
Abstract
Traumatic brain injury (TBI) affects 1.6 million Americans annually. The injury severity impacts the overall outcome and likelihood for survival. Current treatment of acute TBI includes surgical intervention and supportive care therapies. Treatment of elevated intracranial pressure and optimizing cerebral perfusion are cornerstones of current therapy. These approaches do not directly address the secondary neurological sequelae that lead to continued brain injury after TBI. Depending on injury severity, a complex cascade of processes are activated and generate continued endogenous changes affecting cellular systems and overall outcome from the initial insult to the brain. Homeostatic cellular processes governing calcium influx, mitochondrial function, membrane stability, redox balance, blood flow and cytoskeletal structure often become dysfunctional after TBI. Interruption of this cascade has been the target of numerous pharmacotherapeutic agents investigated over the last two decades. Many agents such as selfotel, pegorgotein (PEG-SOD), magnesium, deltibant and dexanabinol were ineffective in clinical trials. While progesterone and ciclosporin have shown promise in phase II studies, success in larger phase III, randomized, multicentre, clinical trials is pending. Consequently, no neuroprotective treatment options currently exist that improve neurological outcome after TBI. Investigations to date have extended understanding of the injury mechanisms and sites for intervention. Examination of novel strategies addressing both pathological and pharmacological factors affecting outcome, employing novel trial design methods and utilizing biomarkers validated to be reflective of the prognosis for TBI will facilitate progress in overcoming the obstacles identified from previous clinical trials.
Similar content being viewed by others

1. Overview of Traumatic Brain Injury
Traumatic brain injury (TBI) continues to be an important cause of morbidity and mortality worldwide. In the US alone, approximately 1.7 million Americans sustain a TBI each year, of which over 50 000 succumb to the injury and at least 35% of the 230 000 hospitalized survivors will be faced with long-term disability.[1–3] This injury also places a significant emotional and economic burden on both the patient and their families as the population most commonly affected is young men whose economic future is generally compromised. Research efforts have dramatically improved our understanding of TBI pathology over the last few decades. Although some progress has been made with prevention measures and clinical care efforts in early resuscitation, the overall neurological recovery and long-term morbidity continues to be a significant healthcare challenge.[4]
Classifications of TBI are used clinically to describe the severity of injury using physical assessment and radiographic measures. Common clinical examples include the Glasgow Coma Scale (GCS) and the Marshall CT score, which is based on the nature of the intracranial injury. Prognostic models have been validated to predict early outcome after severe TBI with some accuracy.[5] The post-resuscitation GCS is often used to classify injury severity and predict outcome. Patients with a GCS of 8 or less are categorized as severe TBI. Mortality approaches 40% in many studies of severe TBI patients and up to 60% of survivors are left with significant disability.[6–8] Moderate TBI is defined as a GCS of 9–13. Mortality rates are much lower in these patients when compared with severe TBI (approximately 10%).[9] However, depression, inattention, learning deficits and loss of executive function are common after moderate TBI, leaving many of these patients with insidious disabilities that impact their ability to resume working or even executing activities of daily living. Mild TBI is defined by a GCS of 14–15.[10] These patients often have complete resolution of symptoms over time, although as many as 90% of patients report dizziness or headache up to 1 month after their injury.[11,12] This presentation is clinically described as post-concussion syndrome and may also include fatigue, irritability, depression, difficulty concentrating, insomnia or emotional lability.[11,13] The precise incidence of these symptoms and their typical time to resolution is ill-defined.
The Marshall CT Score classifies the radiographic findings and provides specific information about the injury type as opposed to the clinical presentation assessed by the GCS (table I). Focal injuries are generally caused by a direct and linear force to the cranium, leading to direct compression of neural tissue and local, more superficial, damage. Haemorrhagic contusions and intraparenchymal haematomas are more common in the frontal and temporal lobes, where the brain comes into contact with a rougher surface of bone. This results in destruction of focal neural tissue and expected vasogenic oedema, but not widespread deterioration of neurons or axons. Epidural and subdural haematomas are further examples of injuries that compress the underlying/adjacent brain tissue with subsequent focal neurological dysfunction. If there is protracted compression, however, diffuse oedema and elevated intracranial pressure (ICP) can evolve, and the patient will present with a much poorer neurological examination. Diffuse injuries also present with a poor neurological examination despite the potentially benign appearance of initial neuroimaging. These include diffuse axonal injury (DAI), global ischaemia and diffuse oedema. DAI is due to the widespread shearing of axons, usually due to acceleration/deceleration or rotational forces, and involves deep white matter structures, including the brainstem. Clinically, it is common to see patients with a combination of TBI classifications (e.g. subdural haematoma with contusion), rather than one isolated subtype of injury. Penetrating injury or blast injury may also be classified as TBI, but for the purposes of this review we will focus primarily on closed head injury. Readers are referred to other reviews for details on different mechanisms of TBI.[14,15]

Marshall CT classification of traumatic brain injury
TBI results in both primary and secondary injuries. Primary injuries, such as contusions and epidural haematomas (described above), are those immediately resultant from the initial impact or insult. This physical damage is immediate, while secondary injury is a delayed response occurring from a complex group of cellular and molecular responses to the primary injury. DAI can cause both primary injury to neurons as well as secondary injuries.[16] This secondary injury is further exacerbated by ongoing ischaemia, elevated ICP and disrupted cerebral blood flow leading to further cell death. Both primary and secondary injury can ultimately result in cell death and irreversible damage. However, the delayed onset of secondary injury can provide the opportunity for possible prevention or attenuation.[17]
Currently, the mainstay of therapy for TBI is removal of haematomas and revision of significant skull fractures along with supportive therapies aimed at maintaining perfusion and oxygenation to tissues. For patients without an operable lesion (or after surgical intervention), control of ICP (goal ≪20 mmHg), cerebral perfusion pressure (CPP, goal >60 mmHg) and systemic, and perhaps local, oxygenation are cornerstones of intensive care unit management.[18] Other necessary medical interventions are commonly employed such as early enteral nutrition, maintenance of fluid volume, and prevention and treatment of complications such as hyperthermia, hyponatraemia, seizures, pneumonia, venous thromboembolism and stress-related mucosal bleeding.[18–20] Many of these therapies can have an impact on the outcome of a TBI patient. However, aside from avoiding events such as hypoxia and hypotension, there is little evidence that these supportive care therapies inherently have a direct effect on brain tissue preservation. Since the primary injury cannot be reversed, the major opportunity for interventions that could prevent further neurological decline is in reversing or preventing the secondary injury. Currently, there is no neuroprotective agent demonstrated in a large, phase III clinical trial to improve neurological outcome. The purpose of this article is to review the pathophysiology of TBI, discuss trials investigating neuroprotective therapies in TBI that have been completed or are underway and to posit some future directions for optimizing clinical trial design in TBI patients.
The methods for this review included a systematic search of PubMed, clinicaltrials.gov and the Cochrane Library database (the Cochrane central register of controlled trials) for studies reporting TBI outcomes related to neuroprotection, both clinical and experimental published from 1966 through January 2012. To identify additional articles, reference sections were reviewed from the related citations in PubMed. Articles included were randomized controlled trials, experimental animal and systematic and meta-analysis reviews reported in English. Search terms, MeSH subject headings and limits were (‘traumatic brain injury’ OR ‘concussion’ OR ‘penetrating injury’) AND ‘neuroprotection’ AND (‘progesterone’ OR ‘glutamate antagonists’) OR ‘excitotoxicity’ AND (‘animal’ OR ‘human’ OR ‘placebo controlled’ OR ‘randomized controlled trial’). Clinical trials and controlled cortical impact animal models were given preference over in vitro and mechanistic studies, one animal study demonstrating a positive outcome was considered appropriate for inclusion.
2. Causes of Secondary Neurological Injury
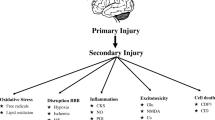
The secondary injury process in TBI is typified by a complex cascade of processes that are simultaneously reacting to the primary injury to the brain and attempting to mitigate the ensuing damage to the brain cells (figure 1).[16,21–23] Many of these processes occur along the spectrum of severity of TBI (mild through severe), although severe TBI models have most often been used to elucidate the various pathological issues in secondary neurological injury. Excitatory neurotransmitters are released in high quantities after injury, promoting elevations in cellular metabolism and cation influx. Cellular membranes are damaged due to oxidation and ion flux. Immune activation occurs in response to injury, sometimes with deleterious consequences to the injured cells. Intracellular homeostasis is disrupted by membrane potential gradient perturbations and the need for energy-expensive repair mechanisms to restore cell structure and function. What follows is a delicate balance of cellular fitness, affected by cellular metabolic competence, oxidative stress and numerous systemic factors. This balance dictates the recovery of the cell or cell death, either by necrosis or apoptosis.

Processes and mediators associated with secondary neurological injury after traumatic brain injury (TBI).
