 Alfonso Represa
Alfonso Represa- INSERM, Institut de Neurobiologie de la Méditerranée, Aix-Marseille University, Marseille, France
Malformations of cortical development (MCDs), a complex family of rare disorders, result from alterations of one or combined developmental steps, including progenitors proliferation, neuronal migration and differentiation. They are an important cause of childhood epilepsy and frequently associate cognitive deficits and behavioral alterations. Though the physiopathological mechanisms of epilepsy in MCD patients remain poorly elucidated, research during the past decade highlighted the contribution of some factors that will be reviewed in this paper and that include: (i) the genes that caused the malformation, that can be responsible for a significant reduction of inhibitory cells (e.g., ARX gene) or be inducing cell-autonomous epileptogenic changes in affected neurons (e.g., mutations on the mTOR pathway); (ii) the alteration of cortical networks development induced by the malformation that will also involve adjacent or distal cortical areas apparently sane so that the epileptogenic focus might be more extended that the malformation or even localized at distance from it; (iii) the normal developmental processes that would influence and determine the onset of epilepsy in MCD patients, particularly precocious in most of the cases.
Introduction
Malformations of cortical development (MCD) are a complex family of rare disorders that result from alterations of one or combined developmental steps, including proliferation of neural progenitors, migration of neuroblasts to the cortical plate, layer organization and neuronal maturation (Barkovich et al., 2012). Thus, in general, alterations of neuronal and glial proliferation associating neuronal dysgenesis are a cause of focal cortical dysplasia type II (FCD-type II); alterations of neuronal migration leading to ectopic localization of neurons are a cause of periventricular nodular heterotopia (neurons accumulate along the ventricular walls) and subcortical band heterotopia (SBH; neurons accumulate in the white matter between the cortex and the ventricular wall); alterations on processes subsequent to neuronal migration are at the origin of polymicrogyria, characterized by a cortex organized in multiple small gyri (Barkovich et al., 2012).
Malformations of cortical development are an important cause of childhood epilepsy. Though the precise incidence of MCDs is not known, it is estimated that they account for up to 40% of cases of intractable or medication-resistant childhood epilepsies (Barkovich et al., 2012; Guerrini and Dobyns, 2014) and that at least 75% of the patients with MCDs will have epilepsy (Leventer et al., 1999). Associated to seizures, the patients display different comorbidities, particularly cognitive deficits, that are more frequent and severe when epilepsy begins early in life (Berg et al., 2017). MCDs consequently represent a severe burden for patients, families and society.
Though the physiopathological mechanisms of epilepsy in MCD patients remain unclear, clinical and experimental data suggest that epileptogenesis results from diverse developmental processes that can be cell autonomous or not, directly imputable to the genetic cause of the malformation or linked to an abnormal development of neuronal networks. In this review, we will discuss the pathophysiological mechanisms involved in MCD epileptogenesis through the analysis of three distinct matters: the genetic causes of MCDs, the localization of the epileptogenic focus and the age at the epilepsy onset in patients and animal models.
Causative Genes and Epileptogenesis
The classification of cortical malformations according to the developmental step involved (proliferation, migration or differentiation) or the type of malformation generated (e.g., gray matter heterotopia, lissencephaly, focal cortical dysplasia) does not provide clear indications about the epileptogenic process. However, the analysis of causative genes and their respective cellular roles, provided in some cases interesting data that would help the understanding of the epileptogenic process.
The pathogenesis of MCDs is multifactorial involving different genes and environmental factors. There is a large and increasing number of genes identified during the past decade as causative of MCD (Guerrini and Dobyns, 2014). This is an exciting research field that is also contributing to our knowledge of genetic factors controlling brain development. However, the objective of this report is not to provide a complete overview of the genetic causes of MCDs, but to discuss some examples that help our understanding of epileptogenesis.
One of the more fascinating genes causing brain malformation is ARX (aristaless related homeobox gene). In vitro and in vivo studies have shown that the Arx gene is both a positive and a negative regulator of gene transcription important for brain development (Collombat et al., 2003; Seufert et al., 2005; McKenzie et al., 2007; Fullenkamp and El-Hodiri, 2008). Among the roles of this gene are the regionalization of the brain, the proliferation of cortical progenitors, the migration of interneurons and early commitment of cholinergic neurons (Colombo et al., 2004; Marsh et al., 2009, 2016; Friocourt and Parnavelas, 2010). Numerous mutations of the ARX gene have been reported in more than a dozen different early neurological disorders, where intellectual disability is associated or not with epileptic seizures (Bienvenu et al., 2002; Kitamura et al., 2002; Stromme et al., 2002). These conditions do or do not associate brain malformations during embryonic development (Shoubridge et al., 2010). Phenotypic heterogeneity may, in part, be explained by the nature and location of ARX mutations (Kato et al., 2004; Olivetti and Noebels, 2012). Indeed, phenotypes without malformation are mainly caused by mutations that are in the polyalanine domains and outside the homeodomain. Conversely, the more severe phenotypes with brain malformation are mostly associated with mutations leading to protein truncation or located in the homeodomain; this is the case for the XLAG syndrome characterized by a severe lissencephaly, agenesis of the corpus callosum and abnormal genitalia. Animal models so far generated have shown that Arx deficient mice or Knockin mice displaying Arx mutations associate a more or less pronounced reduction of cortical GABAergic and cholinergic neurons (reviewed in Olivetti and Noebels, 2012) and the analysis of post-mortem brain tissue reported a three-layered cortex containing exclusively pyramidal neurons in XLAG patients from three different families (Bonneau et al., 2002). In the context of developmental malformations, ARX related syndromes can be thus considered as “interneuronopathies” and the epilepsy and cognitive deficits reported in patients and animal models, would be directly related to the reduction of inhibition. Glutamatergic neurons do not express ARX and are not directly affected by the mutation of ARX but support the consequences and are thus responsible for the expression of epileptic seizures.
Mutations affecting the activation of the mammalian target of rapamycin (mTOR)-signaling pathway have been identified in focal malformations of cortical development associating alterations of progenitor cell proliferation, defective neuronal migration and lamination and the presence of cytomegalic neurons and balloon cells as a result of a defective differentiation program of cortical cells. These malformations include FCD type II and Hemimegalencephaly (Harvey et al., 2008; Blümcke et al., 2011; D’Gama et al., 2017). Tuberous sclerosis, a rare multisystem genetic disease condition that in the brain generates cortical tubers (focal distortions in cellular organization and morphology which extend into the subcortical white matter) is also caused by a hyperactivation of mTORC1, due to mutations in either TSC1 or TSC2 genes (European Chromosome 16 Tuberous Sclerosis Consortium, 1993). This disorder presents intractable epilepsy, cognitive disability, and autism spectrum disorders. Interestingly, tuberal lesion display cellular features similar to FCD type II (i.e., cytomegalic neurons and balloon cells). Therefore, collectively, these disorders might be referred to as “mTORopathies” (Reviewed by Crino, 2015; Marsan and Baulac, 2018). During the past decade somatic activation mutations in mTOR itself have been identified in these syndromes (Lim et al., 2015; Mirzaa et al., 2016; Møller et al., 2016; D’Gama et al., 2017; Ribierre et al., 2018). In addition, positive (e.g., gain of function mutations in Akt1 or AKT3; Lee et al., 2012; Poduri et al., 2012; Jansen et al., 2015) or negative (e.g., TSC2, or DEPDC5; Baulac et al., 2015; D’Gama et al., 2017; Lim et al., 2017) regulators of mTOR have been implicated in FCD type II and Hemimegalencephaly.
Because some patients with FCD are surgically treated, there have been opportunities for investigating on resected tissue neuronal properties (reviewed by Abdijadid et al., 2015). These investigations described for example an abnormal expression of glutamate and GABA receptor in dysplastic and heterotopic neurons (Crino et al., 2001; Lozovaya et al., 2014), a reduction of GABAA -receptor-mediated inhibition (Calcagnotto et al., 2005), an altered pattern of expression and distribution of synaptic protein SV2 (Toering et al., 2009). Carlos Cepeda and coworkers nicely evaluated the electrophysiological properties of cytomegalic neurons and balloon cells and identified in dysplastic areas the presence of neurons with immature cellular and synaptic properties (Cepeda et al., 2007); these observations gave origin to the hypothesis that local interactions of dysmature cells, that would be directly affected by the mutations, with normal postnatal neurons produce seizures (Cepeda et al., 2007). Though these investigations did not elucidate the exact mechanisms of epileptogenesis in FCD, they indicate clearly that different cell types, molecular changes and cellular interactions are contributing factors.
