The calcium-activated slow AHP: cutting through the Gordian knot
- 1Department of Pharmacology, Wayne State University School of Medicine, Detroit, MI, USA
- 2Department of Anatomy and Neurobiology, The University of Tennessee Health Science Center, Memphis, TN, USA
- 3Department of Physiology and Neurobiology, University of Connecticut, Storrs, CT, USA
The phenomenon known as the slow afterhyperpolarization (sAHP) was originally described more than 30 years ago in pyramidal cells as a slow, Ca2+-dependent afterpotential controlling spike frequency adaptation. Subsequent work showed that similar sAHPs were widely expressed in the brain and were mediated by a Ca2+-activated potassium current that was voltage-independent, insensitive to most potassium channel blockers, and strongly modulated by neurotransmitters. However, the molecular basis for this current has remained poorly understood. The sAHP was initially imagined to reflect the activation of a potassium channel directly gated by Ca2+ but recent studies have begun to question this idea. The sAHP is distinct from the Ca2+-dependent fast and medium AHPs in that it appears to sense cytoplasmic [Ca2+]i and recent evidence implicates proteins of the neuronal calcium sensor (NCS) family as diffusible cytoplasmic Ca2+ sensors for the sAHP. Translocation of Ca2+-bound sensor to the plasma membrane would then be an intermediate step between Ca2+ and the sAHP channels. Parallel studies strongly suggest that the sAHP current is carried by different potassium channel types depending on the cell type. Finally, the sAHP current is dependent on membrane PtdIns(4,5)P2 and Ca2+ appears to gate this current by increasing PtdIns(4,5)P2 levels. Because membrane PtdIns(4,5)P2 is essential for the activity of many potassium channels, these finding have led us to hypothesize that the sAHP reflects a transient Ca2+-induced increase in the local availability of PtdIns(4,5)P2 which then activates a variety of potassium channels. If this view is correct, the sAHP current would not represent a unitary ionic current but the embodiment of a generalized potassium channel gating mechanism. This model can potentially explain the cardinal features of the sAHP, including its cellular heterogeneity, slow kinetics, dependence on cytoplasmic [Ca2+], high temperature-dependence, and modulation.
In many types of neurons, Ca2+-activated potassium currents mediate afterhyperpolarizing potentials (AHPs) that play important roles in shaping action potentials and firing patterns (Hille, 2001; Sah and Faber, 2002; Vogalis et al., 2003; Bean, 2007). Work during the last two decades has identified the ion channels carrying some of these currents and elucidated the mechanisms underlying their gating by Ca2+ as well as their modulation. One of these currents, however, the aptly named slow Ca2+-activated potassium current (IsAHP) has remained a conspicuous laggard in both of these regards. This current was originally identified in pyramidal cells of hippocampus and cortex and has been implicated in the control of repetitive firing including spike frequency adaptation (Sah and Faber, 2002; Vogalis et al., 2003), the setting of a neuron's dynamic firing range and the regulation of neuronal gain (Higgs et al., 2006). Yet, in spite of a well appreciated functional importance, the elucidation of its molecular basis has proven remarkably elusive. Most notably, in spite of considerable effort there remains considerable incertitude regarding how Ca2+ gates this current and about the molecular identity of the channels carrying it. In this article, we review past work on the slow afterhyperpolarization (sAHP) and its underlying current and highlight some of the difficulties encountered when trying to understand this current as resulting from the activation of a canonical calcium-activated potassium channel. We then focus on more recent studies that have begun to sketch a possible model capable of explaining the unusual properties of this enigmatic Ca2+-activated potassium current.
Early Studies
In the early 1980's, several studies reported that strong stimuli capable of triggering trains of action potentials elicited a long lasting AHP in many neurons including pyramidal cells of the CA1 and CA3 subfields of the hippocampus, neurons of the locus coeruleus, the nucleus of solitary tract, and myenteric neurons (Alger and Nicoll, 1980; Hotson and Prince, 1980; Schwartzkroin and Stafstrom, 1980; Gustafsson and Wigstrom, 1981; Madison and Nicoll, 1982; Morita et al., 1982; Brown and Griffith, 1983; Haas and Konnerth, 1983; Andrade and Aghajanian, 1984; Dekin and Getting, 1984; Williams et al., 1984; Pennefather et al., 1985; Lancaster and Adams, 1986; Storm, 1990). These AHPs could be shown to be Ca2+-dependent (Alger and Nicoll, 1980; Hotson and Prince, 1980; Schwartzkroin and Stafstrom, 1980; Morita et al., 1982; Andrade and Aghajanian, 1984; Hille, 2001) and to reflect the activation of a K+ selective current (Alger and Nicoll, 1980; Andrade and Aghajanian, 1984; Williams et al., 1984). Most distinctively, they all exhibited remarkably slow activation and decay that distinguished them from other AHPs known at the time from work in muscle cells and invertebrate neurons (Meech, 1978).
In CA1 pyramidal cells, Storm (1987, 1989, 1990) described three AHP components following action potentials that were termed the fast, medium, and slow AHP, respectively. Three distinct AHP components were subsequently described in cat cerebral cortex using combined current- and voltage-clamp recordings (Schwindt et al., 1988a,b). In both pyramidal cell types the fast AHP (fAHP) was defined as the early component that followed the repolarization of an action potential (Figures 1A,B). This fAHP was followed by a more slowly decaying component that could also follow a single action potential and was named the medium AHP (mAHP: Figures 1A,B). Finally, there was a delayed component, the slow AHP (sAHP), which was evident only after a burst of spikes and could be distinguished by its strong regulation by neuromodulators (Figure 1C).
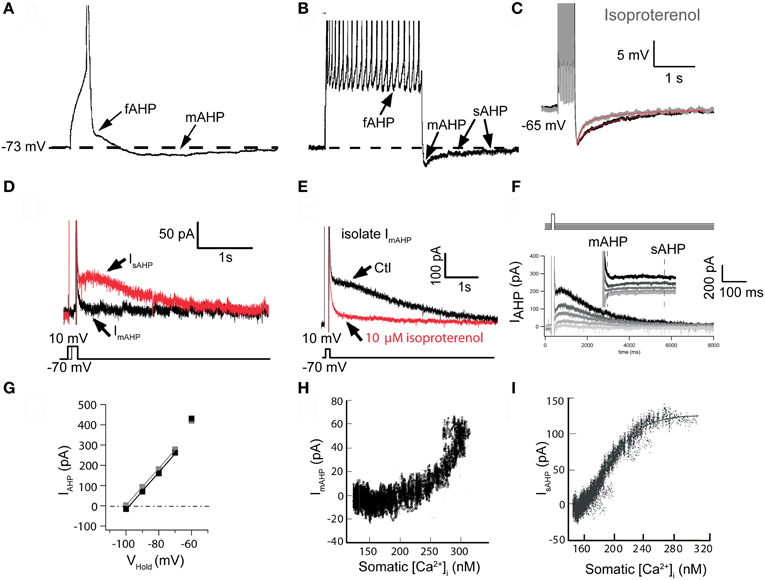
Figure 1. The slow sAHP and underlying current (IsAHP) in neocortical pyramidal neurons from somatosensory cortex. (A) The three AHPs. A single action potential (AP) was elicited with a 10 ms suprathreshold intracellular current injection (spikes truncated by digitization to emphasize afterpotentials). Note the notch following AP repolarization (the fast AHP: fAHP) and subsequent medium AHP (mAHP). (B) Data from the same cell as in (A), except 10 APs were elicited with 10 ms suprathreshold current injections [@ 50 Hz, Panels A and B are redrawn from results presented in Pineda et al. (1998)]. Following the train of APs, two AHP components are evident: the mAHP is the main determinant of the initial peak response. A much slower decaying (τ > 1 s) slow AHP (sAHP) follows (spikes truncated by digitization to emphasize afterpotentials). (C) The sAHP elicited by 1 s repetitive firing is reduced in the presence of the β-agonist isoproterenol [10 μ M: modified from Figure 5A in Abel et al. (2004)]. (D) Tail currents were elicited following voltage steps from −70 mV to 0 mV for different durations. Following the 20 ms step (black trace), only IfAHP and ImAHP were observed upon return to −70 mV. The longer, 150 ms step (red trace) elicited both an initial ImAHP and IsAHP. Note the slow time to peak (the peak occurs well after the voltage step) and slow decay of the sAHP [τ > 1 s: modified from Figure 7B in Abel et al. (2004)]. (E) In a different cell, the IsAHP tail current following a 200 ms voltage step to 0 mV and then returning to −70 mV was blocked by 10 μ M isoproterenol, isolating ImAHP [modified from Figure 7C in Abel et al. (2004)]. (F) Reversal potential for IsAHP. Tail currents were elicited by a 200 ms step to +10 mV and amplitudes were measured upon return to various potentials. ImAHP was measured as the peak response and IsAHP was measured at 500 ms after the peak, when ImAHP had completely decayed [modified from Figure 8A in Abel et al. (2004)]. (G) Plots of IAHP amplitudes from data in (F). Extrapolated reversal potentials approximated EK, as determined by the Nernst equation [EK = −102 mV: modified from Figure 8B in Abel et al. (2004)]. (H) Plot of isolated ImAHP vs. bulk cytoplasmic [Ca2+]i. Since the underlying SK channels respond to a sub-membrane microdomain of [Ca2+], the dose-response relationship is distorted [data from eight cells; modified from Figure 10C in Abel et al. (2004)]. (I) Plot of isolated IsAHP vs. bulk cytoplasmic [Ca2+]i. Note the sigmoidal dose-response curve indicating response to a “well-mixed” bulk [Ca2+]i [data from five cells; estimated KD = ~200 nM, Hill coefficient ~4.5: modified from Figure 9C in Abel et al. (2004)]. Panels A and B were from layer 5A of somatosensory cortex. Panels C–I were from layer 2/3.