2.1 Extracellular Contributors to Secondary Neurological Injury
Blood flow alterations are common after TBI, often due to impaired cerebral autoregulation and substances produced in response to injury.[22] Disruption of cerebral autoregulation leads cerebral blood flow to be reliant upon the CPP for adequate perfusion.[24] Hypotension in these patients can be devastating, as evidenced by the increased risk of mortality (at least 2-fold).[18,25] Maintenance of cerebral blood flow is a cornerstone of early TBI treatment. Systemic hypotension is avoided by administering isotonic or hypertonic intravenous fluids and vasopressors (when necessary).[18,20] The global cerebral blood flow pattern in TBI can be variable, but generally seems to be bimodal, falling to near-ischaemia levels (20 mL/min/g), then rising to near normal levels (approximately 50 mL/min/g).[26,27] Blood flow is reduced immediately after TBI, mediated by vasoconstricting prostaglandins and other local factors, most likely as a result of reduced oxygen demand in the brain in response to injury. Cerebral blood flow increases thereafter due to vasodilation from cellular acidosis, increases in bradykinin and inducible nitric oxide synthetase (iNOS) activity, and increases in excitatory neurotransmitters. This hyperaemia can exacerbate early cerebral oedema and intracranial hypertension.[28]
Ischaemia/reperfusion injury and other blood flow abnormalities are also evident early in TBI.[29,30] Cellular hypoxia has only recently been easily measurable at the bedside, with devices such as the intraparenchymal brain tissue oxygen monitor. Mismatches between cerebral blood flow and cerebral metabolic rate of oxygen often lead to ischaemia.[31,32] Mass-occupying lesions such as a subdural haematoma also may compress local vasculature and cause ischaemia in the region of the haematoma. Decompression of these areas by surgical evacuation of the haematoma leads to ischaemia/reperfusion injury, often resulting in oxidative damage to vulnerable tissue. One cohort study demonstrated that nearly 50% of TBI patients (ranging from mild to severe injury) had cerebral vasospasm during the first 2 weeks after injury.[33] Increasing injury severity seemed to be associated with increased risk of vasospasm (mean GCS on admission of 7 in patients with vasospasm, 10 in patients without vasospasm, p = 0.001). Clinically, it is rarely screened for, despite the fact that some small clinical trials suggest a neuroprotective benefit in preventing vasospasm.[34,35]
2.2 Intracellular Contributors to Secondary Neurological Injury
Oxidative stress is high after TBI and causes direct damage to brain tissues.[36,37] Oxidation of haemoglobin, arachidonic acid metabolic by-products, and disruption in the integrity of mitochondrial membranes leads to elevated concentrations of reactive species such as superoxide (O2 •−) and peroxynitrite (OONO•−). These reactive species may overwhelm endogenous antioxidant stores, such as superoxide dismutase (SOD), causing DNA damage and lipid peroxidation, which can propagate well beyond the site of origin.[38,39]
The inflammatory response to TBI, like in many other types of critical illness, can either be harmful or helpful, depending on the maintenance of homeostasis and ‘normal’ function.[23,40,41] Nuclear factor kappa B (NF-κB) seems to play a role in perpetuating the immune response to TBI. NF-κB is known to increase genetic transcription of pro-inflammatory mediators such as tumour necrosis factor-α (TNFα) and interleukin (IL)-6.[42] The role of IL-1 and IL-6 are poorly understood, but these inflammatory cytokines are present in high concentrations in the injured brain.[23,43] Generally, these cytokines promote oedema formation and immune activation, which may implicate these pro-inflammatory mediators in the early TBI swelling response.[43,44]
Complement activation after TBI results in a multifactorial process including increased neutrophil chemotaxis, increased cerebral oedema, cell lysis, cerebral ischaemia and blood-brain barrier breakdown.[45] The blood-brain barrier appears to open periodically within the first 24 hours of TBI, thereby permitting the passage of proteins and other substances that otherwise would be excluded from the brain.[46] This may result in alterations of brain osmolarity and oedema formation. The dynamic actions of the blood-brain barrier after TBI have been difficult to illustrate, although it is clear that increased permeability of this restrictive surface represents a window of opportunity for neuroprotective agents to be administered and have enhanced penetration and concentration in the brain.
The balance of the inflammatory and immune response is that these processes, under sufficient homeostatic control, promote healing and repair. Macrophage infiltration and microglia activation occur after TBI, primarily to consume and clear remnants of damaged or dead tissue. Blood-brain barrier disruption allows systemic immune cells to enter the CNS compartment, supporting the local inflammatory response. Other evidence suggests that immune activation is associated with anti-apoptotic activity and improved neurological recovery. Taken together, the varied actions of the inflammatory cells support the concept that the immune response is a double-edged sword.[41,42]
Cerebral oedema often leads to elevations in ICP. Different types of oedema have been described in TBI, although clinically vasogenic and cytotoxic oedema are most pertinent. Vasogenic oedema is seen in numerous types of neurological problems such as meningitis and brain tumour and is typified by protein extravasation (so-called ‘third-spacing’).[47] This oedema is usually related to acute inflammation and bradykinin release. Vasogenic oedema may represent some of the reason for blood-brain barrier disruption after TBI. Over-expression of aquaporin (AQP) channels, which permit the movement of water into the lateral ventricles, may exacerbate cerebral oedema as well.[48] Conversely, cytotoxic oedema is related to cellular swelling and is typical of the neurotoxic process that often follows severe TBI. Direct disruption of cellular membranes may cause cellular fluid disturbances and perturb normal cell function. Dysregulation of ion flux is also implicated in cytotoxic oedema. For instance, sodium and calcium are seen in high intracellular concentrations after TBI, whereas potassium is more common in the extracellular space (likely due to malfunction of Na+/H+ exchange pumps and other similar acid/base or electrical gradient transporters).[47]
Calcium is a significant factor in the progression of secondary injury and swelling after TBI. For neurons, calcium is a common element of various apoptosis pathways. In TBI, several reasons for increased intracellular calcium concentrations are present. The principle examples of such factors are excitatory neurotransmitters such as glutamate, which are in high concentration in the brain after TBI.[23] Glutamate acts on the NMDA, kainite and 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl) propanoic acid (AMPA) receptors, which cause calcium and sodium influx into the cell. Excessive NMDA agonism results in tremendously elevated intracellular calcium concentrations. High intracellular concentrations initiates various apoptotic pathways related to the cytochrome C/mitochondrial permeability transition pore and proteases such as calpains and caspases.[23,49,50]
Excessive calcium influx is initially buffered by the sarcoplasmic reticulum. As intracellular calcium homeostasis is lost, regulation by the sarcoplasmic reticulum is saturated. The excess calcium is taken up by the mitochondria, which acts as a ‘calcium sink’ under situations of stress.[51] Several stimuli, such as cellular redox status, pH and calcium concentration, may cause the mitochondrial permeability transition pore (MPTP) to form. This leads to a depolarization of the mitochondria and disrupts the mitochondrial electrical membrane potential.[52] The mitochondria swell with calcium uptake, leak due to intra-organelle oedema, and adenosine triphosphate (ATP) production becomes deficient. Ultimately, the minimal supply of ATP in a highly metabolically active cell leads to cellular energy failure. Furthermore, cytochrome C is released, which also promotes apoptosis.[52]
Elevated intracellular calcium also results in the activation of proteases such as calpains and caspases, which begin disassembling cellular structures such as the cytoskeleton and cytosolic proteins. As calcium concentrations increase inside the cell, calpains are unleashed, leading to proteolysis of spectrin and neurofilament proteins.[53] In addition to destabilizing cellular structure, calpain activation may also impair axonal transmission and neuronal plasticity. As such, cells that survive the secondary injury process may be dysfunctional long-term, which may translate to clinical residual deficits during TBI recovery.[53] Cytoskeletal proteins, DNA repair enzymes and a host of other intracellular proteins may be lysed by various caspases.[54] Caspases also interact with TNF and Fas ligand receptors in the cell membrane, which when stimulated, produce an irrevocable apoptosis response. Intracellular caspases (specifically caspase-3) also combine with mitochondria-derived cytochrome C to create an ‘apoptosome’, which indirectly promotes cell lysis and inhibits repair of DNA.
Poly (adenosine diphosphate [ADP[-ribose) polymerase (PARP) is a DNA repair enzyme that uses nicotinamide adenosine dinucleotide as a substrate to add ADP-ribose molecules to damaged DNA. This ubiquitous enzyme becomes overactive in TBI, resulting in high rates of ATP consumption. Ultimately, in severe TBI, the intracellular ATP supply is depleted and the cell is predisposed to energy failure.[55] PARP also appears to be integral to apoptosis via activation of calpains and movement of cytochrome C and apoptosis-inducing factor into the nucleus.
Endogenous mediators with potential benefit to the injured brain continue to be of interest as pharmacological targets for intervention. Insulin-like growth factor (IGF)-1 was investigated as a modulator of both central and systemic responses following TBI.[56] IGF-1 has neurotrophic effects on many CNS cell types and has recently shown promise in synaptic plasticity.[57–59] Through influences on neurotransmitters, CNS cellular growth and differentiation, metabolic and energy dynamics, and vascular reactivity, this agent has demonstrated a multitude of potential mechanisms for improved neurological outcome following TBI.[60,61] Fluid percussion models of TBI in rats have demonstrated that IGF-1 improves cognitive and motor outcomes, whereas a penetrating brain injury model also suggests benefit of IGF-1 supplementation (increasing brain-derived neurotrophic factor).[62,63] Clinical exposure to this investigational agent is limited in TBI patients but preliminary evidence demonstrated improved systemic metabolic responses and no safety concerns when IGF-1 was combined with aggressive nutrition support and growth hormone in this population.[56] Studies in human TBI were affected by the altered pharmacokinetic disposition of IGF-1 that limited both blood and brain concentrations of the exogenous investigational formulation.[64] Recent advances in drug formulations for IGF-1 may provide opportunity for renewed clinical trials of this agent in patients with TBI.[65]
It is evident that the interdigitation of these processes is quite complex. Several excellent reviews focusing on the secondary injury process and the various mediators involved are available for the reader to acquire more depth of information in this area.[16,21,66,67] What is more difficult for clinicians and scientists to discern is how much of this interwoven process must be disrupted (or at what critical juncture) in order to have an impact on maintaining homeostasis and the overall process of cell damage and death. This is made more difficult by challenges encountered in the pre-clinical and clinical models employed to evaluate agents for utility in neuroprotection.
3. Translating Neuroprotective Agents from the Bench to the Bedside
The pathophysiology of TBI is multi-faceted and rich with pharmacological targets. To date, most of the studies of putative neuroprotective agents have been focused on a single component of a complex cascade of injury. All of these agents showed potential in laboratory and/or animal models. However, failure has been consistent as the agents are transitioned from animals to humans with TBI. The disconnect between the bench and the bedside has been well described in a number of recent reviews.[68,69] There are potential problems with various aspects of the animal models utilized to evaluate TBI in addition to the clinical trial design approach and various other factors related to the variability of care and patient-specific response that may have impacted the success.
First, the injury models used to simulate TBI represent a relatively homogenous injury type. For instance, weight drop or controlled cortical impact involves the use of a defined impact on exposed brain to generate a contusion-type injury accompanied by subdural or epidural haematoma.[70] This model is useful because it is reproducible and predictable with regard to the extent and type of injury it inflicts. However, the heterogeneity of TBI in reality is difficult to replicate.[71] Patients often present with more than one abnormality or injury type due to neurotrauma. For instance, two patients may be admitted with severe TBI and an identical GCS. However, if one patient has DAI and scattered subarachnoid haemorrhage, and the other has a subdural hematoma with a contralateral contusion, the nature of those injuries are distinctly different. In many respects, their response to injury and subsequent treatment may be quite dissimilar. Yet, in large clinical trials, the severity of these injuries may be considered equivalent. The interplay among different injury types when simultaneously present is ill-defined and may be a factor in the difficulty in translating experimental findings to clinical trials.
Second, a robust description of the therapeutic window, the pharmacokinetics, and the pharmacodynamics for each agent is often not available. It is increasingly evident that there exists a window of opportunity for specific agents, depending on their onset of action, ability to access the central compartment and the mechanism of action. Timing is of the essence for most therapies in TBI. Administration of the agent when the brain penetration is suboptimal (i.e. during a time when the blood-brain barrier is likely closed) or after the process targeted by the agent has begun in earnest is likely to lead to failure.[72] Drug efflux pumps may also diminish the brain concentrations of many agents used for neuroprotection.[73,74] Optimization of retaining the pharmacological agent in the brain would be helpful in achieving satisfactory concentrations of the agent for prolonged durations. The increased use of the exception from informed consent (EFIC) process or developing agents with broad therapeutic windows may facilitate more timely administration of neuroprotective agents within a desirable timeframe.
