Brain Ischemia as a Prelude to Alzheimer's Disease
 Ryszard Pluta
Ryszard Pluta Sławomir Januszewski1
Sławomir Januszewski1  Stanisław J. Czuczwar
Stanisław J. Czuczwar- 1Laboratory of Ischemic and Neurodegenerative Brain Research, Mossakowski Medical Research Institute, Polish Academy of Sciences, Warsaw, Poland
- 2Department of Pathophysiology, Medical University of Lublin, Lublin, Poland
Transient ischemic brain injury causes massive neuronal death in the hippocampus of both humans and animals. This was accompanied by progressive atrophy of the hippocampus, brain cortex, and white matter lesions. Furthermore, it has been noted that neurodegenerative processes after an episode of ischemia-reperfusion in the brain can continue well-beyond the acute stage. Rarefaction of white matter was significantly increased in animals at 2 years following ischemia. Some rats that survived 2 years after ischemia developed severe brain atrophy with dementia. The profile of post-ischemic brain neurodegeneration shares a commonality with neurodegeneration in Alzheimer's disease. Furthermore, post-ischemic brain injury is associated with the deposition of folding proteins, such as amyloid and tau protein, in the intracellular and extracellular space. Recent studies on post-ischemic brain neurodegeneration have revealed the dysregulation of Alzheimer's disease-associated genes such as amyloid protein precursor, α-secretase, β-secretase, presenilin 1, presenilin 2, and tau protein. The latest data demonstrate that Alzheimer's disease-related proteins and their genes play a key role in the development of post-ischemic brain neurodegeneration with full-blown dementia in disease types such as Alzheimer's. Ongoing interest in the study of brain ischemia has provided evidence showing that ischemia may be involved in the development of the genotype and phenotype of Alzheimer's disease, suggesting that brain ischemia can be considered as a useful model for understanding the mechanisms responsible for the initiation of Alzheimer's disease.
Introduction
Presently, brain ischemia and Alzheimer's disease create a huge burden to the healthcare system and caregivers due to the lack of causal treatment. Both the diseases are the main causes of irreversible disability and dementia worldwide (Ballard et al., 2011; Pluta et al., 2011, 2020b,c,d; Kim and Lee, 2018; Lo et al., 2019), and there is a risk of stroke or Alzheimer's disease is one in three persons (Seshadri and Wolf, 2007). With increasing numbers of aging in the world, the number of subjects with dementia is forecast to reach 82 million by 2030 and 152 million by 2050 (Ballard et al., 2011). There are no causal treatments that could stop the development of dementia in both patients after stroke and Alzheimer's disease. For this reason, there is a lot of pressure to increase the understanding of the mechanisms of post-ischemic brain neurodegeneration in connection with its recommended relationship with Alzheimer's disease (Pluta, 2007b, 2019; Pluta et al., 2011, 2013a,b; Salminen et al., 2017) and to make causal therapy available (Ułamek-Kozioł et al., 2020a). It is noteworthy that more and more new clinical and experimental studies indicate epidemiological and neuropathological links connecting ischemic brain neurodegeneration with the genotype and phenotype of Alzheimer's disease. Human investigations have revealed that Alzheimer's disease is a risk factor for stroke (Chi et al., 2013; Tolppanen et al., 2013) and vice versa (Gamaldo et al., 2006), indicating that the same or closely related pathological mechanisms may be involved in the development of both disorders. Animal studies have also presented a synergistic link between brain ischemia and Alzheimer's disease, leading to an increased risk of cognitive decline and development of Alzheimer's disease-type dementia (De la Tremblaye and Plamondon, 2011; Kiryk et al., 2011; Li et al., 2011; Cohan et al., 2015; Traylor et al., 2016; Salminen et al., 2017). The main cause of ischemic stroke in humans is atherosclerosis (Roher et al., 2004; Beach et al., 2007). Atherosclerosis is also associated with Alzheimer's disease (Farkas and Luiten, 2001; Thal et al., 2003; Beach et al., 2007). At least 33% cases of Alzheimer's disease have neuropathological changes resulting from small vessel arteriosclerosis (Kalaria, 2002). Atherosclerosis has been found to coexist with cerebral amyloid angiopathy and it also correlates well with cognitive decline (Thal et al., 2003; Pluta et al., 2009). On the other hand, the increased level of amyloid in the post-ischemic brain causes the accumulation of amyloid not only in the brain tissue, but also in the vessel wall, causing the development of cerebral amyloid angiopathy (Pluta et al., 2009; Hecht et al., 2018). Reduction in the length of cerebral vessels post-ischemia or impaired cerebral blood flow in the brain as a result of vasoconstriction (Wisniewski et al., 1995) and/or the development of cerebral amyloid angiopathy (Pluta et al., 2009; Hecht et al., 2018) not only limits the transport of energy substrates and the supply of oxygen and nutrients to the brain through the blood–brain barrier after ischemia, but also reduces the clearance of potential neurotoxins from the brain, such as amyloid (Hecht et al., 2018). This leads to the idea that brain vascular diseases, such as ischemic brain episode, may make the regions in the brain more susceptible to Alzheimer's disease pathology, due to impaired clearance of amyloid from the brain (Farkas and Luiten, 2001) and dysfunctional tau protein. Alternatively, post-ischemic brain neurodegeneration and Alzheimer's disease may finally represent independent but convergent common pathological mechanisms, and can therefore be expected to have common proteomic and genomic risk factors (Traylor et al., 2016; Pluta et al., 2019a,b; Ułamek-Kozioł et al., 2020b).
Neuropathology in Post-Ischemic Neurodegeneration
The death of neurons in the CA1 area of hippocampus develops during 7 days following experimental ischemic episode (Pluta, 2002a). Extended survival, following ischemic brain injury up to 2 years, triggers additional pathology in neuronal cells in the hippocampal CA3 region which is resistant to ischemic injury (Pluta et al., 2009). The same changes are observed in post-stroke cases (Gemmell et al., 2012, 2014). In cortex, layers 3, 5, and 6 presented massive neuronal changes (Pluta, 2000a, 2002a,b), but changes in striatum were related to medium-sized neurons. Two years after ischemia, in addition to localized neuron loss, a variety of pathological stages of neuronal cells were observed (Pluta et al., 2009). The neuronal loss took the form of chronic neuronal damage. The other alterations with acute character were noted in those areas of the brain, which were not involved in primary ischemic pathology, i.e. in CA2, CA3, and CA4 areas of the hippocampus (Pluta et al., 2009).
Brain ischemia increases the permeability of the blood–brain barrier to cellular and non-cellular blood elements i.e., platelets and amyloid (Mossakowski et al., 1993, 1994; Pluta et al., 1994a, 2006; Wisniewski et al., 1995; Pluta, 2003, 2005). In post-ischemic leakage of the blood–brain barrier, two facts deserve attention. The first one is very important in terms of amyloid extravasations (Pluta et al., 1996, 1997b) during the development of brain neurodegeneration, and the second is the movement of platelets containing a huge amount of amyloid, which causes toxic and mechanical damage to the brain parenchyma (Pluta et al., 1994c). The ability of the soluble amyloid to pass through the insufficient blood–brain barrier leads to neurotoxic effects on neurons, which may then lead to subsequent increased accumulation of amyloid in post-ischemic brain. Soluble amyloid is delivered to the brain following ischemia from the circulatory system, and as a consequence, it contributes to brain vasoconstriction, amyloidosis, and cerebral amyloid angiopathy, following brain ischemia (Jendroska et al., 1995, 1997; Wisniewski et al., 1995; Pluta et al., 1996, 1999, 2009; Wisniewski and Maslinska, 1996; Lee et al., 2005; Qi et al., 2007; Zetterberg et al., 2011; Liu et al., 2015; Hecht et al., 2018).
Following brain ischemia in regions with massive neuronal alterations, a powerful inflammatory response was observed (Orzyłowska et al., 1999; Pluta, 2000a, 2002a,b; Sekeljic et al., 2012; Radenovic et al., 2020). These data indicate that the increase of inflammatory mediators in microglia and astrocytes is directly related to the selective vulnerability of neurons to ischemia (Orzyłowska et al., 1999; Radenovic et al., 2020). After ischemia, inflammatory factors can trigger a self-sustaining cycle that leads the post-ischemic brain to neurodegeneration. Interleukin-1 is a key player that stimulates ischemic neurons to amyloidogenic processing of the amyloid protein precursor along with the induction of inflammatory factors. These processes induce changes in neurons and their loss, with irreversible interruption of the neuronal network. Consequently, this neuropathology activates microglial cells, which leads to self-propagation of the inflammatory cycle. Furthermore, it is evident that the amyloid, which is generated following ischemia (Pluta et al., 1994b, 2009; Ishimaru et al., 1996), triggers the release of inflammatory mediators by microglia. In the hippocampus, activation of glial cells precedes neuron loss, and it lasts for a long time after an ischemic episode (Sekeljic et al., 2012; Radenovic et al., 2020). Factors released by astrocytes and microglia cells, i.e., matrix metalloproteinases, interleukin-1, and tumor necrosis factor α increase the leakage of the blood–brain barrier (Amantea et al., 2015). Microglia and astrocytes belong to the first line of defense and are activated a few minutes after ischemia (Dabrowska et al., 2019). Increased activation is noted within 2–3 days after ischemia, persisting for years following ischemic brain injury (Sekeljic et al., 2012; Dabrowska et al., 2019; Radenovic et al., 2020).
An increased influx of monocytes in the brain after ischemia was documented within 1 day after ischemia as a result of additional injury to the blood–brain barrier by neuroglial inflammatory factors. An increased number of monocytes in brain tissue were observed up to 7 days following ischemia (Kim et al., 2014). Sooner or later, anti-inflammatory macrophages begin to dominate the brain after ischemia, as they are essential for the processes of regeneration and healing (Gliem et al., 2016). Another type of cells involved in the immune response induced by ischemia are neutrophils which appear in post-ischemic brain parenchyma immediately after the ischemic insult (Price et al., 2004). The neutrophils focus around damaged areas and release inflammatory factors, proteolytic enzymes, and oxygen free radicals that initiate secondary damage to the already damaged brain tissue (Dabrowska et al., 2019; Radenovic et al., 2020). The number of neutrophils appearing in the brain following ischemia directly corresponds to the size of the brain injury (Ahmad et al., 2014; Leinweber et al., 2021). Also, T and B lymphocytes, natural killer cells, and dendritic cells infiltrate the brain after ischemia (Gelderblom et al., 2018; Dabrowska et al., 2019). In addition, mast cells intensify inflammatory responses by releasing tumor necrosis factor α, histamine, heparin, and proteases, i.e., tryptase, chymase, and matrix metalloproteinases, which cause secondary changes in the blood–brain barrier, edema of the brain, and penetration of neutrophils into the ischemic brain tissue (Pluta et al., 1989; Lindsberg et al., 2010; Dabrowska et al., 2019; Leinweber et al., 2021).