The existence of these three AHP components was later confirmed by multiple studies in rodent and human neocortex (Lorenzon and Foehring, 1992, 1993), and several other cell types (Viana et al., 1993; Pape and Driesang, 1998; Teruyama and Armstrong, 2005), although the relative expression of these components, and their corresponding currents were found to vary between cell types. These studies also revealed that the fAHP and the mAHP, as defined by their kinetics, consisted of Ca2+-dependent as well as calcium-independent components (Storm, 1987, 1989, 1990; Schwindt et al., 1988a,b; Pineda et al., 1992; Miles et al., 2005; Pedarzani and Stocker, 2008). The Ca2+-activated component of the fAHP was found to be mediated by large conductance BK-type channels (Lancaster and Nicoll, 1987; Storm, 1987, 1990; Sah and McLachlan, 1992; Miles et al., 2005; Ghatta et al., 2006) while the Ca2+-activated component of the mAHP, at least in neocortical pyramidal cells, was shown to be apamin sensitive indicating it is mediated by small-conductance calcium activated potassium channels (SK, now known as KCa2; Schwindt et al., 1988a,b; Lorenzon and Foehring, 1992; Pineda et al., 1992). In contrast, the sAHP appeared to be consistently Ca2+-dependent suggesting a unitary mechanism. Interestingly, in CA1 pyramidal neurons, the mAHP does not appear to have a Ca2+- or apamin-sensitive component (Storm, 1989; Gu et al., 2005, 2008), despite the presence of clear SK-mediated currents in response to voltage steps (Sah and Clements, 1999; Stocker et al., 1999). It should also be mentioned that the latter part of the sAHP in cat neocortical pyramidal neurons was not Ca2+-dependent but rather appeared due to a Na+-dependent potassium conductance (Foehring et al., 1989; Schwindt et al., 1989). The basis for this Na+-dependent conductance is beyond the scope of the present review.
In the absence of specific blockers for the sAHP, the strongest indication that this AHP component reflected the activation of a distinct calcium-activated potassium current came from the observation that the sAHP, unlike the fAHP or mAHP, was highly susceptible to neuromodulation. This was initially demonstrated for norepinephrine, acting through β-adrenergic receptors (Madison and Nicoll, 1982), and histamine acting via H2 receptors (Haas and Konnerth, 1983), both of which inhibited the sAHP and decreased spike frequency adaptation in pyramidal neurons of the CA1 region of the hippocampus. Subsequent studies extended these observations to other cell types and for other transmitters that activate Gαs-coupled receptors leading to increases in cAMP and activation of protein kinase A (PKA, Figures 1C,E; e.g., Andrade and Nicoll, 1987; McCormick and Prince, 1988; Foehring et al., 1989; McCormick and Williamson, 1989; Pedarzani and Storm, 1993, 1995; Torres et al., 1995; Pedarzani et al., 1998; Haug and Storm, 2000; Lancaster et al., 2006) or that activate Gαq−11 leading to the activation of phospholipase C and the breakdown of membrane phosphatidylinositol 4,5-biphosphate (PtdIns(4,5)P2, Dutar and Nicoll, 1988; Krause et al., 2002; Villalobos et al., 2011). In fact most known neuromodulators and neurotransmitters acting through receptors coupling to these canonical signaling cascades have been shown to inhibit the sAHP (Benardo and Prince, 1982; Cole and Nicoll, 1984; Lancaster and Nicoll, 1987; Madison et al., 1987; Schwindt et al., 1988a; McCormick and Williamson, 1989; Charpak et al., 1990; Araneda and Andrade, 1991; Lorenzon and Foehring, 1992; Torres et al., 1996; Villalobos et al., 2005).
The Search for the Elusive sAHP Channel
Early studies showed that the reversal potential for the sAHP/IsAHP was dependent on extracellular potassium concentration in a manner predicted by the Nernst equation (Figures 1D–G; Alger and Nicoll, 1980; Andrade and Aghajanian, 1984; Williams et al., 1984; Lancaster and Adams, 1986; Constanti and Sim, 1987; Schwindt et al., 1988b; Lorenzon and Foehring, 1992). The sAHP was also clearly activated by an elevation in intracellular [Ca2+]. Thus, the sAHP was blocked by extracellular application of inorganic calcium channel blockers (e.g., Cd2+ or Co2+; Alger and Nicoll, 1980; Hotson and Prince, 1980; Morita et al., 1982; Andrade and Aghajanian, 1984; Madison and Nicoll, 1984; Schwindt et al., 1988b; Pineda et al., 1998) and by intracellular injection of Ca2+ chelators (Alger and Nicoll, 1980; Schwartzkroin and Stafstrom, 1980; Madison and Nicoll, 1984; Storm, 1987; Schwindt et al., 1988b; Lorenzon and Foehring, 1995; Velumian and Carlen, 1999). Similarly the sAHP was also activated by photolytic release of Ca2+ (Lancaster and Zucker, 1994; Sah and Clements, 1999) and inhibited by photolytic Ca2+ chelation (although not rapidly, Lancaster and Zucker, 1994; Sah and Clements, 1999). These observations indicated that the sAHP is mediated by the activation of a Ca2+-activated potassium current. However, these results provide only limited guidance as to the molecular identity of the channel carrying the IsAHP.
The search for the ion channel mediating the sAHP coincided with the molecular identification of ion channel families during the 1990s. At the time the expectation was that the channel responsible for IsAHP would turn out to be a potassium channel directly gated by Ca2+ and the discovery of the Drosophila Ca2+-activated potassium channel Slo initially supported this idea (Adelman et al., 1992; Bond et al., 2004; Salkoff et al., 2006). The subsequent identification of SK channels seemed initially to identify a plausible candidate channel family capable of carrying IsAHP (Bond et al., 2004). Specifically SK1 channels were initially reported to exhibit a lower sensitivity to apamin (Kohler et al., 1996), raising the possibility that such channels could mediate the sAHP. This observation led to the explicit proposal that SK1 channels, in association with delayed facilitation of L-type calcium channels, could be responsible for the sAHP in CA1 pyramidal neurons (Bowden et al., 2001). Subsequent work, however, questioned the apamin insensitivity of SK1, casting doubts on this possibility (Shah and Haylett, 2000b; Grunnet et al., 2001; Weatherall et al., 2010). Nevertheless, it remained possible that SK channels could be formed with unique stoichiometries or co-assemble with additional subunits to render them insensitive to apamin. To address these uncertainties two independent groups used dominant negative and overexpression approaches (Villalobos et al., 2004) or gene deletion strategies (Bond et al., 2004) to target SK channels in pyramidal neurons. Both studies confirmed the role of SK channels in carrying the current responsible for the mAHP but could find no evidence that these channels participated in the generation of IsAHP. Thus, IsAHP was clearly not carried through BK or SK channels. So what are the molecular underpinnings of IsAHP?
Clues from the Activation of the sAHP by Ca2+
During neuronal firing intracellular Ca2+ increases with the number of spikes until a plateau is attained where there is a balance between Ca2+ entry and extrusion (Regehr et al., 1994; Helmchen et al., 1996; Maravall et al., 2000; Abel et al., 2004). Consequently the amplitude of the Ca2+-activated AHP is strongly dependent on neuronal activity and summates non-linearly as the action potential firing frequency increases. However, there are important differences in how Ca2+ activates the different components of the AHP.
Ca2+ entering a cell through calcium channels during one or more action potentials creates transient nanodomains of high calcium concentration that can sustain the activation of low affinity BK channels (μM EC50; Fakler and Adelman, 2008). As Ca2+ diffuses away from the calcium channel and the plasma membrane it combines with Ca2+ from other channels to create larger microdomains of elevated Ca2+ (Neher, 1998; Fakler and Adelman, 2008). Thus, Ca2+ flowing through multiple calcium channels are thought to contribute to the formation of these microdomains. Following termination of the Ca2+ influx, diffusion and the interaction with intracellular Ca2+ reaction partners leads to the dissipation of the [Ca2+] gradient associated with these microdomains and equilibration with the bulk cytoplasm. Bulk cytoplasmic [Ca2+] remains elevated until Ca2+ is taken up into intracellular stores or extruded from the plasma membrane. An important difference between the mAHP and the sAHP concerns how they are activated by the different Ca2+ pools resulting from these processes.
In expression systems, SK channels respond rapidly to elevations in intracellular [Ca2+] and there is a sigmoidal and cooperative relationship between [Ca2+] and the macroscopic SK current (Kohler et al., 1996; Hirschberg et al., 1998). Wilson and Callaway (2000) considered the relationship between the apamin sensitive (SK-mediated) IAHP versus bulk intracellular [Ca2+] in dopaminergic neurons of the substantia nigra and concluded that a sigmoidal dose-response relationship between IAHP and bulk cytoplasmic [Ca2+] would occur only if cytoplasmic [Ca2+] was well mixed. Since such a situation is unlikely to occur near the membrane immediately after Ca2+ entry, when [Ca2+] would be highest at the membrane and lower in the cytoplasm, or subsequently as pumps lower [Ca2+] near the plasma membrane, this provided an avenue for assessing the location of the activating Ca2+. They observed a distorted sigmoidal relationship between bulk [Ca2+] and the apamin sensitive AHP in dopamine cells, as did Abel et al. (2004) for ImAHP in neocortical pyramidal cells (Figure 1H). These results suggest that apamin-sensitive mAHP responds to restricted microdomains of Ca2+ not accurately reflected by measurement of bulk [Ca2+]. This is consistent with the previous demonstration that SK channels sense Ca2+ through their constitutive association with calmodulin (Xia et al., 1998). In contrast, the sAHP current exhibits a cooperative and sigmoidal dose-response relationship with bulk cytoplasmic calcium (Figure 1I, Abel et al., 2004). In other words, the sAHP channels in neocortical pyramidal cells integrate a Ca2+ signal that is proportional to that measured in the bulk cytoplasm. These results suggest a cytoplasmic localization for the sAHP Ca2+ sensor.
