
Abstract
Addictive drugs have been shown to severely influence many neuronal functions, which are considered as the underlying mechanisms for physiological and psychological dependences. We previously showed that in vivo LTP in rat hippocampal CA1 region is significantly reduced during withdrawal following chronic opiates treatment, and the reduced LTP can be restored by re-exposure of animals to corresponding drugs. Here, we further demonstrated that during opiates withdrawal, the re-exposure of morphine either systemically (subcutaneously) or locally (intracerebroventricularly) could restore the reduced LTP in heroin-dependent rats, but heroin could not restore the reduced LTP, in morphine-dependent rats, indicating differential modulations of hippocampal functions by those two opiates. In contrast, DAMGO, a mu-opioid receptor (MOR) agonist, could restore the reduced LTP, and CTOP, a MOR antagonist, could block the restoration in rats dependent on both opiates, showing that MOR is functional under such conditions. However, the upregulation of hippocampal PKA activity during morphine withdrawal could be suppressed by re-exposure of morphine but not that of heroin, suggesting a likely underlying mechanism of the differential modulation of LTP by two opiates. Taken together, our study clearly demonstrates that chronic abuse of opiates inevitably leads to severe alteration of hippocampal LTP, and reveals the interesting differences between morphine and heroin in their effects on the differential modulation of hippocampal synaptic plasticity.
Similar content being viewed by others


INTRODUCTION
Opiates such as morphine and heroin are among the most widely consumed drugs of abuse (Pouletty, 2002). Morphine currently serves as the most effective pain reliever and is frequently prescribed for moderate to severe pain, but the clinical utility of morphine can often alter the central pain-related systems and is hampered by the development of tolerance, decreased analgesic effect of the drugs, and physical and even psychological dependence, a behavioral state requiring continued dose to avoid a series of aversive withdrawal syndromes following the long-term usage of the drugs (O’Brien, 1997). Heroin (3, 6-diacetylmorphine) was introduced in 1898 and was heralded as a more potent pain killer and remedy for morphine. However, the major pharmacological effects of heroin can be traced back to structural properties of the morphine molecule, and heroin shares a similar pharmacological profile with morphine: both induce analgesia, hypothermia, sedation, inhibition of intestinal motility, and depression of the immune function (Gutstein and Akil, 2001; Thronhill et al, 1976; O’Mahony et al, 2001; Petersen and Fujimoto, 1983; Ghodse and Reed, 1984; Manzanares et al, 1999; Vallejo et al, 2004; Weber et al, 2004).
As a derivative of morphine, heroin also exhibits many properties distinguished from morphine. On the basis of the current findings (Kamendulis et al, 1996; Glare and Walsh, 1991; Milne et al, 1996), heroin may function as a highly lipophilic prodrug for the active metabolites morphine, 6-monoacetylmorphine and morphine-6-glucuronide, which may contribute to the subjective effects of heroin (Inturrisi et al, 1983, 1984; Corrigall and Coen, 1990). Considering the very limited structural differences from heroin to morphine, it is also interesting that heroin is easier to cross the blood–brain barrier owing to its increased hydrophobicity and is faster to be absorbed owing to its higher solubility. Although heroin itself has low affinity for opioid receptors, it is rapidly metabolized to 6-MAM and morphine, which have high affinity for mu-opioid receptors (MOR) (Inturrisi et al, 1983, 1984). Antisense mapping also demonstrates a differential effect on analgesia between heroin and morphine (Rossi et al, 1995a, 1995b, 1996, 1997). Antioligodeoxynucleotides targeting exon 2 of the MOR-1 gene can decrease heroin and M6G analgesia, but not morphine (Rossi et al, 1996), and, conversely, the deletion of exon 1 effectively lowers morphine analgesia but has no effect on heroin and M6G (Matthes et al, 1996; Schuller et al, 1999). Finally, heroin and morphine show an incomplete cross-tolerance owing to the maintenance of heroin analgesic activity in a mouse model of morphine tolerance (Lange et al, 1980; Rossi et al, 1996; Gilbert et al, 2004).
The hippocampus, a well-known subcortical brain structure related to spatial learning and memory (Moser et al, 1998; Bliss and Collingridge, 1993; Malenka and Nicoll, 1999), plays a key role in message encoding and retrieving in the CNS (Riedel et al, 1999; Morris et al, 2003). Cumulating evidence has revealed that the hippocampus is also a crucial region involved in restoration and distribution of the addictive drug-associated information in the brain (Nestler, 2001a), engaging in reward-related response (White, 1996; Rezayof et al, 2003) and drug-seeking behavior (Vorel et al, 2001; Black et al, 2004; Yang et al, 2004). As a brain disease, drug addiction has been considered as a neuronal adaptation with the altered functions of neuronal circuits (Leshner, 1997; Volkow and Li, 2004; Nestler, 2004), including changes in neuronal plasticity (Nestler and Aghajanian, 1997; Zhang et al, 1998; Robbins and Everitt, 1999; Nestler, 2001b). Addictive drugs like morphine and heroin may employ learning and memory mechanisms in brain regions including the hippocampus and may also affect the normal function of those regions leading to a formation of aberrant learning. It has been reported that chronic exposure to opiate could significantly decrease neurogenesis and alter synaptic transmission in the hippocampus (Eisch et al, 2000).
Our previous results demonstrated that chronic morphine and heroin treatment could reduce hippocampal LTP and re-exposure of the animal to the corresponding opiates could restore LTP induction, reflecting an opiate-dependent neuronal plasticity (Pu et al, 2002). As morphine and heroin display many different features, the present study further investigates whether these opiates can have differential effect on the opioid dependent LTP.