Third, TBI is commonly associated with pharmacokinetic alterations in drug distribution, metabolism and elimination.[75] Consequently, the pharmacokinetics of neuroprotective agents may vary among TBI patients. Alterations in blood-brain barrier integrity, albumin and other serum protein concentrations, and the metabolic capacity of the cytochrome P450 (CYP) and other hepatic enzyme systems related to the systemic stress and immune response after TBI may change the concentration-time profile or tissue concentrations that might be typical of a non-TBI patient.[75,76] For instance, the pharmacokinetic profile of ciclosporin, a commonly used immunosuppressant in transplant and autoimmune disease, has been modelled in a number of different populations.[77–80] Ciclosporin pharmacokinetics in the normal individual are complex due to high protein binding, dependence upon CYP3A4 metabolism, and the fact that not only is it a substrate for efflux transporters, but it is also an inhibitor of these same transporters. Certainly, the pharmacokinetics of ciclosporin in TBI merit consideration. As demonstrated in phase II studies, the volume of distribution of ciclosporin may be slightly elevated (although quite variable) in TBI patients compared with normal individuals, possibly due to increased CNS penetration.[81,82] Ciclosporin is usually not detectable in the brain due to high plasma binding and efflux transporters in the brain. However, after TBI, ciclosporin is detectable in the brain, suggesting that the blood-brain barrier may have enhanced permeability post-injury or that high doses or continuous infusions of ciclosporin may yield a brain concentration sufficient to inhibit the efflux pumps. Clearance of ciclosporin is also elevated in the TBI patient, which is reflective of the hepatic enzyme induction commonly seen in the days and weeks after TBI.[76] Dramatic changes in serum or tissue drug exposure may impact the efficacy and safety of investigational agents for TBI and should be considered in the context of TBI during clinical trial evaluation.
Fourth, several clinical trials have included patients with a spectrum of TBI severity, ranging from mild to severe.[9,83–85] Certainly, the need for neuroprotection may be more desperate in severely injured patients, whereas patients with mild TBI may have less obvious benefits. Patients with severe TBI may be assessed by mortality rate and broader outcomes measures like the Glasgow Outcome Score (GOS) or the Disability Ratings Scale. In contrast, mild to moderate TBI patients have a relatively low mortality rate, making this a less desirable target for a clinical trial. The focus for outcomes in these patients needs to be on functional outcomes and subtle cognitive findings, for which there are numerous tests (although some of them are onerous and time consuming).[86,87] Fundamentally, a method of precise definition and classification of TBI that incorporates all of the factors related to the underlying pathology of the injury including patient-specific biomechanics and neurochemical factors has yet to be developed. Biomarkers of TBI may also play a role in screening or determining severity early after injury, although specific applicable examples are only now emerging.[88–91]
Finally, the variety of supportive care and treatment approaches within TBI may impact the outcome of previous studies. Disparities in fluid resuscitation, evaluation and treatment of elevated ICP, CPP targets, timing to nutrition provision and numerous other aspects of therapy may combine to sway the results of a trial in one direction or another. If the studies do not randomize within centre strata, or if the study is not large enough to account for heterogeneity, then bias could be introduced. In addition, centres with high enrolment rates or TBI volume may have different outcomes in large multicentre studies than lower volume centres or slow enrollers.[92] Areas with high concentrations of specific age groups or ethnicities may exhibit different outcomes or pharmacokinetic variability with the neuroprotective agent. Consensus on key points in treatment, adherence to study-endorsed treatment protocols and limiting the heterogeneity of injury type may all be methods to limit the variability that has plagued many of the TBI trials to date.
4. Pharmacotherapeutic Agents Used for Traumatic Brain Injury
Any investigational agent for neuroprotection faces an incredibly difficult challenge in becoming an effective clinical therapy with proven benefit in neurological outcome. The ideal therapy would have a significant clinical impact outside a narrow window of the acute injury to avoid the practical limitations of clinical trials. It would be administered systemically via infusion or by enteral administration, while achieving an elevated concentration at the site of injury, including penetration of the blood-brain barrier, and limiting toxicity in other areas of the body or activation/inhibition of other cellular receptors. It is possible that our current research strategy of a single targeted therapy may be inadequate. Any effective therapy would most likely need to be pluripotent to ameliorate the multiple pathways of ischaemia and cell death in TBI rather than affecting one target in the complicated cascade of events. The National Institutes of Health even issued a request for applications ♯RFA-HD-08-003, to promote pre-clinical research for multi-drug combinations in treating TBI.[93] Moving forward, one might consider if there are numerous agents that have been abandoned, which if combined with other agents with different mechanisms of action, might potentially yield a comprehensive therapeutic inhibition of calcium dysregulation, oxidative stress, apoptosis and cell death. Table II reviews published clinical trials of neuroprotective agents in TBI; table III describes clinical trials of neuroprotective agents currently ongoing; and table IV highlights other trials of neuroprotective agents in animal models.

Clinical trials for neuroprotection in traumatic brain injury (TBI)

On-going clinical trials for neuroprotection in traumatic brain injury (TBI)

Experimental agents with positive or in vitro evidence without published clinical evidence of efficacy
4.1 Calcium-Channel Antagonists
Numerous randomized controlled trials have evaluated nimodipine and nicardipine in TBI.[35] Antagonism of the calcium channel would theoretically abrogate the harmful effects of excitatory neurotransmitters and the initiation of the apoptosis cascade. Nimodipine has shown benefit in other acute neurological injuries such as aneurysmal subarachnoid haemorrhage due to a reduction in vasospasm and possible neuroprotective role.[199] However, calcium channel blockers are also active systemically and can cause a reduction in systemic blood pressure (and subsequently, CPP), which may complicate supportive care efforts in the acutely brain-injured patients. An extensive Cochrane meta-analysis demonstrated that the only subgroup with a significant reduction in death or severe disability were patients with traumatic subarachnoid haemorrhage (odds ratio [OR[ 0.67; 95% CI 0.46, 0.98).[35] Based on these data, these medications are not recommended for the general TBI population.
4.2 Corticosteroids
The role of corticosteroids has been extensively studied in TBI. Attenuation of the vasogenic oedema and ‘swelling’ after TBI generally seems like a prudent target. However, the risks attendant with high doses of corticosteroids (e.g. bleeding, hyperglycaemia) may outweigh the benefits.[200–203] In addition, the resulting increase in cerebral metabolic rate may exacerbate the metabolic derangements that are typical of the secondary injury response.[200,204] The Corticosteroid Randomization After Significant Head Injury (CRASH) study demonstrated an increase in mortality in TBI patients receiving high doses of methylprednisolone compared with placebo (particularly for moderate to severe injuries).[9] A later Cochrane meta-analysis including 12 203 randomized patients demonstrated that corticosteroids may be associated with an increased risk of gastrointestinal bleeding, although not statistically significant (OR 1.23; 95% CI 0.91, 1.67). However, death was significantly increased relative to placebo (OR 1.15; 95% CI 1.07, 1.24).[205] These data support one of the few level I recommendations in the TBI guidelines, suggesting that high doses of corticosteroids for the purposes of treating or preventing secondary injury after TBI should be avoided.[18]
4.3 Ciclosporin A
Since calcium dysregulation has been strongly linked with TBI, the regulation of intracellular calcium by mitochondria is a strong area of scientific research. Ciclosporin A, a well known immunosuppressant in solid organ transplant, has been demonstrated to inhibit cyclophilin A via calcineurin inhibition, which results in immunosuppression. Ciclosporin also inhibits mitochondrial permeability transition pore opening (cyclophilin D binding) and the combination of these actions likely prevents organelle swelling and cell death.[52] Ciclosporin has been investigated for use in TBI through several phase II clinical trials. Thus far, these trials have demonstrated that ciclosporin is safe in the TBI population and may suggest the possibility of good outcomes.
Hatton and colleagues completed a phase IIb single-centre study that compared placebo with ciclosporin in patients with severe TBI within 8 hours of injury.[95] This was primarily a dose-escalation study that described the pharmacokinetics of ciclosporin in the TBI population and provided information on safety.[82] Overall, 40 subjects were enrolled (32 ciclosporin and 8 placebo). No differences in safety measures such as renal dysfunction, infection or seizure were found. The mortality rate was similar between the two groups, although patients who received a higher dose (and continuous infusion) of ciclosporin appeared to have a greater probability of favourable outcome as measured by GOS-extended. This is notable from a pharmacokinetic standpoint, since it is likely that higher serum ciclosporin concentrations increased drug exposure in the brain. CNS concentrations of ciclosporin may be maximized in TBI due to inhibition of efflux transporters that would ordinarily eliminate ciclosporin from the CNS compartment.
Mazzeo and colleagues performed another phase II prospective trial to evaluate safety, haemodynamics and pharmacokinetics in a population with severe TBI.[206] They administered a continuous intravenous infusion of 5 mg/kg/day for 24 hours of ciclosporin or placebo in a 3 : 1 fashion to 50 adult TBI patients. There was no statistical difference in the lactate/pyruvate ratio in the brain. Mean arterial pressure (MAP), ICP and CPP were all significantly elevated in the ciclosporin group versus placebo, which the authors hypothesize was due to a ciclosporin hypertensive effect. Ciclosporin appears to have little effects on T-lymphocyte counts or incidence of infection in the acute phases of TBI.[81,95,207]
4.4 Deltibant
Marmarou et al. evaluated the bradykinin antagonist, deltibant, in a phase II, randomized controlled trial for severe TBI (GCS 3–8).[96] Subjects (n = 115) were randomized to either 3 μg/kg/min or placebo for 5 days and assessed by mortality and GOS at 3 and 6 months. Mortality was numerically lower at 6 months but not statistically significant (20% vs 28%, respectively; p = 0.31). Favourable outcomes were also more common at 3 months but not statistically different in the deltibant group (40.3% vs 30%). A post hoc analysis demonstrated a significant reduction in mean time with ICP >15 mmHg. The trial was stopped prematurely by the company based on emergent toxicity demonstrated in animal models, despite no demonstrated increased risk of adverse events in the study. There are no ongoing trials evaluating deltibant. Marmarou et al. also conducted a small phase I study with another bradykinin antagonist Anatibant (LF16-06087), with favourable safety and pharmacokinetic outcomes, but no other trials have been published with this agent.[94]
4.5 Modulation of Excitotoxicity and Glutamate
Dexanabinol is a synthetic cannabinoid receptor agonist with NMDA antagonist properties. Knoller et al. studied dexanabinol in a small, randomized, phase II safety trial where 67 patients with severe brain trauma were randomized to a single dose of 150 mg of dexanabinol or placebo, and monitored for primary endpoints of ICP and CPP.[208] Secondary endpoints included the disability rating scale and GOS. Mean GCS was 6 in both groups, although the groups were not entirely comparable (basal cistern compression was more common in the placebo group than the dexanabinol group). Drug-treated patients achieved significantly higher MAP, lower ICP and higher CPP than control patients. At 1 month the number of patients achieving good recovery was higher in the treatment arm (20% vs 2.7%), but did not remain statistically different at 6 months (47% vs 32.4%; p = 0.1). The results of this trial led to a phase III efficacy study where 861 patients were randomized to either dexanabinol or placebo, with the primary endpoint being GOS 6 months after injury, with early and late mortality, ICP and CPP as secondary endpoints.[102] Dexanabinol had no clinical or statistical effect on the 6-month extended GOS assessment, mortality, ventilator duration or ICP/CPP values.