If the sAHP activation reflects the rise in bulk calcium, it could be expected to exhibit a loose coupling to calcium channels. Consistent with this idea, the relationship of the sAHP to specific calcium channel subtypes is not strict and the coupling between specific calcium channels and the sAHP appears to be cell type-specific. For example, pyramidal cells of the cerebral cortex express a large sAHP during early postnatal development that is activated, at least in part, by L-type channels and ryanodine-sensitive internal stores (Lorenzon and Foehring, 1993; Pineda et al., 1999). However, in mature neocortical pyramidal neurons the sAHP is activated instead by N- and P/Q-type but not by L-type channels (Pineda et al., 1998). More generally, it is now clear that practically all calcium channel classes can activate the sAHP. Thus, for example, N-type calcium channels have been shown to couple to the sAHP in vagal motoneurons (Sah, 1995), superior cervical ganglion (Maingret et al., 2008), AH-type myenteric neurons from duodenum (Vogalis et al., 2001), and mouse sympathetic neurons (Martinez-Pinna et al., 2000b). Similarly L-type channels have been reported to contribute to the activation of the sAHP in CA1 (Moyer et al., 1992) and CA3 (Tanabe et al., 1998) pyramidal cells of the hippocampus, and in cholinergic interneurons of the striatum (Goldberg and Wilson, 2005; Gamelli et al., 2011). L- and N-type channels both contribute to the activation of the sAHP in CA1 pyramidal cells in culture (Shah and Haylett, 2000a) as well as dentate granule cells (Aradi and Holmes, 1999). Finally, L-, N-, P-type channels have been reported to activate the sAHP in guinea pig sympathetic neurons (Martinez-Pinna et al., 2000a), while T-type calcium channels can activate the sAHP in thalamic paraventricular nucleus (Zhang et al., 2009). Ryanodine-sensitive calcium stores have also been implicated in sAHP activation via calcium-induced calcium release in CA1 and CA3 neurons (Torres et al., 1996; Tanabe et al., 1998; Shah and Haylett, 2000a), guinea pig sympathetic neurons (Jobling et al., 1993) and vagal motoneurons (Sah and McLachlan, 1991). Thus, these results indicate considerable promiscuity in the coupling Ca2+ sources to the sAHP. This is consistent with the idea that IsAHP senses bulk cytoplasmic [Ca2+] and therefore is relatively unselective with respect to the origin of the Ca2+.
The Time Course of the sAHP
A central feature of IsAHP is that it activates very slowly after a spike train (hundred of ms). Specifically the IsAHP rises much slower than the cytoplasmic [Ca2+] (Sah and Clements, 1999; Abel et al., 2004; Gerlach et al., 2004; Goldberg et al., 2009) and continues to rise after the peak of the calcium transient (Lasser-Ross et al., 1997; Jahromi et al., 1999). It also decays very slowly, up to several seconds in some cells. Historically several different possibilities have been proposed to account for these unusually slow kinetics.
The simplest idea that could explain the slow onset kinetics of the sAHP is that it reflects the slow equilibration of free Ca2+ in the cytosol. This explanation, however, is unlikely since free Ca2+ declines rapidly (~99% in first ms) due to binding to its reaction partners (Markram et al., 1998; Goldberg et al., 2009). Furthermore, activation of the sAHP by either neuronal depolarization or rapid Ca2+ uncaging results in similar time courses (Sah and Clements, 1999) and, at least in pyramidal cells and striatal cholinergic interneurons, there is a mismatch between the time course of the decay of IsAHP and bulk [Ca2+] concentration in the soma or dendrites (Lasser-Ross et al., 1997; Jahromi et al., 1999; Abel et al., 2004; Goldberg et al., 2009). Finally changes in Ca2+ buffering can have differential effects on the time course of the calcium transient and IsAHP (Schwindt et al., 1992b; Lorenzon and Foehring, 1995; Lasser-Ross et al., 1997; Jahromi et al., 1999).
Alternatively, the time course of activation could reflect the diffusion of Ca2+ ions from their source to distally located sAHP channels (Lancaster et al., 1991; Lancaster and Zucker, 1994; Zhang et al., 1995; Jahromi et al., 1999). This explanation, however, also seems unlikely. If we assume that Ca2+ diffusion distance determines onset kinetics and use the activation kinetics of BK or SK as benchmarks, it is possible to estimate the expected distance between calcium source and sAHP channels. Because of their low (μM) affinity for Ca2+, BK channels must be located within 10–20 nm of the Ca2+ source to be activated (Muller et al., 2007; Fakler and Adelman, 2008). In contrast, SK channels have a higher Ca2+ affinity (200–500 nM: Kohler et al., 1996; Xia et al., 1998) and thus can be effectively activated 50–100 nm from the Ca2+ source (Fakler and Adelman, 2008). The channels underlying the sAHP have similar affinity for Ca2+ as SK channels (Abel et al., 2004), but activate an order of magnitude slower. This indicates that the sAHP Ca2+ sensor would need to be located prohibitively far (100 s of nms) from the site of calcium entry to account for the slow activation of the current. Also inconsistent with this idea are studies using vagal motoneurons (Sah and McLachlan, 1991) and pyramidal cells (Sah and Isaacson, 1995; Lee et al., 2005) that have shown that the activation of the sAHP exhibits a high temperature sensitivity. Specifically, the sAHP has a Q10 between 2 and 4, a range of values that is inconsistent with aqueous diffusion and is usually associated with enzymatic activity or slow channel gating events. This argues against diffusion of Ca2+ as being the rate-limiting step for sAHP activation.
A third possibility is that the slow activation of the sAHP could reflect delayed facilitation of L-type calcium channels (Cloues et al., 1997; Bowden et al., 2001). In particular, the slow kinetics of the sAHP in CA1 pyramidal neurons has been attributed to delayed facilitation of L-type channels of the α1D (CaV1.3) type (Bowden et al., 2001). However, given the limited and partial role of L type calcium channels as calcium sources for the sAHP, this mechanism again seems unlikely to provide a universal account for the slow time course of activation of this current.
Finally, the slow activation of the sAHP current has also been attributed to slow binding of Ca2+ to its sensor, slow intrinsic kinetics of the sAHP potassium channel (Lancaster et al., 1991; Sah and Clements, 1999) or the involvement Ca2+-induced Ca2+ release (CICR) from internal stores. None of these factors alone, however, appears capable of fully accounting for the time course of the sAHP. For example the time to onset of the sAHP current has been shown to be insensitive to [Ca2+] casting doubts on the idea that slow calcium binding to the sensor could represent the rate-limiting step for activation of IsAHP [(Sah and Clements, 1999; Gerlach et al., 2004), but see below]. Similarly, estimates of the sAHP channel mean open time based upon noise analysis are much too short to fully account for the slow onset kinetics of the current (Sah and Isaacson, 1995). Finally, although CICR does contribute to the sAHP in some neurons (Sah and McLachlan, 1992; Davies et al., 1996; Torres et al., 1996; Shah and Haylett, 2000a; Vogalis et al., 2001), it contributes little to the sAHP in other cell types including mature, repetitively firing neocortical pyramidal neurons (Zhang et al., 1995; Pineda et al., 1998) again casting doubt on the generality of such a mechanism.
The possibilities outlined above all have assumed that the channels underlying the sAHP are gated by Ca2+ in a relatively direct manner. Therefore, the solution to the anomalous properties of the sAHP/IsAHP had to reside in the properties of the Ca2+ signal or the sAHP channel itself. More recent studies have focused on the possibility of more complex intermediate steps between Ca2+ influx and the activation of IsAHP. A recent study combining experimental and modeling approaches in striatal cholinergic interneurons has suggested a key role for intracellular Ca2+ buffering mechanisms in generating the slow time course of AHP currents. Using a computational approach this study revealed that non-equilibrium dynamics of Ca2+ redistribution among cytoplasmic binding sites with different Ca2+ binding kinetics can give rise to multiple timescales within the same cytoplasmic volume (Goldberg et al., 2009). Key to this model is the assumption that the sAHP Ca2+ binding site does not have direct access to cytoplasmic Ca2+ with a time course determined only by Ca2+ entry. Rather, the presence of other Ca2+ reaction partners with faster binding kinetics can shape the time course of calcium available to bind the sAHP sensor (Markram et al., 1998). The kinetics of the various reaction partners, not the rate of Ca2+ entry to the cell, would then determine the delivery of Ca2+ to the sAHP site. The Goldberg et al. (2009) analysis raises the possibility that the temporal properties of the sAHP, including the delay in its onset and its slow decay, may be caused by the kinetics of the sAHP Ca2+ binding site/sensor, interacting with alternate binding sites in the cytoplasm. This explanation is consistent with the sAHP's sensitivity to fast and slow exogenous buffers and its insensitivity to brief Ca2+ transients.
Alternatively, recent findings using molecular approaches have rekindled interest in the possibility that calcium may activate the sAHP indirectly, through a signaling cascade involving one or more intermediate step (Hocherman et al., 1992; Schwindt et al., 1992a; Zhang et al., 1995; Sah and Faber, 2002; Abel et al., 2004; Tzingounis et al., 2007; Villalobos and Andrade, 2010; Villalobos et al., 2011). An attractive feature of interposing one or more molecular steps between Ca2+ binding and channel activation is that such a mechanism can accomodate most of the puzzling features of the sAHP current including its slow kinetics and temperature-dependence, the multiple action potential requirement, and the fact that rather than responding to micro- or nanodomains of calcium, sAHP activation requires an elevation of Ca2+ in the cytoplasm.