Many neuronal changes reported on neurons from FCD type II or tuber resections would be considered as cell autonomous as they indicate that the hyperactivation of the mTOR pathway leads to neuronal and synaptic dysfunctions, that contribute to epilepsy, independently from the cytoarchitectonic alteration. This notion has been confirmed by Hsieh et al. (2016) when investigating a murine model of type II FCDs by increasing mTOR activity in layer 2/3 neurons of the medial prefrontal cortex. When the hyperactivation of mTOR was induced in cortical progenitors by in utero electroporation, animals displayed dyslamination and cytomegalic neurons; associated to these changes, animals developed spontaneous tonic-clonic seizures. However, when mTOR hyperactivation was induced after corticogenesis, thanks to the use of inducible vectors, animals still displayed an epileptic condition without neuronal misplacement and dysmorphogenesis. In agreement with this, my lab has shown that heterozygous mice from a Tsc1-KO line, develop spontaneous seizures during the first month of life in the absence of any apparent cortical dysplasia (Lozovaya et al., 2014). In this study we demonstrated that Tsc1+/−neurons, particularly L4 spiny stellate cells, display an anomalous expression of NR2C receptors resulting in a change on the kinetics of NMDA currents and that this change was sufficient to induce the epileptic condition. Indeed, the treatment of pups with antagonists specific for NR2C transiently abolished seizures (Lozovaya et al., 2014). We also demonstrated in this report that treatment of newborn mice with rapamycin was sufficient to reverse the phenotype confirming the link between mTORC1 hyperactivation and NMDA receptor changes. Interestingly, cortical resections from patients with tuberous sclerosis and FCD type II also demonstrated higher expression levels of NR2C as compared with control fetal or adult samples (Lozovaya et al., 2014); in addition to this, patch-clamp recordings on these cortical resections demonstrated the contribution of NR2C to NMDA currents confirming the potential role of NR2C to epileptogenesis in mTORpathies.
It is also important to remind, however, that human samples investigated were obtained from severely affected, pharmaco-resistant patients with resection of the epileptogenic zone being the only therapeutic option. The epileptic process by itself can be cause of many subsequent alterations, including an excitatory/inhibitory imbalance. Thus, based on investigations of a Tsc1-KO mice line, Bateup et al. (2013) reported that many biochemical, transcriptional and functional changes in Tsc1 neurons arise secondarily, due to increased network activity. To study the epileptogenic process itself, we must develop appropriate experimental paradigms in order to evaluate the changes that increase the excitability of neurons and networks and that take place before epilepsy onset.
In conclusion, the two paradigmatic examples discussed before, suggest that the initial genetic alteration that yields cytoarchitectonic disruptions of cortical development, might also be responsible for the clinical manifestations. However, these investigations also highlighted to a certain extent the importance of cellular interactions and the possibility that developmental changes, that might involve non-mutated neurons during a particularly vulnerable developmental period, could also contribute to the emergence of epilepsy and/or cognitive deficits.
The Epileptogenic Network
Investigations combining EEG and functional imaging have demonstrated that patients with focal cortical malformations display interictal or ictal events not only in the affected area (the malformation itself) but also in more or less distal cortical areas. Thus, reports of patients with SBH or periventricular nodular heterotopia indicate that the epileptogenic network is restricted to the heterotopia, or involves both the heterotopia and the surrounding cortex or localizes out of the heterotopia (Mai et al., 2003; Tassi et al., 2005; Kobayashi et al., 2006; Tyvaert et al., 2008; Valton et al., 2008; Christodoulou et al., 2012; Shafi et al., 2015; Pizzo et al., 2017). In patients with FCD, it has also been reported that epileptogenicity extends beyond the limit of the malformation in many patients (Aubert et al., 2009). Another pathological conditions in which the adjacent or even more distal cortex can be the primary origin of epileptiform activity is the tuberous sclerosis. Although tubers are thought to be the initial heart of the epileptogenic zone, electrocorticographic recordings of some patients revealed epileptiform activities and ictal onsets in the perituberal cortex (Madhavan et al., 2007; Major et al., 2009; Ma et al., 2012).
Though conclusions are hampered by the diversities of clinical courses, that includes variabilities on epilepsy onset, type of seizures, efficiency of AEDs, cortical area affected, etc., these data tend to support the notion that epileptic network frequently involves supposedly “healthy” cortical areas that are affected by the presence of a cortical malformation. We investigated this notion in a rat model of SBH induced by in utero (by embryonic day 15) knockdown (KD) of Dcx, the main causative gene of this condition (des Portes et al., 1998; Gleeson et al., 1998; Pilz et al., 1998). In Dcx-KD rats, affected neurons fail to migrate to the cortical plate and form a band of ectopic neurons in the white matter of the electroporated hemisphere (Bai et al., 2003). Dcx-KD rats display altered neocortical excitability already present at the second postnatal week, resulting in an increased propensity for convulsant-induced seizures and spontaneous absence-like seizures in adulthood (Ackman et al., 2009; Manent et al., 2009; Lapray et al., 2010).
We first investigated in Dcx-KD juvenile rats the phenotype of ectopic neurons and found that they displayed a reduced dendritic tree as compared with control (mismatch) neurons and a reduced density of dendritic spines (Ackman et al., 2009; Martineau et al., 2018). We also performed genetic labeling of scaffolding proteins PSD-95 and gephyrin for quantifying, respectively, glutamatergic and GABAergic synapses in ectopic neurons and observed that they were severely reduced (Martineau et al., 2018). These structural changes were associated with a decrease of the frequency of glutamatergic and GABAergic synaptic currents in patch-clamp recordings (Ackman et al., 2009; Martineau et al., 2018). Though DCX also plays a role on neuron maturation (Martineau et al., 2018), the ectopic position of neurons is mainly responsible for the impaired development and synaptogenesis.
Reports on other animal models of cortical migration defects revealed similar features: neurons displayed a simplification of their dendritic arbors in ectopic gray masses induced by either fetal irradiation (Ferrer et al., 1984) or treatment of pregnant rats with methylazoxymethanol (MAM) (Singh, 1980; Chevassus au Louis et al., 1998; Rafiki et al., 1998; Sancini et al., 1998); a significant diminution in spine numbers was also reported in MAM treated offspring (Colciaghi et al., 2014). All together these observations confirm that the positioning of neocortical neurons significantly impacts the subsequent development of dendrites and synapses impairing their integration in functional networks.
In contrast to these dendritic and synaptic defects, ectopic neurons develop axonal projections toward targets that are considered to be “normal” for cortical neurons. In DCX-KD rats, ectopic neurons form cortico-cortical projections like Layer II/III neurons: they reach layers V-VI in the ipsilateral cortex and send axonal projections to the contralateral cortex (Ackman et al., 2009); they also form cortico-striatal connections like Layer V neurons. Similarly, ectopic neurons in irradiated rats and in Tish rats, respectively, lesional and genetic models of SBH, project as normal Layer V neurons to the spinal cord and thalamus (D’Amato and Hicks, 1980; Jensen and Killackey, 1984; Lee et al., 1997). Therefore, ectopic neurons, though poorly innervated as compared with their normotopic counterparts, are in the position of spreading signals to typical cortical targets and participating in the propagation of epileptic activities.
We then analyzed the synaptic properties of normotopic neurons from DCX-KD rats to evaluate if the SBH altered the development of normal positioned neurons. Our studies revealed that normotopic neurons displayed an increased frequency of spontaneous glutamatergic post-synaptic currents while GABAergic currents were unaffected (Ackman et al., 2009). As a consequence, the excitatory/inhibitory ratio of synaptic inputs was increased in normotopic neurons (Ackman et al., 2009). Calcium imaging analysis also revealed that more neurons were active in the cortex overlaying SBH than in the cortex of control rats and that they displayed higher frequency of spontaneous events. In addition, more neurons exhibited synchronized events (Ackman et al., 2009). More recently, in a rat model of bilateral double cortex we also observed that the strength of excitatory L4 to L2/3 synapses (Figure 1), the intrinsic properties of L4 glutamatergic cells and the excitation/inhibition ratio in L2/3 converge into making the early stage of cortical sensory integration abnormally strong in somatosensory cortex adjacent to heterotopia, thus demonstrating developmental alterations of cortical functional circuits that likely play a major role in the cortical dysfunction of the malformed brain (Plantier et al., 2018). Investigations of animal models of periventricular heterotopia induced by irradiation (Zhu and Roper, 2000) or treatment with carmustine (Benardete and Kriegstein, 2002) also found an excitatory/inhibitory misbalance in normotopic cortex.