Temkin et al.[84] investigated the use of magnesium sulphate for neuroprotection in TBI. Magnesium had been previously demonstrated to be deficient in TBI, and several animal models demonstrated benefit, likely due to the ability of magnesium to inhibit calcium influx at the NMDA receptor.[209–211] A loading dose of 0.30 mmol/kg followed by an initial infusion of 0.05 mmol/kg/h of magnesium sulphate or placebo was administered within 8 hours of moderate or severe head injury. The composite endpoint score included: survival, seizure occurrence and neurobehavioural functioning. Subjects were stratified by GCS and age. After interim safety data demonstrated increased risk of death and lower blood pressures, the target magnesium level was lowered from 1.25–2.5 to 1.0–1.85 mmol/L. The authors demonstrated that magnesium sulphate was not associated with any positive outcome, and may possibly increase mortality (28% vs 14%; p = 0.05) in the high target range group.
Morris et al. investigated the competitive NMDA antagonist selfotel (CGS 19755) for the treatment of severe TBI.[111] Two separate double-blind, randomized, phase III trials were conducted comparing 5 mg/kg of selfotel to placebo with a primary outcome of GOS at 1, 3 and 6 months post-injury. At 6 months, favourable outcomes were achieved in 185 (55%) of the 338 patients who had received selfotel and in 204 (58%) of the 352 patients in the placebo group (p > 0.25). Mortality rates were 23% in the selfotel and 21% in the placebo group (p > 0.25). The study was discontinued early due to futility, as well as because of concomitant stroke trials, which were indicating increased risk of mortality in the selfotel arms. The authors hypothesized that selfotel had no effect on outcome because it was unable to overcome the excessive glutamate levels found in severe TBI.
Based on in vitro and in vivo evidence demonstrating the benefit of glutamate antagonism in experimental TBI, numerous other clinical trials have been conducted with NMDA antagonists. Some agents were competitive NMDA inhibitors, while others were bound to the post-synaptic region of the glutamate receptor. Generally, these agents were safe, except traxoprodil, which did exhibit some QTc prolongation.[100,101,212,213] None of these therapies had statistically significant effects on GOS or mortality. Failures of therapies notwithstanding, glutamate continues to be widely implicated in the secondary injury process and the NMDA receptor remains a target for novel agents.
4.6 Progesterone
The serendipitous discovery that gender and menstrual cycle may have an effect on animal response to experimental TBI has ultimately led to the development of progesterone as an intravenous product (solubilized in egg phospholipid emulsion, much like propofol). Progesterone appears to have pluripotent activity in the injured brain by limiting cerebral oedema through reducing lipid peroxidation, aquaporin expression, pro-inflammatory cytokine release and complement activation.[214] Progesterone is one of the most promising agents currently being investigated and two previous phase II studies have already demonstrated safety and suggested efficacy in TBI.[85,99]
The Progesterone for the Treatment of Traumatic Brain Injury (ProTECT) study was a phase II, single-centre study where moderate to severe TBI patients were randomized to receive progesterone or lipid vehicle within 11 hours of injury.[85] The study was powered to detect predetermined safety measures such as hypotension, pneumonia and hepatotoxicity. Other functional measures such as duration of coma, duration of post-traumatic amnesia and mortality were also recorded. Overall, 100 subjects were enrolled. Among the safety measures, no differences between progesterone and placebo were noted. However, death within 30 days of injury was lower in the progesterone group (13% vs 30.4%; relative risk [RR[ 0.43; 95% CI 0.18, 0.99), primarily in the severe TBI subpopulation.
Another phase II, single-centre, study included only severe TBI patients who were randomized to receive progesterone or matching placebo within 8 hours of injury.[99] The power analysis for this study was not reported, but the primary endpoint was 3-month GOS. Overall, 159 subjects were enrolled in this Chinese trial. Patients who received progesterone had a higher incidence of favourable outcome, as measured by GOS compared with the placebo group at 3 months (47% progesterone vs 31% placebo; p = 0.034). Interestingly, the mortality rate was also different in the progesterone group in this study (18% progesterone vs 32% placebo; p = 0.039), despite the different time window, a more severely injured population and a different dosing regimen (1 mg/kg intramuscularly every 12 hours for 96 hours) when compared with ProTECT.
Currently, the ProTECT III study and the Study of the Neuroprotective Activity of Progesterone in Severe Traumatic Brain Injuries (SyNAPSe), two large, phase III trials, are underway to evaluate the efficacy of progesterone in moderate to severe TBI.[83,215] ProTECT III patients are moderate to severe TBI patients randomized to receive a loading dose of 0.714 mg/kg within 4 hours after injury, then a continuous intravenous infusion at 0.5 mg/kg/h for 71 hours, which is then tapered over an additional 24 hours for a total 96-hour infusion (or matching intralipid vehicle control). The SyNAPSe trial uses a similar loading and continuous infusion dosing, but differs slightly in that the time window for initial dosing is 8 hours and the duration of the infusion is 120 hours. The SyNAPSe trial is also only including patients with severe TBI.
4.7 Statins
HMG-CoA reductase inhibitors (‘statins’) are commonly used in patients with cardiovascular disease and include agents such as simvastatin, atorvastatin and rosuvastatin among others. More recent reports have also focused on the pleiotropic nature of these drugs.[216] Specifically, statins appear to attenuate inducible nitric oxide synthetase (iNOS) activity and stabilize endothelial nitric oxide synthetase (eNOS) activity, as well as exhibiting some immune neutralizing properties. Although a significant amount of in vitro and animal data exist, so far only rosuvastatin has been studied in a small (n = 22) phase II trial for moderate TBI.[110] Patients with moderate TBI (GCS of 9–13) were randomized to rosuvastatin 20 mg or placebo. The primary outcome was a change on the Galveston Orientation Amnesia Test (GOAT) up to 3 months after TBI. Disability rating score upon discharge and at 3 months was also assessed, along with a panel of immunomodulatory cytokines that might be affected by rosuvastatin (IL-6 and TNFα). The treatment group only included eight patients so it is difficult to draw definitive conclusions but rosuvastatin seemed to be associated with improved amnesia scores within the first 2–3 weeks, although scores were similar at 3 months follow-up. Further investigation is warranted before these medications can be applied to clinical practice.
4.8 Pegorgotein (PEG-SOD)
Neutralization of reactive oxygen species (ROS) was investigated as a therapy to prevent neurotoxicity after severe TBI in the 1996 study involving pegorgotein (also known as polyethylene glycol-conjugated superoxide dismutase or PEG-SOD).[97] This was a prospective, double-blind, placebo-controlled, multicentre study that compared two doses of pegorgotein with placebo. The study agents were given within 8 hours of injury and patients were followed for as long as 6 months after TBI. The primary outcome was good outcome as measured by GOS. A total of 463 patients were randomized to the three study arms, with baseline data essentially comparable among the different groups. Overall, there was no difference between either dose of pegorgotein and placebo on the 3- or 6-month GOS. However, when patients admitted with a GCS of 3 were excluded, more patients who received the lower dose of pegorgotein (10 000 units/kg) had a favourable outcome (good recovery or moderate disability) than did patients who received placebo (63% vs 51%, respectively; p = 0.06), although this post hoc analysis was not statistically significant. The study had an a priori power design to detect a 14% absolute improvement in good outcome on the GOS (whereas many other TBI clinical trials have been powered to detect a 10% difference). However, only a 9.4% improvement was demonstrated with pegorgotein, which may have affected statistical significance. Interestingly, to further support the putative benefit of ROS scavenging, the incidence of acute respiratory distress syndrome (ARDS) was significantly lower in patients receiving the lower pegorgotein dose compared with placebo (0% vs 4%; p = 0.015). The mortality rate was not significantly different among the three groups and ranged from 22% to 25%. The potential of pegorgotein was ultimately not pursued for TBI, despite the potentially encouraging results of this under-powered trial.
4.9 Tirilizad
Marshall and colleagues conducted a large, multicentre TBI study on the use of tirilizad, a 21-aminosteroid with antioxidative and lipid peroxidation inhibitor effects.[112] Tirilizad had been previously studied in spinal cord injury, with possible benefit. Primary outcome was GOS score at 6 months. Patients were stratified by GCS and randomized within 4 hours to 10 mg/kg every 6 hours for 5 days or placebo. Baseline hypotension and hypoxia was significantly higher in the tirilizad-treated group despite the large sample size (n = 1131), introducing possible bias. In the severe injury group at 6 months, there was no difference in favourable outcome (35% vs 38%) or mortality (29% or 28%) in the tirilizad or placebo groups, respectively. Although post hoc analysis demonstrated mortality benefit in patients with traumatic subarachnoid haemorrhage, there was no overall benefit of tirilizad demonstrated in the study.
4.10 Zinc Supplementation
Young and colleagues investigated supplementing severe closed-head injury patients (n = 68) with either standard 2.5 mg of zinc or 12.5 mg of zinc in their parenteral nutrition for 15 days.[217] Mean GCS scores in the zinc-supplemented group exceeded the mean GCS score of the standard group at day 28 (p = 0.03). The groups did not have statistically different serum zinc concentrations, weight, energy expenditure or total urinary nitrogen excretion. Mean prealbumin concentrations were significantly higher in the zinc-supplemented group (p = 0.003) at 3 weeks after injury. However, more patients in the standard group underwent craniotomies than the supplementation group possibly creating a bias towards worse GCS scores. It is currently unknown whether zinc supplementation improves outcome in TBI.
