




Abstract
17β-Hydroxysteroid dehydrogenase type 10 (HSD10) is multifunctional protein coded by the X-chromosomal HSD17B10 gene. Mutations in this gene cause HSD10 disease characterized by progressive neurological abnormalities and cardiomyopathy. Disease progression and severity of symptoms is unrelated to the protein's dehydrogenase activity. Recently, it was shown that HSD10 is an essential component of mitochondrial Ribonuclease P (RNase P), an enzyme required for mitochondrial tRNA processing, but little is known about the role of HSD10 in RNase P function. RNase P consists of three different proteins MRPP1, MRPP2 (HSD10) and MRPP3, each of which is essential for RNase P function. Here, we show that HSD10 protein levels are significantly reduced in fibroblasts from patients carrying the HSD17B10 mutation p.R130C. A reduction in HSD10 levels was accompanied by a reduction in MRPP1 protein but not MRPP3 protein. In HSD10 knock-down cells, MRPP1 protein content was also reduced, indicating that HSD10 is important for the maintenance of normal MRPP1 protein levels. Ectopic expression of HSD10 partially restored RNA processing in HSD10 knock-down cells and fibroblasts, and also expression of MRPP1 protein was restored to values comparable to controls. In both, patient fibroblasts and HSD10 knock-down cells, there was evidence of impaired processing of precursor tRNA transcripts of the mitochondrial heavy strand but not the light strand compared with controls. Our findings indicate that HSD10 is important for the maintenance of the MRPP1-HSD10 subcomplex of RNase P and that loss of HSD10 causes impaired mitochondrial precursor transcript processing which may explain mitochondrial dysfunction observed in HSD10 disease.
INTRODUCTION
HSD10 disease (1), originally described as 2-methylhydroxy-butyryl-CoA dehydrogenase (MHBD) deficiency, is a rare neurodegenerative childhood disorder caused by inherited mutations of the HSD17B10 gene located on the X-chromosome (2–4). The encoded protein 17β-hydroxysteroid dehydrogenase type 10 (HSD10) is a mitochondrial NAD+-dependent member of the short-chain dehydrogenase/reductase superfamily which is involved in isoleucine metabolism and other cellular functions (5,6). Clinical features of HSD10 disease (reviewed in 1) form a spectrum including a severe ‘neonatal form’ (associated with mutations p.N247S, p.R226Q and p.D86G) with little neurological development and progressive cardiomyopathy; an ‘infantile form’ (associated with the prevalent mutation p.R130C and mutations p.L122V and p.P210S) with a prominent neurodegenerative disease course after relatively normal development in the first months of life and an ‘atypical form’ (associated with mutation p.Q165H) with normal intellectual and neurological development. Late onset in school age has been found in one patient (mutation p.E249Q) (7) and classified as the juvenile form (see also Supplementary Material, Table S1). Furthermore, an overlap of the infantile form has been described with both, the neonatal presentation pattern, described as an early infantile form (4) as well with the less severe infantile forms denoted as the late infantile presentation pattern (1). In contrast to other metabolic disorders, clinical symptoms in HSD10 disease are not correlated with the dehydrogenase function of this protein. In a series of in vitro studies and animal model experiments analyzing HSD17B10 mutations, we previously showed that mutant HSD10 protein without enzyme function can prevent apoptosis in HSD10 knock-down cells and in HSD10-depleted Xenopus embryos (8). This study indicated that an enzyme-independent function of HSD10 is at least in part responsible for the clinical features in HSD10 disease. HSD10 thus is an example of ‘moonlightning’ proteins which assume multiple independent functions via different mechanisms/pathways (9). However, the exact pathogenesis of HSD10 disease is so far poorly understood.
Holzmann et al. (10) identified HSD10 as a component of mitochondrial Ribonuclease P (RNase P), an RNA-free endonuclease complex that consists of three nuclear-coded proteins denoted MRPP1 (gene RG9MTD1), MRPP2 (=HSD10; gene HSD17B10) and MRPP3 (gene KIAA0391). Transcription of the human mitochondrial genome produces long polycistronic precursor RNA molecules which undergo a series of endonucleolytic cleavages to produce the individual mRNAs, rRNAs and tRNAs. During mitochondrial RNA processing, RNase P executes the cleavage at the 5′-end of precursor tRNAs required for correct mitochondrial protein translation (11). Using purified components, Holzmann et al. (10) reconstituted RNase P activity in vitro and demonstrated that HSD10 is indispensable for a functional RNase P. More recently, MRPP1 has been shown to form a subcomplex with HSD10 (12), in which MRPP1 recognizes mitochondrial tRNA and methylates purine bases at m1G9 or m1A9 via its SPOUT-type methyltransferase domain. The methyltransferase activity is not required for, or dependent on, the tRNA endonuclease activity of RNase P (12). Like RNase P, the MRPP1 methyltransferase activity requires HSD10 as an essential component, but not via its dehydrogenase and NAD(H)-binding functions. Furthermore, HSD10 is not involved in tRNA recognition in the RNase P complex (10,12). MRPP3 is the only subunit of the RNase P which possesses structural features of a metallonuclease that could be responsible for the endonuclease activity in RNase P (10). So far the exact role of HSD10 in the mitochondrial RNase P complex or the methyltransferase subcomplex remains unknown.
Considering that the clinical features and laboratory findings in HSD10 disease resemble a mitochondrial disorder, we hypothesized that they may be at least partly linked to impaired mitochondrial transcript processing caused by deficient RNase P function. Here, we report that HSD10 is required for stable protein expression of MRPP1 and that HSD10 mutations observed in patients causes reduced availability of RNase P components and deficient mitochondrial RNA processing which explain impaired mitochondrial function in HSD10 disease.
