
Abstract
A loss of function mutation in the TRESK K2P potassium channel (KCNK18), has recently been linked with typical familial migraine with aura. We now report the functional characterisation of additional TRESK channel missense variants identified in unrelated patients. Several variants either had no apparent functional effect, or they caused a reduction in channel activity. However, the C110R variant was found to cause a complete loss of TRESK function, yet is present in both sporadic migraine and control cohorts and no variation in KCNK18 copy number was found. Thus despite the previously identified association between loss of TRESK channel activity and migraine in a large multigenerational pedigree, this finding indicates that a single non-functional TRESK variant is not alone sufficient to cause typical migraine and highlights the genetic complexity of this disorder.
Similar content being viewed by others

Introduction
Migraine is a common, disabling neurological disorder with a genetic, environmental and in some cases hormonal component. It is characterized by attacks of severe, usually unilateral and throbbing headache, can be accompanied by nausea, vomiting and photophobia and is clinically divided into two main subtypes, migraine with aura (MA) when a migraine is accompanied by transient and reversible focal neurological symptoms and migraine without aura (MO)1. The multifactorial and clinical heterogeneity of the disorder have considerably hindered the identification of common migraine susceptibility genes and most of our current understanding comes from the studies of familial hemiplegic migraine (FHM), a rare monogenic autosomal dominant form of MA2.
So far, the three susceptibility genes that have been convincingly identified in FHM families all encode ion channels or transporters: CACNA1A encoding the α1 subunit of the Cav2.1 calcium channel3, SCN1A encoding the Nav1.1 sodium channel4 and ATP1A2 encoding the α2 subunit of the Na+/K+ pump5. It is believed that mutations in these genes may lead to increased efflux of glutamate and potassium in the synapse and thereby cause migraine by rendering the brain more susceptible to cortical spreading depression (CSD)6 which is thought to play a role in initiating a migraine attack7,8. However, these genes have not to date been implicated in common forms of migraine9.
Nevertheless, current opinion suggests that typical migraine, like FHM, is also disorder of neuronal excitability, ion homeostasis and neurotransmitter release10,11,12. Mutations in the SLC4A4 gene encoding the sodium-bicarbonate cotransporter NBCe1, have recently been implicated in several different forms of migraine13 and a variety of genes involved in glutamate homeostasis (PGCP, MTDH14 and LRP115) and a cation channel (TRPM8)15 have also recently been implicated in migraine via genome-wide association studies. Ion channels are therefore highly likely to play an important role in the pathogenesis of typical migraine.
TRESK (KCNK18), is a member of the two-pore domain (K2P) family of potassium channels involved in the control of cellular electrical excitability16. Regulation of TRESK activity by the calcium-dependent phosphatase calcineurin17, as well as its expression in dorsal root ganglia (DRG)18 and trigeminal ganglia (TG)19,20 has led to a proposed role for this channel in a variety of pain pathways. In a recent study, a frameshift mutation (F139Wfsx24) in TRESK was identified in a large multigenerational pedigree where it co-segregated perfectly with typical MA and a significant genome-wide linkage LOD score of 3.0. Furthermore, functional analysis revealed that this mutation caused a complete loss of TRESK function and that the truncated subunit was also capable of down regulating wild-type channel function. This therefore highlighted KCNK18 as potentially important candidate gene and suggested that TRESK dysfunction might play a possible role in the pathogenesis of familial migraine with visual aura20.
Additional screening for KCNK18 mutations in unrelated sporadic migraine and control cohorts also identified a number of other missense variants; R10G, A34V, C110R, S231P and A233V20. The A233V variant was found only in the control cohort, whilst A34V was identified in a single Australian migraine proband for which family samples were not available, but it was not detected in controls. By contrast, the R10G, C110R and S231P variants were found in both migraineurs and controls in both cohorts. In this study, we have investigated the functional effect of these variants to further probe the potential association of TRESK dysfunction with typical migraine.
Results
Location of missense variants in the TRESK channel
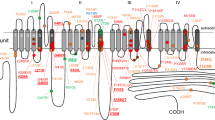
The predicted location of these variants in the TRESK channel is shown in Figure 1; R10G is located within the N-terminus, A34V within the first transmembrane domain (TM1) and C110R close to the selectivity filter in first pore domain (P1). The S231P and A233V variants are both located within the intracellular loop between TM2 and TM4. This domain acts as the site for regulation by calcineurin and both the S231P and A233V variants occur directly between two motifs involved in this process; the nuclear factor of activated T cells (NFAT)-like docking motif, PQIIIS and the 14-3-3 binding motif, RSNSCP17,21,22.