5. The Future of Treatment for Patients with Traumatic Brain Injury
Although the secondary neurological injury process after TBI is complex, the opportunities for pharmacological intervention are robust. The frustrations of limited progress over the last decade have actually provided improved understanding of the obstacles and limitations existing in early study designs and unrecognized drug delivery challenges. Creative intervention strategies based on sound pharmacokinetic and pharmacodynamic principles integrated into clinical trials along with possible combination strategies based on pharmacology are reasonable expectations for future advances in the treatment of TBI. Recognizing that numerous challenges may exist in translating the pre-clinical studies into clinical practice where TBI injury severity and clinical course is highly variable, the knowledge acquired to date will afford improved monitoring using alternative measures such as biomarkers to more closely define drug effect. A more detailed understanding of the permeability of the blood-brain barrier, effect of drug transporters (for both CNS penetration and efflux) and specific pharmacological target regulation over the injury time-course will further refine the optimal treatment approach. Innovative trial designs and statistical methods incorporating TBI-specific outcome assessment tools that target behavioural and cognitive changes, as well as further definition of surrogate markers of therapy to improve study population enrolment, will facilitate future success of clinical trials for TBI.[91,218–220]
References
Faul M, Xu L, Wald MM, et al. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths 2002–2006. Atlanta (GA): Centers for Disease Control and Prevention National Center for Injury Prevention and Control, 2010 [online]. Available from URL: http://www.cdc.gov/traumaticbraininjury/pdf/blue_book.pdf [Accessed 2011 Dec 28]
Thurman D, Guerrero J. Trends in hospitalization associated with traumatic brain injury. JAMA 1999 Sep 8; 282(10): 954–7
Thurman DJ, Alverson C, Dunn KA, et al. Traumatic brain injury in the United States: a public health perspective. J Head Trauma Rehabil 1999 Dec; 14(6): 602–15
Coronado VG, Xu L, Basavaraju SV, et al. Surveillance for traumatic brain injury-related deaths: United States, 1997–2007. MMWR Surveill Summ 2011 May 6; 60(5): 1–32
Roozenbeek B, Chiu YL, Lingsma H, et al. Predicting 14-day mortality after severe traumatic brain injury: application of the IMPACT models in the Brain Trauma Foundation TBI-trac® New York State database. J Neurotrauma 2012; 29(7): 1306–12
Clifton GL, Valadka A, Zygun D, et al. Very early hypothermia induction in patients with severe brain injury (the National Acute Brain Injury Study: Hypothermia II): a randomised trial. Lancet Neurol 2011 Feb; 10(2): 131–9
Patel HC, Bouamra O, Woodford M, et al. Trends in head injury outcome from 1989 to 2003 and the effect of neurosurgical care: an observational study. Lancet 2005 Oct 29–Nov 4; 366(9496): 1538–44
Marshall LF, Gautille T, Klauber MR, et al. The outcome of severe closed head injury. J Neurosurg 1991; 75(11): S28–36
Roberts I, Yates D, Sandercock P, et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRCCRASH trial): randomised placebo-controlled trial. Lancet 2004 Oct 9–15; 364(9442): 1321–8
Mena JH, Sanchez AI, Rubiano AM, et al. Effect of the modified Glasgow Coma Scale score criteria for mild traumatic brain injury on mortality prediction: comparing classic and modified Glasgow Coma Scale score model scores of 13. J Trauma 2011 Nov; 71(5): 1185–92; discussion 93
De Kruijk JR, Leffers P, Menheere PP, et al. Prediction of post-traumatic complaints after mild traumatic brain injury: early symptoms and biochemical markers. J Neurol Neurosurg Psychiatr 2002 Dec; 73(6): 727–32
Ropper AH, Gorson Clinical practice. Concussion. N Engl J Med 2007 Jan 11; 356(2): 166–72
Ryan LM, Warden DL. Post concussion syndrome. Int Rev Psychiatry 2003 Nov; 15(4): 310–6
Guidelines for the management of penetrating brain injury. J Trauma 2001; 51 (2): S1-86
Ling G, Bandak F, Armonda R, et al. Explosive blast neurotrauma. J Neurotrauma 2009 Jun; 26(6): 815–25
Sahuquillo J, Poca MA, Amoros S. Current aspects of pathophysiology and cell dysfunction after severe head injury. Curr Pharm Des 2001; 7(15): 1475–503
Andriessen TM, Jacobs Vos PE. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J Cell Mol Med 2010 Oct; 14(10): 2381–92
Brain Trauma Foundation. Management of severe traumatic brain injury. J Neurotrauma 2007; 24 Suppl. 1: S1–95
Rhoney DH, Parker Jr D. Considerations in fluids and electrolytes after traumatic brain injury. Nutr Clin Pract 2006; 21(5): 462–78
Clifton G, Miller E, Choi S, et al. Fluid thresholds and outcome from severe brain injury. Crit Care Med 2002; 30(4): 739–45
Hatton J. Pharmacological treatment of traumatic brain injury. CNS Drugs 2001; 15(7): 553–81
Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth 2007 Jul; 99(1): 4–9
Enriquez P, Bullock R. Molecular and cellular mechanisms in the pathophysiology of severe head injury. Curr Pharm Des 2004; 10(18): 2131–43
Czosnyka M, Smielewski P, Piechnik S, et al. Cerebral auto-regulation following head injury. J Neurosurg 2001 Nov; 95(5): 756–63
Manley G, Knudson MM, Morabito D, et al. Hypotension, hypoxia, and head injury: frequency, duration, and consequences. Arch Surg 2001 Oct; 136(10): 1118–23
DeWitt DS, Prough DS. Traumatic cerebral vascular injury: the effects of concussive brain injury on the cerebral vasculature. J Neurotrauma 2003 Sep; 20(9): 795–825
Steiner LA, Coles JP, Johnston AJ, et al. Assessment of cerebrovascular autoregulation in head-injured patients: a validation study. Stroke 2003 Oct; 34(10): 2404–9
Cherian L, Hlatky R, Robertson CS. Nitric oxide in traumatic brain injury. Brain Pathol 2004 Apr; 14(2): 195–201
Schroder ML, Muizelaar JP, Fatouros P, et al. Early cerebral blood volume after severe traumatic brain injury in patients with early cerebral ischemia. Acta Neurochir Suppl 1998; 71: 127–30
Schroder ML, Muizelaar JP, Kuta AJ, et al. Thresholds for cerebral ischemia after severe head injury: relationship with late CT findings and outcome. J Neurotrauma 1996 Jan; 13(1): 17–23
Obrist WD, Langfitt TW, Jaggi JL, et al. Cerebral blood flow and metabolism in comatose patients with acute head injury: relationship to intracranial hypertension. J Neu-rosurg 1984 Aug; 61(2): 241–53
Overgaard J, Tweed WA. Cerebral circulation after head injury, 1: cerebral blood flow and its regulation after closed head injury with emphasis on clinical correlations. J Neurosurg 1974 Nov; 41(5): 531–41
Oertel M, Boscardin WJ, Obrist WD, et al. Posttraumatic vasospasm: the epidemiology, severity, and time course of an underestimated phenomenon: a prospective study performed in 299 patients. J Neurosurg 2005 Nov; 103(5): 812–24
Martin NA, Patwardhan RV, Alexander MJ, et al. Characterization of cerebral hemodynamic phases following severe head trauma: hypoperfusion, hyperemia, and vasospasm. J Neurosurg 1997 Jul; 87(1): 9–19
Langham J, Goldfrad C, Teasdale G, et al. Calcium channel blockers for acute traumatic brain injury. Cochrane Database Syst Rev 2003; 4: CD000565
Hall ED, Andrus PK, Yonkers PA. Brain hydroxyl radical generation in acute experimental head injury. J Neu-rochem 1993 Feb; 60(2): 588–94
Shohami E, Beit-Yannai E, Horowitz M, et al. Oxidative stress in closed-head injury: brain antioxidant capacity as an indicator of functional outcome. J Cerebral Blood Flow Metab 1997 Oct; 17(10): 1007–19
Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma 2000 Oct; 17(10): 871–90
Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics 2010 Jan; 7(1): 51–61
Ray SK, Dixon CE, Banik NL. Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol Histo-pathol 2002 Oct; 17(4): 1137–52
Lenzlinger PM, Morganti-Kossmann MC, Laurer HL, et al. The duality of the inflammatory response to traumatic brain injury. Mol Neurobiol 2001 Aug-Dec; 24(1–3): 169–81
Yang L, Tao LY, Chen XP. Roles of NF-kappaB in central nervous system damage and repair. Neurosci Bull 2007 Sep; 23(5): 307–13
Ott L, McClain C, Gillespie M, et al. Cytokines and metabolic dysfunction after severe head injury. J Neurotrauma 1994; 11(5): 447–72
Royo NC, Wahl F, Stutzmann JM. Kinetics of polymorphonuclear neutrophil infiltration after a traumatic brain injury in rat. Neuroreport 1999 Apr 26; 10(6): 1363–7
Stahel PF, Morganti-Kossmann MC, Kossmann T. The role of the complement system in traumatic brain injury. Brain Res Brain Res Rev 1998 Aug; 27(3): 243–56
Baldwin SA, Fugaccia I, Brown DR, et al. Blood-brain barrier breach following cortical contusion in the rat. J Neurosurg 1996 Sep; 85(3): 476–81
Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus 2007; 22(5): 1–10
Venero JL, Machado A, Cano J. Importance of aquaporins in the physiopathology of brain edema. Curr Pharm Des 2004; 10(18): 2153–61
Young W. Role of calcium in central nervous system injuries. J Neurotrauma 1992 Mar; 9 Suppl. 1: S9–25
Kawamata T, Katayama Y, Hovda DA, et al. Lactate accumulation following concussive brain injury: the role of ionic fluxes induced by excitatory amino acids. Brain Res 1995 Mar 20; 674(2): 196–204
Mazzeo AT, Beat A, Singh A, et al. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp Neurol 2009 Aug; 218(2): 363–70
Sullivan PG, Rabchevsky AG, Waldmeier PC, et al. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res 2005; 79(1–2): 231–9
Kampfl A, Posmantur RM, Zhao X, et al. Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: implications for pathology and therapy — a review and update. J Neurotrauma 1997 Mar; 14(3): 121–34
Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol 2004 Apr; 14(2): 215–22
Sarnaik AA, Conley YP, Okonkwo DO, et al. Influence of PARP-1 polymorphisms in patients after traumatic brain injury. J Neurotrauma 2010 Mar; 27(3): 465–71
Hatton J, Kryscio R, Ryan M, et al. Systemic metabolic effects of combined insulin-like growth factor-I and growth hormone therapy in patients who have sustained acute traumatic brain injury. J Neurosurg 2006 Dec; 105(6): 843–52
Neff NT, Prevette D, Houenou LJ, et al. Insulin-like growth factors: putative muscle-derived trophic agents that promote motoneuron survival. J Neurobiol 1993 Dec; 24(12): 1578–88