RESULTS
Normal MRPP1 protein levels depend on the presence of HSD10
Knock-down of HSD10 in human cells inhibited RNase P activity in vitro (10). Therefore, HSD10 knock-downs in different cell lines were performed to establish a reference for the analysis of mitochondrial tRNA processing defects and to correlate the observations with the expected impact of mutated HSD10 on RNase P function. After testing different siRNAs for their ability to reduce HSD10 in HeLa (Fig. 1A), HEK-293 (Supplementary Material, Fig. S1A) and HRT18 cells (data not shown), we selected the siRNA which caused an HSD10 knock-down of 82 ± 7% (mean ± SD) (siRNA 2) for subsequent experiments. HSD10 knock-down resulted in a strong reduction in cell proliferation and cell viability (not shown) indicating a vital role of HSD10 in cell physiology. HSD10 knock-down did not alter the expression of MRPP1 and MRPP3 mRNA (Supplementary Material, Fig. S1A) but was consistently associated with a markedly reduced amount of MRPP1 protein, whereas MRPP3 protein levels were not affected (Fig. 1A). Additionally, quantitative densiometric analysis was performed (Fig. 1A). For siRNA 2, the expression of HSD10 was reduced to 32.8 ± 17% (mean ± SD) and the amount of MRPP1 to 41.1 ± 16.14% (mean ± SD) compared with control. siRNA 3 showed a minor decrease in the HSD10 protein of 70.25 ± 12.8% (mean ± SD), as well as for MRPP1 (78.3 ± 23.6%). MRPP3 protein levels were not affected (97.9 ± 1.09% for siRNA 2). This observation indicates that HSD10 is required for the steady-state levels of MRPP1 but not MRPP3.
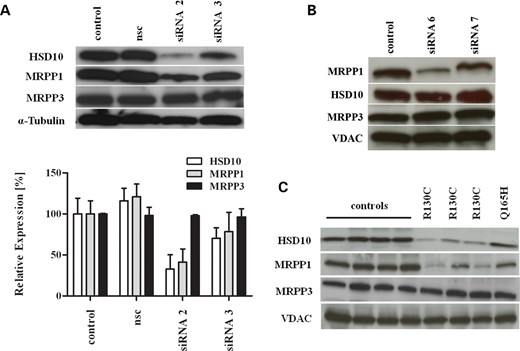
Loss of HSD10 protein causes loss of MRPP1 protein in human patient fibroblasts and HSD10 knock-down cells. (A) Western blot analysis of RNase P proteins in HeLa cells treated with HSD17B10 siRNA 2 and 3 and a non-silencing control (nsc). siRNA 2 reduced HSD10 protein levels to 82% and was associated with a significant lower amount of MRPP1 protein. Expression of MRPP3 was not affected. Densiometric analysis of RNase P proteins normalized to α-Tubulin is shown below. (B) Analysis of RNase P proteins after MRPP1 knock-down demonstrated unaffected expression of HSD10 and MRPP3 protein, whereas MRPP1 expression was down-regulated by siRNA 6 targeting MRPP1. (C) Reduced expression of HSD10 and MRPP1 in patient fibroblasts bearing mutation p.R130C compared with mutation p.Q165H and control fibroblasts. α-Tubulin and voltage-dependent anion channel (VDAC) were used as loading controls.
In further analysis, we tested if MRPP1 knock-down had an effect on the amounts of HSD10 and MRPP3 protein in human cells. Knock-down of MRPP1 was performed in HeLa cells with three different siRNAs (Supplementary Material, Fig. S1B). As shown in Figure 1B, MRPP1 knock-down was found for one siRNA which reduced MRPP1 expression to 78 ± 14% (mean ± SD) compared with controls. HSD10 and MRPP3 protein levels were not affected by MRPP1 knock-down indicating that MRPP1 expression does not modulate the level of the other two proteins of RNase P (Fig. 1B).
We next determined the HSD10 transcript level by quantitative real time-polymerase chain reaction (qRT)–PCR in fibroblasts from three patients carrying mutation p.R130C and one patient with mutation p.Q165H and compared it with control fibroblasts. Mutation p.R130C causes the classical ‘infantile form’, associated with severe neurodegeneration and early death, of HSD10 disease and was found in more than half of the diagnosed patients so far (Supplementary Material, Table S1). In contrast, mutation p.Q165H, associated with the ‘atypical form’ of the disease, causes complete loss of HSD10 enzyme activity but is associated with normal neurological development. HSD17B10 as well as RG9MTD1 and KIAA0391 transcript levels were not significantly different in patient fibroblasts compared with controls (Supplementary Material, Fig. S1C). In contrast, HSD10 protein content was significantly reduced in patient fibroblasts with the mutation p.R130C, compared with four different control cell lines, whereas HSD10 levels in patient fibroblasts with the p.Q165H mutation were similar to control fibroblasts (Fig. 1C). This finding is consistent with recombinant expression in Escherichia coli showing reduced soluble protein for HSD10R130C compared with HSD10wt and HSD10Q165H, an in vitro indicator of a protein stability defect (Supplementary Material, Fig. S2). In the HSD10 crystal structure, Arg130 forms a couple of charge interactions that hold two spatially proximal, but sequentially distant, helices together (Supplementary Material, Fig. S2). Therefore, it seems plausible that this mutation alters these interactions and destabilizes the HSD10 protein. In addition to the reduced HSD10 level, there is a concomitant decrease in the amount of MRPP1, but not MRPP3, in patient fibroblasts with mutation p.R130C (Fig. 1C), resembling the observations in our knock-down studies and lending further support to the notion that HSD10 is necessary to maintain MRPP1 protein levels in the RNase P protein complex.
To investigate the role of HSD10 in MRPP1 and MRPP3 protein expression, we performed rescue experiments after siRNA-mediated knock-down of HSD10 in HeLa cells by subsequent transfection with an HSD10wt expression vector designed to be unaffected by the siRNAs used. HSD10 knock-down without co-transfection caused an 81 ± 3% (mean ± SD, n = 3) reduction in HSD10 protein levels and a 73 ± 2.3% (mean ± SD) reduction in MRPP1 protein levels compared with non-silencing controls (Fig. 2A, middle lane).
