethon, Italy (GGP05236 and GGP09196 to Maurizio Giustetto and Tommaso Pizzorusso); ERA-Net for research programs on rare diseases, E-RARE EuroRETT network (to Maurizio Giustetto and Tommaso Pizzorusso); MIUR-PRIN (to Maurizio Giustetto and Tommaso Pizzorusso), Fondazione SanPaolo, Italy (to Maurizio Giustetto), Non Invasive Nanotransducer for In Vivo gene therapy FP6 European Union, Imaging function and dysfunction of neuronal circuits in the visual cortex (EUROV1SION), Promotion of plasticity as a treatment for neurodegenerative conditions (PLASTICISE) FP7 European Union projects to Tommaso Pizzorusso. We thank Dr. Noemi Morello and Dr. Eleonora Calcagno for critically reading previous versions of this manuscript.
References
Alvarez, V. A., and Sabatini, B. L. (2007). Anatomical and physiological plasticity of dendritic spines. Annu. Rev. Neurosci. 30, 79–97.
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188.
Asaka, Y., Jugloff, D. G., Zhang, L., Eubanks, J. H., and Fitzsimonds, R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 21, 217–227.
Belichenko, N. P., Belichenko P. V., and Mobley, W. C. (2009a). Evidence for both neuronal cell autonomous and nonautonomous effects of methyl-CpG-binding protein 2 in the cerebral cortex of female mice with Mecp2 mutation. Neurobiol. Dis. 34, 71–77.
Belichenko, P. V., Wright, E. E., Belichenko, N. P., Masliah, E., Li, H. H., Mobley, W. C., and Francke, U. (2009b). Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J. Comp. Neurol. 514, 240–258.
Belichenko, P. V., Oldfors, A., Hagberg, B., and Dahlstrom, A. (1994). Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport 5, 1509–1513.
Ben-Shachar, S., Chahrour, M., Thaller, C., Shaw, C. A., and Zoghbi, H. Y. (2009). Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 18, 2431–2442.
Caldeira Araújo, H., Smit, W., Verhoeven, N. M., Salomons, G. S., Silva, S., Vasconcelos, R., Tomás, H., Tavares de Almeida, I., Jakobs, C., and Duran, M. (2005). Guanidinoacetate methyltransferase deficiency identified in adults and a child with mental retardation. Am. J. Med. Genet. A 133A, 122–127.
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T. C., Qin, J., and Zoghbi, H. Y. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229.
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437.
Chang, Q., Khare, G., Dani, V., Nelson, S., and Jaenisch, R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348.
Chao, H., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 1–8.
Chao, H. T., and Zoghbi, H. Y. (2009). The yin and yang of MeCP2 phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106, 4577–4578.
Chapleau, C. A., Calfa, G. D., Lane, M. C., Albertson, A. J., Larimore, J. L., Kudo, S., Armstrong, D. L., Percy, A. K., and Pozzo-Miller, L. (2009). Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 35, 219–233.
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331.
Chen, W. G., Chang, Q., Lin, Y., Meissner, A., West, A. E., Griffith, E. C., Jaenisch, R., and Greenberg, M. E. (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302, 885–889.
Cobb, S., Guy, J., and Bird, A. (2010). Reversibility of functional deficits in experimental models of Rett syndrome. Biochem. Soc. Trans. 38, 498–506.
Collins, A. L., Levenson, J. M., Vilaythong, A. P., Richman, R., Armstrong, D. L., Noebels, J. L., Sweatt, D. J., and Zoghbi, H. Y. (2004). Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 13, 2679–2689.
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 12560–12565.
Dani, V. S., and Nelson, S. B. (2009). Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of Rett syndrome. J. Neurosci. 29, 11263–11270.
Fukuda, T., Itoh, M., Ichikawa, T., Washiyama, K., and Goto, Y. (2005). Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J. Neuropathol. Exp. Neurol. 64, 537–544.
Fyffe, S. L., Neul, J. L., Samaco, R. C., Chao, H. T., Ben-Shachar, S., Moretti, P., McGill, B. E., Goulding, E. H., Sullivan, E., Tecott, L. H., and Zoghbi, H. Y. (2008). Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron 59, 947–958.
Giacometti, E., Luikenhuis, S., Beard, C., and Jaenisch, R. (2007). Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc. Natl. Acad. Sci. U.S.A. 104, 1931–1936.
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147.
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326.
Horike, S., Cai, S., Miyano, M., Cheng, J. F., and Kohwi-Shigematsu, T. (2005). Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37, 31–40.