Saenger S, Goeldner C, Frey JR, et al. PEGylation enhances the therapeutic potential for insulin-like growth factor I in central nervous system disorders. Growth Horm IGF Res 2011 Oct; 21(5): 292–303
Aberg ND, Brywe KG, Isgaard J. Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. Sci World J 2006; 6: 53–80
Taguchi A, White MF. Insulin-like signaling, nutrient homeostasis, and life span. Annu Rev Physiol 2008; 70: 191–212
Zeger M, Popken G, Zhang J, et al. Insulin-like growth factor type 1 receptor signaling in the cells of oligodendrocyte lineage is required for normal in vivo oligodendrocyte development and myelination. Glia 2007 Mar; 55(4): 400–11
Kazanis I, Giannakopoulou M, Philippidis H, et al. Alterations in IGF-I, BDNF and NT-3 levels following experimental brain trauma and the effect of IGF-I administration. Exp Neurol 2004 Apr; 186(2): 221–34
Saatman KE, Contreras PC, Smith DH, et al. Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp Neurol 1997; 147(2): 418–27
Rockich KT, Hatton JC, Kryscio RJ, et al. Effect of recombinant human growth hormone and insulin-like growth factor-1 administration on IGF-1 and IGF-binding protein-3 levels in brain injury. Pharmacotherapy 1999; 19(12): 1432–6
Metzger F, Sajid W, Saenger S, et al. Separation of fast from slow anabolism by site-specific PEGylation of insulinlike growth factor I (IGF-I). J Biol Chem 2011 Jun 3; 286(22): 19501–10
Xi G, Keep RF, Hoff JT. Pathophysiology of brain edema formation. Neurosurg Clin N Am 2002; 13(3): 371–83
Schouten JW. Neuroprotection in traumatic brain injury: a complex struggle against the biology of nature. Curr Opin Crit Care 2007; 13(2): 134–42
Bullock MR, Lyeth BG, Muizelaar JP. Current status of neuroprotection trials for traumatic brain injury: Lessons from animal models and clinical studies. Neurosurgery 1999; 45(2): 207–20
Tolias CM, Bullock MR. Critical appraisal of neuroprotection trials in head injury: what have we learned? Neu-roRx 2004; 1(1): 71–9
Dixon CE, Clifton GL, Lighthall JW, et al. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods 1991 Oct; 39(3): 253–62
Morales DM, Marklund N, Lebold D, et al. Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience 2005; 136(4): 971–89
Sullivan PG, Rabchevsky AG, Hicks RR, et al. Dose-response curve and optimal dosing regimen of cyclosporin A after traumatic brain injury in rats. Neuroscience 2000; 101(2): 289–95
Lee G, Dallas S, Hong M, et al. Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol Rev 2001; 53(4): 569–96
Loscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nature Rev Neurosci 2005; 6(8): 591–602
Boucher BA, Hanes SD. Pharmacokinetic alterations after severe head injury. Clin Pharmacokinet 1998; 35(3): 209–21
McKindley DS, Boucher BA, Hess MM, et al. Effect of acute phase response on phenytoin metabolism in neurotrauma patients. J Clin Pharmacol 1997; 37(2): 129–39
Baraldo M, Ferraccioli G, Pea F, et al. Cyclosporine A pharmacokinetics in rheumatoid arthritis patients after 6 months of methotrexate therapy. Pharmacol Res 1999; 40(6): 483–6
Kovarik JM, Koelle EU. Cyclosporin pharmacokinetics in the elderly. Drugs Aging 1999; 15(3): 197–205
Ptachcinski RJ, Venkataramanan R, Rosenthal JT, et al. Cyclosporine kinetics in renal transplantation. Clin Pharmacol Ther 1985; 38: 296–300
Yee GC, Salomon DR. Cyclosporine. In: Evans WE, Schentag JJ, Jusko WJ, editors. Applied pharmacokinetics: principles of therapeutic drug monitoring. 3rd ed. Baltimore (MD): Lippincott Williams & Wilkins; 1992; 28.1–28.40
Cook AM, Whitlow J, Hatton J, et al. Cyclosporin A neuroprotection: establishing dosing guidelines for safe and effective use. Expert Opin Drug Saf 2009; 8(4): 411–9
Empey PE, McNamara PJ, Young B, et al. Cyclosporin A disposition following acute traumatic brain injury. J Neurotrauma 2006; 23(1): 109–16
Wright DW. Progesterone for traumatic brain injury (Pro-TECT III), 2009 [online]. Available from URL: http://www.clinicaltrials.gov/ct2/show/NCT00822900?term=TRAUMATIC+BRAIN+INJURY&rank=39 [Accessed 2011 Dec 27]
Temkin NR, Anderson GD, Winn HR, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol 2007; 6(1): 29–38
Wright DW, Kellermann AL, Hertzberg VS, et al. Pro-TECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med 2007; 49(4): 391–402
Guilfoyle MR, Seeley HM, Corteen E, et al. Assessing quality of life after traumatic brain injury: examination of the short form 36 health survey. J Neurotrauma 2010 Dec; 27(12): 2173–81
von Steinbuchel N, Wilson L, Gibbons H, et al. Quality of Life after Brain Injury (QOLIBRI): scale validity and correlates of quality of life. J Neurotrauma 2010 Jul; 27(7): 1157–65
Pineda JA, Lewis SB, Valadka AB, et al. Clinical significance of alphaII-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury. J Neurotrauma 2007 Feb; 24(2): 354–66
Manley GT, Diaz-Arrastia R, Brophy M, et al. Common data elements for traumatic brain injury: recommendations from the biospecimens and biomarkers working group. Arch Phys Med Rehabil 2010 Nov; 91(11): 1667–72
Ottens AK, Bustamante L, Golden EC, et al. Neuropro-teomics: a biochemical means to discriminate the extent and modality of brain injury. J Neurotrauma 2010 Oct; 27(10): 1837–52
Kochanek PM, Berger RP, Bayir H, et al. Biomarkers of primary and evolving damage in traumatic and ischemic brain injury: diagnosis, prognosis, probing mechanisms, and therapeutic decision making. Curr Opin Crit Care 2008 Apr; 14(2): 135–41
Maas AI, Murray G, Henney III H, et al. Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol 2006; 5: 38–45
National Institutes of Health. Multi-drug combinations to promote neurological recovery in traumatic brain injury (R01), 2008 [online]. Available from URL: http://grants.nih.gov/grants/guide/rfa-files/RFA-HD-08-003.html [Accessed 2012 Jan 3]
Marmarou A, Guy M, Murphey L, et al. A single dose, three-arm, placebo-controlled, phase I study of the bradykinin B2 receptor antagonist Anatibant (LF160687Ms) in patients with severe traumatic brain injury. J Neurotrauma 2005 Dec; 22(12): 1444–55
Hatton J, Rosbolt B, Empey P, et al. Dosing and safety of cyclosporine in patients with severe brain injury. J Neurosurg 2008; 109: 699–707
Marmarou A, Nichols J, Burgess J, et al. Effects of the bradykinin antagonist Bradycor (deltibant, CP-1027) in severe traumatic brain injury: results of a multi-center, randomized, placebo-controlled trial. American Brain Injury Consortium Study Group. J Neurotrauma 1999 Jun; 16(6): 431–44
Young B, Runge JW, Waxman KS, et al. Effects of pe-gorgotein on neurologic outcome of patients with severe head injury: a multicenter, randomized controlled trial. JAMA 1996 Aug; 276(7): 538–43
Muizelaar JP, Marmarou A, Young HF, et al. Improving the outcome of severe head injury with the oxygen radical scavenger polyethylene glycol-conjugated superoxide dismutase: a phase II trial. J Neurosurg 1993; 78(3): 375–82
Xiao G, Wei J, Yan W, et al. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit Care 2008; 12(2): R61
Merchant RE, Bullock MR, Carmack CA, et al. A doubleblind, placebo-controlled study of the safety, tolerability and pharmacokinetics of CP-101, 606 in patients with a mild or moderate traumatic brain injury. Ann N Y Acad Sci 1999; 890: 42–50
Yurkewicz L, Weaver J, Bullock MR, et al. The effect of the selective NMDA receptor antagonist traxoprodil in the treatment of traumatic brain injury. J Neurotrauma 2005 Dec; 22(12): 1428–43
Maas AI, Murray G, Henney 3rd H, et al. Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol 2006 Jan; 5(1): 38–45
Natale JE, Guerguerian AM, Joseph JG, et al. Pilot study to determine the hemodynamic safety and feasibility of magnesium sulfate infusion in children with severe traumatic brain injury. Pediatr Crit Care Med 2007; 8(1): 1–9
Bailey I, Bell A, Gray J, et al. A trial of the effect of nimodipine on outcome after head injury. Acta Neurochir (Wien) 1991; 110(3–4): 97–105
Teasdale G, Bailey I, Bell A, et al. A randomized trial of nimodipine in severe head injury: HIT I. British/Finnish Co-operative Head Injury Trial Group. J Neurotruama 1992; 9 Suppl. (2): S545–50
Murray GD, Teasdale GM, Schmitz H. Nimodipine in traumatic subarachnoid haemorrhage: a re-analysis of the HIT I and HIT II trials. Acta Neurochir (Wien) 1996; 138(10): 1163–7
The European Study Group on Nimodipine in Severe Head Injury. A multicenter trial of the efficacy of nimodipine on outcome after severe head injury. The European Study Group on Nimodipine in Severe Head Injury. J Neurosurg 1994; 80(5): 797–804
Pillai SV, Kolluri VR, Mohanty A, et al. Evaluation of nimodipine in the treatment of severe diffuse head injury: a double-blind placebo-controlled trial. Neurol India 2003; 51(3): 361–3
Zhou XE, Wang XY, Xu RX, et al. Effects of nimodipine on the cerebrovascular hemodynamics in patients with severe head injuries. Di Yi Jun Yi Da Xue Xue Bao 2002; 22(6): 527–9
Tapia-Perez JH, Sanchez-Aguilar M, Torres-Corzo JG, et al. Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758). J Neurotrauma 2008; 25(8): 1011–7
Morris GF, Bullock R, Marshall SB, et al. Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J Neurosurg 1999; 91(5): 737–43
Marshall LF, Maas AI, Marshall SB, et al. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J Neurosurg 1998; 89(4): 519–25
LaPlaca MC, Zhang J, Raghupathi R, et al. Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma 2001; 18(4): 369–76
Hoane MR, Pierce JL, Kaufman NA, et al. Variation in chronic nicotinamide treatment after traumatic brain injury can alter components of functional recovery independent of histological damage. Oxid Med Cell Longev 2008; 1(1): 46–53
Goffus AM, Anderson GD, Hoane M. Sustained delivery of nicotinamide limits cortical injury and improves functional recovery following traumatic brain injury. Oxid Med Cell Longev 2010; 3(2): 145–52
Swan AA, Chandrashekar R, Beare J, et al. Preclinical efficacy testing in middle-aged rats: nicotinamide, a novel neuroprotectant, demonstrates diminished preclinical efficacy after controlled cortical impact. J Neurotrauma 2011; 28(3): 431–40
Hoane MR, Tan AA, Pierce JL, et al. Nicotinamide treatment reduces behavioral impairments and provides cortical protection after fluid percussion injury in the rat. J Neurotrauma 2006; 23(10): 1535–48