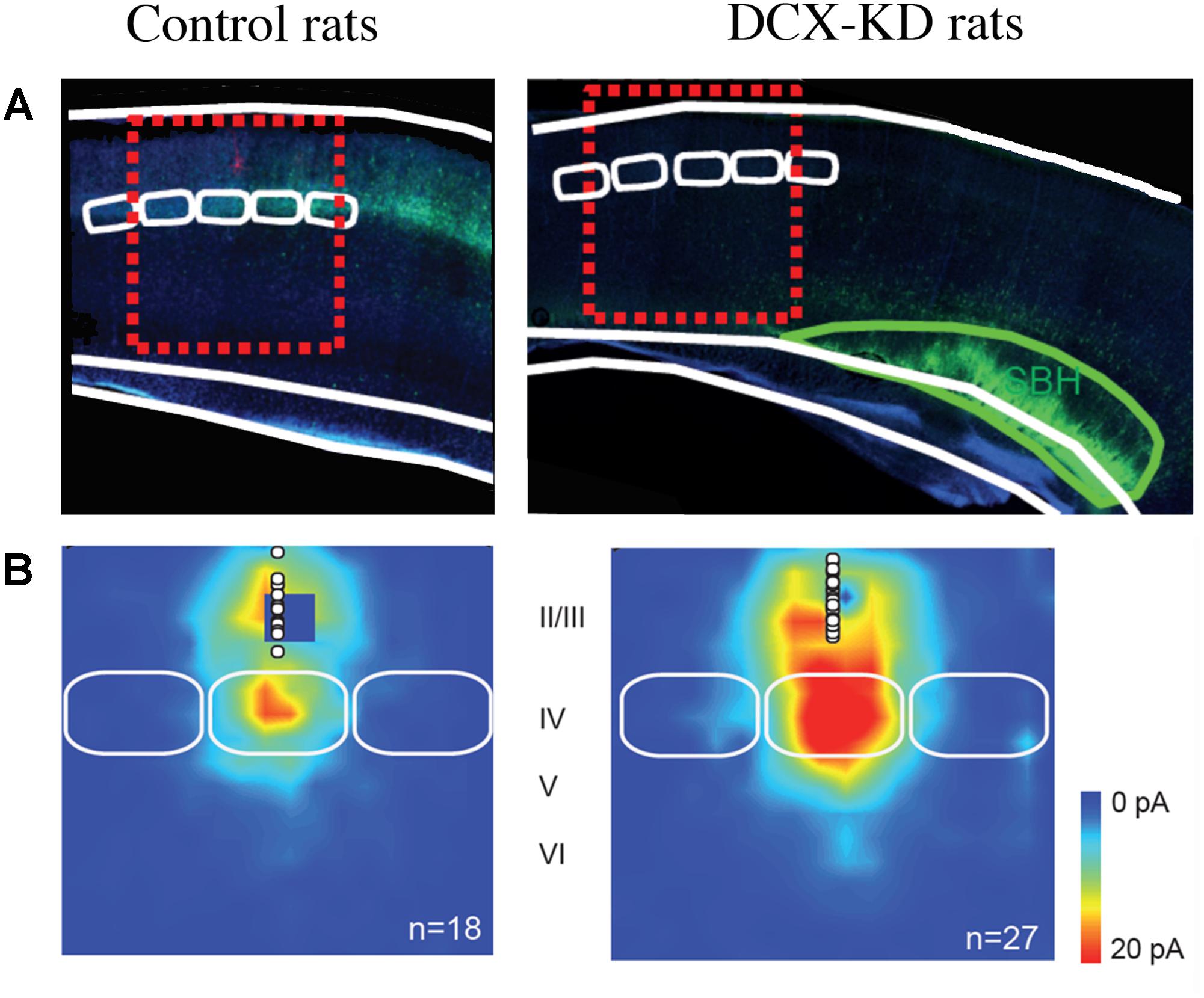
Figure 1. Collateral Alterations of Functional Cortical Circuits in a Rat Model of Subcortical Band Heterotopia. (A) Representative examples of across-barrel brain slices from control (Mismatch) and Dcx-KD rats. Fluorescence microphotographs of electroporated cells are superimposed on schematized views of barrels (in white) and SBH (in green). Red dashed line rectangles illustrate the cortical columns evaluated in the study. (B) Excitatory synaptic input maps for L2/3 neurons in mismatch and Dcx-KD rats. Colors indicate the mean amplitude of excitatory synaptic responses in a 100-ms time window. White circles show soma positions of recorded L2/3 neurons. Solid white lines delineate barrels. Note that in DCX-KD rats the strength of excitatory inputs to L2/3 neurons are dramatically increased (for more details see Plantier et al., 2018).
Taken together these data support the notion that normotopic cortex becomes hyper excitable during postnatal development and that it would be responsible for epileptogenesis in DCX-KD rats. Interestingly, the suppression of excitability of ectopic neurons by their transfection with potassium channels Kir2.1, did not alter the high propensity of DCX-KD rats to experience seizures, while the transfection of ectopic and normotopic neurons significantly reduces it (Petit et al., 2014). These observations prompted us to evaluate the origin of interictal events on acute slices from DCX-KD rats using 60-channels microelectrode arrays (Petit et al., 2014). Our data demonstrated that most of interictal discharges initiated in normotopic cortex and propagated secondarily to the SBH. In vivo recording with deep electrodes in Tish rats (Chen et al., 2000) also indicated that normotopic neurons were more prone to exhibit epileptiform activities than heterotopic neurons and that blocking the connectivity between the two fields by a local TTX injection inhibited the firing of ectopic but not that of normotopic neurons.
Another pathological condition in which the dysplastic or malformed brain area is not the primary origin of epileptiform activities is provided by the rat model of microgyria induced in rats by freeze-lesioning of deep layer neurons at neonatal stage. Though injured animals did not display spontaneous epileptic events, it has been proven that there is a focal region of hyperexcitability around the lesion (Jacobs et al., 1996); In the cortex lateral to the microgyria, an increased excitation of L5 neurons and an increased inhibition of L5 and L2/3 neurons were observed (Jacobs and Prince, 2005; Brill and Huguenard, 2010; Jin et al., 2014). In addition to this, a rather widespread cortical reduction of GABAA receptor subunits α1, α2, α3, α5, and γ2 expression has been reported (Redecker et al., 2000). These alterations together would account for the increased excitability of an apparently normal cortex lateral to the microgyria.
Collectively, these clinical and experimental observations support the notion that anatomically unaltered cortical regions surrounding the malformation are included in a large epileptogenic network. We propose that developmental changes in these areas play a major role in the generation of more or less large epileptogenic networks.
The Time of Onset of Epilepsy
The age at epilepsy onset in patients with MCD is largely variable ranging from newborn to adulthood. This is observed even within the same type of cortical malformations. For example, in a series of 132 patients with polymicrogyria (Leventer et al., 2010), the mean age of epilepsy onset was 4.9 ± 6.7 years but ranging from 1 day to 34 years of age. However, 43% of the patients had the first seizures during the first year of life, coinciding with an important period of synaptogenesis. In a similar way, the analysis of 86 female patients with mutations of DCX (Bahi-Buisson et al., 2013) indicate an early onset, during the first year of life, in 55% of cases, but the age at onset varied between the first month of life to 17 years.
These data indicate that the majority of patients with MCD develop the first clinical manifestations of epilepsy during the first year of life, a period of brain development characterized by an intense neuronal maturation and synaptogenesis. It is thus plausible that epilepsy onset is facilitated in these patients by the increasing weigh of maturing synaptic inputs and/or the maturation of the axonal initial segment responsible for the genesis of action potentials and/or other molecular and synaptic changes linked to neuronal maturation. Interestingly, in Dravet syndrome (Dravet, 1978), a severe encephalopathy due to de novo loss-of-function mutations in the SCN1A gene, leading to haploinsufficiency of NaV1.1 channel (reviewed by Brunklaus and Zuberi, 2014), epilepsy typically presents around 6 months of age. Expression analysis on human temporal cortex and hippocampus demonstrated that Na(v)1.1 immunoreactivity increased significantly during the late fetal and postnatal periods, reaching peaks 7–9 months after birth (Wang et al., 2011). Hence, epilepsy onset in Dravet patients compares with the developmental course of the affected channel. It is not unlikely that such developmental pattern plays a role on the onset of epileptic manifestations in Dravet and would be also participating in other epileptic syndromes.