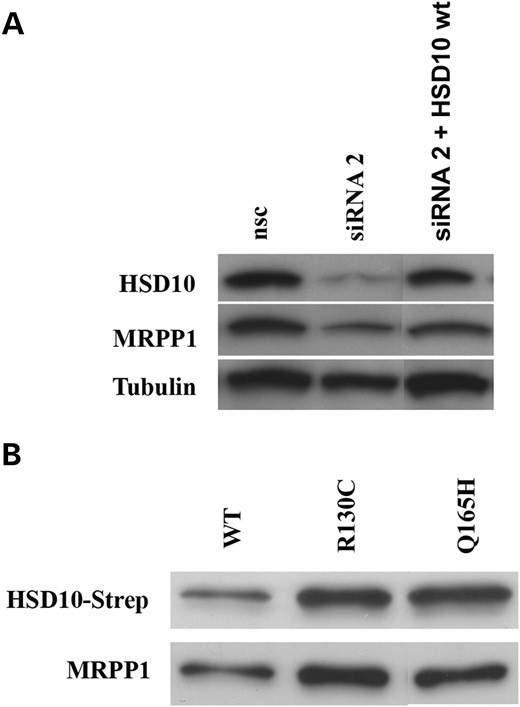
Ectopic expression of HSD10wt partly restored MRPP1 protein level in human cells. (A) Expression of HSD10 and MRPP1 was significantly reduced in HSD10 knock-down cells. Ectopic expression of HSD10wt in knock-down cells partly restored expression of both proteins. (B) Co-immunoprecipitation of HSD10-strep and MRPP1 after transient transfection of strep-tagged HSD10 proteins (wt, p.R130C and p.Q165H). In each sample, expression of MRPP1 was of similar intensity than found for HSD10 Strep-tag protein, indicating that MRPP1 binding to HSD10 was not reduced by HSD10 mutations.
Ectopic expression of HSD10wt increased HSD10 protein to approximately control levels (Fig. 2A). This also rescued the level of MRPP1 to 64 ± 16% (mean ± SD) of control (Fig. 2A, right lane).
In order to determine whether MRPP1 binding to HSD10 is impaired by the p.R130C or p.Q165H mutations, we performed co-immunoprecipitation experiments in HeLa cells. Ectopically expressed strep-tag labeled HSD10wt, HSD10R130C and HSD10Q165H proteins were purified with anti-strep beads and bound MRPP1 was determined by immunoblotting with specific antibodies against MRPP1 and strep-tag, respectively. As shown in Figure 2B, HSD10wt as well as HSD10R130C and HSD10Q165H bind to MRPP1 protein, indicating that the HSD10–MRPP1 interaction is not blocked by these HSD10 mutations.
Loss of HSD10 protein is associated with reduced mitochondrial tRNA processing
To investigate mitochondrial RNA processing in HSD10 knock-down cells and patient fibroblasts, we used a quantitative real-time PCR (qPCR) approach. We designed primers flanking the RNase P processing site of mRNA-tRNA and rRNA-tRNA junctions on the heavy strand, and the non-coding RNA-tRNA junctions on the light strand (Fig. 3A). Except for tRNAPhe, all gene-tRNA junctions were analyzed. For reverse transcription of RNA, tRNA-specific primers fused to an adaptor sequence were used. The adaptor sequence served as a reverse primer in the subsequent qPCR and allowed strand-specific quantification of unprocessed mitochondrial transcripts (Fig. 3B). The qPCR analysis in HSD10 knock-down HeLa and HRT18 cells revealed a significant increase in almost all unprocessed transcripts from the heavy strand RNA up to >25-fold (Fig. 3C). Interestingly, HSD10 knock-down had little or no effect on the processing of light strand transcripts, raising the possibility that a reduction in HSD10–MRPP1 expression primarily affects processing of heavy strand transcripts. Accumulation of precursor transcripts after HSD10 knock-down was partly rescued by the ectopic expression of wild-type (wt) HSD10, resulting in an accumulation of precursor transcripts ∼2.4 times higher for pre-Val and 3.76 more for pre-Lys compared with control cells (exemplarily shown for tRNAs pre-Val, pre-Leu and pre-Lys, Fig. 4). Although co-transfection with HSD10wt reduced the accumulation of the precursor tRNAs compared with the knock-down, a complete rescue was not observed, which could be explained by lower transfection efficiencies (<90%).
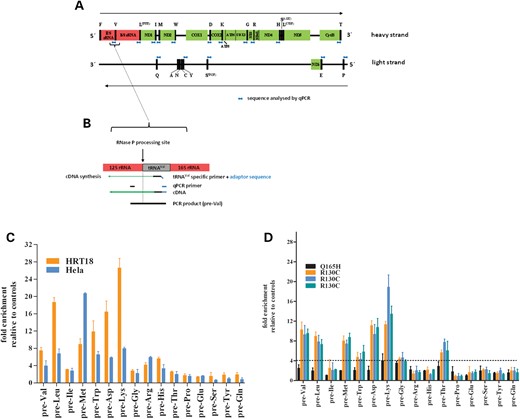
Effect of reduced HSD10 expression on mitochondrial tRNA processing. (A) Schematic representation of the mitochondrial genome with coding regions presented as colored boxes and tRNAs as black boxes. The location of quantified gene-tRNA sequences are indicated in blue. (B) Schematic drawing of 12S rRNA-tRNAVal junction to illustrate the strand-specific quantification method. (C) Quantification of tRNA precursor transcripts in HSD10 knock-down HeLa and HRT18 cell lines. Accumulation of precursor transcripts was found for heavy strand but not for light strand transcripts, indicating that impaired processing by loss of HSD10 is strand specific. Furthermore the extent of precursor accumulation is different for each individual pre-tRNA transcript. (D) Quantification of pre-tRNA transcripts in fibroblasts with p.R130C and p.Q165H mutation, respectively. The pattern of accumulation of pre-tRNA transcripts is quite similar to HSD10 knock-down cell lines with enrichment of only heavy strand pre-tRNA transcripts. However, pre-tRNA processing of pre-Ile, pre-Arg and pre-His was not significantly different in patient fibroblasts compared with control cells. Dotted line represents cutoff.