Lesions in white matter with the proliferation of neuroglial cells were documented in human and animal brains, after ischemia (Pluta, 2000a, 2002a,b; Fernando et al., 2006; Pluta et al., 2006, 2008, 2009; Scherr et al., 2012; Sekeljic et al., 2012; Thiebaut de Schotten et al., 2014; Zamboni et al., 2017; Radenovic et al., 2020). An episode of cerebral ischemia in animals causes massive damage to the subcortical white matter and corpus callosum (Wakita et al., 1994; Pluta et al., 2006, 2008, 2009). Increased permeability of the blood–brain barrier, after ischemia, allows the movement of inflammatory cells and amyloid from the circulatory system to the brain, which additionally leads to the progression of changes in white matter (Pluta et al., 1996, 1997b, 1999, 2000; Anfuso et al., 2004; Lee et al., 2005; Zetterberg et al., 2011; Liu et al., 2015).
Data suggest that brain ischemia triggers massive neuronal death, especially in brain structures sensitive to ischemia (Pluta, 2000a; Pluta et al., 2009; Bivard et al., 2018). These processes develop both in the early and later stages following ischemia (Pluta, 2002a; Pluta et al., 2009; Bivard et al., 2018; Radenovic et al., 2020). In the years following ischemia, ischemic neurodegenerative processes cause general brain atrophy (Hossmann et al., 1987; Pluta, 2000a; Pluta et al., 2009; Jabłoński et al., 2011; Bivard et al., 2018). Brain autopsy after experimental ischemia, with survival up to 2 years, showed the hallmarks of brain hydrocephalus and dilatation of the subarachnoid space around the brain hemispheres (Hossmann et al., 1987; Pluta, 2000a; Pluta et al., 2009; Jabłoński et al., 2011). Hippocampal atrophy was also observed in humans and animals after ischemia (Pluta, 2000a; Pluta et al., 2009; Gemmell et al., 2012, 2014).
Amyloid-Related Genes in Post-Ischemic Neurodegeneration
Following experimental brain ischemia, with survival up to 2 years after ischemia, amyloid staining was documented in intracellular and extracellular space (Pluta et al., 1994b, 1997a, 1998, 2012a; Hall et al., 1995; Tomimoto et al., 1995; Ishimaru et al., 1996; Yokota et al., 1996; Pluta, 1997, 2000a,b; Lin et al., 1999, 2001; Sinigaglia-Coimbra et al., 2002; Fujioka et al., 2003; Jabłoński et al., 2011; Pluta and Jabłoński, 2012). Accumulation of amyloid in the extra-cellular space ranged from small dots to diffuse and senile amyloid plaques (Pluta et al., 1994b, 1998, 2000, 2009, 2010; Pluta, 2000a,b; Pluta, 2002a,b; Pluta, 2003, 2005, 2007a; Van Groen et al., 2005). Amyloid plaques were noted in the hippocampus, thalamus, brain cortex, corpus callosum, and around the lateral ventricles. The deposition of amyloid inside neurons and neuroglial cells underscores the possible importance of amyloid in the progress of ischemic brain neurodegeneration (Pluta et al., 1994b; Banati et al., 1995; Palacios et al., 1995; Yokota et al., 1996; Nihashi et al., 2001; Pluta, 2002a,b; Badan et al., 2003, 2004). These observations indicate that following experimental ischemia, amyloid generation may be responsible for additional neurodegenerative mechanisms, which could worsen the outcome after ischemia due to continuous neuronal loss (Pluta et al., 1997a,b; Pluta et al., 1998, 2009, 2011, 2012a,b,c; Jabłoński et al., 2011; Kiryk et al., 2011; Pluta and Jabłoński, 2012). Some data indicate that after ischemic episode, amyloid was generated as a product of neuronal death (Ishimaru et al., 1996). The amyloid is a neurotoxic substance that induces intracellular processes in post-ischemic neurons, astrocytes, and microglia, which further causes extra neurons and neuroglia injury and/or death following ischemia (Giulian et al., 1995; Pluta et al., 2012a).
After ischemia, both diffuse and senile amyloid plaques in the hippocampus and cortex were observed in the human brain (Jendroska et al., 1995, 1997; Wisniewski and Maslinska, 1996; Qi et al., 2007). According to Qi et al. (2007), β-amyloid peptides, 1–40 and 1–42 were found in post-ischemic hippocampus. Hippocampal and cortical neuronal cells were the most intensely stained cells in the post-ischemic human brains. Additionally, evidence from clinical investigations showed that plasma amyloid was raised in cases with ischemic brain injury (Lee et al., 2005; Zetterberg et al., 2011; Liu et al., 2015). The increase of amyloid in serum correlated negatively with the clinical outcome following an ischemic brain episode (Zetterberg et al., 2011).
Following focal brain ischemia, messenger RNA (mRNA) of the amyloid protein precursor increased from 150 to 200% after 7 days of survival (Shi et al., 1998, 2000). The amyloid protein precursor is cleaved via α-secretase along a non-amyloidogenic pathway. Following experimental ischemic brain episode, the α-secretase mRNA was reduced (Nalivaeva et al., 2004; Yan et al., 2007; Pluta et al., 2020a). In the amyloidogenic pathway, the amyloid protein precursor is cleaved via β- and γ-secretase to form amyloid (Wen et al., 2004a; Tabaton and Tamagno, 2007; Pluta et al., 2013a; Guo et al., 2021). Available data suggest that ischemia activates mRNA of β-secretase (Blasko et al., 2004; Chen et al., 2004; Wen et al., 2004a; Chuang et al., 2008; Ye et al., 2009). Presenilins mRNA, which are induced via brain ischemia (Tanimukai et al., 1998; Pennypacker et al., 1999), are involved in the generation of amyloid by the γ-secretase complex (Polavarapu et al., 2008). The above evidence helps us understand the progressive neuronal death after an episode of brain ischemia, accumulation of amyloid, and the slow progressive development of Alzheimer's disease type neurodegeneration (Pluta et al., 2009, 2010, 2020b,c,d). In studies on post-ischemic brain changes, elevated presenilin 1 mRNA in the CA3 area and dentate gyrus of hippocampal neurons was noted at 3 days (Tanimukai et al., 1998). In another study, increased presenilin mRNAs were noted in the hippocampus, striatum, and the brain cortex following focal ischemia (Pennypacker et al., 1999). Presenilin mRNAs showed a maximum increase in the hippocampus and cortex. The cortex exhibited an increase of presenilin 1 and 2 mRNAs within 1–8 days after local ischemia with recirculation (Pennypacker et al., 1999), whereas, the hippocampus exhibited upregulation of presenilin 1 and 2 mRNAs within 4–8 days after ischemia (Pennypacker et al., 1999). The elevated level was greater on the contralateral side than on the side of focal ischemic brain injury. This difference is most likely associated with the loss of brain neurons expressing presenilin mRNAs on the ischemic side (Pennypacker et al., 1999).
In the CA1 subfield of the hippocampus, 2 days after ischemia, the expression of the amyloid protein precursor gene was below the control value (Table 1) (Kocki et al., 2015). Within 7–30 days after ischemia with reperfusion, the expression of the amyloid protein precursor gene was above the control value (Table 1) (Kocki et al., 2015). The expression of the β-secretase gene was above the control value, 2–7 days after ischemia in the CA1 region (Table 1) (Kocki et al., 2015). Thirty days after ischemia, the expression of β-secretase gene was below the control value (Table 1) (Kocki et al., 2015). In the CA1 area of the hippocampus, the expression of presenilin 1 and 2 genes increased within 2–7 days after ischemia (Table 1) (Kocki et al., 2015). In contrast, 30 days following ischemia, the expression of presenilin 1 and 2 genes was below the control value (Table 1) (Kocki et al., 2015).
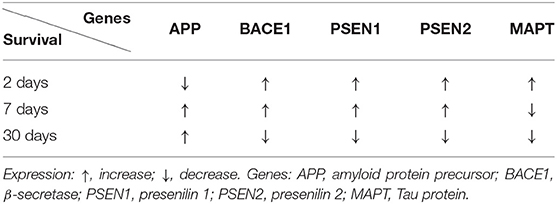
Table 1. Changes in the expression of the Alzheimer's disease-linked genes in the CA1 region of the hippocampus at various times after experimental ischemic brain injury.
At 2, 7, and 30 days following ischemia, the expression of the amyloid protein precursor gene was found to be above the control value in the CA3 region (Table 2) (Pluta et al., 2020a). In this area, the α-secretase gene expression was below the control value at 2, 7, and 30 days after ischemia (Table 2) (Pluta et al., 2020a). The expression of the β-secretase gene was below the control value following ischemia in the CA3 area, in 2–7 days (Table 2). In contrast, 30 days after ischemia, the β-secretase gene expression was above the control value (Table 2) (Pluta et al., 2020a). In the CA3 subfield, the expression of the presenilin 1 gene increased in 2–7 days following ischemia (Table 2). In 30 days after ischemia, the expression of the presenilin 1 gene was below the control value (Table 2) (Pluta et al., 2020a). In contrast, the expression of the presenilin 2 gene was reduced 2–7 days after ischemia (Table 2). But 30 days after ischemia, the expression of the presenilin 2 gene was above the control value (Table 2) (Pluta et al., 2020a).
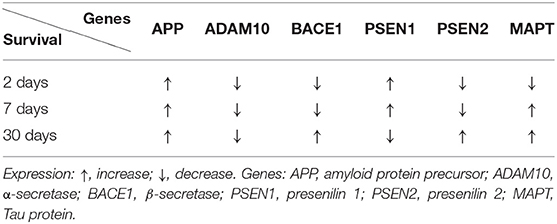
Table 2. Changes in the expression of the Alzheimer's disease-linked genes in the CA3 region of the hippocampus at various times after experimental ischemic brain injury.
In the medial temporal cortex, the expression of the amyloid protein precursor gene was below the control value, 2 days after ischemia (Table 3) (Pluta et al., 2016a). In the above area, in 7–30 days after ischemia, the expression of the amyloid protein precursor gene was above the control value (Table 3) (Pluta et al., 2016a). The β-secretase gene expression was above the control value at 2 days after ischemia (Table 3) (Pluta et al., 2016a). But in 7–30 days after ischemia, the expression of the β-secretase gene was reduced (Table 3) (Pluta et al., 2016a). The expression of the presenilin 1 gene was lowered below the control value, while the presenilin 2 gene was above the control value 2 days after ischemia (Table 3) (Pluta et al., 2016b). Seven days after ischemia, the expression of the presenilin 1 gene was reduced, while that of the presenilin 2 gene was increased (Table 3) (Pluta et al., 2016b). Thirty days after ischemia, the expression of the presenilin 1 gene was above the control value, while that of the presenilin 2 gene was below the control value (Table 3) (Pluta et al., 2016b).
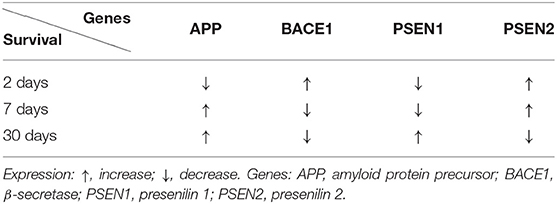
Table 3. Changes in the expression of the Alzheimer's disease-linked genes in the medial temporal cortex at various times after experimental ischemic brain injury.