It is well established that GABAergic synapses play a major pathophysiological role in epilepsy and thus GABAergic transmission is targeted by many antiepileptic drugs. Synaptic currents induced by the activation of GABAA receptors are carried by chloride and consequently the intracellular concentrations of this anion determine the type of response evoked by the transmitter. In adult neurons, the potassium-chloride cotransporter KCC2 usually extrudes chloride promoting hyperpolarizing, inhibitory, responses. In immature neurons the Na+-K+-2Cl− cotransporter NKCC1 loads them with chloride and favors depolarizing responses to GABA so that the transmitter might have in developmental brain an excitatory effect (Ben-Ari et al., 1989). However, though the topic remains controversial, the action of GABA on developing cortical neurons in vivo seems to be inhibitory (Tyzio et al., 2008; Kirmse et al., 2015; Valeeva et al., 2016), maybe because of its shunting effect (Staley and Mody, 1992) that is independent from the polarity of GABAergic signals. Interestingly, in epileptic tissue from patients with temporal lobe epilepsy it has been shown that changes in chloride homeostasis switch GABAergic signaling from hyperpolarizing to depolarizing (Cohen et al., 2002; Khalilov et al., 2003; Huberfeld et al., 2007). It is therefore likely that GABA may play an important role in childhood epilepsy and that its depolarizing actions in immature neurons would contribute to epilepsy onset. However, in cortical resections from pediatric (6 to 14 months old) Sturge-Weber patients, a severe epileptogenic neurocutaneous syndrome, we found that GABA played mainly an inhibitory and anticonvulsive role (Tyzio et al., 2009). On the other hand, investigations on resections from pediatric patients with FCD, Hemimegalencephaly and tuberous sclerosis (Aronica et al., 2007; Talos et al., 2012) suggest a possible dysregulation on the expression of cation-chloride co-transporters. These changes may be activity-dependent and secondary to the epileptic process itself (Puskarjov et al., 2012); they would thus participate more in the expression and evolution of the epileptic disease than at its onset.
The occurrence of a large time window between the initial insult and the onset of clinical manifestations is a common feature for many neurological disorders, including Parkinson and Alzheimer diseases. In these neurodegenerative diseases the latency window can be related to the evolution of the disease, for example the progressive degeneration of dopaminergic neurons in Parkinson disease. Following this reasoning, the late onset of epilepsy reported in some patients might be due to developmental apoptotic processes and/or synaptic (Bourgeois and Rakic, 1993) and dendritic (Zehr et al., 2006) pruning. It is interesting to note that synaptic pruning is particularly important around puberty. For example, electron microscopy studies in primates (Bourgeois and Rakic, 1993) showed a significant reduction in the density of synapses in cortex and hippocampus, during this period of time and imaging analysis of human cortex depicted a reduction in cortical thickness during equivalent periods (Giedd et al., 2006; Raznahan et al., 2011). Would these changes contribute to the peak of incidence of epilepsy that was observed at this period of life (Doherty et al., 2003)? this including juvenile myoclonic epilepsy, emerging mainly in mid-to-late childhood (Martínez-Juárez et al., 2006; Kasteleijn-Nolst Trenité et al., 2013; Ochoa-Gómez et al., 2017)? We do not have an obvious answer on this, but it is clear that we are facing a particular vulnerable period of brain life and that a deregulation of axonal pruning processes, for example affecting more inhibitory synapses or stabilizing recurrent axonal loops, could reveal at last a neuronal excitability status concealed until then.
Alternatively, the intervention of additional precipitating factors during specific time windows, can be at play. Among the factors that would increase neuronal excitability one can mention hormones like estrogens (Zehr et al., 2006) or progesterone (Smith et al., 2002) and inflammation (Bartolini et al., 2018). There is an increasing interest for the role of inflammation and glia in neurodevelopmental disorders like autism spectrum disorders (ASD), schizophrenia, cerebral palsy, cognitive impairment, epilepsy and depression (Galic et al., 2012; Devinsky et al., 2013; Vezzani, 2013; Knuesel et al., 2014; Marchi et al., 2014; Rosenblat et al., 2014; Jiang et al., 2018). Indeed, microglia could have a significant physiological role, contributing to the regulation of cell death/survival, synapse pruning and neurogenesis (Stolp et al., 2011; Vukovic et al., 2012; Kettenmann et al., 2013; Wake et al., 2013). Microglia thus contribute to the regulation of maturation and plasticity of developing neuronal circuits. Furthermore, it has been suggested that microglia would act as a versatile modulator of neurogenesis depending on its activation state: pro-inflammatory microglia would reduce neurogenesis while anti-inflammatory microglia could increase neurogenesis through release of trophic factors (e.g., Kyritsis et al., 2012) and it is suspected that similar dual action of microglia applies to synaptic functioning, plasticity and stability (Marin and Kipnis, 2013; Nisticò et al., 2017).
The contribution of inflammation and glial cells in epilepsy has been particularly investigated (reviewed by Devinsky et al., 2013; Vezzani, 2013) and it has been proposed that inflammation plays an important role in the onset of pediatric seizures (Bartolini et al., 2018). While some infections (e.g., bacterial meningitis, herpes virus, toxoplasmosis) are known to cause acute seizures (Lowenstein et al., 2014; Vezzani et al., 2016) an actual epileptogenic process would take place in some patients after an initial infection. The precise mechanisms remain to be elucidated but they seem to depend on “the pathogen itself, the developmental stage, the degree of cytokine-mediated inflammatory response and the genotype-phenotype of the person concerned” (Vezzani, 2013). It can be thus proposed that in a patient with a susceptible brain condition like MCDs, inflammation-induced responses would act as a second-hit, a trigger or an aggravating factor. Interestingly, signs of activation of both innate and adaptative immunities have been found in dysplastic tissue from FCD type II patients (Iyer et al., 2010), suggesting that at least in this type of malformation an inflammatory process is engaged. To note, however, that some of these changes can be related to the action of mTOR on glial cells or be a consequence of the epileptic activity per se. Future research is required for better understanding this important question.
Conclusion
In conclusion, epileptogenesis in MCDs occur during a period of brain development characterized by many molecular, cellular and structural changes (Figure 2) that determine the features of brain operation and functioning and impact epileptogenic processes and epilepsy expression. Epileptogenesis in MCDs involves complex multifactorial causes that would relate to the type of gene or insult involved in the malformation, normal developmental processes and developmental adaptative or reactive changes in cortical circuitry. The emergence of new animal models reproducing focal mosaic lesions associating manifestation reminiscent of human clinical symptoms, open promising vistas for better understanding the physiopathology of these disorders and testing new therapeutic options.
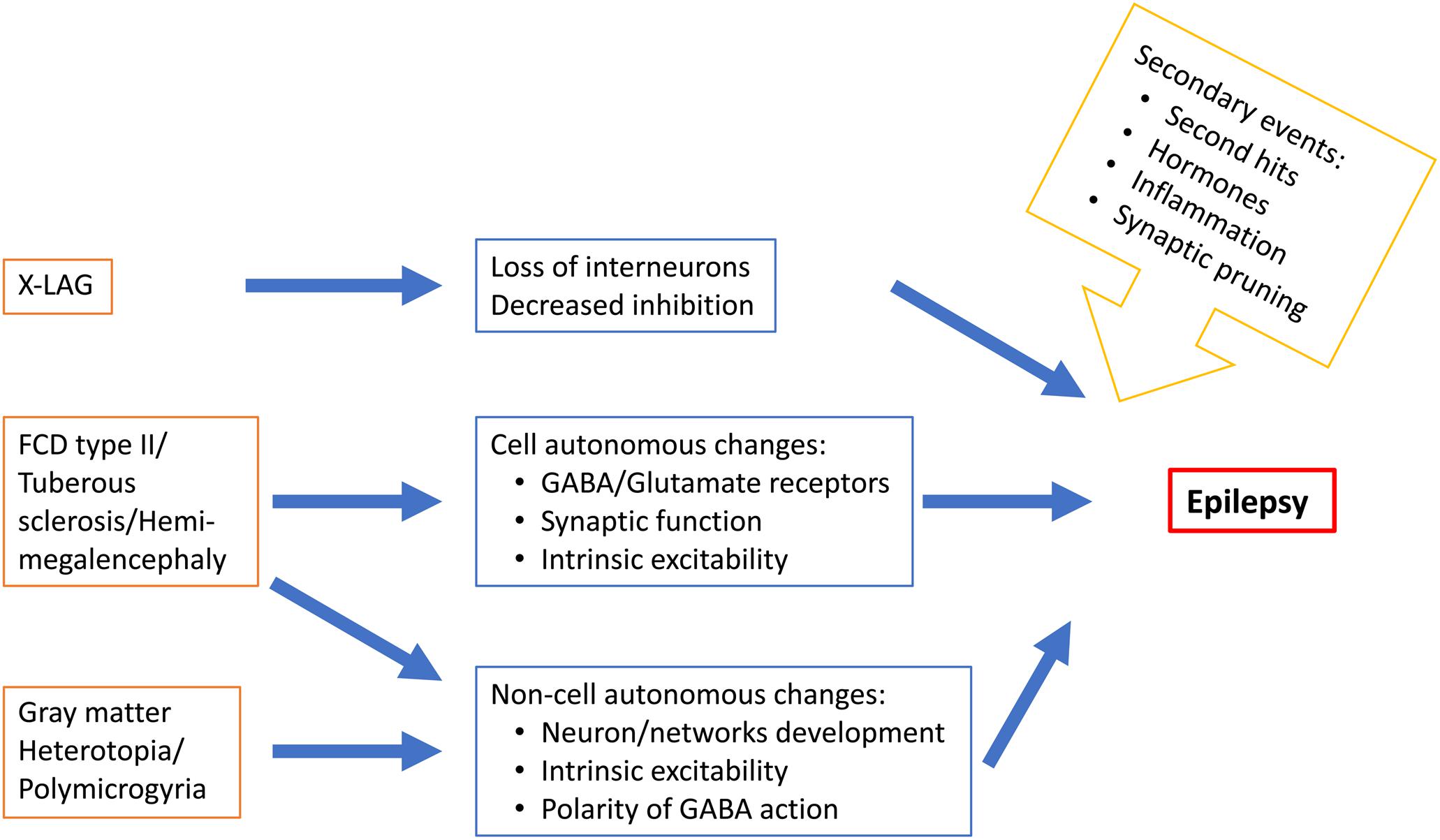
Figure 2. Schematic representation of some MCD-related epileptogenic changes in neurons and neuronal networks.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
The financial support from European Community 7th Framework programs [Development and Epilepsy – Strategies for Innovative Research to improve diagnosis, prevention and treatment in children with difficult to treat Epilepsy (DESIRE)], EraNet Neuron [Deciphering hyperexcitable networks associated with neurodevelopmental lesions (DeCipher), #ANR-15-NEUR-0001-03], INSERM and CNRS are acknowledged.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks Drs. P. P. Lenck-Santini, V. Crepel and M. Milh for critically reading the manuscript.