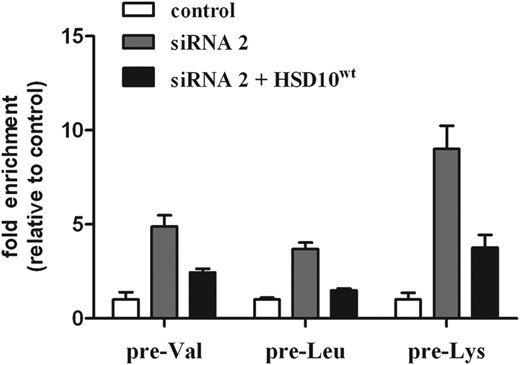
qRT-PCR analysis of three precursor tRNAs demonstrating that mt-tRNA processing can be partly rescued by the ectopic expression of HSD10wt. HSD10 knock-down increased expression of unprocessed pre-tRNAs pre-Val, pre-Leu and pre-Lys. After transfection of HSD10 knock-down cells with HSD10wt gene, the accumulation of unprocessed pre-tRNAs was ∼50% (pre-Val, pre-Leu) and 30% (pre-Lys) the amount of knock-down cells. The data demonstrated that HSD10wt expression restored at least in part RNase P function.
We next determined the expression of precursor tRNA transcripts in our patient fibroblasts. Ten different control fibroblast cell lines from donors of different age (newborn to 40 years) between passages 5 and 12 of cell culture were used as the reference. The relative expression was calculated according to the 2ΔΔCt method using the mean of the Ct-values of these control fibroblasts (13). Having determined the variance of mitochondrial precursor transcripts in control fibroblasts (Supplementary Material, Fig. S3), we defined a 4-fold larger relative expression of unprocessed transcripts from patients as significantly different from controls. qPCR analysis of patient fibroblasts revealed a similar pattern as in HSD10 knock-down cells (Fig. 3D), with accumulation of unprocessed precursor transcripts of pre-Val, pre-Leu, pre-Met, pre-Trp, pre-Asp and pre-Lys. In contrast to HSD10 knock-down cells, precursors for pre-Ile, pre-Arg and pre-His were in the normal range, whereas pre-Thr was significantly increased in patient fibroblasts but not in HSD10 knock-down cells (Fig. 3D), compatible with a partially different RNase P activity in tumor cells compared with fibroblasts. Accumulation of unprocessed transcripts was significantly higher in fibroblasts bearing the p.R130C mutation than the p.Q165H mutation where the processing was close to normal. This observation correlates well with the protein level of HSD10 and MRPP1 in p.Q165H and control fibroblasts as determined by western blots (Fig. 1C).
We next used northern blot analysis to confirm that low amounts of RNase P proteins have an effect on the steady-state levels of mitochondrial precursor transcripts. Using strand-specific probes for individual genes and tRNAs, we expected that reduced processing of mitochondrial transcripts should lead to the appearance of new RNA species of higher molecular weight than the native RNAs (Fig. 5A). Strand-specific northern analysis with MT-ND1 probe demonstrated strong expression of the normal MT-ND1 transcript (RNA 1.3 in Fig. 5A) at ∼950 bp in all fibroblast cell lines as well in HeLa cells (Fig. 5B). However, there was a marked increase in the specific RNA19 transcript which covers the 16S rRNA–tRNALeu(UUA)–MT-ND1 sequence (RNA 1.2, 2.59 kb) in patient cells with mutation p.R130C compared with control fibroblasts and with fibroblasts with mutation p.Q165H. The RNA19 transcript is also found in normal cells and has been reported to be increased in many human mitochondrial diseases (14); still, the pathogenetic relevance of this observation is unknown. Furthermore, in patient fibroblasts and HeLa HSD10 knock-down cells, an additional RNA molecule at 3.60 kb (RNA 1.1) was found which corresponds to the (tRNAPhe)–12S rRNA–tRNAVal–16S rRNA–tRNALeu–MT-ND1 sequence and indicates a reduced processing of tRNAVal and tRNALeu in line with a reduced cleavage of the respective tRNAs observed by qPCR (Fig. 3C and D). Northern analysis using a probe specific for 12S rRNA revealed a strong signal for 12S rRNA (950 bp) and a larger RNA species of ∼3.5 kb equal to (tRNAPhe)–12S rRNA–tRNAVal–16S rRNA–tRNALeu–MT-ND1 transcript (RNA 1.1) in fibroblasts with mutation p.R130C (Supplementary Material, Fig. S4B).