Data show that brain ischemia triggers neuronal death in the hippocampus and in the medial temporal cortex in conjunction with amyloid, defining a new process that regulates the survival of neurons and/or death after-ischemia (Pluta et al., 1994b, 1997a, 2009, 2016a,b; Hall et al., 1995; Jendroska et al., 1995, 1997; Palacios et al., 1995; Ishimaru et al., 1996; Wisniewski and Maslinska, 1996; Yokota et al., 1996; Qi et al., 2007; Kocki et al., 2015; Pluta et al., 2020a; Pluta and Ułamek-Kozioł, 2021).
Tau Protein in Post-Ischemic Neurodegeneration
Strong neuronal staining of tau protein was found following ischemia in the hippocampus and the brain cortex (Dewar et al., 1993, 1994; Geddes et al., 1994; Sinigaglia-Coimbra et al., 2002). Also, tau protein staining was documented in post-ischemic microglia, astrocytes, and oligodendrocytes (Dewar and Dawson, 1995; Irving et al., 1997; Uchihara et al., 2004; Majd et al., 2016; Fujii et al., 2017). Evidence shows that after ischemia, the hyperphosphorylated tau protein dominates in neuronal cells and goes along with apoptosis (Wen et al., 2004b,c; Wen et al., 2007; Majd et al., 2016; Fujii et al., 2017; Basurto-Islas et al., 2018). The above data point out that neuronal apoptosis following ischemia is connected with hyperphosphorylation of tau protein. In addition, it is also evident that ischemic brain injury was engaged in paired helical filaments (Khan et al., 2018), neurofibrillary tangle-like (Wen et al., 2004b,c; Wen et al., 2007), and neurofibrillary tangles development (Kato et al., 1988; Hatsuta et al., 2019). Tau protein was detected in the plasma samples of humans after ischemic brain injury and it most probably indicated the progress of neuronal changes after ischemia (Bitsch et al., 2002; Kurzepa et al., 2010; Bielewicz et al., 2011; Mörtberg et al., 2011; Randall et al., 2013; Lasek-Bal et al., 2016; De Vos et al., 2017).
Dysregulation of Tau Protein Gene in Post-Ischemic Neurodegeneration
In the CA1 area of the hippocampus, the expression of the tau protein gene was found to be increased above the control value, 2 days after ischemia (Table 1) (Pluta et al., 2018). In contrast, at 7–30 days after brain ischemia, gene expression was found to be below the control value (Table 1) (Pluta et al., 2018). In the CA3 subfield, the expression of the tau protein gene was found to be below the control value, 2 days after ischemia (Table 2) (Pluta et al., 2020a). But, within 7–30 days following ischemia, the expression of the tau protein gene was higher than the control value (Table 2) (Pluta et al., 2020a). The data indicate that post-ischemic brain injury triggers neuronal damage and death in the hippocampus in a tau protein-dependent mechanism, defining a new process, which in the long run regulates neuronal survival and/or death following ischemia (Geddes et al., 1994; Dewar and Dawson, 1995; Wen et al., 2004b, 2007; Majd et al., 2016; Fujii et al., 2017; Basurto-Islas et al., 2018; Khan et al., 2018; Pluta et al., 2018, 2020a; Pluta and Ułamek-Kozioł, 2021).
Dementia in Post-Ischemic Neurodegeneration
Many studies have documented the development of dementia in animals after ischemic brain injury with recirculation (De la Tremblaye and Plamondon, 2011; Kiryk et al., 2011; Li et al., 2011; Pluta et al., 2011; Cohan et al., 2015). Locomotor hyperactivity was noted after experimental brain ischemia (Kuroiwa et al., 1991; Karasawa et al., 1994) as in Alzheimer's disease. A lengthening of ischemic episode causes a longer duration of motor hyperactivity, and this is positively correlated with an increased number of damaged and lost neurons, especially in the hippocampus and progressive inflammation in the brain (Pluta et al., 2010; Kiryk et al., 2011; Sekeljic et al., 2012; Radenovic et al., 2020). Additionally, post-ischemic brain damage causes loss of reference and working memory with the progress of a spatial memory deficit (Kiryk et al., 2011). The progress of cognitive deficit develops systematically along with the lengthening of post-ischemic time (Kiryk et al., 2011). After repeated experimental brain ischemia with recirculation, durable motor hyperactivity with cognitive deficits and reduced anxiety was presented (Ishibashi et al., 2006). The development of dementia was related to general brain atrophy (Hossmann et al., 1987; Pluta, 2000a, 2002a,b; Pluta et al., 2009, 2012b,c; Jabłoński et al., 2011). In these cases learning and memory deficits in experimental post-ischemic brain neurodegeneration irreversibly progressed and persisted forever (Kiryk et al., 2011).
The progressive development of dementia is a patients dangerous consequence of post-ischemic pathology (Gemmell et al., 2012, 2014; Brainin et al., 2015; Mok et al., 2016; Portegies et al., 2016; Kim and Lee, 2018). The occurrence of dementia after the first ischemic stroke and recurrent stroke is calculated roughly at 10 and 41%, respectively (Pendlebury and Rothwell, 2009). Within a 25-year follow-up, the incidence of dementia has been calculated approximately at 48% (Kokmen et al., 1996). Worldwide, dementia after ischemic stroke occurs between 5 and 50% of cases, depending on diagnostic criteria, population demographics, and geographical location (Surawan et al., 2017). In fact, it is certain that dementia in post-ischemia brain neurodegeneration has many risk factors in common with the development of dementia in sporadic cases of Alzheimer's disease (Thal et al., 2003). It is highly likely that post-ischemic brain neurodegeneration may precede the final development of Alzheimer's disease.
Conclusion
This review presents the phenotype and genotype of Alzheimer's disease in post-ischemic brain neurodegeneration, such as neuropathology, amyloid, dysfunctional tau protein, and their genes, which altogether play an important role in the development of full-blown dementia (Figure 1). It provides Alzheimer's disease-linked gene changes of the amyloid protein precursor, α-secretase, β-secretase, presenilin 1, presenilin 2, and tau protein in experimental post-ischemic injury in the CA1 and CA3 areas of the hippocampus and medial temporal cortex. The evidence demonstrates that post-ischemic brain damage causes the death of neuronal cells in the hippocampus and temporal cortex in a manner that is dependent on the amyloid and dysfunctional tau protein. The above changes are associated with the accumulation of the amyloid in brain cells and in the extracellular spaces, such as diffuse and senile amyloid plaques, and the massive loss of neuronal cells with general brain atrophy, especially in the hippocampus, which finally leads to full-blown dementia in Alzheimer's disease (Figure 1). It is likely that following ischemia, the development of amyloid plaques in the brain is caused by an increased amyloid generation, an intensified inflow from blood, and worsening of amyloid clearance from the brain tissue. Based on the presented evidence, it can be concluded that post-ischemic brain neurodegeneration influences the processing of the amyloid protein precursor, at both the gene and protein level, and leads to the generation of amyloid plaques in brain parenchyma.
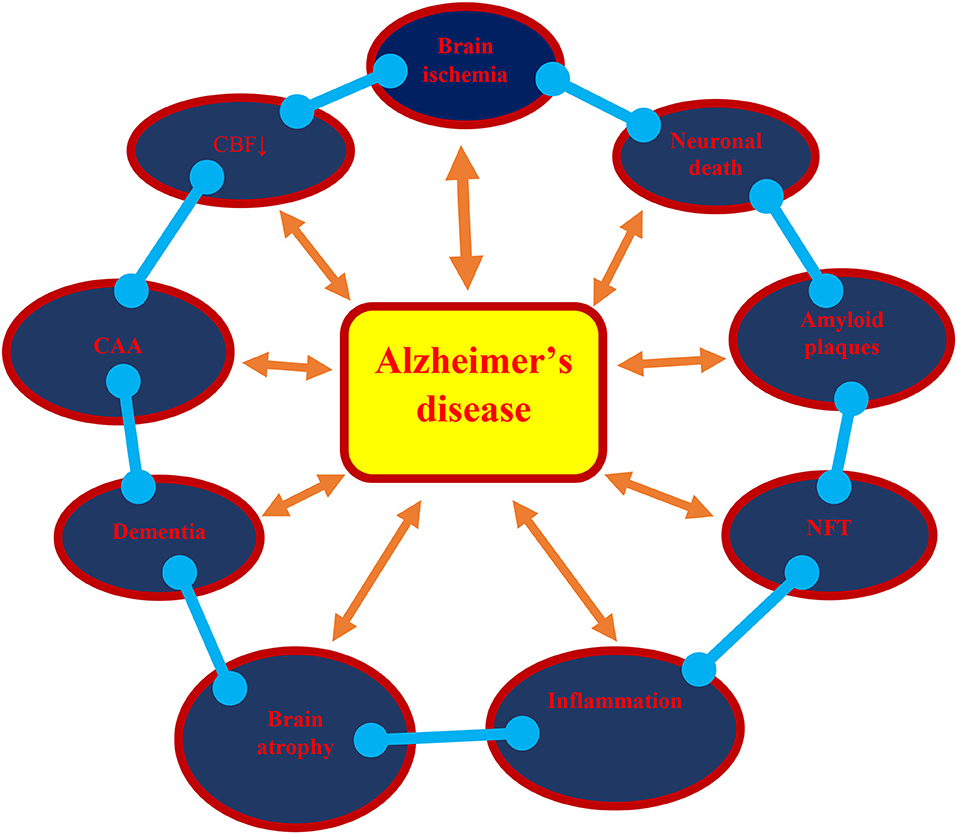
Figure 1. A possible vicious circle in the development of Alzheimer's disease as a result of brain ischemia. ↓, decrease; CAA, cerebral amyloid angiopathy; CBF, cerebral blood flow; NFT, neurofibrillary tangles.
In addition, observations showed that ischemic injury of the CA1 and CA3 areas of the hippocampus influence the expression of the tau protein gene. It can also be noted that dysregulated tau protein takes part in neuronal cell death in the CA1 and CA3 areas of the hippocampus. The existing data documented the development of neurofibrillary tangles following ischemia (Figure 1). In support of the above fact, elevated levels of cyclin-dependent kinase 5, which are involved in the development of neurofibrillary tangle-like tauopathy, were found after experimental ischemic brain injury (Wen et al., 2007). This may suggest the linking of dysfunctional tau protein with the onset of neuronal cell death in the hippocampus after ischemia. The above evidence also indicates the regulation of the ischemic death of neuronal cells in the CA1 and CA3 areas of the hippocampus in a manner dependent on the structure of the tau protein.
The parallelism between post-ischemic brain neurodegeneration and Alzheimer's disease at the molecular level (Figure 1) is remarkable. The conclusions drawn from this investigation of ischemia-triggered Alzheimer's disease-like neuropathology, proteins, and their genes indicates that they contribute to the death of neuronal cells, the generation of the amyloid plaques, the development of neurofibrillary tangles, and finally, neurodegeneration with full-blown dementia (Figure 1). These findings may significantly contribute to finding new causal treatments of post-ischemic brain neurodegeneration and Alzheimer's disease.