References
Abdijadid, S., Mathern, G. W., Levine, M. S., and Cepeda, C. (2015). Basic mechanisms of epileptogenesis in pediatric cortical dysplasia. CNS Neurosci. Ther. 21, 92–103. doi: 10.1111/cns.12345
Ackman, J. B., Aniksztejn, L., Crepel, V., Becq, H., Pellegrino, C., Cardoso, C., et al. (2009). Abnormal network activity in a targeted genetic model of human double cortex. J. Neurosci. 29, 313–327. doi: 10.1523/JNEUROSCI.4093-08.2009
Aronica, E., Boer, K., Redeker, S., Spliet, W. G., van Rijen, P. C., Troost, D., et al. (2007). Differential expression patterns of chloride transporters, nNa+-K+-2Cl–cotransporter and K+-Cl–cotransporter, in epilepsy-associated malformations of cortical development. Neuroscience 145, 185–196. doi: 10.1016/j.neuroscience.2006.11.041
Aubert, S., Wendling, F., Regis, J., McGonigal, A., Figarella-Branger, D., Peragut, J. C., et al. (2009). Local and remote epileptogenicity in focal cortical dysplasias and neurodevelopmental tumours. Brain 132, 3072–3086. doi: 10.1093/brain/awp242
Bahi-Buisson, N., Souville, I., Fourniol, F. J., Toussaint, A., Moores, C. A., Houdusse, A., et al. (2013). New insights into genotype-phenotype correlations for the doublecortin-related lissencephaly spectrum. Brain 136, 223–244. doi: 10.1093/brain/aws323
Bai, J., Ramos, R. L., Ackman, J. B., Thomas, A. M., Lee, R. V., and LoTurco, J. J. (2003). RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci. 6, 1277–1283. doi: 10.1038/nn1153
Barkovich, A. J., Guerrini, R., Kuzniecky, R. I., Jackson, G. D., and Dobyns, W. B. (2012). A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135, 1348–1369. doi: 10.1093/brain/aws019
Bartolini, L., Libbey, J. E., Ravizza, T., Fujinami, R. S., Jacobson, S., and Gaillard, W. D. (2018). Viral triggers and inflammatory mechanisms in pediatric epilepsy. Mol. Neurobiol. 56, 1897–1907. doi: 10.1007/s12035-018-1215-5
Bateup, H. S., Johnson, C. A., Denefrio, C. L., Saulnier, J. L., Kornacker, K., and Sabatini, B. L. (2013). Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522. doi: 10.1016/j.neuron.2013.03.017
Baulac, S., Ishida, S., Marsan, E., Miquel, C., Biraben, A., Nguyen, D. K., et al. (2015). Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann. Neurol. 77, 675–683. doi: 10.1002/ana.24368
Benardete, E. A., and Kriegstein, A. R. (2002). Increased excitability and decreased sensitivity to GABA in an animal model of dysplastic cortex. Epilepsia 43, 970–982. doi: 10.1046/j.1528-1157.2002.40901.x
Ben-Ari, Y., Cherubini, E., Corradetti, R., and Gaiarsa, J. L. (1989). Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325. doi: 10.1113/jphysiol.1989.sp017762
Berg, A. T., Tarquinio, D., and Koh, S. (2017). Early life epilepsies are a comorbidity of developmental brain disorders. Semin. Pediatr. Neurol. 24, 251–263. doi: 10.1016/j.spen.2017
Bienvenu, T., Poirier, K., Friocourt, G., Bahi, N., Beaumont, D., Fauchereau, F., et al. (2002). ARX, a novel Prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum. Mol. Genet. 11, 981–991. doi: 10.1093/hmg/11.8.981
Blümcke, I., Thom, M., Aronica, E., Armstrong, D. D., Vinters, H. V., Palmini, A., et al. (2011). The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia 52, 158–174. doi: 10.1111/j.1528-1167.2010.02777.x
Bonneau, D., Toutain, A., Laquerriere, A., Marret, S., Saugier-Veber, P., Barthez, M., et al. (2002). X-linked lissencephaly with absent corpus callosum and ambiguous genitalia (XLAG): clinical, magnetic resonance imaging, and neuropathological findings. Ann. Neurol. 51, 340–349. doi: 10.1002/ana.10119
Bourgeois, J. P., and Rakic, P. (1993). Changes of synaptic density in the primary visual cortex of the macaque monkey from fetal to adult stage. J. Neurosci. 13, 2801–2820. doi: 10.1523/JNEUROSCI.13-07-02801.1993
Brill, J., and Huguenard, J. R. (2010). Enhanced infragranular and supragranular synaptic input onto layer 5 pyramidal neurons in a rat model of cortical dysplasia. Cereb. Cortex 20, 2926–2938. doi: 10.1093/cercor/bhq040
Brunklaus, A., and Zuberi, S. M. (2014). Dravet syndrome–from epileptic encephalopathy to channelopathy. Epilepsia 55, 979–984. doi: 10.1111/epi.12652
Calcagnotto, M. E., Paredes, M. F., Tihan, T., Barbaro, N. M., and Baraban, S. C. (2005). Dysfunction of synaptic inhibition in epilepsy associated with focal cortical dysplasia. J. Neurosci. 25, 9649–9657. doi: 10.1523/JNEUROSCI.2687-05.2005
Cepeda, C., André, V. M., Wu, N., Yamazaki, I., Uzgil, B., Vinters, H. V., et al. (2007). Immature neurons and GABA networks may contribute to epileptogenesis in pediatric cortical dysplasia. Epilepsia 48(Suppl. 5), 79–85. doi: 10.1111/j.1528-1167.2007.01293.x
Chen, Z. F., Schottler, F., Bertram, E., Gall, C. M., Anzivino, M. J., and Lee, K. S. (2000). Distribution and initiation of seizure activity in a rat brain with subcortical band heterotopia. Epilepsia 41, 493–501. doi: 10.1111/j.1528-1157.2000.tb00201.x
Chevassus au Louis, N., Rafiki, A., Jorquera, I., Ben-Ari, Y., and Represa, A. (1998). Neocortex in the hippocampus: morphofunctional analysis of CA1 heterotopiae in the hippocampus of MAM treated rats. J. Comp. Neurol. 394,520–536.