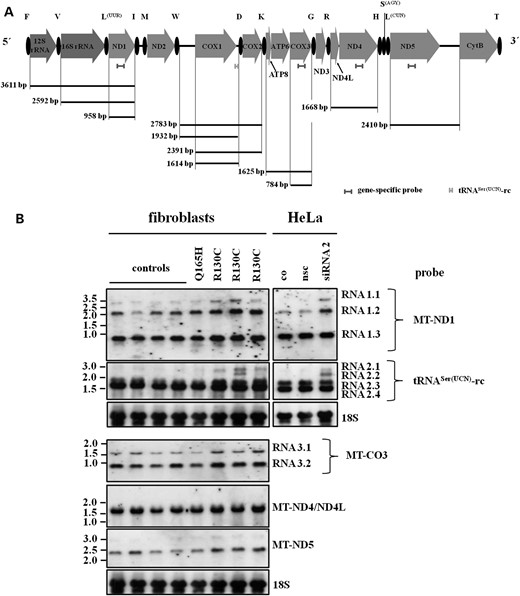
Northern blot analysis of mitochondrial transcripts in human fibroblasts and HSD10 knock-down HeLa cells. (A) Schematic representation of the mitochondrial heavy strand genome. Indicated are the strand-specific probes for northern analysis and the predicted size of RNA molecules expected to detect on northern blots. (B) Expression pattern of RNA transcripts in human control fibroblasts (controls), in fibroblasts from HSD10 diseased patients and in HSD10 knock-down HeLa cells. An accumulation of larger RNA molecules was found in fibroblasts with HSD10R130C mutation and in HSD10 knock-down cells. Probing with MT-ND1 revealed an accumulation of a transcript of approx. 3.6 kb size (RNA 1.1) which covers (tRNAPhe) –12SrRNA–tRNAVal–16SrRNA–tRNALeu-ND1 indicating reduced processing of tRNAVal and tRNALeu as also found by real-time PCR. In a further analysis, we used a heavy strand-specific probe corresponding to light strand tRNASer(UCN) (tRNASer(UCN)-rc). The data demonstrated an accumulation of larger transcripts in HSD10R130C patient fibroblasts and HSD10 knock-down cells. These larger transcripts cover MT-CO1–tRNAAsp–MT-CO2 sequence with 5′-UTR of MT-CO1 (transcript at 2.6 kb, RNA 2.1) or without (transcript at 2.2 kb, RNA 2.2) and indicated an impaired tRNAAsp processing. Northern blots with strand-specific probes for MT-ND4, MT-CO3 and MT-ND5 revealed no accumulation of high molecular weight precursor transcripts in patient fibroblasts.
Analysis with an H-strand-specific probe corresponding to tRNASer(UCN) coded on the light strand (tRNASer rc) revealed an accumulation of a larger molecular weight RNA of ∼2.8 kb in fibroblasts from patients with mutation p.R130C and in HeLa HSD10 knock-down cells (Fig. 5B, RNA 2.1). This large RNA molecule corresponds to the MT-CO1–tRNAAsp–MT-CO2 sequence, which includes the 5′-UTR of the MT-CO1 gene and represents impaired processing of tRNAAsp as previously shown by qPCR (Fig. 3C and D). Precursor transcripts corresponding to RNA 2.1 and RNA 2.2, respectively, were also found by using an MT-CO2-specific probe supporting the observation of reduced tRNAAsp processing in HSD10 deficient cells (Fig. 3 and Supplementary Material, Fig. S3B). Two additional RNA molecules were found in all cell lines: one strongly expressed RNA species of 1.61 kb corresponding to the MT-CO1 gene (RNA 2.4), and 1.93 kb species that also include the 5′-UTR of this gene (RNA 2.3). MT-CO1 transcripts with unprocessed 5′-UTR are significantly enriched in fibroblasts from patients with HSD10 disease. This is in line with a previous study that used MRPP1 knock-down in tumor cells to show that RNase P is required for the removal of the MT-CO1 5′-UTR (15) and indicates that this function is also impaired in HSD10 disease. Finally, an RNA species of ∼2.4 kb predicted to include MT-CO1 without 5′-UTR, tRNAAsp and MT-CO2 (RNA 2.2) was found in fibroblasts from one patient and in HSD10 knock-down cells, providing further evidence of reduced pre-tRNAAsp processing in HSD10 disease. Interestingly, our Northern analysis revealed a previously unknown 3′UTR for the MT-CO1 gene, since our specific probe detects this sequence and therefore was included in the RNA species detected. Similar transcript species were found when northern blots were probed with MT-CO1-specific probe (data not shown).
There are additional RNA cleavage sites in mitochondrial transcripts which are not adjacent to tRNAs and therefore assumed to be processed by a different mechanism. These include processing of the junction between ATPase 8/6 and MT-CO3, the ND5-CytB mRNA and the 3′-end of ND6 mRNA. Northern blots with strand-specific probes for MT-CO3 and MT-ND5 revealed no accumulation of precursor transcripts in patient fibroblasts (Fig. 5B). Using a specific probe for MT-ND4, a specific signal corresponding to MT-ND4/ND4L mRNA was found in all fibroblasts (Fig. 5B). A similar lack of precursors was previously observed in tumor cells with MRPP1 and MRPP3 knock-down, respectively (15).
Northern blot analysis of potential precursor RNAs of the light strand transcripts by using specific probes for tRNAGln and tRNASer(UCN) on the light strand, respectively (Supplementary Material, Fig. S4A) showed normal tRNA signals and no evidence of high-molecular RNA species (Supplementary Material, Fig. S4B). This finding supports the notion of normal light strand transcript processing in patient fibroblasts and is in agreement with the results of qPCR analyses (Fig. 3D).
DISCUSSION
HSD10 protein is part of the mitochondrial RNase P enzyme complex which is necessary for the 5′ cleavage of mitochondrial tRNAs in the polycistronic precursor transcripts (10). The exact function of HSD10 in this process is unknown. Here, we report that the cellular amount of MRPP1, the RNase P component that recognizes mitochondrial tRNA sequences and catalyzes the methylation of specific tRNA bases (12), is strongly associated with cellular HSD10 levels. A reduction in HSD10 either by knock-down or by HSD17B10 mutation in fibroblasts of patients with HSD10 disease causes a marked reduction in MRPP1, whereas MRPP3 levels are unaffected. In contrast, MRPP1 knock-down has no recognizable effect on HSD10 levels. This finding is in line with the known MRPP1-independent functions of HSD10: the MHBD reaction in isoleucine breakdown is catalyzed in the mitochondrial matrix by HSD10 in the homotetrameric form (16), which may act as a stable HSD10 reservoir independent of MRPP1. Loss of HSD10 causes the accumulation of mitochondrial tRNA transcript precursors as shown by both qPCR and northern blot analysis, an effect limited in our experimental setting to the mitochondrial heavy chain with the light chain transcript processing unaffected.