Holland MA, Tan AA, Smith DC, et al. Nicotinamide treatment provides acute neuroprotection and GFAP regulation following fluid percussion injury. J Neurotruama 2008; 25(2): 140–52
Hoane MR, Pierce JL, Holland MA, et al. Nicotinamide treatment induces behavioral recovery when administered up to 4 hours following cortical contusion injury in the rat. Neuroscience 2008; 154(3): 861–8
Su X, Wang H, Zhao J, et al. Beneficial effects of ethyl pyruvate through inhibiting high-mobility group box 1 expression and TLR4/NF-kappaB pathway after traumatic brain injury in the rat. Mediators Inflamm. Epub 2011 Jun 16
Abdel Baki SG, Schwab Haber M, et al. Minocycline synergizes with N-acetylcysteine and improves cognition and memory following traumatic brain injury in rats. PLoS ONE 2010; 5(8): e12490
Chen G, Shi J, Hu Z, et al. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm 2008; 2008: 716458
Khan M, Sakakima H, Dhammu TS, et al. S-Nitro-soglutathione reduces oxidative injury and promotes mechanisms of neurorepair following traumatic brain injury in rats. J Neuroinflammation 2011 July 6; 8: 78
Khan M, Im YB, Shunmugavel A, et al. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. J Neuroinflammation 2009 Nov 4; 6: 32
Qu Mahmood A, Ning R, et al. The treatment of traumatic brain injury with velcade. J Neurotrauma 2010; 27(9): 1625–34
Arun P, Ariyannur PS, Moffett JR, et al. Metabolic acetate therapy for the treatment of traumatic brain injury. J Neurotruama 2010; 27(1): 293–8
Appelberg KS, Hovda DA, Prins ML. The effects of a ketogenic diet on behavioral outcome after controlled cortical impact injury in the juvenile and adult rat. J Neurotrauma 2009; 26(4): 497–506
Pike BR, Hamm RJ. Post-injury administration of BIBN 99, a selective muscarinic M2 receptor antagonist, improves cognitive performance following traumatic brain injury in rats. Brain Res 1995; 686(1): 37–43
Durmaz R, Ertilav K, Akyüz F, et al. Lazaroid U-74389G attenuates edema in rat brain subjected to post-ischemic reperfusion injury. J Neurol Sci 2003 Nov 15; 215(1–2): 87–93
Durmaz R, Kanbak G, Akyüz F, et al. Lazaroid attenuates edema by stabilizing ATPase in the traumatized rat brain. Can J Neurol Sci 2003 May; 30(2): 143–9
Lu XC, Chen RW, Yao et al. NNZ-2566, a glypromate analog, improves functional recovery and attenuates apoptosis and inflammation in a rat model of penetrating ballistic-type brain injury. J Neurotrauma 2009; 26(1): 141–54
Wei HH, Lu XC, Shear DA, et al. NNZ-2566 treatment inhibits neuroinflammation and pro-inflammatory cytokine expression induced by experimental penetrating bal-listic-like brain injury in rats. J Neuroinflammation 2009 August 7; 6: 19
Berman RF, Verweij BH, Muizelaar JP. Neurobehavioral protection by the neuronal calcium channel blocker ziconotide in a model of traumatic diffuse brain injury in rats. J Neurosurg 2000; 93(5): 821–8
Verweij BH, Muizelaar JP, Vinas FC, et al. Improvement in mitochondrial dysfunction as a new surrogate efficiency measure for preclinical trials: dose-response and time-window profiles for administration of the calcium channel blocker ziconotide in experimental brain injury. J Neurosurg 2000; 93(5): 829–34
Faden AI, O’Leary DM, Fan L, et al. Selective blockade of the mGluR1 receptor reduces traumatic neuronal injury in vitro and improves outcome after brain trauma. Exp Neurol 2001; 167(2): 435–44
Movsesyan VA, O’Leary DM, Fan L, et al. mGluR5 antagonists 2-methyl-6-(phenylethynyl)-pyridine and (E)-2-methyl-6-(2-phenylethenyl)-pyridine reduce traumatic neuronal injury in vitro and in vivo by antagonizing N-methyl-D-aspartate receptors. J Pharmacol Exp Ther 2001; 296(1): 41–7
Barbre AB, Hoane MR. Magnesium and riboflavin combination therapy following cortical contusion injury in the rat. Brain Res Bull 2006; 69(6): 639–46
Hoane MR, Wolyniak JG, Akstulewicz SL. Administration of riboflavin improves behavioral outcome and reduces edema formation and glial fibrillary acidic protein expression after traumatic brain injury. J Neurotrauma 2005; 22(10): 1112–22
Belayev L, Becker DA, Alonso OF, et al. Stilbazulenyl nitrone, a novel azulenyl nitrone antioxidant: improved neurological deficit and reduced contusion size after traumatic brain injury in rats. J Neurosurg 2002; 96(6): 1077–83
Guluma KZ, Saatman KE, Brown A, et al. Sequential pharmacotherapy with magnesium chloride and basic fibroblast growth factor after fluid percussion brain injury results in less neuromotor efficacy than that achieved with magnesium alone. J Neurotrauma 1999; 16(4): 311–21
Dietrich WD, Alonso OF, Busto R, et al. Posttreatment with intravenous basic fibroblast growth factor reduces histo-pathological damage following fluid-percussion brain injury in rats. J Neurotrauma 1996; 13(6): 309–16
McDermott KL, Raghupathi R, Fernandez SC, et al. Delayed administration of basic fibroblast growth factor (bFGF) attenuates cognitive dysfunction following parasagittal fluid percussion brain injury in the rat. J Neurotrauma 1997; 14(4): 191–200
Sun D, Bullock MR, McGinn MJ, et al. Basic fibroblast growth factor-enhanced neurogenesis contributes to cognitive recovery in rats following traumatic brain injury. Exp Neurol 2009; 216(1): 56–65
Cernak I, O’Connor C, Vink R. Inhibition of cyclooxy-genase 2 by nimesulide improves cognitive outcome more than motor outcome following diffuse traumatic brain injury in rats. Exp Brain Res 2002; 147(2): 193–9
Wahl F, Grosjean-Piot O, Bareyre F, et al. Enoxaparin reduces brain edema, cerebral lesions, and improves motor and cognitive impairments induced by a traumatic brain injury in rats. J Neurotrauma 2000 Nov; 17(11): 1055–65
Stutzmann JM, Mary V, Wahl F, et al. Neuroprotective profile of enoxaparin, a low molecular weight heparin, in in vivo models of cerebral ischemia or traumatic brain injury in rats: a review. CNS Drug Rev 2002 Spring; 8(1): 1–30
Bye N, Habgood MD, Callaway JK, et al. Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp Neurol 2007 Mar; 204(1): 220–33
Crack PJ, Gould J, Bye N, et al. The genomic profile of the cerebral cortex after closed head injury in mice: effects of minocycline. J Neural Transm 2009 Jan; 116(1): 1–12
Homsi S, Federico F, Croci N, et al. Minocycline effects on cerebral edema: relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res 2009 Sep 29; 1291: 122–32
Homsi S, Piaggio T, Croci N, et al. Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J Neurotrauma 2010 May; 27(5): 911–21
Stover JF, Beyer TF, Unterberg AW. Riluzole reduces brain swelling and contusion volume in rats following controlled cortical impact injury. J Neurotrauma 2000; 17(12): 1171–8
Zhang C, Raghupathi R, Saatman KE, et al. Riluzole attenuates cortical lesion size, but not hippocampal neuronal loss, following traumatic brain injury in the rat. J Neurosci Res 1998; 52(3): 342–9
Zhang Y, Xiong Y, Mahmood A, et al. Therapeutic effects of erythropoietin on histological and functional outcomes following traumatic brain injury in rats are independent of hematocrit. Brain Res 2009; 1294: 153–64
Zhang Y, Xiong Y, Mahmood A, et al. Sprouting of corticospinal tract axons from the contralateral hemisphere into the denervated side of the spinal cord is associated with functional recovery in adult rat after traumatic brain injury and erythropoietin treatment. Brain Res 2010; 1353: 249–57
Liao ZB, Zhi XG, Shi QH, et al. Recombinant human erythropoietin administration protects cortical neurons from traumatic brain injury in rats. Eur J Neurol 2008; 15(2): 140–9
Zhao J, Li G, Zhang Y, et al. The potential role of JAK2/STAT3 pathway on the anti-apoptotic effect of recombinant human erythropoietin (rhEPO) after experimental traumatic brain injury of rats. Cytokine 2011; 56(2): 343–50
Hartley CE, Varma M, Fischer JP, et al. Neuroprotective effects of erythropoietin on acute metabolic and pathological changes in experimentally induced neurotrauma. J Neurosurg 2008; 109(4): 708–14
Grasso G, Sfacteria A, Meli F, et al. Neuroprotection by erythropoietin administration after experimental traumatic brain injury. Brain Res 2007; 1182: 99–105
Chen G, Shi JX, Hang CH, et al. Inhibitory effect on cerebral inflammatory agents that accompany traumatic brain injury in a rat model: a potential neuroprotective mechanism of recombinant human erythropoietin (rhEPO). Neurosci Lett 2007; 425(3): 177–82
Liao ZB, Jiang GY, Tang ZH, et al. Erythropoietin can promote survival of cerebral cells by downregulating Bax gene after traumatic brain injury in rats. Neurol India 2009; 57(6): 722–8
Mammis A, McIntosh TK, Maniker AH. Erythropoietin as a neuroprotective agent in traumatic brain injury. Surg Neurol 2009; 71(5): 527–31
Xiong Y, Mahmood A, Zhang Y, et al. Effects of posttraumatic carbamylated erythropoietin therapy on reducing lesion volume and hippocampal cell loss, enhancing angiogenesis and neurogenesis, and improving functional outcome in rats following traumatic brain injury. J Neurosurg 2011; 114(2): 549–59
Xiong Y, Mahmood A, Meng Y, et al. Delayed administration of erythropoietin reducing hippocampal cell loss, enhancing angiogenesis and neurogenesis, and improving functional outcome following traumatic brain injury in rats: comparison of treatment with single and triple dose. J Neurosurg 2010; 113(3): 598–608
Brines ML, Ghezzi P, Keenan S, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 2000; 97(19): 10526–31
Kuypers NJ, Hoane MR. Pyridoxine administration improves behavioral and anatomical outcome after unilateral contusion injury in the rat. J Neurotrauma 2010; 27(7): 1275–82
Kleindienst A, Harvey HB, Rice AC, et al. Intraventricular infusion of the neurotrophic protein S100B improves cognitive recovery after fluid percussion injury in the rat. J Neurotrauma 2004; 21(5): 541–7
Kleindienst A, McGinn MJ, Harvey HB, et al. Enhanced hippocampal neurogenesis by intraventricular S100B infusion is associated with improved cognitive recovery after traumatic brain injury. J Neurotruama 2005; 22(6): 645–55
Kline AE, Massucci JL, Marion DW, et al. Attenuation of working memory and spatial acquisition deficits after a delayed and chronic bromocriptine treatment regimen in rats subjected to traumatic brain injury by controlled cortical impact. J Neurotrauma 2002; 19(4): 415–25