Basal currents of missense variants identified in TRESK.
(A) Schematic topology of the human K2P TRESK subunit showing the position of the identified missense variants. (B) Side view of homology model of the TM/pore domains of TRESK with the A34 and C110 residues shown in blue and red respectively. For clarity only the P1 domain is shown. (C) Representative whole-cell currents from an oocyte expressing either WT TRESK or the variants. Oocytes were bathed in lowK solution, the membrane potential held at −80 mV and voltage steps from −120 to +60 mV applied for 1 sec in 20 mV increments. (D) Normalised basal currents for the TRESK variants. Steady state currents were measured at the end of the 1s pulse to +60 mV from the whole cell currents (as shown in C). For each batch of oocytes, the currents were normalised to the average of control group. Data were pooled from 2 to 4 batches. Oocytes were injected with 0.9 ng of RNA except for the C110R mutant where 4.5 ng was injected. N-numbers are given above the bars.
There are currently no crystal structures available for K2P channels and so we mapped the position of these variants using a homology model of the TRESK channel pore (Figure 1B). Only the A34 and C110 residues are included in this structure because there are no templates for the intracellular domain. Interestingly, the model predicts that A34 in TM1 is in contact with C110 in the pore-helix and that both residues are adjacent to the selectivity filter which has recently been implicated in the control of K2P channel gating23.
Functional properties of missense variants
We next examined the effect of these different variants on the functional properties of TRESK. Figure 1C shows representative whole-cell basal currents recorded from oocytes expressing either wild-type (WT) or mutant TRESK. WT TRESK exhibited large outwardly-rectifying whole cell K+ currents similar to those previously reported24,25. Currents recorded from the R10G, S231P and A233V variants appeared almost indistinguishable to WT currents and were similar in magnitude (Figure 1D). By contrast, the A34V mutant expressed markedly reduced currents compared to WT TRESK, whilst currents recorded from oocytes expressing the C110R mutant channel were indistinguishable from those of uninjected controls (Figure 1D).
Even though the S231P and A233V mutants had no apparent effect on the magnitude of the basal current levels, their close proximity to important regulatory sites suggests that they may interfere with the calcineurin-dependent regulation of TRESK. Similarly, even though the A34V and C110R variants expressed little or no basal currents, they may still be capable of responding to calcineurin activation. It has been shown that an increase of intracellular calcium produced by the calcium ionophore ionomycin, can produce calcineurin-dependent activation of TRESK expressed in Xenopus oocytes17. We therefore determined the effect of ionomycin on these different TRESK variants (Figure 2). As shown previously, 500 nM ionomycin induced a large, robust and reversible activation of WT TRESK17. Ionomycin also activated the R10G, S231P and A233V variants and the extent of this activation did not differ significantly from that of the WT channel (Figure 2B). By contrast, the response of the A34V and C110R variants to ionomycin was essentially undetectable and similar to the background currents activated by ionomycin in control oocytes26.

Response of TRESK variants to ionomycin.
(A) Representative current responses to 500 nM ionomycin. Plotted current values represent measurements taken at the end of 300-ms voltage steps to −100 mV applied every 3 s from a 0-mV holding potential. As shown above the trace, oocytes were switched from lowK to highK solution prior to the application of ionomycin. (B) Normalised responses to ionomycin. The current measured after ionomycin activation was normalised to the current measured before activation in each oocyte. Oocytes were injected with 0.13 ng RNA. N-numbers are given within the bars.
Loss of TRESK function in the A34V and C110R variants
Although very small, some residual current could be detected for the A34V variant (Figure 1) and we found that when greater quantities of mRNA (4.5 ng) were injected, larger currents could be recorded very similar in profile to WT TRESK (Figure 3A). We also found that despite the absence of calcineurin activation seen in Figure 1, when these larger basal currents were recorded, ionomycin induced a reversible activation similar to that of WT TRESK (Figure 3B). This suggests that the A34V variant can still produce functional channels and that the mechanisms which underlie calcineurin activation must therefore remain intact. By contrast, no basal currents or any response to ionomycin could be recorded from the C110R variant even when larger quantities of mRNA (4.5 ng) were injected (Figure 3C).

Loss of function in the A34V and C110R variants.