HSD10 disease was first reported as an organic aciduria with unusual disease course resembling a primary mitochondrial disorder (2). We recently showed that the clinical presentation was unrelated to the protein's enzymatic function as 2-methyl-3-hydroxybutyryl-CoA dehydrogenase. The HSD17B10 mutation p.Q165H, which completely removes dehydrogenase function (8), was found in children with normal development. In contrast, mutations p.R130C and p.D86G which are associated with the classical or severe forms of HSD10 disease have substantial residual 2-methyl-3-hydroxybutyryl-CoA-dehydrogenase function when analyzed using recombinant proteins. The enzyme activity of recombinant p.R130C protein rapidly declines at room temperature (RT), most likely due to protein instability (8). Low enzyme activity has been reported in fibroblasts from patients with mutation p.R130C (Supplementary Material, Table S1); however, this was associated with low HSD10 protein levels in the study where this was investigated (17). Rescue experiments in Xenopus embryos confirmed that a non-enzymatic function of HSD10 is required for structural and functional integrity of mitochondria, and impairment of this function causes apoptotic cell death (8). In our present study, a strong HSD10 knock-down by specific lentiviral shRNA resulted in loss of cell proliferation and cell death in HeLa cells (data not shown). Additionally, we analyzed fibroblasts from a patient with HSD10D86G mutation and found no detectable HSD10 levels in western blots. Fibroblasts from this patient stopped cell proliferation after 2–3 passages and were therefore not included in our experiments.
Here, we studied in more detail mutation p.R130C which has been found in the majority of patients diagnosed with HSD10 disease. In line with the previous observation that this mutation is relatively unstable (8), we observed reduced HSD10 protein levels in fibroblasts from patients with this mutation despite normal RNA levels. This is consistent with the equivalent recombinant data, where HSD10R130C exhibits reduced protein solubility compared with HSD10wt and HSD10Q165H (Supplementary Material, Fig. S2) and could therefore be recognized and degraded by the mitochondrial protein quality machinery. This is a cellular mechanism maintained by chaperones and proteases to target the removal of non-native polypeptides during mitochondrial biogenesis (18). Additional genetic or external factors may contribute to the variable protein levels of HSD10R130C as well as MRPP1 in patient fibroblasts. Compared with HSD10R130C, the HSD10Q165H mutant shows similar turnover and protein level as wild-type HSD10 in both, patient fibroblasts and recombinant expression. Our observation that neither p.R130C nor p.Q165H had a noticeable impact on the HSD10-MRPP1 interaction raises the possibility that the severe disease course associated with p.R130C is best explained by more rapid degradation, with a concomidant reduction in MRPP1. In contrast, Garcia-Villoria et al. (17) reported normal HSD10 levels for mutations p.N247S, p.R226Q and p.P210S, respectively, which are all associated with a severe disease course. We hypothesize that at least some of these mutations cause reduced interaction with MRPP1 resulting in low MRPP1 level and impaired RNase P function. Due to the lack of fibroblasts from patients, this hypothesis could not yet be tested by us.
A reduced amount of HSD10 protein to 60–70% of controls was found recently in several patients with mild mental retardation, choreoathetosis and abnormal behavior from the same family (19). Affected males carried the hemizygous mutation c.574C>A in exon 5 of HSD17B10 gene which causes skipping of exon 5 and a premature stop codon in exon 6 (19). Due to this alternative splicing process, the amount of HSD10 protein is reduced, whereas MHBD activity is similar to controls. How a relatively minor reduction in apparently normal HSD10 protein causes the clinical phenotype was unknown. Considering the correlation between HSD10 and MRPP1, it is possible that a reduced cellular amount of MRPP1 protein may also be a prominent pathogenetic mechanism in these patients.
MRPP1 forms a subcomplex with HSD10 that interacts with mitochondrial tRNAs and catalyzes the methylation of G9 or A9 purine bases, independent of HSD10 dehydrogenase function and RNase P cleavage activity (12). The MRPP1-HSD10 subcomplex also recruits MRPP3 to form the functional RNase P complex (10). MRPP3 is predicted to harbor a nuclease domain that could perform the actual phosphodiester bond hydrolysis in the 5′-tRNA cleavage. It is therefore reasonable to assume that a reduced formation of the HSD10–MRPP1 subcomplex, caused by reduced availability of HSD10, also affects MRPP3 function in RNase P. Several studies on different other protein complexes illustrate that mutations in individual constituent subunits can impact on intact complex formation and function (20–22). Sanchez et al. (15) recently showed that MRPP1 or MRPP3 knock-down in HeLa cells resulted in a strong increase in unprocessed precursor RNAs of all gene–tRNA junctions of the heavy chain transcript. These data are similar to our present findings, indicating that loss of any component of RNase P may have the same effect on mitochondrial RNA processing.
Unexpectedly, loss of HSD10 (and consequently MRPP1) in our study had differential effects on the transcripts of the two mtDNA strands. Although there was clear evidence of impaired tRNA processing on the mtDNA heavy chain transcript, light chain transcript processing appeared to be relatively unaffected by both, HSD10 knock-down in vitro as well as HSD10 mutation in patient fibroblasts. There is no easy explanation for this observation. It is possible that specific tRNA substrates have different tRNA processing requirements or RNase P kinetics or that there are differential compensatory mechanisms. Added to this complexity, the full range of proteins involved in mtRNA processing is far from understood. Recently, it was reported that MRPP1 is a binding partner of protein 32 which in mitochondria forms a complex with RNase H1, physically interacts with mitochondrial DNA and pre-rRNA, and is required for transcription and processing of ribosomal 12S/16S RNA in mitochondria (23). Potentially more relevant to this work is the recent identification of ‘mitochondrial RNA granules’, foci of newly synthesized mtRNA next to mitochondrial nucleoids which contain the RNA-binding protein GRSF1, RNase P and other proteins (24). These granules seem to be crucial for post-transcriptional storage and processing of mitochondrial RNAs. However, the individual contribution of GRSF1 and RNase P in processing of mitochondrial RNAs remains to be solved (24,25).