The dominant hypothesis of the etiology of Alzheimer's disease, neuropathological guidelines for the diagnosis of the disease, and most of the wide-ranging therapeutic efforts, both in research and clinical practice, have been built around the amyloid and tau proteins as causal factors (Ittner and Ittner, 2018; Busche and Hyman, 2020; Uddin et al., 2020). However, the causal link between amyloid and the development of Alzheimer's disease remains unproven. Currently, in the context of a comprehensive evaluation of the past and contemporary research, critical questions are being raised regarding the role of amyloid and tau protein in the diagnosis, etiology, and definition of Alzheimer's disease. It is argued that a holistic review of the available data does not allow a clear conclusion for the fact that amyloid plays a central or unique role in the development of Alzheimer's disease. A new analysis of data proposes that deposits of amyloid and dysfunctional tau protein may not be the primary cause in the pathogenesis of Alzheimer's disease; further research is needed in this field (Tse and Herrup, 2017; Jack et al., 2018; Morris et al., 2018). Regarding the two potentially dangerous substances blamed for the green light in Alzheimer's disease, recent data show that amyloid and tau protein pathology should be triggered by some factors and then interact with each other, exerting a synergistic deleterious effect on the neural network, which is believed to initiate the progression of Alzheimer's disease (Morris et al., 2018). It is certain that alterations of amyloid and tau protein are currently ruled out as the sole cause of dementia, as it does not explain why about half of the world's population over the age of 45 have amyloid plaques and neurofibrillary tangles without dementia (Katzman, 1988; Price et al., 1992; Rowe et al., 2007; Jagust et al., 2009; Knopman et al., 2013; Atlante et al., 2020). The presence of amyloid plaques, which do not cause any disturbance, has been found in older adults who identify themselves with excellent cognitive functions (Katzman, 1988; Price et al., 1992; Rowe et al., 2007; Jagust et al., 2009; Knopman et al., 2013; Atlante et al., 2020). In these subjects, the average neurofibrillary tangle concentration was observed to increase exponentially with age (Price and Morris, 1999). To sum up, there are no older adults who do not exhibit the presence of amyloid plaques and neurofibrillary tangles in their brains (Garrett, 2018). Additionally, the appearance of the hippocampal atrophy in cognitively normal elderly may not be dependent on amyloidosis (Chételat, 2013). On the other hand, a multicenter study reported that patients diagnosed with Alzheimer's disease were found to be negative for brain amyloid; for example, amyloid was noted to be absent in 32% of cases studied (Doraiswamy et al., 2012).
After analyzing past and contemporary studies about on the amyloid theory of Alzheimer's disease by Morris et al. (2018), the following four important conclusions were drawn: (1) Amyloid plaques and neurofibrillary tangles may be present in the brain without cognitive impairment. (2) The clinical diagnosis of Alzheimer's disease does not include the structure of amyloid and tau protein in the brain. (3) The number, size, and enlargement of amyloid plaques are not related to cognitive impairment. (4) Soluble amyloid and amyloid plaques in the brain are not a warning sign of impending dementia. Novel discoveries have demonstrated that amyloid and neurofibrillary tangles do not initiate Alzheimer's disease as these are only two among a multitude of degenerative changes that occur in this disease (Gauthier et al., 2018; Kalvach and Vogner, 2019). Our review aims to challenge these shared views and suggests that continuing this approach may be counterproductive. Instead, the data suggest that we should look for alternative views on the etiology of Alzheimer's disease that are currently potentially under consideration. We propose that a thorough understanding of the etiology of Alzheimer's disease and the implementation of the final, effective treatment requires a new objective approach in this topic, beyond the current amyloid-centric approach, without excluding the role of amyloid. The continued interest in the research on cerebral ischemia provides evidence that ischemia may be involved in the development of the genotype and phenotype of Alzheimer's disease, suggesting that cerebral ischemia can be considered a useful model for understanding the mechanisms underlying the development of Alzheimer's disease. In spite of reasonable doubts about the role of brain ischemia in the etiology of Alzheimer's disease, the mounting evidence on the ischemic development of the disease should not be ignored. Ignoring the huge number of experimental and clinical evidence on the connection between ischemic brain injury and Alzheimer's disease will not only hamper proper understanding of the disease pathways, but also development strategies for diagnosis, management, and therapy of Alzheimer's disease. Although significant progress has been made in the last few years, it is clear from a review of the available publications that much remains to be clarified regarding the relationship between cerebral ischemia and Alzheimer's disease. The fact that even the combined association of amyloid and tau protein pathology does not necessarily lead to Alzheimer's disease, in addition to the fact that other factors like ischemia might be playing a role in triggering or accelerating the Alzheimer's disease type cognitive decline, has prompted a handful of investigators to conclude that studies should aim to understand the interactions among these factors during the progression of injury following brain ischemia, and that the genetic, molecular, and cognitive profiles of patients must be analyzed on an individual basis (Gauthier et al., 2018). In conclusion, the behavior of Alzheimer's disease-related genes in the human brain after ischemia should be investigated in the near future as the lack of such data is a significant limitation in our presentation.
Author Contributions
RP: idea of MS, preparing, and editing. SJ: searching for literature and preparing figure and tables. SC: preparing and editing MS. All authors: contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge financial support from the following institutions: the Mossakowski Medical Research Institute, Polish Academy of Sciences, Warsaw, Poland (T3-RP), and the Medical University of Lublin, Lublin, Poland (DS 475/20-SC).
References
Ahmad, M., Dar, N. J., Bhat, Z. S., Hussain, A., Shah, A., Liu, H., et al. (2014). Inflammation in ischemic stroke: mechanisms, consequences and possible drug targets. CNS Neurol. Disord. Drug. Targets 13, 1378–1396. doi: 10.2174/1871527313666141023094720
Amantea, D., Micieli, G., Tassorelli, C., Cuartero, M. I., Ballesteros, I., Certo, M., et al. (2015). Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front. Neurosci. 9:147. doi: 10.3389/fnins.2015.00147
Anfuso, C. D., Assero, G., Lupo, G., Nicota, A., Cannavo, G., Strosznajder, R. P., et al. (2004). Amyloid beta(1-42) and its beta(25-35) fragment induce activation and membrane translocation of cytosolic phospholipase A(2) in bovine retina capillary pericytes. Biochim. Biophys. Acta 1686, 125–138. doi: 10.1016/j.bbalip.2004.09.006
Atlante, A., Amadoro, G., Bobba, A., and Latina, V. (2020). Functional foods: an approach to modulate molecular mechanisms of Alzheimer's disease. Cells 9:2347. doi: 10.3390/cells9112347
Badan, I., Dinca, I., Buchhold, B., Suofu, Y., Walker, L., Gratz, M., et al. (2004). Accelerated accumulation of N- and C-terminal beta APP fragments and delayed recovery of microtubule-associated protein 1B expression following stroke in aged rats. Eur. J. Neurosci. 19, 2270–2280. doi: 10.1111/j.0953-816X.2004.03323.x
Badan, I., Platt, D., Kessler, C., and Popa-Wagner, A. (2003). Temporal dynamics of degenerative and regenerative events associated with cerebral ischemia in aged rats. Gerontology 49, 356–365. doi: 10.1159/000073763
Ballard, C., Gauthier, S., Corbett, A., Brayne, C., Aarsland, D., and Jones, E. (2011). Alzheimer's disease. Lancet 377, 1019–1031. doi: 10.1016/S0140-6736(10)61349-9
Banati, R. B., Gehrmann, J., Wießner, C., Hossmann, K. A., and Kreutzberg, G. W. (1995). Glial expression of the β-amyloid precursor protein (APP) in global ischemia. J. Cereb. Blood Flow Metab. 15, 647–654. doi: 10.1038/jcbfm.1995.80
Basurto-Islas, G., Gu, J. H., Tung, Y. C., Liu, F., and Iqbal, K. (2018). Mechanism of tau hyperphosphorylation involving lysosomal enzyme asparagine endopeptidase in a mouse model of brain ischemia. J. Alzheimers Dis. 63, 821–833. doi: 10.3233/JAD-170715
Beach, T. G., Wilson, J. R., Sue, L. I., Newell, A., Poston, M., Cisneros, R., et al. (2007). Circle of Willis atherosclerosis: association with Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 113, 13–21. doi: 10.1007/s00401-006-0136-y
Bielewicz, J., Kurzepa, J., Czekajska-Chehab, E., Stelmasiak, Z., and Bartosik-Psujek, H. (2011). Does serum tau protein predict the outcome of patients with ischemic stroke? J. Mol. Neurosci. 43, 241–245. doi: 10.1007/s12031-010-9403-4
Bitsch, A., Horn, C., Kemmling, Y., Seipelt, M., Hellenbrand, U., Stiefel, M., et al. (2002). Serum tau protein level as a marker of axonal damage in acute ischemic stroke. Eur. Neurol. 47, 45–51. doi: 10.1159/000047946
Bivard, A., Lillicrap, T., Maréchal, B., Garcia-Esperon, C., Holliday, E., Krishnamurthy, V., et al. (2018). Transient ischemic attack results in delayed brain atrophy and cognitive decline. Stroke 49, 384–390. doi: 10.1161/STROKEAHA.117.019276
Blasko, I., Beer, R., Bigl, M., Apelt, J., Franz, G., Rudzki, D., et al. (2004). Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease β-secretase (BACE-1). J. Neural. Transm. 111, 523–536. doi: 10.1007/s00702-003-0095-6
Brainin, M., Tuomilehto, J., Heiss, W. D., Bornstein, N. M., Bath, P. M., Teuschl, Y., et al. (2015). Post-stroke cognitive decline: an update and perspectives for clinical research. Eur. J. Neurol. 22, 229–238. doi: 10.1111/ene.12626
Busche, M. A., and Hyman, B. T. (2020). Synergy between amyloid-β and tau in Alzheimer's disease. Nat. Neurosci. 23, 1183–1193. doi: 10.1038/s41593-020-0687-6
Chen, X. H., Siman, R., Iwata, A., Meaney, D. F., Trojanowski, J. Q., and Smith, D. H. (2004). Long-term accumulation of amyloid-β, β-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 165, 357–371. doi: 10.1016/S0002-9440(10)63303-2
Chételat, G. (2013). Alzheimer disease: Aβ-independent processes-rethinking preclinical AD. Nat. Rev. Neuron. 9, 123–124. doi: 10.1038/nrneurol.2013.21
Chi, N. F., Chien, L. N., Ku, H. L., Hu, C. J., and Chiou, H. Y. (2013). Alzheimer disease and risk of stroke: a population-based cohort study. Neurology 80, 705–711. doi: 10.1212/WNL.0b013e31828250af
Chuang, C. M., Hsieh, C. L., Lin, H. Y., and Lin, J. G. (2008). Panax Notoginseng Burk attenuates impairment of learning and memory functions and increases ED1, BDNF and beta-secretase immunoreactive cells in chronic stage ischemia-reperfusion injured rats. Am. J. Chin. Med. 36, 685–693. doi: 10.1142/S0192415X08006156
Cohan, C. H., Neumann, J. T., Dave, K. R., Alekseyenko, A., Binkert, M., Stransky, K., et al. (2015). Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS ONE 10:e0124918. doi: 10.1371/journal.pone.0124918