Neuronal Calcium Sensor Proteins and the sAHP
Recent experiments have provided strong evidence that hippocalcin, a member of the neuronal calcium sensor (NCS) protein family, is at least a partial Ca2+ sensor for the sAHP in pyramidal neurons (Tzingounis et al., 2007; Villalobos and Andrade, 2010). Hippocalcin is located in the cytoplasm but translocates to the plasma membrane upon Ca2+ binding with a relatively slow time course (Markova et al., 2008). This translocation is thought to result from a Ca2+-induced conformational change that leads to the exposure of a myristoyl group and repartition of the NCS to the plasma membrane. Tzingounis et al. (2007) found that IsAHP was greatly reduced in a hippocalcin null mutant mouse and that expression of hippocalcin into cultured neurons enhanced the sAHP (Figure 2A). In these experiments hippocalcin appeared to act as a true Ca2+ sensor (as opposed to acting as a Ca2+ buffer) since IsAHP was not enhanced by introduction of mutated hippocalcin with impaired myristoylation, and thus impaired translocation to the plasma membrane (Figure 2A). This last finding suggests that while hippocalcin may act as a mobile Ca2+ buffer relative to SK channels, its role in the sAHP is more as a true sensor. Collectively, these findings are consistent with the hypothesis that the Ca2+ sensor for the sAHP channels is not part of the channel complex but rather gates the sAHP upon translocation to the membrane. The need for such a mobile calcium sensor to translocate from the cytosol to the plasma membrane to activate the sAHP could help explain the slow time course of activation and dependence on bulk Ca2+.
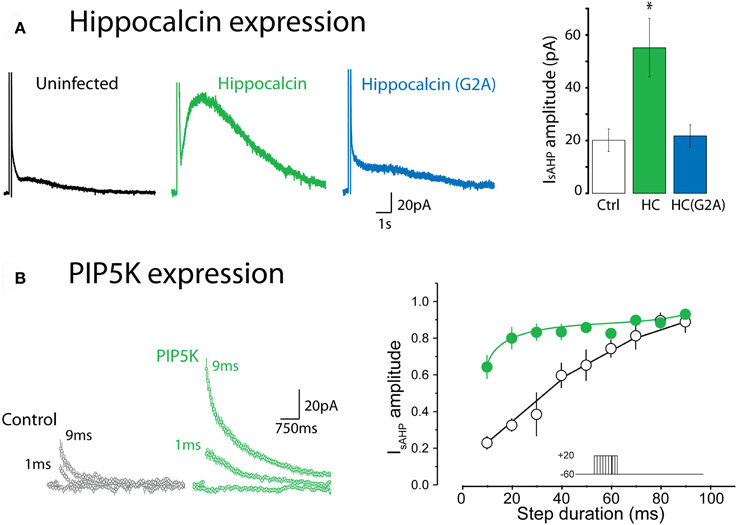
Figure 2. Expression of hippocalcin and the phosphatidylinositol 4-phosphate 5-kinase (PIP5K) regulate IsAHP. (A) Expression of wild type hippocalcin in hippocampal pyramidal cells in primary culture greatly enhances the amplitude of IsAHP. This enhancement is not seen with the G2A mutant, which cannot be myristoylated, thus pointing to an essential role for the translocation of hippocalcin to the plasma membrane. *Indicates p < 0.001. Redrawn from data in Figure 4 of Tzingounis et al. (2007). (B) Expression of PIP5K enhances the apparent ability of calcium to elicit IsAHP. In this experiment calcium influx was titrated using depolarizing steps of increasing duration. Under control conditions IsAHP is activated in a graded manner by depolarizing steps ranging from 10 to 100 ms. In contrast, in cells transfected with PIP5K IsAHP is activated by much shorter steps. Redrawn from data in Figure 7 of Villalobos et al. (2011).
In the Tzingounis et al. (2007) study, the sAHP was not completely eliminated in the hippocalcin knockout mouse. Furthermore, the distribution of hippocalcin in the brain only partly overlaps with the distribution of neurons exhibiting a pronounced sAHP [Allen Brain Atlas; (Villalobos and Andrade, 2010)]. A recent study indicated that in pyramidal cells of the prefrontal cortex, neurocalcin δ, a related neuronal Ca2+ sensor protein, acts similarly and perhaps in combination with hippocalcin to activate IsAHP (Villalobos and Andrade, 2010). These results suggest that two or more NCS family members can act as calcium sensors linking rises in cytoplasmic Ca2+ to sAHP channel activation. A limitation of these studies is that they have relied upon constitutive gene deletions or overexpression strategies. This leaves open the possibility that the observed changes in the sAHP may reflect indirect effects of modifying the cells' ability to sense Ca2+.
The Elusive sAHP Channel
A major impediment to identifying the channel carrying the sAHP has been the scarcity of pharmacological agents capable of selectively targeting this afterpotential. For instance, the sAHP in hippocampus and neocortical neurons is resistant to most known potassium channel blockers or toxins (e.g., apamin, TEA, 4-AP, cesium, quinine, ruthenium red). An important development was the discovery that clotrimazole (Shah et al., 2001) and especially its analog UCL2027 appear to be reasonably selective inhibitors of the sAHP, at least in some cell types (Shah et al., 2006; Lee et al., 2010). Besides the sAHP, the only known targets of UCL2027 are KCNQ-mediated currents (Soh and Tzingounis, 2010) suggesting the possible involvement of these channels in mediating the sAHP (see below). However, a more detailed investigation of the potassium channel selectivity of UCL2077 will be needed before UCL2077 can act as a screening tool for the sAHP channels.
In the absence of strong pharmacological leads several studies have used non-stationary noise analysis and single channel recordings to gain insight into the properties of the ion channels mediating the sAHP. Noise analysis in pyramidal cells of the CA1 region of the hippocampus (Sah and Isaacson, 1995), granule cells of the dentate gyrus (Valiante et al., 1997), and the dorsal motor nucleus of the vagus (Sah, 1995) revealed that potassium channels exhibiting a small conductance mediate the sAHP but there was considerable variability in their estimates for single channel conductance (γ: 2–10 pS). Potassium channels with γ in this range include SK (KCNN), some Kv1 (KCNA), Kv4 (KCND), some Kir (KCNJ) channels, and Kv7 (KCNQ) channels (Coetzee et al., 1999). It has recently been reported that in granule cells of the dentate gyrus, KATP channels open in response to action potential bursts and the resulting sAHP is reduced by the KATP inhibitor glibenclamide (Tanner et al., 2011). This suggests that these channels may contribute to the sAHP in granule cells. However, since γ is typically > >10 pS for KATP channels (Coetzee et al., 1999) such a mechanism is unlikely to be widely generalizable. Thus, collectively, these pharmacological and single channel studies fail to converge on a defined set of properties for the channels mediating the sAHP. The results suggest that IsAHP may not be a unitary current due to a single molecular entity but rather may be mediated by a variety of ion channels depending on the cellular background.
An alternative approach to identify the channels responsible for the sAHP has been to examine the effect of ion channel subunit gene deletions on the sAHP. The KCNQ1–5 genes code for the Kv7 potassium channels that underlie the “M current” in a variety of central and peripheral neurons (Delmas and Brown, 2005). Surprisingly, the genetic deletion of KCNQ2 or KCNQ3 was found to result in a significant decrease in the amplitude of the sAHP current in granule cells of the dentate gyrus. Similarly, expression of a KCNQ2/3 pore-dead dominant negative in slice culture or of a KCNQ5 pore-dead dominant negative in a knock-in mouse both inhibited IsAHP in CA3 pyramidal neurons (Tzingounis and Nicoll, 2008; Tzingounis et al., 2010). These results, along with the inhibition of the sAHP by UCL2077 (Soh and Tzingounis, 2010), suggest a significant role for KCNQ channels in the generation of the sAHP, at least in CA3 pyramidal and dentate granule cells.
The involvement of KCNQ channels in the generation of the sAHP has been controversial, at least in part, because KCNQ channels are inhibited, rather than activated by intracellular Ca2+. Previous studies, however, have shown that the calcium inhibition of KCNQ channels is mediated a Ca2+/calmodulin (CaM)-dependent mechanism (Selyanko and Brown, 1996; Gamper and Shapiro, 2003, reviewed by Delmas and Brown, 2005) while the activation of IsAHP is mediated by NCS proteins of the hippocalcin family, which have much lower Ca2+ operating ranges than calmodulin [reviewed by Burgoyne (2007)]. The IsAHP is activated by calcium with an EC50 ~ 300 nM, well within the operating range of hippocalcin but below that of calmodulin (O'Callaghan et al., 2003; Burgoyne, 2007). Therefore, the reported Ca2+-CaM inhibition of KCNQ channels is unlikely to operate during the IsAHP as the Ca2+ levels necessary to activate the IsAHP are well below those required for calmodulin to inhibit KCNQ channels. The idea that KCNQ channels may contribute to IsAHP has also been questioned because the IsAHP appears to be largely insensitive to KCNQ channel blockers in some of the prototypical cell types expressing this current. For example, in the CA1 region of the hippocampus, administration of KCNQ blockers (e.g., linopirdine or XE-991) has led to inconsistent results with some studies reporting partial block of IsAHP (Schnee and Brown, 1998; Tzingounis and Nicoll, 2008), while others found no effect of these blockers to this current (Aiken et al., 1995; Gerlach et al., 2004; Gu et al., 2005). Similar studies on pyramidal cells from neocortex have also failed to find any effect of KCNQ channel blockers on IsAHP (Abel et al., 2004; Guan et al., 2011). However, these observations are consistent with the possibility outlined above that IsAHP may be mediated by different complement of channels in different cell types.