(A) Representative whole cell currents from an oocyte expressing a larger quantity of A34V mRNA (4.5 ng). (B) Representative current responses to 500 nM ionomycin of an oocyte expressing a larger quantity A34V mRNA (4.5 ng). (C) Normalised peak currents measured at +20 mV for WT TRESK coexpressed with either the A34V or C110R variants. Molar ratios, indicated above the bars, are 1∶1 = 0.45 ng:0.45 ng; 5∶5 = 2.5 ng:2.5 ng; 1∶5 = 0.45 ng:2.5 ng. N-numbers are given within the bars.
In our previous study we demonstrated that 1∶1 coexpression of the F139Wfsx24 mutant with WT TRESK (i.e. a situation that mimics the heterozygous state) reduced WT channel activity20. We therefore examined whether the loss-of-∶1 ratio reduced the whole-cell currents by about 40% and further reduction was observed when the ratio of C110R mRNA was increased (Figure 3C). This suggests the C110R variant produces a completely non-functional subunit, similar to the F139Wfsx24 mutation that shows a strong genetic linkage to migraine with aura. Similar results were also obtained upon coexpression of WT and A34V mRNAs (Figure 3C). But the effect was reduced when the relative amount of A34V mRNA was increased, most likely because the A34V variant does not produce a complete loss of function and can probably form functional heteromeric channels.
A34V forms functional heteromeric channels
To further explore the properties of a heteromeric WT-A34V channel population we created a tandemly linked dimer. The concatenation of subunits has successfully been employed to control subunit stoichiometry in a variety of different types of K+ channels, including several K2P channels27,28. As a control, we first created a WT-WT dimer and found that the basal currents and ionomycin activation of this WT-WT tandem construct were virtually indistinguishable from those of homomeric WT TRESK (Figure 4). Expression of the WT-A34V tandem construct also resulted in TRESK-like currents that could still be activated by ionomycin, but they were of significantly smaller amplitude than the WT-WT dimer. This indicates that the A34V variant is also capable of down-regulating WT TRESK activity through coassembly as a dysfunctional heteromeric channel.

A34V forms functional heteromeric channels with WT TRESK.
(A) Representative basal current and (B) Representative response to 1 μM for the WT-WT tandem channel. (C) Representative basal current and (D) Representative response to 1 μM for the WT-A34V tandem channel.
KCNK18 gene copy number
Both A34V and C110R variants result in a severe loss of TRESK function. Yet, while the A34V variant has thus far been found only in one patient with migraine, the C110R variant shows no genetic association with MA. In an Australian case-control cohort, the C110R variant was present in 12 unaffected individuals of a total 496, compared with 8 affected individuals of a total 47920. Variations in gene copy number have been implicated in several genetic diseases and pathological conditions, including several K2P channel genes29. We therefore assessed KCNK18 copy number variation (CNV) in individuals with and without migraine. However, we found that all individuals with the C110R variant have the expected two copies of KCNK18 irrespective of their migraine status (Figure 5).

Predicted copy number values for KCNK18.
All samples showed a normal predicted copy number value of 2. No differences were seen between C110R migraineurs (red bars) and C110R non-migraineurs (blue bars). Error bars represent the maximum and minimum limits based on the technical replicates for each sample.
Discussion
This functional analysis of missense variants in the KCNK18 gene provides an important insight in the pathogenesis of typical migraine. Whilst our previous report supports a causative role for the F139Wfsx24 loss of function mutation in the KCNK18 gene in familial migraine MA20, the data presented here suggest a more complex association of other loss of function variants with typical migraine.
The R10G, S231P and A233V variants had no obvious effect on TRESK activity consistent with their identification in both control and migraine cohorts. Indeed, the A233V variant has already been reported in the Single Nucleotide Polymorphism Database (rs363360) and has been shown to be common (minor allele frequency = 0.15) in African populations. However, for the two mutants (A34V and C110R) which did show a loss of TRESK function, their association with typical migraine is unclear. The A34V variant was indeed found in an unrelated proband diagnosed with typical migraine and was absent in all controls tested20. However, the C110R variant appears to show no genetic association with typical migraine.