Dabrowska, S., Andrzejewska, A., Lukomska, B., and Jankowski, M. (2019). Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J. Neuroinflam. 16:178. doi: 10.1186/s12974-019-1571-8
De la Tremblaye, P. B., and Plamondon, H. (2011). Impaired conditioned emotional response and object recognition are concomitant to neuronal damage in the amygdale and perirhinal cortex in middle-aged ischemic rats. Behav. Brain Res. 219, 227–233. doi: 10.1016/j.bbr.2011.01.009
De Vos, A., Bjerke, M., Brouns, R., De Roeck, N., Jacobs, D., Van den Abbeele, L., et al. (2017). Neurogranin and tau in cerebrospinal fluid and plasma of patients with acute ischemic stroke. BMC Neurol. 17:170. doi: 10.1186/s12883-017-0945-8
Dewar, D., and Dawson, D. (1995). Tau protein is altered by focal cerebral ischaemia in the rat: an immunohistochemical and immunoblotting study. Brain Res. 684, 70–78. doi: 10.1016/0006-8993(95)00417-O
Dewar, D., Graham, D. I., Teasdale, G. M., and McCulloch, J. (1993). Alz-50 and ubiquitin immunoreactivity is induced by permanent focal cerebral ischaemia in the cat. Acta Neuropathol. 86, 623–629. doi: 10.1007/BF00294302
Dewar, D., Graham, D. I., Teasdale, G. M., and McCulloch, J. (1994). Cerebral ischemia induces alterations in tau and ubiquitin proteins. Dementia 5, 168–173. doi: 10.1159/000106716
Doraiswamy, P. M., Sperling, R. A., Coleman, R. E., Johnson, K. A., Reiman, E. M., Davis, M., et al. (2012). Amyloid-β assessed by florbetapir F18 PET and 18 months cognitive decline: a multicenter study. Neurology 79, 1636–1644. doi: 10.1212/WNL.0b013e3182661f74
Farkas, E., and Luiten, P. G. (2001). Cerebral microvascular pathology in aging and Alzheimer's disease. Prog. Neurobiol. 64, 575–611. doi: 10.1016/S0301-0082(00)00068-X
Fernando, M. S., Simpson, J. E., Matthews, F., Brayne, C., Lewis, C. E., Barber, R., et al. (2006). White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 37, 1391–1398. doi: 10.1161/01.STR.0000221308.94473.14
Fujii, H., Takahashi, T., Mukai, T., Tanaka, S., Hosomi, N., Maruyama, H., et al. (2017). Modifications of tau protein after cerebral ischemia and reperfusion in rats are similar to those occurring in Alzheimer's disease - hyperphosphorylation and cleavage of 4- and 3-repeat tau. J. Cereb. Blood Flow Metab. 37, 2441–2457. doi: 10.1177/0271678X16668889
Fujioka, M., Taoka, T., Matsuo, Y., Mishima, K., Ogoshi, K., Kondo, Y., et al. (2003). Magnetic resonance imaging shows delayed ischemic striatal neurodegeneration. Ann. Neurol. 54, 732–747. doi: 10.1002/ana.10751
Gamaldo, A., Moghekar, A., Kilada, S., Resnick, S. M., Zonderman, A. B., and O'Brien, R. (2006). Effect of a clinical stroke on the risk of dementia in a prospective cohort. Neurology 67, 1363–1369. doi: 10.1212/01.wnl.0000240285.89067.3f
Garrett, M. D. (2018). A critique of the 2018 National Institute on aging's research framework: toward the biological definition of Alzheimer's disease. Curr. Neurobiol. 9, 49–58.
Gauthier, S., Zhang, H., Ng, K. P., Pascoal, T. A., and Rosa-Neto, P. (2018). Impact of the biological definition of Alzheimer's disease using amyloid, tau and neurodegeneration (ATN): what about the role of vascular changes, inflammation, Lewy body pathology? Transl. Neurodegener. 7:1. doi: 10.1186/s40035-018-0117-9
Geddes, J. W., Schwab, C., Craddock, S., Wilson, J. L., and Pettigrew, L. C. (1994). Alterations in tau immunostaining in the rat hippocampus following transient cerebral ischemia. J. Cereb. Blood Flow Metab. 14, 554–564. doi: 10.1038/jcbfm.1994.69
Gelderblom, M., Gallizioli, M., Ludewig, P., Thom, V., Arunachalam, P., Rissiek, B., et al. (2018). IL-23 (interleukin-23)-producing conventional dendritic cells control the detrimental IL-17 (interleukin-17) response in stroke. Stroke 49, 155–164. doi: 10.1161/STROKEAHA.117.019101
Gemmell, E., Bosomworth, H., Allan, L., Hall, R., Khundakar, A., Oakley, A. E., et al. (2012). Hippocampal neuronal atrophy and cognitive function in delayed poststroke and aging-related dementias. Stroke 43, 808–814. doi: 10.1161/STROKEAHA.111.636498
Gemmell, E., Tam, E., Allan, L., Hall, R., Khundakar, A., Oakley, A. E., et al. (2014). Neuron volumes in hippocampal subfields in delayed poststroke and aging-related dementias. J. Neuropathol. Exp. Neurol. 73, 305–311. doi: 10.1097/NEN.0000000000000054
Giulian, D., Haverkamp, L. J., Li, J., Karshin, W. L., Yu, J., Tom, D., et al. (1995). Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem. Int. 27, 119–137. doi: 10.1016/0197-0186(95)00067-I
Gliem, M., Schwaninger, M., and Jander, S. (2016). Protective features of peripheral monocytes/ macrophages in stroke. Biochim. Biophys. Acta 1862, 329–338. doi: 10.1016/j.bbadis.2015.11.004
Guo, Y., Wang, Q., Chen, S., and Xu, C. (2021). Functions of amyloid precursor protein in metabolic diseases. Metab. Clin. Exp. 115:154454. doi: 10.1016/j.metabol.2020.154454
Hall, E. D., Oostveen, J. A., Dunn, E., and Carter, D. B. (1995). Increased amyloid protein precursor and apolipoprotein E immunoreactivity in the selectively vulnerable hippocampus following transient forebrain ischemia in gerbils. Exp. Neurol. 135, 17–27. doi: 10.1006/exnr.1995.1062
Hatsuta, H., Takao, M., Nogami, A., Uchino, A., Sumikura, H., Takata, T., et al. (2019). Tau and TDP-43 accumulation of the basal nucleus of Meynert in individuals with cerebral lobar infarcts or hemorrhage. Acta Neuropathol. Commun. 7:49. doi: 10.1186/s40478-019-0700-z
Hecht, M., Kramer, L. M., von Arnim, C. A. F., Otto, M., and Thal, D. R. (2018). Capillary cerebral amyloid angiopathy in Alzheimer's disease: association with allocortical/hippocampal microinfarcts and cognitive decline. Acta Neuropathol. 135, 681–694. doi: 10.1007/s00401-018-1834-y
Hossmann, K. A., Schmidt-Kastner, R., and Ophoff, B. G. (1987). Recovery of integrative central nervous function after one hour global cerebro-circulatory arrest in normothermic cat. J. Neurol. Sci. 77, 305–320. doi: 10.1016/0022-510X(87)90130-4
Irving, E. A., Yatsushiro, K., McCulloch, J., and Dewar, D. (1997). Rapid alteration of tau in oligodendrocytes after focal ischemic injury in the rat: involvement of free radicals. J. Cereb. Blood Flow Metab. 17, 612–622. doi: 10.1097/00004647-199706000-00003
Ishibashi, S., Kuroiwa, T., LiYuan, S., Katsumata, N., Li, S., Endo, S., et al. (2006). Long-term cognitive and neuropsychological symptoms after global cerebral ischemia in Mongolian gerbils. Acta Neurochir. 96(Suppl.), 299–302. doi: 10.1007/3-211-30714-1_64
Ishimaru, H., Ishikawa, K., Haga, S., Shoji, M., Ohe, Y., Haga, C., et al. (1996). Accumulation of apolipoprotein E and β-amyloid-like protein in a trace of the hippocampal CA1 pyramidal cell layer after ischaemic delayed neuronal death. NeuroReport 7, 3063–3067. doi: 10.1097/00001756-199611250-00054
Ittner, A., and Ittner, L. M. (2018). Dendritic tau in Alzheimer's disease. Neuron 99, 13–27. doi: 10.1016/j.neuron.2018.06.003
Jabłoński, M., Maciejewski, R., Januszewski, S., Ułamek, M., and Pluta, R. (2011). One year follow up in ischemic brain injury and the role of Alzheimer factors. Physiol. Res. 60, 113–119. doi: 10.33549/physiolres.932186
Jack, C. R., Bennett, D. A., Blennow, K., Carrillo, M. C., Dunn, B., Haeberlein, S. B., et al. (2018). NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 14, 535–562. doi: 10.1016/j.jalz.2018.02.018
Jagust, W. J., Landau, S. M., Shaw, L. M., Trojanowski, J. Q., Koeppe, R. A., Reiman, E. M., et al. (2009). Relationships between biomarkers in aging and dementia. Neurology 73, 1193–1199. doi: 10.1212/WNL.0b013e3181bc010c
Jendroska, K., Hoffmann, O. M., and Patt, S. (1997). Amyloid β peptide and precursor protein (APP) in mild and severe brain ischemia. Ann. N.Y. Acad. Sci. 826, 401–405. doi: 10.1111/j.1749-6632.1997.tb48492.x
Jendroska, K., Poewe, W., Daniel, S. E., Pluess, J., Iwerssen-Schmidt, H., Paulsen, J., et al. (1995). Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 90, 461–466. doi: 10.1007/BF00294806
Kalaria, R. N. (2002). Small vessel disease and Alzheimer's dementia: pathological considerations. Crebrovasc. Dis. 13, 48–52. doi: 10.1159/000049150
Kalvach, P., and Vogner, M. (2019). Parting with the concept of Alzheimer's disease in senium. OBM Geriatr. 3:18. doi: 10.21926/obm.geriatr.1903060
Karasawa, Y., Araki, H., and Otomo, S. (1994). Changes in locomotor activity and passive avoidance task performance induced by cerebral ischemia in mongolian gerbils. Stroke 25, 645–650. doi: 10.1161/01.STR.25.3.645
Kato, T., Hirano, A., Katagiri, T., Sasaki, H., and Yamada, S. (1988). Neurofibrillary tangle formation in the nucleus basalis of Meynert ipsilateral to a massive cerebral infarct. Ann. Neurol. 23, 620–623. doi: 10.1002/ana.410230617
Katzman, R. (1988). “Alzheimer's disease as an age-dependent disorder,” in Research and the Ageing Population. CIBA Foundation Symposium 134 (Chichester: John Wiley and Sons), 69–85. Available online at: http://onlinelibrary.wiley.com/doi/10.1002/9780470513583.fmatter/pdf
Khan, S., Yuldasheva, N. Y., Batten, T. F. C., Pickles, A. R., Kellett, K. A. B., and Saha, S. (2018). Tau pathology and neurochemical changes associated with memory dysfunction in an optimized murine model of global cerebral ischaemia – a potential model for vascular dementia? Neurochem. Int. 118, 134–144. doi: 10.1016/j.neuint.2018.04.004
Kim, E., Yang, J., Beltran, C. D., and Cho, S. (2014). Role of spleen-derived monocytes/macrophages in acute ischemic brain injury. J. Cereb. Blood Flow Metab. 34, 1411–1419. doi: 10.1038/jcbfm.2014.101
Kim, J. H., and Lee, Y. (2018). Dementia and death after stroke in older adults during a 10-year follow-up: results from a competing risk model. J. Nutr. Health Aging 22, 297–301. doi: 10.1007/s12603-017-0914-3
Kiryk, A., Pluta, R., Figiel, I., Mikosz, M., Ułamek, M., Niewiadomska, G., et al. (2011). Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav. Brain Res. 219, 1–7. doi: 10.1016/j.bbr.2010.12.004
Knopman, D. S., Jack, C. R., Wiste, H. J., Weigand, S. D., Vemuri, P., Lowe, V. J., et al. (2013). Brain injury biomarkers are not dependent on β-amyloid in normal elderly. Ann. Neurol. 73, 472–480. doi: 10.1002/ana.23816
Kocki, J., Ułamek-Kozioł, M., Bogucka-Kocka, A., Januszewski, S., Jabłoński, M., Gil-Kulik, P., et al. (2015). Dysregulation of amyloid precursor protein, β-secretase, presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J. Alzheimers Dis. 47, 1047–1056. doi: 10.3233/JAD-150299
Kokmen, E., Whisnant, J. P., O'Fallon, W. M., Chu, C. P., and Beard, C. M. (1996). Dementia after ischemic stroke: a population-based study in Rochester, Minnesota (1960–1984). Neurology 46,154–159. doi: 10.1212/WNL.46.1.154
Kuroiwa, T., Bonnekoh, P., and Hossmann, K. A. (1991). Locomotor hyperactivity and hippocampal CA1 injury after transient forebrain ischemia in gerbils. Neurosci. Lett. 122, 141–144. doi: 10.1016/0304-3940(91)90842-H
Kurzepa, J., Bielewicz, J., Grabarska, A., Stelmasiak, Z., Stryjecka-Zimmer, M., and Bartosik-Psujek, H. (2010). Matrix metalloproteinase-9 contributes to the increase of tau protein in serum during acute ischemic stroke. J. Clin. Neurosci. 17, 997–999. doi: 10.1016/j.jocn.2010.01.005
Lasek-Bal, A., Jedrzejowska-Szypulka, H., Rozycka, J., Bal, W., Kowalczyk, A., Holecki, M., et al. (2016). The presence of tau protein in blood as a potential prognostic factor in stroke patients. J. Physiol. Pharmacol. 67, 691–696.