PtdIns(4,5)P2 and the Ca2+-Dependent Regulation of Potassium Channels
Most of the channels considered above, including KCNQ, are not Ca2+-activated and some are also voltage activated. Therefore, if such channels were to underlie the sAHP a mechanism must exist to allow Ca2+ to indirectly activate them and/or alter their apparent voltage sensitivity. Recent work indicates that the functional expression of the sAHP current is dependent on membrane PtdIns(4,5)P2 and that increasing membrane PtdIns(4,5)P2 greatly facilitates the ability of Ca2+ to activate the sAHP (Figure 2B). This has been interpreted to suggest that Ca2+ acts upstream from PtdIns(4,5)P2 to activate IsAHP (Villalobos et al., 2011). Since results from a variety of model systems indicate that Ca2+ can regulate the local availability of PtdIns(4,5)P2 in the membrane, a simple interpretation of these results is that Ca2+ gates the sAHP channels by increasing the availability of PtdIns(4,5)P2 near the channel. Previous studies have shown that PtdIns(4,5)P2 can control potassium channels at multiple levels. For example this phosphoinositide is required for the activity of many potassium channels of the Kir and Kv families (Delmas and Brown, 2005; Hansen et al., 2011), can regulate the inactivation of “A type” potassium channels (Oliver et al., 2004) and modulate the affinity of KATP channels for ATP (Baukrowitz et al., 1998; Shyng and Nichols, 1998). Since many PtdIns(4,5)P2-sensitive potassium channels are subsaturated at rest, a transient Ca2+-triggered increase in PtdIns(4,5)P2 could be expected to result in an increase in the activity of multiple classes of potassium channels. At the macroscopic level this increase in channel activity would result in a slow potassium aftercurrent that could correspond to IsAHP (Villalobos et al., 2011).
One of the attractive features of this mechanism is that it can explain with economy some of the most puzzling aspects of IsAHP. For example, the monoexponential decay of the sAHP (Lancaster and Adams, 1986) has been generally interpreted to indicate the involvement of a single type of channel in the generation of sAHP/IsAHP, even as growing evidence suggest considerable molecular diversity depending on the cell type examined (see above). A monoexponential decay implies a single mechanism functioning as the rate limiting step, which could reflect the involvement of a single ion channel subtype or a single (essential) biochemical step. Thus, a monoexponential decay that is independent of amplitude is equally well predicted by a model where activation of multiple types of potassium channels follows a rate-limiting intermediate step, in the current hypothesis the availability of PtdIns(4,5)P2. While these considerations suggest a broad range of potassium channels could participate in the generation of IsAHP, we believe that there still must be some molecular specificity as the sAHP channels have small single channel conductance and lack sensitivity to multiple known potassium channel blockers and toxins, features that are not shared by most potassium channels.
PtdIns(4,5)P2 by virtue of its effect on potassium channel gating may also help explain the involvement of channels that, based upon their voltage dependence, may appear unlikely candidates to carry IsAHP (which is voltage-insensitive). Previous studies have shown that PtdIns(4,5)P2 stabilizes the open conformation of potassium channels including KCNQ channels (Enkvetchakul et al., 2000; Loussouarn et al., 2003; Park et al., 2005; Hernandez et al., 2009; Falkenburger et al., 2010; Rodriguez et al., 2010). Current models also suggest that KCNQ channels are gated allosterically by voltage, in other words that voltage sensor activation is not obligatory for channel opening. Consequently, a transient PtdIns(4,5)P2 increase might promote KCNQ channel opening at hyperpolarized potentials bypassing the need for multiple voltage sensor activation. A facilitation of such voltage independent transitions by PtdIns(4,5)P2 would manifest itself as a shift of the KCNQ channel half-activation voltage (V0.5) to more hyperpolarized values, leading to an apparent voltage insensitivity at the voltages where the sAHP is measured. Consistent with this possibility, recent work from Suh and Hille (2007) has shown that overexpression of the phosphatidylinositol 4-phosphate 5-kinase (PIP5K) in heterologous cells, which can be expected to increase basal PtdIns(4,5)P2 levels, shifts the KCNQ2/3 V0.5 to more hyperpolarized membrane potentials. Although this model is only a hypothesis it might provide a starting point for understanding the mechanism by which KCNQ channel or other voltage-activated potassium channels can contribute to the sAHP. A similar argument for the modulation of KATP channels by PtdIns(4,5)P2 can be based on a previously described model by Enkvetchakul et al. (2000). More broadly, this brief discussion highlights how the ability of PtdIns(4,5)P2 to regulate potassium channel gating could help explain some the properties of IsAHP.
Finally the PtdIns(4,5)P2 hypothesis also has the potential to help clarify the mechanisms underlying the modulation of IsAHP. The inhibition of a molecularly heterogeneous IsAHP by receptors coupling to Gαq−11/PLCβ would simply follow from the lowering of membrane PtdIns(4,5)P2 levels (Villalobos et al., 2011). The inhibition of IsAHP by activation of the Gαs/adenylate cyclase/cAMP/PKA signaling cascade is thought to involve a poorly understood phosphorylation step downstream from PKA (Pedarzani and Storm, 1993). Since PKA phosphorylation strongly inhibits PIP5K (Park et al., 2001), the rate limiting enzyme for the formation of PtdIns(4,5)P2, it seems possible that PKA may also inhibit IsAHP by reducing membrane PtdIns(4,5)P2 levels. If this conjecture is correct it could explain how PKA activation could inhibit a current carried by ion channels, such as KCNQ, that are not directly modulated by cAMP/PKA.
The ideas outlined above are summarized in Figure 3. While this model offers a way forward in our thinking about the molecular physiology of the sAHP it is important to note that numerous important questions still remain even if this model proves correct. For example, which potassium channels are more likely to mediate the sAHP? Does the modulation of the PtdIns(4,5)P2 generating enzymes by kinases and phosphatases hold the answer to the neuromodulation of the sAHP by cAMP and PKA? How do NCS proteins gate the sAHP, do they bind directly to the sAHP channels or do they shuttle PtdIns(4,5)P2 generating enzymes to the plasma membranes? Fortunately we now have the molecular and conceptual tools for addressing these issues and thus it seems reasonable to expect quick progress on these and other questions central to our understanding the sAHP.
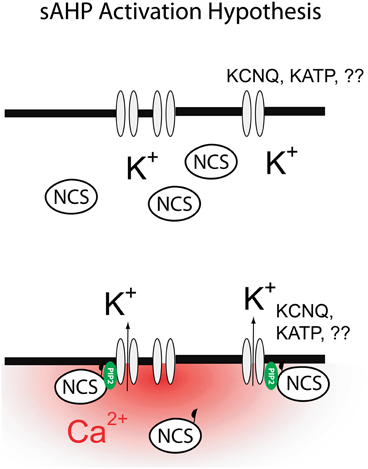
Figure 3. Mechanism for the activation of the sAHP as proposed in this review. Global increases in cytosolic calcium lead to activation of diffusible neuronal calcium sensors (NCS: hippocalcin, neurocalcin δ). Binding of calcium to NCS exposes a previously sequestered myristoyl moiety allowing NCS to bind to the plasma membrane. Binding of NCS to plasma membrane leads to a transient increase in PtdIns(4,5)P2 levels and subsequent activation of the sAHP.
In summary, recent studies have begun to sketch a possible mechanism for IsAHP involving the idea that Ca2+ gates IsAHP indirectly, via a diffusible Ca2+ sensor and PtdIns(4,5)P2. While this idea still remains conjectural at this time, this conceptualization offers an economical way to reconcile some of the most puzzling effects of IsAHP including its anomalous dependence on Ca2+, its slow kinetics and its apparent molecular heterogeneity depending on the cellular background. If these ideas are correct, perhaps after 30 years we may finally be cutting though the knot concealing the sAHP.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the author's laboratories is supported by NIH grants MH43985 (Rodrigo Andrade), NS044163 (Robert C. Foehring) NS060890 (Anastasios V. Tzingounis), NS073981 (Anastasios V. Tzingounis), and Citizens United for Research in Epilepsy (CURE, Anastasios V. Tzingounis) grant.
References
Abel, H. J., Lee, J. C., Callaway, J. C., and Foehring, R. C. (2004). Relationships between intracellular calcium and afterhyperpolarizations in neocortical pyramidal neurons. J. Neurophysiol. 91, 324–335.
Adelman, J. P., Shen, K. Z., Kavanaugh, M. P., Warren, R. A., Wu, Y. N., Lagrutta, A., et al. (1992). Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron 9, 209–216.
Aiken, S. P., Lampe, B. J., Murphy, P. A., and Brown, B. S. (1995). Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br. J. Pharmacol. 115, 1163–1168.
Alger, B. E., and Nicoll, R. A. (1980). Epileptiform burst afterhyperolarization: calcium-dependent potassium potential in hippocampal CA1 pyramidal cells. Science 210, 1122–1124.
Andrade, R., and Aghajanian, G. K. (1984). Locus coeruleus activity in vitro: intrinsic regulation by a calcium-dependent potassium conductance but not alpha 2-adrenoceptors. J. Neurosci. 4, 161–170.
Andrade, R., and Nicoll, R. A. (1987). Pharmacologically distinct actions of serotonin on single pyramidal neurones of the rat hippocampus recorded in vitro. J. Physiol. 394, 99–124.
Aradi, I., and Holmes, W. R. (1999). Role of multiple calcium and calcium-dependent conductances in regulation of hippocampal dentate granule cell excitability. J. Comput. Neurosci. 6, 215–235.