The complete loss of function seen in C110R variant is perhaps not surprising as this residue is located adjacent to the selectivity filter where substitution with an arginine is likely to be disruptive to channel function. Interestingly, both the A34 and C110 residues are located close to one another and the selectivity filter has recently been implicated in the control of K2P channel gating30. Whether these mutations lead to an intrinsic loss of function and/or impair channel trafficking, the functional consequences appear the same i.e. a reduction in TRESK channel activity. Furthermore, both A34V and C110R variants, like the F139Wfsx24 variant, appear to have the ability to interact with and reduce the activity of coexpressed WT TRESK. Therefore in the heterozygous state, in addition to any intrinsic loss of function, both variants could further reduce TRESK activity through coassembly with WT subunits. In other K+ channelopathies e.g. Episodic Ataxia Type 1, non-functional mutant subunits also reduce channel activity through ‘dominant-negative’ coassembly with WT subunits and often result in >90% suppression of channel activity31,32,33. However, in contrast to tetrameric channels, the assembly of K2P channels as a dimer of dimers means that any dominant negative effect will always be far less severe, because even if the mutant subunit can coassemble with WT subunits at least 25% homomeric WT channels will remain. Consequently, for K2P channels in particular, any potential relationship between a dominant inheritance pattern and a dominant negative effect on channel function may be difficult to interpret.
Our observation that some unaffected individuals appear to carry quite deleterious mutations obviously questions a simple direct causative link between defective TRESK function and typical migraine. Differences in KCNK18 copy number variation cannot account for our original observation, because all individuals with the C110R mutation have the expected two copies of KCNK18 irrespective of their migraine status. Genomic imprinting is also unlikely as both maternal and paternal transmission is observed20. Perhaps the most likely explanation is that migraine is now widely accepted as a complex polygenic disorder, even in the case of apparently Mendelian segregation34 and is therefore undoubtedly dependent upon the interplay of multiple genes as well as gene-environment interactions9. In other complex disorders of neuronal excitability e.g. epilepsy, a recent report suggested that even deleterious ion channel mutations conferred uncertain risk to an individual and this risk was highly-dependent on other variants with which they are combined35. Such considerations are now becoming typical of the challenges required to link a disease-causing role to an inherited variant in a single ion channel, despite the overwhelming evidence that many common neurological disorders are associated with ion channel dysfunction36,37.
In conclusion, if the influence of TRESK on TG neuronal excitability does indeed play a role in the pathogenesis of migraine then its effects are undoubtedly complex. These results demonstrate that despite the significant genetic linkage of the F139Wfsx24 variant in an extended MA family, the presence of a single non-functional variant is probably not alone sufficient to determine whether an individual develops migraine. This further highlights the importance of understanding the broader genetic background of any affected individual.
Methods
Molecular Biology
Human TRESK was subcloned between the 5’ and 3’ UTR of the Xenopus β-globin gene in the oocyte expression vector, pBF. The R10G, A34V, C110R, S231P and A233V mutants were made by site directed mutagenesis and confirmed by automated sequencing. Wild type and mutant tandem linked dimers of TRESK were made using extension overlap PCR which deleted the stop codon of the first subunit and introduced a Gly(Gln)10Gly linker immediately prior to the first methionine of the second TRESK subunit. mRNA for wild-type, mutant and tandem linked dimeric channels was synthesized using the SP6 mMESSAGE mMACHINE kit (Ambion) and mRNA concentrations were quantified by spectrophotometric analysis prior to injection.
Electrophysiological recordings
Unless otherwise stated, equal quantities of either wild-type or mutant mRNAs were microinjected into defolliculated Xenopus oocytes according to standard protocols. Whole-cell currents were recorded 18–24 hours after injection, at room temperature (20–22°C), using the two-electrode voltage clamp method (Geneclamp 500B, Axon Instruments). Oocytes were held in a small recording chamber and were continuously perfused with control extracellular solution, lowK (in mM: 95.4 NaCl, 2 KCl, 1.8 CaCl2, 5 HEPES pH 7.5 with NaOH) using a pump driven perfusion system. Recording electrodes were back-filled with 3M KCl and their resistances varied from 0.3 to 1 MΩ. Currents were filtered at 100 Hz and digitised at 1 kHz for analysis. Oocytes were held at −80 mV, voltage commands were applied and currents were recorded using pClamp 9 software (Axon Instruments). TRESK currents were usually measured in lowK solution, at the end of 1 second long voltage steps from a holding potential of −80 mV delivered in 20 mV increments from −120 mV to +60 mV. For the experiments with ionomycin, TRESK currents were measured in highK solution (in mM: 17.4 NaCl, 80 KCl, 1.8 CaCl2, 5 HEPES, pH 7.5 with NaOH) at the end of 300 ms-long voltage steps from 0 mV to −100 mV. Ionomycin (free acid form), was made as a stock solution of 5 mM in dimethyl sulphoxide and diluted in the highK solution to the appropriate test concentration (0.5 μM or 1 µM for activation of tandem dimers) on the day of the experiment. Data were analysed with pCLAMP9 (Axon instruments).