Lee, P. H., Bang, O. Y., Hwang, E. M., Lee, J. S., Joo, U. S., Mook-Jung, I., et al. (2005). Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J. Neural. Transm. 112, 1371–1379. doi: 10.1007/s00702-004-0274-0
Leinweber, J., Mizurini, D. M., Francischetti, I. M. B., Fleischer, M., Hermann, D. M., Kleinschnitz, C., et al. (2021). Elastase inhibitor agaphelin protects from acute ischemic stroke in mice by reducing thrombosis, blood-brain barrier damage, and inflammation. Brain Behav. Immun. doi: 10.1016/j.bbi.2020.12.027. [Epub ahead of print].
Li, J., Wang, Y. J., Zhang, M., Fang, C. Q., and Zhou, H. D. (2011). Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer's disease. Life Sci. 89, 86–92. doi: 10.1016/j.lfs.2011.04.024
Lin, B., Ginsberg, M. D., and Busto, R. (2001). Hyperglycemic but not normoglycemic global ischemia induces marked early intraneuronal expression of β-amyloid precursor protein. Brain Res. 888, 107–116. doi: 10.1016/S0006-8993(00)03023-7
Lin, B., Schmidt-Kastner, R., Busto, R., and Ginsberg, M. D. (1999). Progressive parenchymal deposition of β-amyloid precursor protein in rat brain following global cerebral ischemia. Acta Neuropathol. 97, 359–368. doi: 10.1007/s004010050999
Lindsberg, P. J., Strbian, D., and Karjalainen-Lindsberg, M. L. (2010). Mast cells as early responders in the regulation of acute blood-brain barrier changes after cerebral ischemia and hemorrhage. J. Cereb. Blood Flow Metab. 30, 689–702. doi: 10.1038/jcbfm.2009.282
Liu, Y. H., Cao, H. Y., Wang, Y. R., Jiao, S. S., Bu, X. L., Zeng, F., et al. (2015). Serum Aβ is predictive for short-term neurological deficits after acute ischemic stroke. Neurotox. Res. 27, 292–299. doi: 10.1007/s12640-015-9518-z
Lo, J. W., Crawford, J. D., Desmond, D. W., Godefroy, O., Jokinen, H., Mahinrad, S., et al. (2019). Profile of and risk factors for post-stroke cognitive impairment in diverse ethno-regional groups. Neurology 93, e2257–e2271. doi: 10.1212/WNL.0000000000008612
Majd, S., Power, J. H., Koblar, S. A., and Grantham, H. J. (2016). Early glycogen synthase kinase-3 and protein phosphatase 2A independent tau dephosphorylation during global brain ischaemia and reperfusion following cardiac arrest and the role of the adenosine monophosphate kinase pathway. Eur. J. Neurosci. 44, 1987–1997. doi: 10.1111/ejn.13277
Mok, V. C. T., Lam, B. Y. K., Wang, Z., Liu, W., Au, L., Leung, E. Y. L., et al. (2016). Delayed-onset dementia after stroke or transient ischemic attack. Alzheimers Dement. 12, 1167–1176. doi: 10.1016/j.jalz.2016.05.007
Morris, G. P., Clark, I. A., and Vissel, B. (2018). Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer's disease. Acta Neuropathol. 136, 663–689. doi: 10.1007/s00401-018-1918-8
Mörtberg, E., Zetterberg, H., Nordmark, J., Blennow, K., Catry, C., Decraemer, H., et al. (2011). Plasma tau protein in comatose patients after cardiac arrest treated with therapeutic hypothermia. Acta Anaesthesiol. Scand. 55, 1132–1138. doi: 10.1111/j.1399-6576.2011.02505.x
Mossakowski, M. J., Lossinsky, A. S., Pluta, R., and Wisniewski, H. M. (1993). “Changes in cerebral microcirculation system following experimentally induced cardiac arrest: a SEM and TEM study,” in Microcirculatory Stasis in the Brain, ed M. Tomita (Amsterdam: Elsevier Science Publishers B.V.), 99–106.
Mossakowski, M. J., Lossinsky, A. S., Pluta, R., and Wisniewski, H. M. (1994). Abnormalities of the blood-brain barrier in global cerebral ischemia in rats due to experimental cardiac arrest. Acta Neurochir. 60(Suppl.), 274–276. doi: 10.1007/978-3-7091-9334-1_73
Nalivaeva, N. N., Fisk, L., Kochkina, E. G., Plesneva, S. A., Zhuravin, I. A., Babusikova, E., et al. (2004). Effect of hypoxia/ischemia and hypoxic preconditioning/reperfusion on expression of some amyloid-degrading enzymes. Ann. N. Y. Acad. Sci. 1035, 21–33. doi: 10.1196/annals.1332.002
Nihashi, T., Inao, S., Kajita, Y., Kawai, T., Sugimoto, T., Niwa, M., et al. (2001). Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir. 143, 287–295. doi: 10.1007/s007010170109
Orzyłowska, O., Oderfeld-Nowak, B., Zaremba, M., Januszewski, S., and Mossakowski, M. J. (1999). Prolonged and concomitant induction of astroglial immunoreactivity of interleukin-1 beta and interleukin-6 in the rat hippocampus after transient global ischemia. Neurosci. Lett. 263, 72–76. doi: 10.1016/S0304-3940(99)00043-9
Palacios, G., Mengod, G., Tortosa, A., Ferrer, I., and Palacios, J. M. (1995). Increased β-amyloid precursor protein expression in astrocytes in the gerbil hippocampus following ischaemia: association with proliferation of astrocytes. Eur. J. Neurosci. 7, 501–510. doi: 10.1111/j.1460-9568.1995.tb00346.x
Pendlebury, S. T., and Rothwell, P. M. (2009). Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 8, 1006–1018. doi: 10.1016/S1474-4422(09)70236-4
Pennypacker, K. R., Hernandez, H., Benkovic, S., Morgan, D. G., Willing, A. E., and Sanberg, P. R. (1999). Induction of presenilins in the rat brain after middle cerebral arterial occlusion. Brain Res. Bull. 48, 539–543. doi: 10.1016/S0361-9230(99)00031-3
Pluta, R. (1997). Experimental model of neuropathological changes characteristic for Alzheimer's disease. Folia Neuropathol. 35, 94–98.
Pluta, R. (2000a). The role of apolipoprotein E in the deposition of β-amyloid peptide during ischemia–reperfusion brain injury. A model of early Alzheimer's disease. Ann. N.Y. Acad. Sci. 903, 324–334. doi: 10.1111/j.1749-6632.2000.tb06383.x
Pluta, R. (2000b). No effect of anti-oxidative therapy on cerebral amyloidosis following ischemia–reperfusion brain injury. Folia Neuropathol. 38, 188–190.
Pluta, R. (2002a). Glial expression of the β-amyloid peptide in cardiac arrest. J. Neurol. Sci. 203–204, 277–280. doi: 10.1016/S0022-510X(02)00305-2
Pluta, R. (2002b). Astroglial expression of the beta-amyloid in ischemia-reperfusion brain injury. Ann. N.Y. Acad. Sci. 977, 102–108. doi: 10.1111/j.1749-6632.2002.tb04803.x
Pluta, R. (2003). Blood–brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia–reperfusion brain injury with 1-year survival. Acta Neurochir. 86, 117–122. doi: 10.1007/978-3-7091-0651-8_26
Pluta, R. (2005). Pathological opening of the blood–brain barrier to horseradish peroxidase and amyloid precursor protein following ischemia–reperfusion brain injury. Chemotherapy 51, 223–226. doi: 10.1159/000086924
Pluta, R. (2007a). Role of ischemic blood–brain barrier on amyloid plaques development in Alzheimer's disease brain. Curr. Neurovasc. Res. 4, 121–129. doi: 10.2174/156720207780637207
Pluta, R. (2007b). Ischemia-Reperfusion Pathways in Alzheimer's Disease. New York, NY: Nova Science Publishers, Inc.