Araneda, R., and Andrade, R. (1991). 5-Hydroxytryptamine2 and 5-hydroxytryptamine 1A receptors mediate opposing responses on membrane excitability in rat association cortex. Neuroscience 40, 399–412.
Baukrowitz, T., Schulte, U., Oliver, D., Herlitze, S., Krauter, T., Tucker, S. J., et al. (1998). PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 282, 1141–1144.
Bean, B. P. (2007). The action potential in mammalian central neurons. Nat. Rev. Neurosci. 8, 451–465.
Benardo, L. S., and Prince, D. A. (1982). Cholinergic excitation of mammalian hippocampal pyramidal cells. Brain Res. 249, 315–331.
Bond, C. T., Herson, P. S., Strassmaier, T., Hammond, R., Stackman, R., Maylie, J., et al. (2004). Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J. Neurosci. 24, 5301–5306.
Bowden, S. E., Fletcher, S., Loane, D. J., and Marrion, N. V. (2001). Somatic colocalization of rat SK1 and D class (Ca(v)1.2) L-type calcium channels in rat CA1 hippocampal pyramidal neurons. J. Neurosci. 21, RC175.
Brown, D. A., and Griffith, W. H. (1983). Calcium-activated outward current in voltage-clamped hippocampal neurones of the guinea-pig. J. Physiol. 337, 287–301.
Burgoyne, R. D. (2007). Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193.
Charpak, S., Gahwiler, B. H., Do, K. Q., and Knopfel, T. (1990). Potassium conductances in hippocampal neurons blocked by excitatory amino-acid transmitters. Nature 347, 765–767.
Cloues, R. K., Tavalin, S. J., and Marrion, N. V. (1997). Beta-adrenergic stimulation selectively inhibits long-lasting L-type calcium channel facilitation in hippocampal pyramidal neurons. J. Neurosci. 17, 6493–6503.
Coetzee, W. A., Amarillo, Y., Chiu, J., Chow, A., Lau, D., McCormack, T., et al. (1999). Molecular diversity of K+ channels. Ann. N.Y. Acad. Sci. 868, 233–285.
Cole, A. E., and Nicoll, R. A. (1984). The pharmacology of cholinergic excitatory responses in hippocampal pyramidal cells. Brain Res. 305, 283–290.
Constanti, A., and Sim, J. A. (1987). Calcium-dependent potassium conductance in guinea-pig olfactory cortex neurones in vitro. J. Physiol. 387, 173–194.
Davies, P. J., Ireland, D. R., and McLachlan, E. M. (1996). Sources of Ca2+ for different Ca(2+)-activated K+ conductances in neurones of the rat superior cervical ganglion. J. Physiol. 495(Pt 2), 353–366.
Dekin, M. S., and Getting, P. A. (1984). Firing pattern of neurons in the nucleus tractus solitarius: modulation by membrane hyperpolarization. Brain Res. 324, 180–184.
Delmas, P., and Brown, D. A. (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 6, 850–862.
Dutar, P., and Nicoll, R. A. (1988). Classification of muscarinic responses in hippocampus in terms of receptor subtypes and second-messenger systems: electrophysiological studies in vitro. J. Neurosci. 8, 4214–4224.
Enkvetchakul, D., Loussouarn, G., Makhina, E., Shyng, S. L., and Nichols, C. G. (2000). The kinetic and physical basis of K(ATP) channel gating: toward a unified molecular understanding. Biophys. J. 78, 2334–2348.
Fakler, B., and Adelman, J. P. (2008). Control of K(Ca) channels by calcium nano/microdomains. Neuron 59, 873–881.
Falkenburger, B. H., Jensen, J. B., and Hille, B. (2010). Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J. Gen. Physiol. 135, 99–114.
Foehring, R. C., Schwindt, P. C., and Crill, W. E. (1989). Norepinephrine selectively reduces slow Ca2+- and Na+-mediated K+ currents in cat neocortical neurons. J. Neurophysiol. 61, 245–256.
Gamelli, A. E., McKinney, B. C., White, J. A., and Murphy, G. G. (2011). Deletion of the L-type calcium channel Ca(V) 1.3 but not Ca(V) 1.2 results in a diminished sAHP in mouse CA1 pyramidal neurons. Hippocampus 21, 133–141.
Gamper, N., and Shapiro, M. S. (2003). Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J. Gen. Physiol. 122, 17–31.
Gerlach, A. C., Maylie, J., and Adelman, J. P. (2004). Activation kinetics of the slow afterhyperpolarization in hippocampal CA1 neurons. Pflugers Arch. 448, 187–196.
Ghatta, S., Nimmagadda, D., Xu, X., and O'Rourke, S. T. (2006). Large-conductance, calcium-activated potassium channels: structural and functional implications. Pharmacol. Ther. 110, 103–116.
Goldberg, J. A., Teagarden, M. A., Foehring, R. C., and Wilson, C. J. (2009). Nonequilibrium calcium dynamics regulate the autonomous firing pattern of rat striatal cholinergic interneurons. J. Neurosci. 29, 8396–8407.
Goldberg, J. A., and Wilson, C. J. (2005). Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J. Neurosci. 25, 10230–10238.
Grunnet, M., Jensen, B. S., Olesen, S. P., and Klaerke, D. A. (2001). Apamin interacts with all subtypes of cloned small-conductance Ca2+-activated K+ channels. Pflugers Arch. 441, 544–550.
Gu, N., Hu, H., Vervaeke, K., and Storm, J. F. (2008). SK (KCa2) channels do not control somatic excitability in CA1 pyramidal neurons but can be activated by dendritic excitatory synapses and regulate their impact. J. Neurophysiol. 100, 2589–2604.
Gu, N., Vervaeke, K., Hu, H., and Storm, J. F. (2005). Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J. Physiol. 566, 689–715.
Guan, D., Higgs, M. H., Horton, L. R., Spain, W. J., and Foehring, R. C. (2011). Contributions of Kv7-mediated potassium current to sub- and suprathreshold responses of rat layer II/III neocortical pyramidal neurons. J. Neurophysiol. 106, 1722–1733.
Gustafsson, B., and Wigstrom, H. (1981). Evidence for two types of afterhyperpolarization in CA1 pyramidal cells in the hippocampus. Brain Res. 206, 462–468.
Haas, H. L., and Konnerth, A. (1983). Histamine and noradrenaline decrease calcium-activated potassium conductance in hippocampal pyramidal cells. Nature 302, 432–434.
Hansen, S. B., Tao, X., and MacKinnon, R. (2011). Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498.
Haug, T., and Storm, J. F. (2000). Protein kinase A mediates the modulation of the slow Ca(2+)-dependent K(+) current, I(sAHP), by the neuropeptides CRF, VIP, and CGRP in hippocampal pyramidal neurons. J. Neurophysiol. 83, 2071–2079.
Helmchen, F., Imoto, K., and Sakmann, B. (1996). Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys. J. 70, 1069–1081.
Hernandez, C. C., Falkenburger, B., and Shapiro, M. S. (2009). Affinity for phosphatidylinositol 4, 5-bisphosphate determines muscarinic agonist sensitivity of Kv7 K+ channels. J. Gen. Physiol. 134, 437–448.
Higgs, M. H., Slee, S. J., and Spain, W. J. (2006). Diversity of gain modulation by noise in neocortical neurons: regulation by the slow afterhyperpolarization conductance. J. Neurosci. 26, 8787–8799.
Hirschberg, B., Maylie, J., Adelman, J. P., and Marrion, N. V. (1998). Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J. Gen. Physiol. 111, 565–581.
Hocherman, S. D., Werman, R., and Yarom, Y. (1992). An analysis of the long-lasting after-hyperpolarization of guinea-pig vagal motoneurones. J. Physiol. 456, 325–349.
Hotson, J. R., and Prince, D. A. (1980). A calcium-activated hyperpolarization follows repetitive firing in hippocampal neurons. J. Neurophysiol. 43, 409–419.
Jahromi, B. S., Zhang, L., Carlen, P. L., and Pennefather, P. (1999). Differential time-course of slow afterhyperpolarizations and associated Ca2+ transients in rat CA1 pyramidal neurons: further dissociation by Ca2+ buffer. Neuroscience 88, 719–726.
Jobling, P., McLachlan, E. M., and Sah, P. (1993). Calcium induced calcium release is involved in the afterhyperpolarization in one class of guinea pig sympathetic neurone. J. Auton. Nerv. Syst. 42, 251–257.
Kohler, M., Hirschberg, B., Bond, C. T., Kinzie, J. M., Marrion, N. V., Maylie, J., et al. (1996). Small-conductance, calcium-activated potassium channels from mammalian brain. Science 273, 1709–1714.
Krause, M., Offermanns, S., Stocker, M., and Pedarzani, P. (2002). Functional specificity of G alpha q and G alpha 11 in the cholinergic and glutamatergic modulation of potassium currents and excitability in hippocampal neurons. J. Neurosci. 22, 666–673.
Lancaster, B., and Adams, P. R. (1986). Calcium-dependent current generating the afterhyperpolarization of hippocampal neurons. J. Neurophysiol. 55, 1268–1282.
Lancaster, B., Hu, H., Gibb, B., and Storm, J. F. (2006). Kinetics of ion channel modulation by cAMP in rat hippocampal neurones. J. Physiol. 576, 403–417.
Lancaster, B., and Nicoll, R. A. (1987). Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J. Physiol. 389, 187–203.
Lancaster, B., Nicoll, R. A., and Perkel, D. J. (1991). Calcium activates two types of potassium channels in rat hippocampal neurons in culture. J. Neurosci. 11, 23–30.
Lancaster, B., and Zucker, R. S. (1994). Photolytic manipulation of Ca2+ and the time course of slow, Ca(2+)-activated K+ current in rat hippocampal neurones. J. Physiol. 475, 229–239.