Almost 2/3 of pathogenic mtDNA mutations have been found in tRNA genes, consistent with the key role of tRNA in translation. The consequence of tRNA point mutations is traditionally assumed to be impaired mitochondrial translation due to a deficiency of functional tRNA which results in impaired respiratory chain function and in production of ROS as an early event in neurodegeneration (26,27). Recently, a tRNA mutation which affects RNase P activity and 5′-cleavage has been described in a case of maternally transmitted hypertension due to a mutation in the mitochondrial tRNAIle gene (28). It has also been shown that the function of the tRNA cleaving enzyme PROP1 in Arabidopsis is strongly influenced by the tRNA sequence (29). PROP1 resembles human mitochondrial RNase P in that it is devoid of RNA component and, like MRPP3, contains pentatricopeptide (PPR) repeats as well as a highly conserved metallonuclease domain. The authors speculate that PPR repeats in PRORP specifically recognize individual nucleotides in tRNA loops, in particular unstacked bases or residues not involved in basepairing which are highly conserved among tRNA sequences (29).
It is interesting to note that patients with ELAC2 mutations causing a deficiency of RNase Z, the enzyme complex that cleaves the 3′-end of mitochondrial tRNAs, have clinical features that resemble those in patients with HSD10 disease (30). Symptoms include marked hypertrophic cardiomyopathy and impaired psychomotor development. Although these clinical features were also observed with many other mitochondrial disorders, it is possible that they represent typical functional consequences of impaired mitochondrial transcript processing. Interestingly, normal steady-state levels of mitochondrial mRNAs and tRNAs were found in these patient fibroblasts despite the accumulation of precursor transcripts and reduced mitochondrial protein synthesis (30). The authors suggest that the accumulation of precursor transcripts interferes with ribosome function, thereby blocking mitochondrial protein translation. Sanchez et al. showed similar data in an in vitro model of MRPP1 and MRPP3 knock-down. They demonstrated that only loss of MRPP1 causes an accumulation of mitochondrial precursor transcripts and subsequently reduced expression of mitochondrial ribosomal proteins (15). Similar mechanisms may be responsible for the disease course in HSD10 patients.
Except mutations in ELAC2, no other proteins involved in mitochondrial transcript cleavage have been reported to be associated with disease in humans. Moreover, the exact pathomechanism that explains the specific pattern of organ dysfunction in HSD10 disease remains to be clarified.
MATERIALS AND METHODS
Ethics statement
The study was approved by the institutional review board in Heidelberg, and informed consent was obtained for all patient and control cell lines used.
Cell culture
Fibroblasts from HSD10 diseased patients as well as control fibroblasts from healthy donors were cultivated in Minimum Essential Medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin and 2 mm glutamine in 5% CO2 at 37°C. HeLa, HEK-293 and HRT18 (human rectum adenocarcinoma) cell lines were purchased from the American Type Culture Collection and cultivated as described. Fibroblasts from patients were obtained from the children's hospital in Heidelberg, control fibroblasts originated from our local cell repository. Media and additives were purchased from PAA Laboratories. All cell lines were tested once a month for mycoplasma infection using a PCR detection kit (Minerva Biolabs).
Quantitative RT-PCR
Quantitative analysis of HSD17B10, RG9MTD1 and KIAA0391 expression was determined in all cell lines. Therefore, total RNA was isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer's instruction. RNA was quantified using the NanoDrop™ and 1 µg cDNA was prepared using the Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Scientific) with random hexamer primers and used as a template in SYBR Green (Invitrogen) based PCRs performed on ABI Prism® Real time PCR system (Applied Biosystems). Relative gene expression was calculated according to the 2ΔΔCt method using GAPDH as a reference gene. Assays contained 10 ng cDNA template and 20 mm primer. A 10 min hot-start activation at 94°C was followed by 40 cycles of 20 s denaturation at 94°C and 1 min annealing/extension at 60°C and a dissociation analysis (60–95°C). Quantification of gene expression was carried out by the comparative 2ΔΔCt method. Primer sequences are listed in Supplementary Material, Table S2.
Strand-specific qRT-PCR
Total RNA from confluent cell monolayer's were isolated using the RNeasy Kit (Qiagen) according to the supplier's instructions. RNA concentration was determined by NanoDrop™ (ThermoFisher) and RNA integrity was confirmed by gel electrophoresis. 2 µg of total RNA was treated with Turbo™ DNase (Ambion) for 60 min at 37°C. DNase was inactivated by addition of ethylenediaminetetraacetic acid (EDTA) (15 mm) and incubation at 75°C for 10 min. First-strand cDNA synthesis was carried out using the Maxima H minus first-strand cDNA synthesis kit (Fermentas). 1 µg of DNase-treated total RNA and 2 µl of a primer mix in a total volume of 15 µl were incubated at 70°C for 10 min and chilled on ice. After adding of 4 µl of first-strand buffer and 1 µl Revert-Aid reverse transcriptase, the reaction was incubated at 60°C for 1 h and finally, for 5 min at 85°C. The cDNA was diluted 1:4 for use in real-time PCR. The primer mix used in first-strand synthesis contained reverse primers specific for four tRNAs and Ubiquitin B (UBB) as reference gene, respectively. In total, four different primer mixes for first-strand synthesis were prepared. An adaptor sequence was fused to each gene-specific primer (Fig. 3B and Supplementary Material, Table S3) which served as a reverse primer in real-time PCR analysis.