Pluta, R. (2019). Brain Ischemia: Alzheimer's Disease Mechanisms. New York, NY: Nova Science Publishers, Inc.
Pluta, R., Barcikowska, M., Debicki, G., Ryba, M., and Januszewski, S. (1997a). Changes in amyloid precursor protein and apolipoprotein E immunoreactivity following ischemic brain injury in rat with long-term survival: influence of idebenone treatment. Neurosci. Lett. 232, 95–98. doi: 10.1016/S0304-3940(97)00571-5
Pluta, R., Barcikowska, M., Januszewski, S., Misicka, A., and Lipkowski, A. W. (1996). Evidence of blood–brain barrier permeability/leakage for circulating human Alzheimer's β-amyloid-(1–42)-peptide. NeuroReport 7, 1261–1265. doi: 10.1097/00001756-199605170-00008
Pluta, R., Barcikowska, M., Misicka, A., Lipkowski, A. W., Spisacka, S., and Januszewski, S. (1999). Ischemic rats as a model in the study of the neurobiological role of human β-amyloid peptide. Time-dependent disappearing diffuse amyloid plaques in brain. NeuroReport 10, 3615–3619. doi: 10.1097/00001756-199911260-00028
Pluta, R., Barcikowska, M., Mossakowski, M. J., and Zelman, I. (1998). Cerebral accumulation of beta-amyloid following ischemic brain injury with long-term survival. Acta Neurochir. 71(Suppl.), 206–208. doi: 10.1007/978-3-7091-6475-4_59
Pluta, R., Bogucka-Kocka, A., Ułamek-Kozioł, M., Bogucki, J., and Czuczwar, S. J. (2018). Ischemic tau protein gene induction as an additional key factor driving development of Alzheimer's phenotype changes in CA1 area of hippocampus in an ischemic model of Alzheimer's disease. Pharmacol. Rep. 70, 881–884. doi: 10.1016/j.pharep.2018.03.004
Pluta, R., Furmaga-Jabłońska, W., Maciejewski, R., Ułamek-Kozioł, M., and Jabłoński, M. (2013a). Brain ischemia activates β- and γ- secretase cleavage of amyloid precursor protein: significance in sporadic Alzheimer's disease. Mol. Neurobiol. 47, 425–434. doi: 10.1007/s12035-012-8360-z
Pluta, R., and Jabłoński, M. (2012). “Alzheimer's factors in ischemic brain injury,” in Brain Injury, Pathogenesis, Monitoring, Recovery and Management, ed A. Agrawal (Rijeka: InTech, Open Book), 97–138.
Pluta, R., Jabłoński, M., and Czuczwar, S. J. (2012c). Postischemic dementia with Alzheimer phenotype: selectively vulnerable versus resistant areas of the brain and neurodegeneration versus β-amyloid peptide. Folia Neuropathol. 50, 101–109.
Pluta, R., Jabłoński, M., Ułamek-Kozioł, M., Kocki, J., Brzozowska, J., Januszewski, S., et al. (2013b). Sporadic Alzheimer's disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer's disease genes. Mol. Neurobiol. 48, 500–515. doi: 10.1007/s12035-013-8439-1
Pluta, R., Januszewski, S., Jabłoński, M., and Ułamek, M. (2010). Factors in creepy delayed neuronal death in hippocampus following brain ischemia-reperfusion injury with long-term survival. Acta Neurochir. 106, 37–41. doi: 10.1007/978-3-211-98811-4_5
Pluta, R., Januszewski, S., and Ułamek, M. (2008). Ischemic blood–brain barrier and amyloid in white matter as etiological factors in leukoaraiosis. Acta Neurochir. 102, 353–356. doi: 10.1007/978-3-211-85578-2_67
Pluta, R., Jolkkonen, J., Cuzzocrea, S., Pedata, F., Cechetto, D., and PopaWagner, A. (2011). Cognitive impairment with vascular impairment and degeneration. Curr. Neurovasc. Res. 8, 342–350. doi: 10.2174/156720211798120981
Pluta, R., Kida, E., Lossinsky, A. S., Golabek, A. A., Mossakowski, M. J., and Wisniewski, H. M. (1994b). Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer's β-amyloid protein precursor in the brain. Brain Res. 649, 323–328. doi: 10.1016/0006-8993(94)91081-2
Pluta, R., Kocki, J., Maciejewski, R., Ułamek-Kozioł, M., Jabłoński, M., Bogucka-Kocka, A., et al. (2012b). Ischemia signaling to Alzheimer-related genes. Folia Neuropathol. 50, 322–329. doi: 10.5114/fn.2012.32362
Pluta, R., Kocki, J., Ułamek-Kozioł, M., Bogucka-Kocka, A., Gil-Kulik, P., Januszewski, S., et al. (2016b). Alzheimer-associated presenilin 2 gene is dysregulated in rat medial temporal lobe cortex after complete brain ischemia due to cardiac arrest. Pharmacol. Rep. 68, 155–161. doi: 10.1016/j.pharep.2015.08.002
Pluta, R., Kocki, J., Ułamek-Kozioł, M., Petniak, A., Gil-Kulik, P., Januszewski, S., et al. (2016a). Discrepancy in expression of β-secretase and amyloid-β protein precursor in Alzheimer-related genes in the rat medial temporal lobe cortex following transient global brain ischemia. J. Alzheimers Dis. 51, 1023–1031. doi: 10.3233/JAD-151102
Pluta, R., Lossinsky, A. S., Walski, M., Wiśniewski, H. M., and Mossakowski, M. J. (1994c). Platelet occlusion phenomenon after short- and long-term survival following complete cerebral ischemia in rats produced by cardiac arrest. J. Hirnforsch. 35, 463–471.
Pluta, R., Lossinsky, A. S., Wiśniewski, H. M., and Mossakowski, M. J. (1994a). Early blood–brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res. 633, 41–52. doi: 10.1016/0006-8993(94)91520-2
Pluta, R., Misicka, A., Barcikowska, M., Spisacka, S., Lipkowski, A. W., and Januszewski, S. (2000). Possible reverse transport of β-amyloid peptide across the blood-brain barrier. Acta Neurochir. 76, 73–77. doi: 10.1007/978-3-7091-6346-7_15
Pluta, R., Misicka, A., Januszewski, J., Barcikowska, M., and Lipkowski, A. W. (1997b). Transport of human β-amyloid peptide through the rat blood-brain barrier after global cerebral ischemia. Acta Neurochir. 70, 247–249. doi: 10.1007/978-3-7091-6837-0_76
Pluta, R., Tomida, S., Ikeda, J., Nowak, T. S. Jr., and Klatzo, I. (1989). Cerebral vascular volume after repeated ischemic insults in the gerbil: comparison with changes in CBF and brain edema. J. Cereb. Blood Flow Metab. 9, 163–170. doi: 10.1038/jcbfm.1989.24
Pluta, R., Ułamek, M., and Jabłoński, M. (2009). Alzheimer's mechanisms in ischemic brain degeneration. Anat. Rec. 292, 1863–1881. doi: 10.1002/ar.21018
Pluta, R., Ułamek, M., and Januszewski, S. (2006). Micro-blood–brain barrier openings and cytotoxic fragments of amyloid precursor protein accumulation in white matter after ischemic brain injury in long-lived rats. Acta Neurochir. 96, 267–271. doi: 10.1007/3-211-30714-1_57
Pluta, R., and Ułamek-Kozioł, M. (2021). Genes associated with Alzheimer's disease affecting ischemic neurodegeneration of the hippocampal CA3 region. Neural. Regen. Res. 16, 1392–1393. doi: 10.4103/1673-5374.300982
Pluta, R., Ułamek-Kozioł, M., and Czuczwar, S. J. (2019a). “Shared genomic and proteomic contribution to brain ischemia and Alzheimer's disease: ischemic etiology of Alzheimer's disease,” in Brain Ischemia: Alzheimer's Disease Mechanisms, ed R. Pluta (New York, NY: Nova Science Publishers, Inc.), 209–249.
Pluta, R., Ułamek-Kozioł, M., Januszewski, S., and Czuczwar, S. J. (2019b). “Common proteomic and genomic contribution to ischemic brain damage and Alzheimer's disease,” in Alzheimer's Disease, ed T. Wisniewski (Brisbane, QLD: Codon Publications), 53–68.
Pluta, R., Ułamek-Kozioł, M., Januszewski, S., and Czuczwar, S. J. (2020b). Gut microbiota and pro/prebiotics in Alzheimer's disease. Aging 12, 5539–5550. doi: 10.18632/aging.102930
Pluta, R., Ułamek-Kozioł, M., Januszewski, S., and Czuczwar, S. J. (2020c). Shared genomic and proteomic contribution of amyloid and tau protein characteristic of Alzheimer's disease to brain ischemia. Int. J. Mol. Sci. 21:3186. doi: 10.3390/ijms21093186
Pluta, R., Ułamek-Kozioł, M., Januszewski, S., and Czuczwar, S. J. (2020d). Participation of amyloid and tau protein in neuronal death and neurodegeneration after brain ischemia. Int. J. Mol. Sci. 21:4599. doi: 10.3390/ijms21134599
Pluta, R., Ułamek-Kozioł, M., Januszewski, S., Sciślewska, M., and Bogucka-Kocka A, Kocki, J. (2012a). Alzheimer's factors in postischemic dementia. Rom. J. Morphol. Embryol. 53, 461–466.
Pluta, R., Ułamek-Kozioł, M., Kocki, J., Bogucki, J., Januszewski, S., Bogucka-Kocka, A., et al. (2020a). Expression of the tau protein and amyloid protein precursor processing genes in the CA3 area of the hippocampus in the ischemic model of Alzheimer's disease in the rat. Mol. Neurobiol. 57, 1281–1290. doi: 10.1007/s12035-019-01799-z
Polavarapu, R., An, J., Zhang, C., and Yepes, M. (2008). Regulated intramembrane proteolysis of the low-density lipoprotein receptor-related protein mediates ischemic cell death. Am. J. Pathol. 172, 1355–1362. doi: 10.2353/ajpath.2008.070975
Portegies, M. L., Wolters, F. J., Hofman, A., Ikram, M. K., Koudstaal, P. J., and Ikram, M. A. (2016). Prestroke vascular pathology and the risk of recurrent stroke and poststroke dementia. Stroke 47, 2119–2122. doi: 10.1161/STROKEAHA.116.014094
Price, C. J. S., Menon, D. K., Peters, A. M., Ballinger, J. R., Barber, R. W., Balan, K. K., et al. (2004). Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging-based study. Stroke 35, 1659–1664. doi: 10.1161/01.STR.0000130592.71028.92
Price, D. L., Walker, L. C., Martin, L. J., and Sisodia, S. S. (1992). Amyloidosis in aging and Alzheimer's disease. Am. J. Pathol. 141, 762–772.