Christodoulou, J. A., Walker, L. M., Del Tufo, S. N., Katzir, T., Gabrieli, J. D., Whitfield-Gabrieli, S., et al. (2012). Abnormal structural and functional brain connectivity in gray matter heterotopia. Epilepsia 53, 1024–1032. doi: 10.1111/j.1528-1167.2012.03466.x
Cohen, I., Navarro, V., Clemenceau, S., Baulac, M., and Miles, R. (2002). On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298, 1418–1421. doi: 10.1126/science.1076510
Colciaghi, F., Finardi, A., Nobili, P., Locatelli, D., Spigolon, G., and Battaglia, G. S. (2014). Progressive brain damage, synaptic reorganization and NMDA activation in a model of epileptogenic cortical dysplasia. PLoS One 9:e89898. doi: 10.1371/journal.pone.0089898
Collombat, P., Mansouri, A., Hecksher-Sorensen, J., Serup, P., Krull, J., Gradwohl, G., et al. (2003). Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 17, 2591–2603. doi: 10.1101/gad.269003
Colombo, E., Galli, R., Cossu, G., Gecz, J., and Broccoli, V. (2004). Mouse orthologue of ARX, a gene mutated in several X-linked forms of mental retardation and epilepsy, is a marker of adult neural stem cells and forebrain GABAergic neurons. Dev. Dyn. 231, 631–639. doi: 10.1002/dvdy.20164
Crino, P. B. (2015). mTOR signaling in epilepsy: insights from malformations of cortical development. Cold Spring Harb. Perspect. Med. 5:a022442. doi: 10.1101/cshperspect.a022442
Crino, P. B., Duhaime, A. C., Baltuch, G., and White, R. (2001). Differential expression of glutamate and GABA-A receptor subunit mRNA in cortical dysplasia. Neurology 56, 906–913. doi: 10.1212/WNL.56.7.906
D’Amato, C. J., and Hicks, S. P. (1980). Development of the motor system: effects of radiation on developing corticospinal neurons and locomotor function. Exp. Neurol. 70, 1–23. doi: 10.1016/0014-4886(80)90002-3
des Portes, V., Francis, F., Pinard, J. M., Desguerre, I., Moutard, M. L., Snoeck, I., et al. (1998). Doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH). Hum. Mol. Genet. 7, 1063–1070. doi: 10.1093/hmg/7.7.1063
Devinsky, O., Vezzani, A., Najjar, S., De Lanerolle, N. C., and Rogawski, M. A. (2013). Glia and epilepsy: excitability and inflammation. Trends Neurosci. 36, 174–184. doi: 10.1016/j.tins.2012.11.008
D’Gama, A. M., Woodworth, M. B., Hossain, A. A., Bizzotto, S., Hatem, N. E., LaCoursiere, C. M., et al. (2017). Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 21, 3754–3766. doi: 10.1016/j.celrep.2017.11.106
Doherty, M. J., Simon, E., De Menezes, M. S., Kuratani, J. D., Saneto, R. P., Homles, M. D., et al. (2003). When might hemispheric favouring of epileptiform discharges begin? Seizure 12, 595–598.
European Chromosome 16 Tuberous Sclerosis Consortium (1993). Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75, 1305–1315.
Ferrer, I., Xumetra, A., and Santamaría, J. (1984). Cerebral malformation induced by prenatal X-irradiation, an autoradiographic and Golgi study. J. Anat. 138, 81–93.
Friocourt, G., and Parnavelas, J. G. (2010). Mutations in ARX result in several defects involving GABAergic neurons. Front. Cell. Neurosci. 4:4. doi: 10.3389/fncel.2010.00004
Fullenkamp, A. N., and El-Hodiri, H. M. (2008). The function of the Aristaless-related homeobox (Arx) gene product as a transcriptional repressor is diminished by mutations associated with X-linked mental retardation (XLMR). Biochem. Biophys. Res. Commun. 377, 73–78. doi: 10.1016/j.bbrc.2008.09.116
Galic, M. A., Riazi, K., and Pittman, Q. J. (2012). Cytokines and brain excitability. Front. Neuroendocrinol. 33, 116–125. doi: 10.1016/j.yfrne.2011.12.002
Giedd, J. N., Clasen, L. S., Lenroot, R., Greenstein, D., Wallace, G. L., Ordaz, S., et al. (2006). Puberty-related influences on brain development. Mol. Cell. Endocrinol. 254–255, 154–162. doi: 10.1016/j.mce.2006.04.016
Gleeson, J. G., Allen, K. M., Fox, J. W., Lamperti, E. D., Berkovic, S., Scheffer, I., et al. (1998). Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72. doi: 10.1016/S0092-8674(00)80899-5
Guerrini, R., and Dobyns, W. B. (2014). Malformations of cortical development: clinical features and genetic causes. Lancet Neurol. 13, 710–726. doi: 10.1016/S1474-4422(14)70040-7
Harvey, A. S., Cross, J. H., Shinnar, S., Mathern, G. W., and ILAE Pediatric Epilepsy Surgery Survey Taskforce (2008). Defining the spectrum of international practice in pediatric epilepsy surgery patients. Epilepsia 49, 146–155. doi: 10.1111/j.1528-1167.2007.01421.x
Hsieh, L. S., Wen, J. H., Claycomb, K., Huang, Y., Harrsch, F. A., Naegele, J. R., et al. (2016). Convulsive seizures from experimental focal cortical dysplasia occur independently of cell misplacement. Nat. Commun. 7:11753. doi: 10.1038/ncomms11753
Huberfeld, G., Wittner, L., Clemenceau, S., Baulac, M., Kaila, K., Miles, R., et al. (2007). Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 27, 9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007
Iyer, A., Zurolo, E., Spliet, W. G., van Rijen, P. C., Baayen, J. C., Gorter, J. A., et al. (2010). Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 51, 1763–1773. doi: 10.1111/j.1528-1167.2010.02547.x
Jacobs, K. M., Gutnick, M. J., and Prince, D. A. (1996). Hyperexcitability in a model of cortical maldevelopment. Cereb. Cortex 6, 514–523. doi: 10.1093/cercor/6.3.514
Jacobs, K. M., and Prince, D. A. (2005). Excitatory and inhibitory postsynaptic currents in a rat model of epileptogenic microgyria. J. Neurophysiol. 93, 687–696. doi: 10.1152/jn.00288.2004
Jansen, L. A., Mirzaa, G. M., Ishak, G. E., O’Roak, B. J., Hiatt, J. B., Roden, W. H., et al. (2015). PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 138, 1613–1628. doi: 10.1093/brain/awv045
Jensen, K. F., and Killackey, H. P. (1984). Subcortical projections from ectopic neocortical neurons. Proc. Natl. Acad. Sci. U.S.A. 81, 964–968. doi: 10.1073/pnas.81.3.964
Jiang, N. M., Cowan, M., Moonah, S. N., and Petri, W. A. Jr. (2018). The impact of systemic inflammation on neurodevelopment. Trends Mol. Med. 24, 794–804. doi: 10.1016/j.molmed.2018.06.008
Jin, X., Jiang, K., and Prince, D. A. (2014). Excitatory and inhibitory synaptic connectivity to layer V fast-spiking interneurons in the freeze lesion model of cortical microgyria. J. Neurophysiol. 112, 1703–1713. doi: 10.1152/jn.00854.2013
Kasteleijn-Nolst Trenité, D. G., Schmitz, B., Janz, D., Delgado-Escueta, A. V., Thomas, P., Hirsch, E., et al. (2013). Consensus on diagnosis and management of JME: from founder’s observations to current trends. Epilepsy Behav. 28(Suppl. 1), S87–S90. doi: 10.1016/j.yebeh.2012.11.051
Kato, M., Das, S., Petras, K., Kitamura, K., Morohashi, K., Abuelo, D. N., et al. (2004). Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat. 23, 147–159. doi: 10.1002/humu.10310
Kettenmann, H., Kirchhoff, F., and Verkhratsky, A. (2013). Microglia: new roles for the synaptic stripper. Neuron 77, 10–18. doi: 10.1016/j.neuron.2012.12.023
Khalilov, I., Holmes, G. L., and Ben-Ari, Y. (2003). In vitro formation of a secondary epileptogenic mirror focus by interhippocampal propagation of seizures. Nat. Neurosci. 6, 1079–1085. doi: 10.1038/nn1125
Kirmse, K., Kummer, M., Kovalchuk, Y., Witte, O. W., Garaschuk, O., and Holthoff, K. (2015). GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat. Commun. 6:7750. doi: 10.1038/ncomms8750
Kitamura, K., Yanazawa, M., Sugiyama, N., Miura, H., Iizuka-Kogo, A., Kusaka, M., et al. (2002). Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet. 32, 359–369. doi: 10.1038/ng1009