Lasser-Ross, N., Ross, W. N., and Yarom, Y. (1997). Activity-dependent [Ca2+]i changes in guinea pig vagal motoneurons: relationship to the slow afterhyperpolarization. J. Neurophysiol. 78, 825–834.
Lee, J. C., Callaway, J. C., and Foehring, R. C. (2005). Effects of temperature on calcium transients and Ca2+-dependent afterhyperpolarizations in neocortical pyramidal neurons. J. Neurophysiol. 93, 2012–2020.
Lee, K., Duan, W., Sneyd, J., and Herbison, A. E. (2010). Two slow calcium-activated afterhyperpolarization currents control burst firing dynamics in gonadotropin-releasing hormone neurons. J. Neurosci. 30, 6214–6224.
Lorenzon, N. M., and Foehring, R. C. (1992). Relationship between repetitive firing and afterhyperpolarizations in human neocortical neurons. J. Neurophysiol. 67, 350–363.
Lorenzon, N. M., and Foehring, R. C. (1993). The ontogeny of repetitive firing and its modulation by norepinephrine in rat neocortical neurons. Brain Res. Dev. Brain Res. 73, 213–223.
Lorenzon, N. M., and Foehring, R. C. (1995). Alterations in intracellular calcium chelation reproduce developmental differences in repetitive firing and afterhyperpolarizations in rat neocortical neurons. Brain Res. Dev. Brain Res. 84, 192–203.
Loussouarn, G., Park, K. H., Bellocq, C., Baro, I., Charpentier, F., and Escande, D. (2003). Phosphatidylinositol-4, 5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 22, 5412–5421.
Madison, D. V., Lancaster, B., and Nicoll, R. A. (1987). Voltage clamp analysis of cholinergic action in the hippocampus. J. Neurosci. 7, 733–741.
Madison, D. V., and Nicoll, R. A. (1982). Noradrenaline blocks accommodation of pyramidal cell discharge in the hippocampus. Nature 299, 636–638.
Madison, D. V., and Nicoll, R. A. (1984). Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. J. Physiol. 354, 319–331.
Maingret, F., Coste, B., Hao, J., Giamarchi, A., Allen, D., Crest, M., et al. (2008). Neurotransmitter modulation of small-conductance Ca2+-activated K+ channels by regulation of Ca2+ gating. Neuron 59, 439–449.
Maravall, M., Mainen, Z. F., Sabatini, B. L., and Svoboda, K. (2000). Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys. J. 78, 2655–2667.
Markova, O., Fitzgerald, D., Stepanyuk, A., Dovgan, A., Cherkas, V., Tepikin, A., et al. (2008). Hippocalcin signaling via site-specific translocation in hippocampal neurons. Neurosci. Lett. 442, 152–157.
Markram, H., Roth, A., and Helmchen, F. (1998). Competitive calcium binding: implications for dendritic calcium signaling. J. Comput. Neurosci. 5, 331–348.
Martinez-Pinna, J., Davies, P. J., and McLachlan, E. M. (2000a). Diversity of channels involved in Ca(2+) activation of K(+) channels during the prolonged AHP in guinea-pig sympathetic neurons. J. Neurophysiol. 84, 1346–1354.
Martinez-Pinna, J., McLachlan, E. M., and Gallego, R. (2000b). Distinct mechanisms for activation of Cl- and K+ currents by Ca2+ from different sources in mouse sympathetic neurones. J. Physiol. 527(Pt 2), 249–264.
McCormick, D. A., and Prince, D. A. (1988). Noradrenergic modulation of firing pattern in guinea pig and cat thalamic neurons, in vitro. J. Neurophysiol. 59, 978–996.
McCormick, D. A., and Williamson, A. (1989). Convergence and divergence of neurotransmitter action in human cerebral cortex. Proc. Natl. Acad. Sci. U.S.A. 86, 8098–8102.
Meech, R. W. (1978). Calcium-dependent potassium activation in nervous tissues. Annu. Rev. Biophys. Bioeng. 7, 1–18.
Miles, G. B., Dai, Y., and Brownstone, R. M. (2005). Mechanisms underlying the early phase of spike frequency adaptation in mouse spinal motoneurones. J. Physiol. 566, 519–532.
Morita, K., North, R. A., and Tokimasa, T. (1982). The calcium-activated potassium conductance in guinea-pig myenteric neurones. J. Physiol. 329, 341–354.
Moyer, J. R. Jr., Thompson, L. T., Black, J. P., and Disterhoft, J. F. (1992). Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J. Neurophysiol. 68, 2100–2109.
Muller, A., Kukley, M., Uebachs, M., Beck, H., and Dietrich, D. (2007). Nanodomains of single Ca2+ channels contribute to action potential repolarization in cortical neurons. J. Neurosci. 27, 483–495.
Neher, E. (1998). Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20, 389–399.
O'Callaghan, D. W., Tepikin, A. V., and Burgoyne, R. D. (2003). Dynamics and calcium sensitivity of the Ca2+/myristoyl switch protein hippocalcin in living cells. J. Cell Biol. 163, 715–721.
Oliver, D., Lien, C. C., Soom, M., Baukrowitz, T., Jonas, P., and Fakler, B. (2004). Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science 304, 265–270.
Pape, H. C., and Driesang, R. B. (1998). Ionic mechanisms of intrinsic oscillations in neurons of the basolateral amygdaloid complex. J. Neurophysiol. 79, 217–226.
Park, K. H., Piron, J., Dahimene, S., Merot, J., Baro, I., Escande, D., et al. (2005). Impaired KCNQ1-KCNE1 and phosphatidylinositol-4, 5-bisphosphate interaction underlies the long QT syndrome. Circ. Res. 96, 730–739.
Park, S. J., Itoh, T., and Takenawa, T. (2001). Phosphatidylinositol 4-phosphate 5-kinase type I is regulated through phosphorylation response by extracellular stimuli. J. Biol. Chem. 276, 4781–4787.
Pedarzani, P., Krause, M., Haug, T., Storm, J. F., and Stuhmer, W. (1998). Modulation of the Ca2+-activated K+ current sIAHP by a phosphatase-kinase balance under basal conditions in rat CA1 pyramidal neurons. J. Neurophysiol. 79, 3252–3256.
Pedarzani, P., and Stocker, M. (2008). Molecular and cellular basis of small–and intermediate-conductance, calcium-activated potassium channel function in the brain. Cell. Mol. Life Sci. 65, 3196–3217.
Pedarzani, P., and Storm, J. F. (1993). PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron 11, 1023–1035.
Pedarzani, P., and Storm, J. F. (1995). Dopamine modulates the slow Ca(2+)-activated K+ current IAHP via cyclic AMP-dependent protein kinase in hippocampal neurons. J. Neurophysiol. 74, 2749–2753.
Pennefather, P., Lancaster, B., Adams, P. R., and Nicoll, R. A. (1985). Two distinct Ca-dependent K currents in bullfrog sympathetic ganglion cells. Proc. Natl. Acad. Sci. U.S.A. 82, 3040–3044.
Pineda, J. C., Galarraga, E., Bargas, J., Cristancho, M., and Aceves, J. (1992). Charybdotoxin and apamin sensitivity of the calcium-dependent repolarization and the afterhyperpolarization in neostriatal neurons. J. Neurophysiol. 68, 287–294.
Pineda, J. C., Galarraga, E., and Foehring, R. C. (1999). Different Ca2+ source for slow AHP in completely adapting and repetitive firing pyramidal neurons. Neuroreport 10, 1951–1956.
Pineda, J. C., Waters, R. S., and Foehring, R. C. (1998). Specificity in the interaction of HVA Ca2+ channel types with Ca2+-dependent AHPs and firing behavior in neocortical pyramidal neurons. J. Neurophysiol. 79, 2522–2534.
Regehr, W. G., Delaney, K. R., and Tank, D. W. (1994). The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J. Neurosci. 14, 523–537.
Rodriguez, N., Amarouch, M. Y., Montnach, J., Piron, J., Labro, A. J., Charpentier, F., et al. (2010). Phosphatidylinositol-4, 5-bisphosphate (PIP(2)) stabilizes the open pore conformation of the Kv11.1 (hERG) channel. Biophys. J. 99, 1110–1118.
Sah, P. (1995). Properties of channels mediating the apamin-insensitive afterhyperpolarization in vagal motoneurons. J. Neurophysiol. 74, 1772–1776.
Sah, P., and Clements, J. D. (1999). Photolytic manipulation of [Ca2+]i reveals slow kinetics of potassium channels underlying the afterhyperpolarization in hippocampal pyramidal neurons. J. Neurosci. 19, 3657–3664.
Sah, P., and Faber, E. S. (2002). Channels underlying neuronal calcium-activated potassium currents. Prog. Neurobiol. 66, 345–353.
Sah, P., and Isaacson, J. S. (1995). Channels underlying the slow afterhyperpolarization in hippocampal pyramidal neurons: neurotransmitters modulate the open probability. Neuron 15, 435–441.
Sah, P., and McLachlan, E. M. (1991). Ca(2+)-activated K+ currents underlying the afterhyperpolarization in guinea pig vagal neurons: a role for Ca(2+)-activated Ca2+ release. Neuron 7, 257–264.
Sah, P., and McLachlan, E. M. (1992). Potassium currents contributing to action potential repolarization and the afterhyperpolarization in rat vagal motoneurons. J. Neurophysiol. 68, 1834–1841.
Salkoff, L., Butler, A., Ferreira, G., Santi, C., and Wei, A. (2006). High-conductance potassium channels of the SLO family. Nat. Rev. Neurosci. 7, 921–931.