Homology Modelling
To build a structure of the TRESK channel pore, the extracellular and intracellular loops were omitted due to a lack of homology with any known structural template. The secondary structure and transmembrane topology for TRESK (Q7Z418) was predicted using the PSI-Prediction server (http://bioinf.cs.ucl.ac.uk/psipred/). KvAP (PDB 1ORQ) was chosen as the primary structural templates for the TRESK homology models and ClustalX used to align two copies of the KvAP sequence modified to resemble a dimer. Alignments were generated conserving key features such as the selectivity filter, glycine-hinge (in M2 and M4) and glutamate residues at the top of M1 and M3 helices. The homology models of TRESK were created using Modeller 9v8. For each set of ten TRESK models generated, the lowest energy model was selected.
Copy number values for KCNK18
A known copy number variation (CNV) identified in a Micronesian control cohort overlaps the second exon and first and second introns of KCNK18 (Gusev et al. 2009 Genome Res. 19:318–26.). The presence of this CNV was analyzed in genomic DNA from 20 individuals with and without migraine with the C110R variant from the Australian case-control cohort using an Applied Biosystems TaqMan Copy Number Assay. The KCNK18 CNV assay (Assay ID: Hs00964672_cn) was run along with the recommended control assay taken from a region devoid of CNVs (RnaseP). Triplicate assays were run on the AB 7900HT System under standard conditions according to the manufacturer’s protocol. Data was analysed using CopyCaller Software (v1.0 Applied Biosystems).
Statistics
Data are given as mean values ± standard error of the mean (SEM), where n represents the number of oocytes. Results were reproducible in at least 2–3 different batches of oocytes. Statistical significance was determined using a student’s t-test. When error bars are not shown they are smaller than the size of the symbol.
References
Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders: 2nd edition. Cephalalgia 24 Suppl 1, 9–160 (2004).
de Vries, B., Frants, R. R., Ferrari, M. D. & van den Maagdenberg, A. M. Molecular genetics of migraine. Hum. Genet. 126, 115–132 (2009).
Ophoff, R. A. et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552 (1996).
Dichgans, M. et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366, 371–377 (2005).
De Fusco, M. et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 33, 192–196 (2003).
Pietrobon, D. Familial hemiplegic migraine. Neurotherapeutics 4, 274–284 (2007).
Ayata, C. Cortical spreading depression triggers migraine attack: pro. Headache 50, 725–730 (2010).
Eikermann-Haerter, K. & Ayata, C. Cortical spreading depression and migraine. Curr. Neurol. Neurosci. Rep. 10, 167–173 (2010).
Maher, B. H. & Griffiths, L. R. Identification of molecular genetic factors that influence migraine. Mol. Genet. Genomics (2011).
Pietrobon, D. & Striessnig, J. Neurobiology of migraine. Nat. Rev. Neurosci. 4, 386–398 (2003).
Goadsby, P. J., Charbit, A. R., Andreou, A. P., Akerman, S. & Holland, P. R. Neurobiology of migraine. Neuroscience 161, 327–341 (2009).
Andreou, A. P. & Goadsby, P. J. Therapeutic potential of novel glutamate receptor antagonists in migraine. Expert Opin. Investig. Drugs 18, 789–803 (2009).
Suzuki, M. et al. Defective membrane expression of the Na(+)-HCO(3)(−) cotransporter NBCe1 is associated with familial migraine. Proc. Natl. Acad. Sci. U. S. A. 107, 15963–15968 (2010).
Anttila, V. et al. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat. Genet. 42, 869–873 (2010).
Chasman, D. I. et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat. Genet. 43, 695–698 (2011).
Enyedi, P. & Czirjak, G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol. Rev. 90, 559–605 (2010).
Czirjak, G., Toth, Z. E. & Enyedi, P. The two-pore domain K+ channel, TRESK, is activated by the cytoplasmic calcium signal through calcineurin. J. Biol. Chem. 279, 18550–18558 (2004).
Yoo, S. et al. Regional expression of the anesthetic-activated potassium channel TRESK in the rat nervous system. Neurosci. Lett. 465, 79–84 (2009).
Bautista, D. M. et al. Pungent agents from Szechuan peppers excite sensory neurons by inhibiting two-pore potassium channels. Nat. Neurosci. 11, 772–779 (2008).