Huang, Z. J., Kirkwood, A., Pizzorusso, T., Porciatti, V., Morales, B., Bear, M. F., Maffei, L., and Tonegawa, S. (1999). BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell 98, 739–755.
Jellinger, K., Armstrong, D., Zoghbi, H. Y., and Percy, A. K. (1988). Neuropathology of Rett syndrome. Acta Neuropathol. 76, 142–158.
Johnston, M. V., Blue, M. E., and Naidu, S. (2005). Rett syndrome and neuronal development. J. Child Neurol. 20, 759–763.
Kerr, A. M., and Witt Engerström I. (2001). Rett Disorder and the Developing Brain. Oxford: Oxford University Press.
Kishi, N., and Macklis, J. D. (2004). MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 27, 306–321.
Klein, M. E., Lioy, D. T., Ma, L., Impey, S., Mandel, G., and Goodman, R. H. (2007). Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 10, 1513–1514.
Kondo, M., Gray, L. J., Pelka, G. J., Christodoulou, J., Tam, P. P., and Hannan, A. J. (2008). Environmental enrichment ameliorates a motor coordination deficit in a mouse model of Rett syndrome – Mecp2 gene dosage effects and BDNF expression. Eur. J. Neurosci. 27, 3342–3350.
Lonetti, G., Angelucci, A., Morando, L., Boggio, E. M., Giustetto, M., and Pizzorusso, T. (2010). Early environmental enrichment moderates the behavioral and synaptic phenotype of MeCP2 null mice. Biol. Psychiatry 67, 657–665.
Makedonski, K., Abuhatzira, L., Kaufman, Y., Razin, A., and Shemer, R. (2005). MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affects UBE3A expression. Hum. Mol. Genet. 14, 1049–1058.
Markowska, A. L., Mooney, M., and Sonntag, W. E. (1998). Insulin-like growth factor-1 ameliorates age- related behavioral deficits. Neuroscience 87, 559–569.
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., Fan, G., and Sun, Y. E. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893.
Matarazzo, V., and Ronnett, G. V. (2004). Temporal and regional differences in the olfactory proteome as a consequence of MeCP2 deficiency. Proc. Natl. Acad. Sci. U.S.A. 101, 7763–7768.
Medrihan, L., Tantalaki, E., Aramuni, G., Sargsyan, V., Dudanova, I., Missler, M., and Zhang, W. (2008). Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol. 99, 112–121.
Monteggia, L. M., Barrot, M., Powell, C. M., Berton, O., Galanis, V., Gemelli, T., Meuth, S., Nagy, A., Greene, R. W., and Nestler, E. J. (2004). Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc. Natl. Acad. Sci. U.S.A. 101, 10827–10832.
Moretti, P., Levenson, J. M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B., Armstrong, D., Arancio, O., Sweatt, J. D., and Zoghbi, H. Y. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327.
Nag, N., Moriuchi, J. M., Peitzman, C. G., Ward, B. C., Kolodny, N. H., and Berger-Sweeney, J. E. (2009). Environmental enrichment alters locomotor behaviour and ventricular volume in Mecp2(1lox) mice. Behav. Brain Res. 196, 44–48.
Nelson, E. D., Kavalali, E. T., and Monteggia, L. M. (2006). MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr. Biol. 16, 710–716.
Nithianantharajah, J., and Hannan, A. J. (2006). Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat. Rev. Neurosci. 7, 697–709.
Perera, M., Merlo, G. R., Verardo, S., Paleari, L., Corte, G., and Levi, G. (2004). Defective neuronogenesis in the absence of Dlx5. Mol. Cell. Neurosci. 25, 153–161.
Reiss, A. L., Faruque, F., Naidu, S., Abrams, M., Beaty, T., Bryan, R. N., and Moser, H. (1993). Neuroanatomy of Rett syndrome: a volumetric imaging study. Ann. Neurol. 34, 227–243.
Sale, A., Maya Vetencourt, J. F., Medini, P., Cenni, M. C., Baroncelli, L., De Pasquale, R., and Maffei, L. (2007). Environmental enrichment in adulthood promotes amblyopia recovery through a reduction of intracortical inhibition. Nat. Neurosci. 10, 679–681.
Samaco, R. C., Hogart, A., and LaSalle, J. M. (2005). Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 14, 483–492.