Knuesel, I., Chicha, L., Britschgi, M., Schobel, S. A., Bodmer, M., Hellings, J. A., et al. (2014). Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 10, 643–660. doi: 10.1038/nrneurol.2014.187
Kobayashi, E., Hawco, C. S., Grova, C., Dubeau, F., and Gotman, J. (2006). Widespread and intense BOLD changes during brief focal electrographic seizures. Neurology 66, 1049–1055. doi: 10.1212/01.wnl.0000204232.37720.a4
Kyritsis, N., Kizil, C., Zocher, S., Kroehne, V., Kaslin, J., Freudenreich, D., et al. (2012). Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 338, 1353–1356. doi: 10.1126/science.1228773
Lapray, D., Popova, I. Y., Kindler, J., Jorquera, I., Becq, H., Manent, J. B., et al. (2010). Spontaneous epileptic manifestations in a DCX knockdown model of human double cortex. Cereb. Cortex 20, 2694–2701. doi: 10.1093/cercor/bhq014
Lee, J. H., Huynh, M., Silhavy, J. L., Kim, S., Dixon-Salazar, T., Heiberg, A., et al. (2012). De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 44, 941–945. doi: 10.1038/ng.2329
Lee, K. S., Schottler, F., Collins, J. L., Lanzino, G., Couture, D., Rao, A., et al. (1997). A genetic animal model of human neocortical heterotopia associated with seizures. J. Neurosci. 17, 6236–6242. doi: 10.1523/JNEUROSCI.17-16-06236.1997
Leventer, R. J., Jansen, A., Pilz, D. T., Stoodley, N., Marini, C., Dubeau, F., et al. (2010). Clinical and imaging heterogeneity of polymicrogyria: a study of 328 patients. Brain 133, 1415–1427. doi: 10.1093/brain/awq078
Leventer, R. J., Phelan, E. M., Coleman, L. T., Kean, M. J., Jackson, G. D., and Harvey, A. S. (1999). Clinical and imaging features of cortical malformations in childhood. Neurology 53, 715–722. doi: 10.1212/WNL.53.4.715
Lim, J. S., Gopalappa, R., Kim, S. H., Ramakrishna, S., Lee, M., Kim, W. I., et al. (2017). Somatic mutations in TSC1 and TSC2 cause focal cortical dysplasia. Am. J. Hum. Genet. 100, 454–472. doi: 10.1016/j.ajhg.2017.01.030
Lim, J. S., Kim, W. I., Kang, H. C., Kim, S. H., Park, A. H., Park, E. K., et al. (2015). Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med. 21, 395–400. doi: 10.1038/nm.3824
Lowenstein, D. H., Walker, M., and Waterhouse, E. (2014). Status epilepticus in the setting of acute encephalitis. Epilepsy Curr. 14, 43–49. doi: 10.5698/1535-7511-14.s2.43
Lozovaya, N., Gataullina, S., Tsintsadze, T., Tsintsadze, V., Pallesi-Pocachard, E., Minlebaev, M., et al. (2014). Selective suppression of excessive GluN2C expression rescues early epilepsy in a tuberous sclerosis murine model. Nat. Commun. 5:4563. doi: 10.1038/ncomms5563
Ma, T. S., Elliott, R. E., Ruppe, V., Devinsky, O., Kuzniecky, R., Weiner, H. L., et al. (2012). Electrocorticographic evidence of perituberal cortex epileptogenicity in tuberous sclerosis complex. J. Neurosurg. Pediatr. 10, 376–382. doi: 10.3171/2012.8.PEDS1285
Madhavan, D., Weiner, H. L., Carlson, C., Devinsky, O., and Kuzniecky, R. (2007). Local epileptogenic networks in tuberous sclerosis complex: a case review. Epilepsy Behav. 11, 140–146. doi: 10.1016/j.yebeh.2007.03.017
Mai, R., Tassi, L., Cossu, M., Francione, S., Lo Russo, G., Garbelli, R., et al. (2003). A neuropathological, stereo-EEG, and MRI study of subcortical band heterotopia. Neurology 60, 1834–1838. doi: 10.1212/01.WNL.0000065884.61237.24
Major, P., Rakowski, S., Simon, M. V., Cheng, M. L., Eskandar, E., Baron, J., et al. (2009). Are cortical tubers epileptogenic? Evidence from electrocorticography. Epilepsia 50, 147–154. doi: 10.1111/j.1528-1167.2008.01814.x
Manent, J. B., Wang, Y., Chang, Y., Paramasivam, M., and LoTurco, J. J. (2009). Dcx reexpression reduces subcortical band heterotopia and seizure threshold in an animal model of neuronal migration disorder. Nat. Med. 15, 84–90. doi: 10.1038/nm.1897
Marchi, N., Granata, T., and Janigro, D. (2014). Inflammatory pathways of seizure disorders. Trends Neurosci. 37, 55–65. doi: 10.1016/j.tins.2013.11.002
Marin, I., and Kipnis, J. (2013). Learning and memory... and the immune system. Learn. Mem. 20, 601–606. doi: 10.1101/lm.028357.112
Marsan, E., and Baulac, S. (2018). Mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol. Appl. Neurobiol. 44, 6–17. doi: 10.1111/nan.12463
Marsh, E., Fulp, C., Gomez, E., Nasrallah, I., Minarcik, J., Sudi, J., et al. (2009). Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain 132, 1563–1576. doi: 10.1093/brain/awp107
Marsh, E. D., Nasrallah, M. P., Walsh, C., Murray, K. A., Nicole Sunnen, C., McCoy, A., et al. (2016). Developmental interneuron subtype deficits after targeted loss of Arx. BMC Neurosci. 17:35. doi: 10.1186/s12868-016-0265-8
Martineau, F. S., Sahu, S., Plantier, V., Buhler, E., Schaller, F., Fournier, L., et al. (2018). Correct laminar positioning in the neocortex influences proper dendritic and synaptic development. Cereb. Cortex 28, 2976–2990. doi: 10.1093/cercor/bhy113
Martínez-Juárez, I. E., Alonso, M. E., Medina, M. T., Durón, R. M., Bailey, J. N., López-Ruiz, M., et al. (2006). Juvenile myoclonic epilepsy subsyndromes: family studies and long-term follow-up. Brain 129, 1269–1280. doi: 10.1093/brain/awl048
McKenzie, O., Ponte, I., Mangelsdorf, M., Finnis, M., Colasante, G., Shoubridge, C., et al. (2007). Aristaless-related homeobox gene, the gene responsible for West syndrome and related disorders, is a Groucho/transducin-like enhancer of split dependent transcriptional repressor. Neuroscience 146, 236–247. doi: 10.1016/j.neuroscience.2007.01.038
Mirzaa, G. M., Campbell, C. D., Solovieff, N., Goold, C., Jansen, L. A., Menon, S., et al. (2016). Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol. 73, 836–845. doi: 10.1001/jamaneurol.2016.0363
Møller, R. S., Weckhuysen, S., Chipaux, M., Marsan, E., Taly, V., Bebin, E. M., et al. (2016). Germline and somatic mutations in the MTOR gene in focal cortical dysplasia and epilepsy. Neurol. Genet. 2:e118.
Nisticò, R., Salter, E., Nicolas, C., Feligioni, M., Mango, D., Bortolotto, Z. A., et al. (2017). Synaptoimmunology - roles in health and disease. Mol. Brain 10:26. doi: 10.1186/s13041-017-0308-9
Ochoa-Gómez, L., López-Pisón, J., Lapresta Moros, C., Fuertes Rodrigo, C., Fernando Martínez, R., Samper-Villagrasa, P., et al. (2017). A study of epilepsy according to the age at onset and monitored for 3 years in a regional reference paediatric neurology unit. Ann. Pediatr. 86, 11–19. doi: 10.1016/j.anpedi.2016.05.002
Olivetti, P. R., and Noebels, J. L. (2012). Interneuron, interrupted: molecular pathogenesis of ARX mutations and X-linked infantile spasms. Curr. Opin. Neurobiol. 22, 859–865. doi: 10.1016/j.conb.2012.04.006
Petit, L. F., Jalabert, M., Buhler, E., Malvache, A., Peret, A., Chauvin, Y., et al. (2014). Normotopic cortex is the major contributor to epilepsy in experimental double cortex. Ann. Neurol. 76, 428–442. doi: 10.1002/ana.24237
Pilz, D. T., Matsumoto, N., Minnerath, S., Mills, P., Gleeson, J. G., Allen, K. M., et al. (1998). LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum. Mol. Genet. 7, 2029–2037. doi: 10.1093/hmg/7.13.2029
Pizzo, F., Roehri, N., Catenoix, H., Medina, S., McGonigal, A., Giusiano, B., et al. (2017). Epileptogenic networks in nodular heterotopia: a stereoelectroencephalography study. Epilepsia 58, 2112–2123. doi: 10.1111/epi.13919
Plantier, V., Watrin, F., Buhler, E., Martineau, F. S., Sahu, S., Manent, J. B., et al. (2018). Direct and collateral alterations of functional cortical circuits in a rat model of subcortical band heterotopia. Cereb. Cortex doi: 10.1093/cercor/bhy307 [Epub ahead of print].