Lafreniere, R. G. et al. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat. Med. 16, 1157–1160 (2010).
Czirjak, G. & Enyedi, P. Targeting of calcineurin to an NFAT-like docking site is required for the calcium-dependent activation of the background K+ channel, TRESK. J. Biol. Chem. 281, 14677–14682 (2006).
Czirjak, G., Vuity, D. & Enyedi, P. Phosphorylation-dependent binding of 14-3-3 proteins controls TRESK regulation. J. Biol. Chem. 283, 15672–15680 (2008).
Piechotta, P. L. et al. The pore structure and gating mechanism of K2P channels. EMBO J. 30, 3607–3619 (2011).
Sano, Y. et al. A novel two-pore domain K+ channel, TRESK, is localized in the spinal cord. J. Biol. Chem. 278, 27406–27412 (2003).
Kang, D., Mariash, E. & Kim, D. Functional expression of TRESK-2, a new member of the tandem-pore K+ channel family. J. Biol. Chem. 279, 28063–28070 (2004).
Yoshida, S. & Plant, S. Mechanism of release of Ca2+ from intracellular stores in response to ionomycin in oocytes of the frog Xenopus laevis. J. Physiol. 458, 307–318 (1992).
Pessia, M., Tucker, S. J., Lee, K., Bond, C. T. & Adelman, J. P. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J. 15, 2980–2987 (1996).
Czirjak, G. & Enyedi, P. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J. Biol. Chem. 277, 5426–5432 (2002).
Es-Salah-Lamoureux, Z., Steele, D. F. & Fedida, D. Research into the therapeutic roles of two-pore-domain potassium channels. Trends Pharmacol. Sci. 31, 587–595 (2010).
Mathie, A., Al-Moubarak, E. & Veale, E. L. Gating of two pore domain potassium channels. J. Physiol. 588, 3149–3156 (2010).
Adelman, J. P., Bond, C. T., Pessia, M. & Maylie, J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron 15, 1449–1454 (1995).
Zerr, P., Adelman, J. P. & Maylie, J. Episodic ataxia mutations in Kv1.1 alter potassium channel function by dominant negative effects or haploinsufficiency. J. Neurosci. 18, 2842–2848 (1998).
D'Adamo, M. C., Liu, Z., Adelman, J. P., Maylie, J. & Pessia, M. Episodic ataxia type-1 mutations in the hKv1.1 cytoplasmic pore region alter the gating properties of the channel. EMBO J. 17, 1200–1207 (1998).
Ulrich, V., Russell, M. B., Ostergaard, S. & Olesen, J. Analysis of 31 families with an apparently autosomal-dominant transmission of migraine with aura in the nuclear family. Am. J. Med. Genet. 74, 395–397 (1997).
Klassen, T. et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 145, 1036–1048 (2011).
Schorge, S. & Kullmann, D. M. Sodium channel mutations and epilepsy: association and causation. Exp. Neurol. 226, 8–10 (2010).
Kullmann, D. M. & Waxman, S. G. Neurological channelopathies: new insights into disease mechanisms and ion channel function. J. Physiol. 588, 1823–1827 (2010).
Acknowledgements
This work was supported by research grants from the Wellcome Trust, the Medical Research Council and Pfizer.
Author information
Authors and Affiliations
Contributions
I.A–E., L.S., P.J.S and J.M.M performed research, M.Z.C., M.S.P.S and S.J.T. designed and supervised the project. R.G.L., B.R., L.R.G, G.A.R. and G.C.E. provided important genotypic information. I.A–E. and S.J.T wrote the manuscript with input from other authors.
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Andres-Enguix, I., Shang, L., Stansfeld, P. et al. Functional analysis of missense variants in the TRESK (KCNK18) K+ channel. Sci Rep 2, 237 (2012). https://doi.org/10.1038/srep00237
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00237
This article is cited by
-
Genetics of migraine: where are we now?
The Journal of Headache and Pain (2023)
-
Current Update on Categorization of Migraine Subtypes on the Basis of Genetic Variation: a Systematic Review
Molecular Neurobiology (2023)
-
K2P18.1 translates T cell receptor signals into thymic regulatory T cell development
Cell Research (2022)
-
Common variants in KCNK5 and FHL5 genes contributed to the susceptibility of migraine without aura in Han Chinese population
Scientific Reports (2021)
-
Genetics of migraine aura: an update
The Journal of Headache and Pain (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