Samaco, R. C., Mandel-Brehm, C., Chao, H. T., Ward, C. S., Fyffe-Maricich, S. L., Ren, J., Hyland, K,, Thaller, C., Maricich, S. M., Humphreys, P., Greer, J. J., Percy, A., Glaze, D. G., Zoghbi, H. Y., and Neul, J. L. (2009). Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc. Natl. Acad. Sci. U.S.A. 106, 21966–21971.
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., Armstrong, D., Paylor, R., and Zoghbi, H. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254.
Smrt, R. D., Eaves-Egenes, J., Barkho, B. Z., Santistevan, N. J., Zhao, C., Aimone, J. B., Gage, F. H., and Zhao, X. (2007). Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol. Dis. 27, 77–89.
Stearns, N. A., Schaevitz, L. R., Bowling, H., Nag, N., Berger, U. V., and Berger-Sweeney, J. (2007). Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience 146, 907–921.
Stühmer, T., Anderson, S. A., Ekker, M., and Rubenstein, J. L. (2002). Ectopic expression of the Dlx genes induces glutamic acid decarboxylase and Dlx expression. Development 129, 245–252.
Subramaniam, B., Naidu, S., and Reiss, A. L. (1997). Neuroanatomy in Rett syndrome: cerebral cortex and posterior fossa. Neurology 48, 399–407.
Sykut-Cegielska, J., Gradowska, W., Mercimek-Mahmutoglu, S., and Stöckler-Ipsiroglu, S. (2004). Biochemical and clinical characteristics of creatine deficiency syndromes. Acta Biochim. Pol. 51, 875–882 (review).
Tao, J., Hu, K., Chang, Q., Wu, H., Sherman, N. E., Martinowich, K., Klose, R. J., Schanen, C., Jaenisch, R., Wang,, W., and Sun, Y. E. (2009). Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc. Natl. Acad. Sci. U.S.A. 106, 4882–4887.
Trejo, J. L., Carro, E., Lopez-Lopez, C., and Torres-Aleman, I. (2004). Role of serum insulin-like growth factor I in mammalian brain aging. Growth Horm. IGF Res. 14(Suppl. A), 39–43.
Trejo, J. L., Piriz, J., Llorens-Martin, M. V., Fernandez, A. M., Bolós, M., LeRoith, D., Nuñez, A., and Torres-Aleman, I. (2007). Central actions of liver-derived insulin-like growth factor I underlying its pro-cognitive effects. Mol. Psychiatry 12, 1118–1128.
Tropea, D., Giacometti, E., Wilson, N. R., Beard, C., McCurry, C., Fu, D. D., Flannery, R., Jaenisch, R., and Sur, M. (2009). Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. U.S.A. 106, 2029–2034.
Wang, H., Chan, S. A., Ogier, M., Hellard, D., Wang, Q., Smith, C., and Katz, D. M. (2006). Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J. Neurosci. 26, 10911–10915.
Zakharenko, S. S., Patterson, S. L., Dragatsis, I., Zeitlin, S. O., Siegelbaum, S. A., Kandel, E. R., and Morozov, A. (2003). Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron 39, 975–990.
Zhang, Z. W., Zak, J. D., and Liu, H. (2010). MeCP2 is required for normal development of GABAergic circuits in the thalamus. J. Neurophysiol. 103, 2470–2481.
Zhou, Z., Hong, E. J., Cohen, S., Zhao, W. N., Ho, H. Y., Schmidt, L., Chen, W. G., Lin, Y., Savner, E., Griffith, E. C., Hu, L., Steen, J. A., Weitz, C. J., and Greenberg, M. E. (2006). Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52, 255–269.
Keywords: MeCP2, autism spectrum disorders, LTP, GABA, inhibitory synapses, environmental enrichment
Citation: Boggio EM, Lonetti G, Pizzorusso T and Giustetto M (2010) Synaptic determinants of Rett syndrome. Front. Syn. Neurosci. 2:28. doi: 10.3389/fnsyn.2010.00028
Received: 02 April 2010;
Paper pending published: 23 April 2010;
Accepted: 16 June 2010;
Published online: 06 August 2010
Edited by:
Susana Cohen-Cory, University of California, USAReviewed by:
Angel Barco, Instituto de Neurociencias de Alicante, SpainGraziella DiCristo, CHU Sainte Justine-University of Montreal, Canada
Copyright: © 2010 Boggio, Lonetti, Pizzorusso and Giustetto. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Maurizio Giustetto, Dipartimento di Anatomia, Farmacologia e Medicina Legale, Università di Torino, C.so Massimo d’Azeglio, 52, I-10126 Torino, Italy. e-mail: maurizio.giustetto@unito.it