Schnee, M. E., and Brown, B. S. (1998). Selectivity of linopirdine (DuP 996), a neurotransmitter release enhancer, in blocking voltage-dependent and calcium-activated potassium currents in hippocampal neurons. J. Pharmacol. Exp. Ther. 286, 709–717.
Schwartzkroin, P. A., and Stafstrom, C. E. (1980). Effects of EGTA on the calcium-activated afterhyperpolarization in hippocampal CA3 pyramidal cells. Science 210, 1125–1126.
Schwindt, P. C., Spain, W. J., and Crill, W. E. (1989). Long-lasting reduction of excitability by a sodium-dependent potassium current in cat neocortical neurons. J. Neurophysiol. 61, 233–244.
Schwindt, P. C., Spain, W. J., and Crill, W. E. (1992a). Calcium-dependent potassium currents in neurons from cat sensorimotor cortex. J. Neurophysiol. 67, 216–226.
Schwindt, P. C., Spain, W. J., and Crill, W. E. (1992b). Effects of intracellular calcium chelation on voltage-dependent and calcium-dependent currents in cat neocortical neurons. Neuroscience 47, 571–578.
Schwindt, P. C., Spain, W. J., Foehring, R. C., Chubb, M. C., and Crill, W. E. (1988a). Slow conductances in neurons from cat sensorimotor cortex in vitro and their role in slow excitability changes. J. Neurophysiol. 59, 450–467.
Schwindt, P. C., Spain, W. J., Foehring, R. C., Stafstrom, C. E., Chubb, M. C., and Crill, W. E. (1988b). Multiple potassium conductances and their functions in neurons from cat sensorimotor cortex in vitro. J. Neurophysiol. 59, 424–449.
Selyanko, A. A., and Brown, D. A. (1996). Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron 16, 151–162.
Shah, M., and Haylett, D. G. (2000a). Ca(2+) channels involved in the generation of the slow afterhyperpolarization in cultured rat hippocampal pyramidal neurons. J. Neurophysiol. 83, 2554–2561.
Shah, M., and Haylett, D. G. (2000b). The pharmacology of hSK1 Ca2+-activated K+ channels expressed in mammalian cell lines. Br. J. Pharmacol. 129, 627–630.
Shah, M. M., Javadzadeh-Tabatabaie, M., Benton, D. C., Ganellin, C. R., and Haylett, D. G. (2006). Enhancement of hippocampal pyramidal cell excitability by the novel selective slow-afterhyperpolarization channel blocker 3-(triphenylmethylaminomethyl)pyridine (UCL2077). Mol. Pharmacol. 70, 1494–1502.
Shah, M. M., Miscony, Z., Javadzadeh-Tabatabaie, M., Ganellin, C. R., and Haylett, D. G. (2001). Clotrimazole analogues: effective blockers of the slow afterhyperpolarization in cultured rat hippocampal pyramidal neurones. Br. J. Pharmacol. 132, 889–898.
Shyng, S. L., and Nichols, C. G. (1998). Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282, 1138–1141.
Soh, H., and Tzingounis, A. V. (2010). The specific slow afterhyperpolarization inhibitor UCL2077 is a subtype-selective blocker of the epilepsy associated KCNQ channels. Mol. Pharmacol. 78, 1088–1095.
Stocker, M., Krause, M., and Pedarzani, P. (1999). An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 96, 4662–4667.
Storm, J. F. (1987). Intracellular injection of a Ca2+ chelator inhibits spike repolarization in hippocampal neurons. Brain Res. 435, 387–392.
Storm, J. F. (1989). An after-hyperpolarization of medium duration in rat hippocampal pyramidal cells. J. Physiol. 409, 171–190.
Storm, J. F. (1990). Potassium currents in hippocampal pyramidal cells. Prog. Brain Res. 83, 161–187.
Suh, B. C., and Hille, B. (2007). Electrostatic interaction of internal Mg2+ with membrane PIP2 Seen with KCNQ K+ channels. J. Gen. Physiol. 130, 241–256.
Tanabe, M., Gahwiler, B. H., and Gerber, U. (1998). L-Type Ca2+ channels mediate the slow Ca2+-dependent afterhyperpolarization current in rat CA3 pyramidal cells in vitro. J. Neurophysiol. 80, 2268–2273.
Tanner, G. R., Lutas, A., Martinez-Francois, J. R., and Yellen, G. (2011). Single K ATP channel opening in response to action potential firing in mouse dentate granule neurons. J. Neurosci. 31, 8689–8696.
Teruyama, R., and Armstrong, W. E. (2005). Enhancement of calcium-dependent afterpotentials in oxytocin neurons of the rat supraoptic nucleus during lactation. J. Physiol. 566, 505–518.
Torres, G. E., Arfken, C. L., and Andrade, R. (1996). 5-Hydroxytryptamine4 receptors reduce afterhyperpolarization in hippocampus by inhibiting calcium-induced calcium release. Mol. Pharmacol. 50, 1316–1322.
Torres, G. E., Chaput, Y., and Andrade, R. (1995). Cyclic AMP and protein kinase A mediate 5-hydroxytryptamine type 4 receptor regulation of calcium-activated potassium current in adult hippocampal neurons. Mol. Pharmacol. 47, 191–197.
Tzingounis, A. V., Heidenreich, M., Kharkovets, T., Spitzmaul, G., Jensen, H. S., Nicoll, R. A., et al. (2010). The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc. Natl. Acad. Sci. U.S.A. 107, 10232–10237.
Tzingounis, A. V., Kobayashi, M., Takamatsu, K., and Nicoll, R. A. (2007). Hippocalcin gates the calcium activation of the slow afterhyperpolarization in hippocampal pyramidal cells. Neuron 53, 487–493.
Tzingounis, A. V., and Nicoll, R. A. (2008). Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc. Natl. Acad. Sci. U.S.A. 105, 19974–19979.
Valiante, T. A., Abdul-Ghani, M. A., Carlen, P. L., and Pennefather, P. (1997). Analysis of current fluctuations during after-hyperpolarization current in dentate granule neurones of the rat hippocampus. J. Physiol. 499(Pt 1), 121–134.
Velumian, A. A., and Carlen, P. L. (1999). Differential control of three after-hyperpolarizations in rat hippocampal neurones by intracellular calcium buffering. J. Physiol. 517(Pt 1), 201–216.
Viana, F., Bayliss, D. A., and Berger, A. J. (1993). Multiple potassium conductances and their role in action potential repolarization and repetitive firing behavior of neonatal rat hypoglossal motoneurons. J. Neurophysiol. 69, 2150–2163.
Villalobos, C., and Andrade, R. (2010). Visinin-like neuronal calcium sensor proteins regulate the slow calcium-activated afterhyperpolarizing current in the rat cerebral cortex. J. Neurosci. 30, 14361–14365.
Villalobos, C., Beique, J. C., Gingrich, J. A., and Andrade, R. (2005). Serotonergic regulation of calcium-activated potassium currents in rodent prefrontal cortex. Eur. J. Neurosci. 22, 1120–1126.
Villalobos, C., Foehring, R. C., Lee, J. C., and Andrade, R. (2011). Essential role for phosphatidylinositol 4, 5-bisphosphate in the expression, regulation, and gating of the slow afterhyperpolarization current in the cerebral cortex. J. Neurosci. 31, 18303–18312.
Villalobos, C., Shakkottai, V. G., Chandy, K. G., Michelhaugh, S. K., and Andrade, R. (2004). SKCa channels mediate the medium but not the slow calcium-activated afterhyperpolarization in cortical neurons. J. Neurosci. 24, 3537–3542.
Vogalis, F., Furness, J. B., and Kunze, W. A. (2001). Afterhyperpolarization current in myenteric neurons of the guinea pig duodenum. J. Neurophysiol. 85, 1941–1951.
Vogalis, F., Storm, J. F., and Lancaster, B. (2003). SK channels and the varieties of slow after-hyperpolarizations in neurons. Eur. J. Neurosci. 18, 3155–3166.
Weatherall, K. L., Goodchild, S. J., Jane, D. E., and Marrion, N. V. (2010). Small conductance calcium-activated potassium channels: from structure to function. Prog. Neurobiol. 91, 242–255.
Williams, J. T., North, R. A., Shefner, S. A., Nishi, S., and Egan, T. M. (1984). Membrane properties of rat locus coeruleus neurones. Neuroscience 13, 137–156.
Wilson, C. J., and Callaway, J. C. (2000). Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J. Neurophysiol. 83, 3084–3100.
Xia, X. M., Fakler, B., Rivard, A., Wayman, G., Johnson-Pais, T., Keen, J. E., et al. (1998). Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395, 503–507.
Zhang, L., Pennefather, P., Velumian, A., Tymianski, M., Charlton, M., and Carlen, P. L. (1995). Potentiation of a slow Ca(2+)-dependent K+ current by intracellular Ca2+ chelators in hippocampal CA1 neurons of rat brain slices. J. Neurophysiol. 74, 2225–2241.
Keywords: Ca2+-activated afterhyperpolarization, sAHP, KCNQ, PtdIns(4,5)P2, neuromodulation, pyramidal cell
Citation: Andrade R, Foehring RC and Tzingounis AV (2012) The calcium-activated slow AHP: cutting through the Gordian knot. Front. Cell. Neurosci. 6:47.doi: 10.3389/fncel.2012.00047
Received: 29 June 2012; Accepted: 05 October 2012;
Published online: 25 October 2012.
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyReviewed by:
Johan F. Storm, University of Oslo, NorwayRaffaella Tonini, Istituto Italiano di Tecnologia, Italy
Copyright © 2012 Andrade, Foehring and Tzingounis. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Rodrigo Andrade, Department of Pharmacology, Wayne State University School of Medicine, 540 E Canfield, Detroit, MI 48201, USA. e-mail: randrade@med.wayne.edu