Poduri, A., Evrony, G. D., Cai, X., Elhosary, P. C., Beroukhim, R., Lehtinen, M. K., et al. (2012). Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 74, 41–48. doi: 10.1016/j.neuron.2012.03.010
Puskarjov, M., Ahmad, F., Kaila, K., and Blaesse, P. (2012). Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J. Neurosci. 32, 11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012
Rafiki, A., Chevassus-au-Louis, N., Ben-Ari, Y., Khrestchatisky, M., and Represa, A. (1998). Glutamate receptors in dysplasic cortex: an in situ hybridization and immunohistochemistry study in the brain of rats with prenatal treatment with methylazoxymethanol. Brain Res. 782, 142–152. doi: 10.1016/S0006-8993(97)01273-0
Raznahan, A., Lerch, J. P., Lee, N., Greenstein, D., Wallace, G. L., Stockman, M., et al. (2011). Patterns of coordinated anatomical change in human cortical development: a longitudinal neuroimaging study of maturational coupling. Neuron 72, 873–884. doi: 10.1016/j.neuron.2011.09.028
Redecker, C., Luhmann, H. J., Hagemann, G., Fritschy, J. M., and Witte, O. W. (2000). Differential downregulation of GABAA receptor subunits in widespread brain regions in the freeze-lesion model of focal cortical malformations. J. Neurosci. 20, 5045–5053. doi: 10.1523/JNEUROSCI.20-13-05045.2000
Ribierre, T., Deleuze, C., Bacq, A., Baldassari, S., Marsan, E., Chipaux, M., et al. (2018). Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Invest. 128, 2452–2458. doi: 10.1172/JCI99384
Rosenblat, J. D., Cha, D. S., and Mansur, R. B. (2014). McIntyre RS. Inflamed moods: a review of the interactions between inflammation and mood disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry. 53, 23–34. doi: 10.1016/j.pnpbp.2014.01.013
Sancini, G., Franceschetti, S., Battaglia, G., Colacitti, C., Di Luca, M., Spreafico, R., et al. (1998). Dysplastic neocortex and subcortical heterotopias in methylazoxymethanol-treated rats: an intracellular study of identified pyramidal neurones. Neurosci. Lett. 246, 181–185. doi: 10.1016/S0304-3940(98)00258-4
Seufert, D. W., Prescott, N. L., and El-Hodiri, H. M. (2005). Xenopus aristaless-related homeobox (xARX) gene product functions as both a transcriptional activator and repressor in forebrain development. Dev. Dyn. 232, 313–324. doi: 10.1002/dvdy.20234
Shafi, M. M., Vernet, M., Klooster, D., Chu, C. J., Boric, K., Barnard, M. E., et al. (2015). Physiological consequences of abnormal connectivity in a developmental epilepsy. Ann. Neurol. 77, 487–503. doi: 10.1002/ana.24343
Shoubridge, C., Fullston, T., and Gecz, J. (2010). ARX spectrum disorders: making inroads into the molecular pathology. Hum. Mutat. 31, 889–900. doi: 10.1002/humu.21288
Singh, S. C. (1980). Deformed dendrites and reduced spine numbers on ectopic neurones in the hippocampus of rats exposed to methylazoxymethanol-acetate. A Golgi-Cox study. Acta Neuropathol. 49, 193–198. doi: 10.1007/BF00707106
Smith, M. J., Adams, L. F., Schmidt, P. J., Rubinow, D. R., and Wassermann, E. M. (2002). Effects of ovarian hormones on human cortical excitability. Ann. Neurol. 51, 599–603. doi: 10.1002/ana.10180
Staley, K. J., and Mody, I. (1992). Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABAA receptor-mediated post- synaptic conductance. J. Neurophysiol. 68, 197–212. doi: 10.1152/jn.1992.68.1.197
Stolp, H. B., Turnquist, C., Dziegielewska, K. M., Saunders, N. R., Anthony, D. C., and Molnár, Z. (2011). Reduced ventricular proliferation in the foetal cortex following maternal inflammation in the mouse. Brain 134, 3236–3248. doi: 10.1093/brain/awr237
Stromme, P., Mangelsdorf, M. E., Shaw, M. A., Lower, K. M., Lewis, S. M. E., Bruyere, H., et al. (2002). Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet. 30, 441–445. doi: 10.1038/ng862
Talos, D. M., Sun, H., Kosaras, B., Joseph, A., Folkerth, R. D., Poduri, A., et al. (2012). Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol. 71, 539–551. doi: 10.1002/ana.22696
Tassi, L., Colombo, N., Cossu, M., Mai, R., Francione, S., Lo Russo, G., et al. (2005). Electroclinical, MRI and neuropathological study of 10 patients with nodular heterotopia, with surgical outcomes. Brain 128, 321–337. doi: 10.1093/brain/awh357
Toering, S. T., Boer, K., de Groot, M., Troost, D., Heimans, J. J., Spliet, W. G., et al. (2009). Expression patterns of synaptic vesicle protein 2A in focal cortical dysplasia and TSC-cortical tubers. Epilepsia 50, 1409–1418. doi: 10.1111/j.1528-1167.2008.01955.x
Tyvaert, L., Hawco, C., Kobayashi, E., LeVan, P., Dubeau, F., and Gotman, J. (2008). Different structures involved during ictal and interictal epileptic activity in malformations of cortical development: an EEG-fMRI study. Brain 131, 2042–2060. doi: 10.1093/brain/awn145
Tyzio, R., Khalilov, I., Represa, A., Crepel, V., Zilberter, Y., Rheims, S., et al. (2009). Inhibitory actions of the gamma-aminobutyric acid in pediatric Sturge-Weber syndrome. Ann. Neurol. 66, 209–218. doi: 10.1002/ana.21711
Tyzio, R., Minlebaev, M., Rheims, S., Ivanov, A., Jorquera, I., Holmes, G. L., et al. (2008). Postnatal changes in somatic gamma-aminobutyric acid signalling in the rat hippocampus. Eur. J. Neurosci. 27, 2515–2528. doi: 10.1111/j.1460-9568.2008.06234.x
Valeeva, G., Tressard, T., Mukhtarov, M., Baude, A., and Khazipov, R. (2016). An optogenetic approach for investigation of excitatory and inhibitory network GABA actions in mice expressing channelrhodopsin-2 in GABAergic neurons. J. Neurosci. 36, 5961–5673. doi: 10.1523/JNEUROSCI.3482-15.2016
Valton, L., Guye, M., McGonigal, A., Marquis, P., Wendling, F., Regis, J., et al. (2008). Functional interactions in brain networks underlying epileptic seizures in bilateral diffuse periventricular heterotopia. Clin. Neurophysiol. 119,212–223. doi: 10.1016/j.clinph.2007.09.118
Vezzani, A. (2013). Fetal brain inflammation may prime hyperexcitability and behavioral dysfunction later in life. Ann. Neurol. 74, 1–3. doi: 10.1002/ana.23930
Vezzani, A., Fujinami, R. S., White, H. S., Preux, P. M., Blümcke, I., Sander, J. W., et al. (2016). Infections, inflammation and epilepsy. Acta Neuropathol. 131, 211–234. doi: 10.1007/s00401-015-1481-5
Vukovic, J., Colditz, M. J., Blackmore, D. G., Ruitenberg, M. J., and Bartlett, P. F. (2012). Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J. Neurosci. 32, 6435–6443. doi: 10.1523/JNEUROSCI.5925-11.2012
Wake, H., Moorhouse, A. J., Miyamoto, A., and Nabekura, J. (2013). Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 36, 209–217. doi: 10.1016/j.tins.2012.11.007
Wang, W., Takashima, S., Segawa, Y., Itoh, M., Shi, X., Hwang, S. K., et al. (2011). Nabeshima K, Takeshita M, Hirose S. The developmental changes of Na(v)1.1 and Na(v)1.2 expression in the human hippocampus and temporal lobe. Brain Res. 1389, 61–70. doi: 10.1016/j.brainres.2011.02.083
Zehr, J. L., Todd, B. J., Schulz, K. M., McCarthy, M. M., and Sisk, C. L. (2006). Dendritic pruning of the medial amygdala during pubertal development of the male Syrian hamster. J. Neurobiol. 66, 578–590. doi: 10.1002/neu.20251
Keywords: cortical malformation, epileptogenesis, developmental disorder, ARX, mTOR, focal cortical dysplasia, gray matter heterotopia
Citation: Represa A (2019) Why Malformations of Cortical Development Cause Epilepsy. Front. Neurosci. 13:250. doi: 10.3389/fnins.2019.00250
Received: 21 November 2018; Accepted: 04 March 2019;
Published: 29 March 2019.
Edited by:
Kazunori Nakajima, Keio University, JapanReviewed by:
Carlos Cepeda, Independent Researcher, Los Angeles, CA, United StatesMitsuhiro Kato, Showa University, Japan
Stéphanie Baulac, INSERM U1127 Institut du Cerveau et de la Moelle épinière, France
Copyright © 2019 Represa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alfonso Represa, alfonso.represa@inserm.fr