Mauler F, Hinz V, Augstein KH, et al. Neuroprotective and brain edema-reducing efficacy of the novel cannabinoid receptor agonist BAY 38-7271. Brain Res 2003; 989(1): 99–111
Mauler F, Horváth E, De Vry J, et al. BAY 38-7271: a novel highly selective and highly potent cannabinoid receptor agonist for the treatment of traumatic brain injury. CNS Drug Rev 2003; 9(4): 343–58
Mesenge C, Verrecchia C, Allix M, et al. Reduction of the neurological deficit in mice with traumatic brain injury by nitric oxide synthase inhibitors. J Neurotrauma 1996; 13(1): 11–6
Hogg S, Sanger DJ, Moser PC. Mild traumatic lesion of the right parietal cortex in the rat: characterisation of a conditioned freezing deficit and its reversal by dizocilpine. Behav Brain Res 1998; 93(1–2): 157–65
Bernert H, Turski L. Traumatic brain damage prevented by the non-N-methyl-D-aspartate antagonist 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f] quinoxaline. Proc Natl Acad Sci USA 1996; 93(11): 5235–40
Ikonomidou C, Turski L. Prevention of trauma-induced neurodegeneration in infant and adult rat brain: glutamate antagonists. Metab Brain Dis 1996; 11(2): 125–41
Pohl D, Bittigau P, Ishimaru MJ, et al. N-Methyl-D-aspartate antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc Natl Acad Sci USA 1999; 96(5): 2508–13
McIntosh TK, Vink R, Soares H, et al. Effect of noncompetitive blockade of N-methyl-D-aspartate receptors on the neurochemical sequelae of experimental brain injury. J Neurochem 1999; 55(4): 1170–9
Okiyama K, Smith DH, White WF, et al. Effects of the NMDA antagonist CP-98,113 on regional cerebral edema and cardiovascular, cognitive, and neurobehavioral function following experimental brain injury in the rat. Brain Res 1998; 792(2): 291–8
Kroppenstedt SN, Schneider GH, Thomale UW, et al. Protective effects of aptiganel HCl (Cerestat) following controlled cortical impact injury in the rat. J Neurotrauma 1998; 15(3): 191–7
Toulmond S, Serrano A, Benavides J, et al. Prevention by eliprodil (SL 82.0715) of traumatic brain damage in the rat: existence of a large (18 h) therapeutic window. Brain Res 1993; 620(1): 32–41
Hogg S, Perron C, Barnéoud P, et al. Neuroprotective effect of eliprodil: attenuation of a conditioned freezing deficit induced by traumatic injury of the right parietal cortex in the rat. J Neurotrauma 1998; 15(7): 545–53
Mueller AL, Artman LD, Balandrin MF, et al. NPS 1506, a moderate affinity uncompetitive NMDA receptor antagonist: preclinical summary and clinical experience. Amino Acids 2000; 19(1): 177–9
Dohi K, Satoh K, Mihara Y, et al. Alkoxyl radical-scavengin activity of edaravone in patients with traumatic brain injury. J Neurotrauma 2006; 23(11): 1591–9
Itoh T, Satou T, Nishida S, et al. Edaravone protects against apoptotic neuronal cell death and improves cerebral function after traumatic brain injury in rats. Neurochem Res 2010; 35(2): 348–55
Wang GH, Jiang ZL, Li YC, et al. Free-radical scavenger edaravone treatment confers neuroprotection against traumatic brain injury in rats. J Neurotrauma 2011; 28(10): 2123–34
Belayev L, Alonso OF, Liu Y, et al. Talampanel, a novel noncompetitive AMPA antagonist, is neuroprotective after traumatic brain injury in rats. J Neurotrauma 2001; 18(10): 1031–8
Kline AE, Yu J, Horváth E, et al. The selective 5-HT(1A) receptor agonist repinotan HCl attenuates histopathology and spatial learning deficits following traumatic brain injury in rats. Neuroscience 2001; 106(3): 547–55
Mauler F, Horváth E. Neuroprotective efficacy of repinotan HCl, a 5-HT1A receptor agonist, in animal models of stroke and traumatic brain injury. J Cereb Blood Flow Metab 2005; 25(4): 451–9
Cheng JP, Aslam HA, Hoffman AN, et al. The neurobe-havioral benefit conferred by a single systemic administration of 8-OH-DPAT after brain trauma is confined to a narrow therapeutic window. Neurosci Lett 2007; 416(2): 165–8
Cheng JP, Hoffman AN, Zafonte RD, et al. A delayed and chronic treatment regimen with the 5-HT1A receptor agonist 8-OH-DPAT after cortical impact injury facilitates motor recovery and acquisition of spatial learning. Behav Brain Res 2008; 194(1): 79–85
Kline AE, Wagner AK, Westergom BP, et al. Acute treatment with the 5-HT(1A) receptor agonist 8-OH-DPAT and chronic environmental enrichment confer neurobe-havioral benefit after experimental brain trauma. Behav Brain Res 2007; 177(2): 186–94
Kline AE, Massucci JL, Dixon CE, et al. The therapeutic efficacy conferred by the 5-HT(1A) receptor agonist 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) after experimental traumatic brain injury is not mediated by concomitant hypothermia. J Neurotrauma 2004; 21(2): 175–85
Kline AE, McAloon RL, Henderson KA, et al. Evaluation of a combined therapeutic regimen of 8-OH-DPAT and environmental enrichment after experimental traumatic brain injury. J Neurotruama 2010; 27(11): 2021–32
Kline AE, Yu J, Massucci JL, et al. Protective effects of the 5-HT1A receptor agonist 8-hydroxy-2-(di-n-propylamino) tetralin against traumatic brain injury-induced cognitive deficits and neuropathology in adult male rats. Neurosci Lett 2002; 333(3): 179–82
Ma H, Yu B, Kong L, et al. Transplantation of neural stem cells enhances expression of synaptic protein and promotes functional recovery in a rat model of traumatic brain injury. Mol Med Rep 2011; 4(5): 849–56
Wallenquist U, Brännvall K, Clausen F, et al. Grafted neural progenitors migrate and form neurons after experimental traumatic brain injury. Restor Neurol Neurosci 2009; 2009(27): 323–34
Harting MT, Sloan LE, Jimenez F, et al. Subacute neural stem cell therapy for traumatic brain injury. J Surg Res 2009; 153(2): 188–94
Shohami E, Bass R, Wallach D, et al. Inhibition of tumor necrosis factor alpha (TNFalpha) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab 1996; 16(3): 378–84
Chio CC, Lin JW, Chang MW, et al. Therapeutic evaluation of etanercept in a model of traumatic brain injury. J Neurochem 2010; 115(4): 921–9
Treggiari-Venzi MM, Suter PM, Romand JA. Review of medical prevention of vasospasm after aneurysmal subarachnoid hemorrhage: a problem of neurointensive care. Neurosurgery 2001 Feb; 48(2): 249–61; discussion 61–2
Kinoshita K, Kraydieh S, Alonso O, et al. Effect of posttraumatic hyperglycemia on contusion volume and neutrophil accumulation after moderate fluid-percussion brain injury in rats. J Neurotrauma 2002; 19(6): 681–92
Liu-DeRyke X, Collingridge DS, Orme J, et al. Clinical impact of early hyperglycemia during acute phase of traumatic brain injury. Neurocrit Care 2009; 11(2): 151–7
Rovlias A, Kotsou S. The Influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery 2000; 46(2): 342–3
Salim A, Hadjizacharia P, Dubose J, et al. Persistent hyperglycemia in severe traumatic brain injury: an independent predictor of outcome. Am Surg 2009; 75(1): 25–9
Diaz-Parejo P, Stahl N, Xu W, et al. Cerebral energy metabolism during transient hyperglycemia in patients with severe brain trauma. Intensive Care Med 2003; 29(4): 544–50
Alderson P, Roberts I. Corticosteroids for acute traumatic brain injury. Cochrane Database Syst Rev 2005 Jan 25; 1: CD000196
Mazzeo AT, Alves OL, Gilman CB, et al. Brain metabolic and hemodynamic effects of cyclosporin A after human severe traumatic brain injury: a microdialysis study. Acta Neurochir (Wien) 2008; 150(10): 1019–31
Mazzeo AT, Brophy GM, Gilman CB, et al. Safety and tolerability of cyclosporin a in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma 2009 Dec; 26(12): 2195–206
Knoller N, Levi L, Shoshan I, et al. Dexanabinol (HU-211) in the treatment of severe closed head injury: a randomized, placebo-controlled, phase II clinical trial. Crit Care Med 2002; 30(3): 548–54
McIntosh TK, Faden AI, Yamakami I, et al. Magnesium deficiency exacerbates and pretreatment improves outcome following traumatic brain injury in rats: 31P magnetic resonance spectroscopy and behavioral studies. J Neurotrauma 1988; 5(1): 17–31
Lee JS, Han YM, Yoo DS, et al. A molecular basis for the efficacy of magnesium treatment following traumatic brain injury in rats. J Neurotrauma 2004; 21(5): 549–61
Hoane MR. Treatment with magnesium improves reference memory but not working memory while reducing GFAP expression following traumatic brain injury. Restor Neurol Neurosci 2005; 23(2): 67–77
Wagstaff A, Teasdale GM, Clifton G, et al. The cerebral hemodynamic and metabolic effects of the noncompetitive NMD A antagonist CNS 1102 in humans with severe head injury. Ann N Y Acad Sci 1995 Sep 15; 15: 332–3
Bullock MR, Merchant RE, Carmack CA, et al. An open-label study of CP-101, 606 in subjects with a severe traumatic head injury or spontaneous intracerebral hemorrhage. Ann N Y Acad Sci 1999: 51–8
Stein DG. Progesterone exerts neuroprotective effects after brain injury. Brain Res Rev 2008; 57(2): 386–97
BHR Pharma LLC. Efficacy and safety study of intravenous progesterone in patients with severe traumatic brain injury (SyNAPSe), 2011 [online]. Available from URL: http://www.clinicaltrials.gov/ct2/show/NCT01143064?term=progesterone&recr=Open&cond=brain+injury&rank=2 [Accessed 2011 Dec 27]
Weant KA, Cook AM. Potential roles for statins in critically ill patients. Pharmacotherapy 2007; 27(9): 1279–96
Young B, Ott L, Kasarskis E, et al. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J Neurotrauma 1996; 13(1): 25–34
Menon DK. Unique challenges in clinical trials in traumatic brain injury. Crit Care Med 2009 Jan; 37(1 Suppl.): S129–35
Narayan RK, Michel ME, Ansell B, et al. Clinical trials in head injury. J Neurotrauma 2002 May; 19(5): 503–57
McHugh GS, Butcher I, Steyerberg EW, et al. Statistical approaches to the univariate prognostic analysis of the IMPACT database on traumatic brain injury. J Neurotrauma 2007 Feb; 24(2): 251–8
Acknowledgements
The authors have no potential conflicts of interest or sources of funding related to the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
McConeghy, K.W., Hatton, J., Hughes, L. et al. A Review of Neuroprotection Pharmacology and Therapies in Patients with Acute Traumatic Brain Injury. CNS Drugs 26, 613–636 (2012). https://doi.org/10.2165/11634020-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11634020-000000000-00000