Real-time PCR analysis was performed using the primers shown in Supplementary Material, Table S3 on ABI PRISM 7000 Sequence Detection System using the Maxima SYBR Green qPCR Master Mix (Fermentas). PCR contained 12.5 µl SYBR Green qPCR master mix, 350 nm of forward and adaptor primers, and 2 µl of 1:4 diluted template cDNA in a total volume of 25 µl. PCR was performed using following conditions: 10 min at 95°C followed by 40 cycles of of 15 s at 95°C and 1 min at 60°C. To check that the primers used produced only a single product, a dissociation protocol (65–95°C) was added after the PCR. Additionally, PCR products were loaded onto a 2% agarose gel to verify single PCR products at the calculated molecular size. Each assay included a non-template control, and for each sample, Turbo-DNase-treated RNA without first-strand synthesis was assayed to control DNase digestion. Raw data were analyzed in Sequence Detection System software v.1.2.3 and exported to Excel for final analysis. Ct values were normalized against UBB and relative expression levels were calculated according to the 2ΔΔCt method (13). In first experiments, the expression of ubiquitin B (UBB), glyceraldehydes-3-phosphate dehydrogenase (GAPDH) and hypoxanthine phosphoribosyltransferase 1 (HPRT1) was analyzed by qPCR and UBB was found to be the most stable reference genes using Bestkeeper software (31).
Western blot analysis
Cell extracts were prepared from cells using lysis puffer [50 mm Tris–HCl (pH 7.4), 154 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, 0.05% sodium dodecyl sulfate (SDS)], and protein concentration was determined using Bradford Assay (Biorad). 30 µg of protein was separated on a 10% SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (GE Healthcare). Membrane blots were blocked with tris phosphate-buffered saline containing 0.1% Tween-20 and 5% skim milk powder (Fluka) for 1 h at RT. Membranes were incubated with antibodies directed against HSD10 anti-ERAB [5F3] (1:500, Abcam), anti-RG9MTD1 (1:250, Sigma-Aldrich) as well as anti-MRPP3 (1:2000, Abcam) and detected with horseradish peroxidase conjugated secondary antibodies (Dako). Expression of anti-alpha Tubulin (1:8000, Abcam) and VDAC (N-18) (1:500, Santa Cruz Biotechnology, Inc.) was determined as loading controls, respectively. The blots were developed with enhanced chemiluminescence using the ECL Western Blotting Analysis Detection system (GE Healthcare). For densiometric analysis, protein bands on different blots (n = 7) were quantified with the Image J Software version 3.1 (National Institutes of Health, USA) and data analyzed in GraphPad Prism (GraphPad Software, version 5.04, La Jolla, CA, USA).
Transient knock-down experiments
Cells were plated at 80% confluence in 6-well plates or 25 cm2 flasks and transfected with siRNAs targeting HSD17B10 or RG9MTD1 (Qiagen) and a non-silencing control in MEM without any supplements. Transfections were performed using Turbofect Transfection Reagent (Thermo Scientific) with a ratio of 1:3 to siRNAs following an incubation of 3–5 days. To analyze the efficiency of siRNA transfections, gene silencing was determined by qRT-PCR analysis. siRNA sequences targeting HSD17B10 were CAGCGAGTTCTTGATGTGAAT (siRNA 2) and AAGACTTCCAGCGAGTTCTTG (siRNA 3) and for RG9MTD1 TACCAGCTGTTACTTATCCTA (siRNA 6) and TGGAGATTGCTTGGCAGAGAA (siRNA 7).
Rescue experiment
HeLa cells were seeded the day before transfection in 25 cm2 flasks at a density of 2.5 × 105 cells per flask in 4 ml MEM medium. Transfections were performed by using 12 µg siRNA 2 targeting HSD10 and 12 µl Turbofect Transfection Reagent for 3 days. For ectopic expression, the coding sequence of HSD10wt was cloned in a vector with a strong EF1α promotor (32) and the nucleotide sequence was verified by Sanger sequencing. Cells were then transfected for additional 3 days. In HSD10 genes cloned into the expression vector, the binding site for siRNA 2 was mutated to avoid siRNA-mediated RNA degradation of transfected genes. Cells were harvested after 6 days and analyzed as described above (expression vector was a kindly gift from Prof G. Untergasser, Innsbruck).
HSD10–MRPP1 interaction
HSD10 binding studies were performed using a pTO-HA expression vector, kindly provided by Prof. G. Baier, Innsbruck, containing either the sequence of wt HSD10, mutation p.Q165H or p.R130C in the frame of the Strep-tag III sequence. 4 µg vector DNA was used for transient transfection generating wt or mutated HSD10-Strep-tag expressing cells and harvested after 48 h. HSD10-Strep-tag was purified using a batch system based on the affinity of the strep-tag to strep-Tactin-beads. Elution was performed using 50 µl destobiotinbuffer for 20 min at RT. Beads were tapped to enhance the dissociation of bound protein; after centrifugation, supernatant was used for western blot analysis and probed either with anti-strep-tag (1:1000, IBA) or anti-MRPP1 antibody.
Northern blot analysis
Northern analysis was performed using the NorthernMax kit from Ambion according to the manufacturer's instructions. Briefly, 2–5 µg of total RNA was separated on a 1% denaturing agarose gel. After electrophoresis, RNA was transferred to nylon membrane (Hybond-N+ Amersham, GE Healthcare) by capillary transfer, UV cross-linked and subjected to hybridization with biotinylated probes (10 pmol/l in Ultrahyb-Oligo hybidization buffer, Ambion). Signals were detected using the BrightStar BioDetect kit (Ambion) according to the manufacturer's instructions. Strand-specific 5′ biotinylated probes for the mitochondrial tRNAs, rRNAs and mRNAs were purchased from Microsynth (Switzerland). A biotinylated RNA size marker (BrightStar RNA Millenium Marker, Ambion) was used to determine the size of RNA species. Probe sequences are available in Supplementary Material,Table S4.
Statistics
Data were analyzed with standard statistical methods performed in GraphPad Prism (GraphPad Software, version 5.04, La Jolla, CA, USA). Data are presented as the means ± SD and are representatives of at least three independent experiments.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
The study was performed with free research funds from the institute.
ACKNOWLEDGEMENTS
The authors want to thank Pia Traunfellner for her excellent technical work.
Conflict of Interest statement. None declared.
REFERENCES
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}