Price, J. L., and Morris, C. (1999). Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann. Neurol. 45, 358–368. doi: 10.1002/1531-8249(199903)45:3<358::AID-ANA12>3.0.CO;2-X
Qi, J., Wu, H., Yang, Y., Wand, D., Chen, Y., Gu, Y., et al. (2007). Cerebral ischemia and Alzheimer's disease: the expression of amyloid-β and apolipoprotein E in human hippocampus. J. Alzheimers Dis. 12, 335–341. doi: 10.3233/JAD-2007-12406
Radenovic, L., Nenadic, M., Ułamek-Kozioł, M., Januszewski, S., Czuczwar, S. J., Andjus, P. R., and Pluta, R. (2020). Heterogeneity in brain distribution of activated microglia and astrocytes in a rat ischemic model of Alzheimer's disease after 2 years of survival. Aging 12, 12251–12267. doi: 10.18632/aging.103411
Randall, J., Mörtberg, E., Provuncher, G. K., Fournier, D. R., Duffy, D. C., Rubertsson, S., et al. (2013). Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation 84, 351–356. doi: 10.1016/j.resuscitation.2012.07.027
Roher, A. E., Esh, C., Rahman, A., Kokjohn, T. A., and Beach, T. G. (2004). Atherosclerosis of cerebral arteries in Alzheimer disease. Stroke 35(11 Suppl. 1), 2623–2627. doi: 10.1161/01.STR.0000143317.70478.b3
Rowe, C. C., Ng, S., Ackermann, U., Gong, S. J., Pike, K., Savage, G., et al. (2007). Imaging β-amyloid burden in aging and dementia. Neurology 68, 1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea
Salminen, A., Kauppinen, A., and Kaarniranta, K. (2017). Hypoxia/ischemia activate processing of amyloid precursor protein: impact of vascular dysfunction in the pathogenesis of Alzheimer's disease. J. Neurochem. 140, 536–549. doi: 10.1111/jnc.13932
Scherr, M., Trinka, E., Mc Coy, M., Krenn, Y., Staffen, W., Kirschner, M., et al. (2012). Cerebral hypoperfusion during carotid artery stenosis can lead to cognitive deficits that may be independent of white matter lesion load. Curr. Neurovasc. Res. 9, 193–199. doi: 10.2174/156720212801619009
Sekeljic, V., Bataveljic, D., Stamenkovic, S., Ułamek, M., Jabłoński, M., Radenovic, L., et al. (2012). Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct. Funct. 217, 411–420. doi: 10.1007/s00429-011-0336-7
Seshadri, S., and Wolf, P. A. (2007). Lifetime risk of stroke and dementia: current concepts, and estimates from the Framingham Study. Lancet Neurol. 6, 1106–1114. doi: 10.1016/S1474-4422(07)70291-0
Shi, J., Panickar, K. S., Yang, S. H., Rabbani, O., Day, A. L., and Simpkins, J. W. (1998). Estrogen attenuates over-expression of beta-amyloid precursor protein messenger RNA in an animal model of focal ischemia. Brain Res. 810, 87–92. doi: 10.1016/S0006-8993(98)00888-9
Shi, J., Yang, S. H., Stubley, L., Day, A. L., and Simpkins, J. W. (2000). Hypoperfusion induces overexpression of β-amyloid precursor protein mRNA in a focal ischemic rodent model. Brain Res. 853, 1–4. doi: 10.1016/S0006-8993(99)02113-7
Sinigaglia-Coimbra, R., Cavalheiro, E. A., and Coimbra, C. G. (2002). Postischemic hypertermia induces Alzheimer-like pathology in the rat brain. Acta Neuropathol. 103, 444–452. doi: 10.1007/s00401-001-0487-3
Surawan, J., Areemit, S., Tiamkao, S., Sirithanawuthichai, T., and Saensak, S. (2017). Risk factors associated with post-stroke dementia: a systematic review and meta-analysis. Neurol. Int. 9:7216. doi: 10.4081/ni.2017.7216
Tabaton, M., and Tamagno, E. (2007). The molecular link between β- and γ-secretase activity on the amyloid β precursor protein. Cell Mol. Life Sci. 64, 2211–2218. doi: 10.1007/s00018-007-7219-3
Tanimukai, H., Imaizumi, K., Kudo, T., Katayama, T., Tsuda, M., Takagi, T., et al. (1998). Alzheimer-associated presenilin-1 gene is induced in gerbil hippocampus after transient ischemia. Mol. Brain Res. 54, 212–218. doi: 10.1016/S0169-328X(97)00337-9
Thal, D. R., Ghebremedhin, E., Orantes, M., and Wiestler, O. D. (2003). Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J. Neuropathol. Exp. Neurol. 62, 1287–1301. doi: 10.1093/jnen/62.12.1287
Thiebaut de Schotten, M., Tomaiuolo, F., Aiello, M., Merola, S., Silvetti, M., Lecce, F., et al. (2014). Damage to white matter pathways in subacute and chronic spatial neglect: a group study and 2 single-case studies with complete virtual “in vivo” tractography dissection. Cereb. Cortex. 24, 691–706. doi: 10.1093/cercor/bhs351
Tolppanen, A. M., Lavikainen, P., Solomon, A., Kivipelto, M., Soininen, H., and Hartikainen, S. (2013). Incidence of stroke in people with Alzheimer disease: a national register-based approach. Neurology 80, 353–358. doi: 10.1212/WNL.0b013e31827f08c5
Tomimoto, H., Akiguchi, I., Wakita, H., Nakamura, S., and Kimura, J. (1995). Ultrastructural localization of amyloid protein precursor in the normal and postischemic gerbil brain. Brain Res. 672, 187–195. doi: 10.1016/0006-8993(94)01160-J
Traylor, M., Adib-Samii, P., Harold, D., Alzheimer's Disease Neuroimaging Initiative, International Stroke Genetics Consortium (ISGC), UK Young Lacunar Stroke DNA resource, et al. (2016). Shared genetic contribution to ischaemic stroke and Alzheimer's disease. Ann. Neurol. 79, 739–747. doi: 10.1002/ana.24621
Tse, K.-H., and Herrup, K. (2017). Re-imagining Alzheimer's disease - the diminishing importance of amyloid and a glimpse of what lies ahead. J. Neurochem. 143, 432–444. doi: 10.1111/jnc.14079
Uchihara, T., Nakamura, A., Arai, T., Ikeda, K., and Tsuchiya, K. (2004). Microglial tau undergoes phosphorylation-independent modification after ischemia. Glia 45, 180–187. doi: 10.1002/glia.10318
Uddin, M. S., Kabir, M. T., Rahman, M. S., Behl, T., Jeandet, P., Ashraf, G. M., et al. (2020). Revisiting the amyloid cascade hypothesis: from anti-Aβ therapeutics to auspicious new ways for Alzheimer's disease. Int. J. Mol. Sci. 21:5858. doi: 10.3390/ijms21165858
Ułamek-Kozioł, M., Czuczwar, S. J., Januszewski, S., and Pluta, R. (2020a). Substantiation for the use of curcumin during the development of neurodegeneration after brain ischemia. Int. J. Mol. Sci. 21:517. doi: 10.3390/ijms21020517
Ułamek-Kozioł, M., Czuczwar, S. J., Januszewski, S., and Pluta, R. (2020b). Proteomic and genomic changes in tau protein, which are associated with Alzheimer's disease after ischemia-reperfusion brain injury. Int. J. Mol. Sci. 21:892. doi: 10.3390/ijms21030892
Van Groen, T., Puurunen, K., Maki, H. M., Sivenius, J., and Jolkkonen, J. (2005). Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 36, 1551–1556. doi: 10.1161/01.STR.0000169933.88903.cf
Wakita, H., Tomimoto, H., Akiguchi, I., and Kimura, J. (1994). Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: an immunohistochemical study. Acta Neuropathol. 87, 484–492. doi: 10.1007/BF00294175
Wen, Y., Onyewuchi, O., Yang, S., Liu, R., and Simpkins, J. W. (2004a). Increased beta-secretase activity and expression in rats following transient cerebral ischemia. Brain Res. 1009, 1–8. doi: 10.1016/j.brainres.2003.09.086
Wen, Y., Yang, S., Liu, R., Brun-Zinkernagel, A. M., Koulen, P., and Simpkins, J. W. (2004c). Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer's disease-like tauopathy in female rats. J. Bio.l Chem. 279, 22684–22692. doi: 10.1074/jbc.M311768200
Wen, Y., Yang, S., Liu, R., and Simpkins, J. W. (2004b). Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 1022, 30–38. doi: 10.1016/j.brainres.2004.05.106
Wen, Y., Yang, S. H., Liu, R., Perez, E. J., Brun-Ziukemagel, A. M., Koulen, P., et al. (2007). Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim. Biophys. Acta 1772, 473–483. doi: 10.1016/j.bbadis.2006.10.011
Wisniewski, H. M., and Maslinska, D. (1996). Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol. 34, 65–71.
Wisniewski, H. M., Pluta, R., Lossinsky, A. S., and Mossakowski, M. J. (1995). Ultrastructural studies of cerebral vascular spasm after cardiac arrest-related global cerebral ischemia in rats. Acta Neuropathol. 90, 432–440. doi: 10.1007/BF00294802
Yan, F. L., Zhang, J., Guan, X. N., and Hong, Z. (2007). mRNA expression and activity of ADAM17 in hippocampus after chronic cerebral hypoperfusion: experiment with aged rats. Zhonghua Yi. Xue. Za. Zhi. 87, 2515–2517.
Ye, J. T., Pi, R., Mao, X., Chen, X., Qin, J., Xu, S., et al. (2009). Alterations in mRNA expression of BACE1, cathepsin B, and glutaminyl cyclase in mice ischemic brain. Neurol. Rep. 20, 1456–1460. doi: 10.1097/WNR.0b013e328332024a
Yokota, M., Saido, T. C., Tani, E., Yamaura, I., and Minami, N. (1996). Cytotoxic fragment of amyloid precursor protein accumulates in hippocampus after global forebrain ischemia. J. Cereb. Blood Flow Metab. 16, 1219–1223. doi: 10.1097/00004647-199611000-00016
Zamboni, G., Griffanti, L., Jenkinson, M., Mazzucco, S., Li, L., Küker, W., et al. (2017). White matter imaging correlates of early cognitive impairment detected by the Montreal Cognitive Assessment after transient ischemic attack and minor stroke. Stroke 48, 1539–1547. doi: 10.1161/STROKEAHA.116.016044
Keywords: brain ischemia, neurodegeneration, amyloid protein precursor, secretases, presenilins, tau protein, folding proteins, Alzheimer's disease
Citation: Pluta R, Januszewski S and Czuczwar SJ (2021) Brain Ischemia as a Prelude to Alzheimer's Disease. Front. Aging Neurosci. 13:636653. doi: 10.3389/fnagi.2021.636653
Received: 01 December 2020; Accepted: 25 January 2021;
Published: 18 February 2021.
Edited by:
Jolanta Dorszewska, Poznan University of Medical Sciences, PolandReviewed by:
Qi Wan, Qingdao University, ChinaSantosh Jadhav, Institute of Neuroimmunology (SAS), Slovakia
Copyright © 2021 Pluta, Januszewski and Czuczwar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryszard Pluta, pluta@imdik.pan.pl