MATERIALS AND METHODS
Animals
Male Sprague–Dawley rats (200–250 g) were obtained from the Laboratory Animal Center, Chinese Academy of Sciences (Shanghai, China). Rats were housed in groups and maintained on a 12 h light/dark cycle with food and water available ad libitum. All animal treatments were strictly in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Drugs and Injection
Animals were chronically treated by subcutaneous injection of morphine or heroin twice per day at 12 h intervals for 10 days as previously described (Trujillo and Akil, 1991; Fan et al, 1999). Control rats were treated similarly, except that normal saline (NS) was used. The drugs used in the present study were morphine hydrochloride (Shenyang Pharmaceutical Ltd, China), diacetylmorphine hydrochloride (heroin) (National Laboratory of Narcotics, Beijing, China), [D-Ala(2),N-methyl-Phe(4),Gly(5)-ol]-Enkephalin (DAMGO; Sigma), D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP; Sigma), [D-Pen2,D-Pen5]-Enkephalin (DPDPE, Sigma), deltorphin I (DELT, Sigma), and Naltrindole (NTI, Sigma). All of the above drugs were dissolved in saline. DAMGO, CTOP, DPDPE, DELT, and NTI were administrated intracerebroventricularly (i.c.v.). Vehicle or drugs were injected into the left lateral ventricle (AP from bregma: −0.9 mm; LR: −1.2 mm, DV from skull: −4.5 mm) using a modified Hamilton microliter syringe in a volume of 2 μl per rat.
Electrophysiological Recording In Vivo
Recording was performed under urethane (1.3 g/kg, i.p.) anesthetization. Recordings of field EPSPs (fEPSPs) were made from the CA1 stratum radiatum of the right hippocampal hemisphere in response to stimulation of the Schaffer collateral–commissural pathway. The recording electrode was inserted 3.4 mm posterior to bregma and 2.5 mm right of the midline, and the stimulating electrode was inserted 4.2 mm posterior to bregma and 3.8 mm right of the midline. The dura mater was pierced, and the electrodes were lowered slowly through the cortex and the upper layers of the hippocampus into the CA1 region until the appearance of a negative-deflecting EPSP. The right placement of the electrodes in the stratum radiatum of the CA1 region of the dorsal hippocampus was verified by postmortem examination.
In all experiments, fEPSPs were evoked by stimulating with a square-wave constant current pulse of 50 μs duration at a frequency of 0.033 Hz. At the beginning of each experiment, input–output curves were generated to determine the maximal fEPSP slope, and then the intensity of stimulus was set at a level that evoked a fEPSP slope of 55–65% of the maximum. The slope of fEPSP was measured and averaged every 3 min. LTP was induced by high-frequency stimulation using 20 pulses at 200 Hz, repeated three times at a 30 s interval. All recording and stimulation was performed using an online computerized oscilloscope stimulator and data analysis interface system. Statistical comparisons between baseline and poststimulation values were made using Student's t-test. Values are mean percentage of the baseline fEPSP amplitude±SEM.
Protein Kinase Activity Assay
PKA activity was determined essentially according to the method described by Lou and Pei (1997). The animals were acutely decapitated, and the hippocampi were dissected rapidly and homogenized on ice in homogenization buffer (25 mM Tris-HCl, 1 mM EDTA, 1 mM DTT, and 100 μM leupeptin). The homogenate was centrifuged at 20 000g for 5 min at 4°C. The resulting supernatant was assayed for PKA activity using a PepTag nonradioactive PKA assay kit (Promega, Madison, WI), as described in the Promega Technical Bulletin. All reaction components were added on ice in a final volume of 25 μl of the following mixture: 5 μl of PepTag PKA reaction buffer, 5 μl of PepTag A1 Peptide (0.4 μg/μl), 5 μl of cAMP (5 μM), and 5 μl of sample homogenate. The mixture was incubated for 30 min at 30°C. Then, the reaction was stopped by placing the tube in a boiling water bath for 10 min, and the samples were loaded onto the gel for electrophoresis. Before loading samples, 1 μl of 80% glycerol was added to the sample to ensure that it remained in the well. PKA-specific peptide substrate used in this experiment was PepTag A1 Peptide, L-R-R-A-S-L-G (kemptide). The assay was based on the changes in the net charge of the fluorescent PKA substrates before and after phosphorylation. This change allowed the phosphorylated and unphosphorylated versions of the substrate to be rapidly separated on an agarose gel at neutral pH. The phosphorylated species migrated toward the positive electrode, whereas the nonphosphorylated substrate migrated toward the negative electrode. The intensity of fluorescence of phosphorylated peptides, which reflected the activity of PKA, was quantified using a bioimaging system (Syngene, Cambridge, UK). Data in the graphs are presented as the mean±SEM.
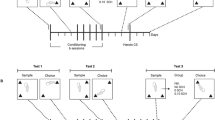
Conditioned Place Preference Test
The conditioned place preference (CPP) test box consisted of a shuttle chamber made of Plexiglas (30 × 60 × 30 cm) and was divided into two equal-sized compartments by insertion of a removable Plexiglas wall. The test box was black. One compartment had white stripes on the wall and a textured floor and the other had walls with white dots and a smooth floor. The test box was placed under conditions of dim illumination (40 lux) and masking white noise. A single-trial CPP model was carried out here. Before the experiment, rats had free access to both the compartments in CPP test box for 2 h per day for 2 days in order to be familiar with the environments. Then, on day 1 (preconditioning), rats were placed in the boxes and were allowed to roam freely from side to side for 15 min, and time spent in each side was recorded. These data were used to separate the animals into groups of approximately equal bias. On days 2–10, rats received chronic saline or morphine injection in their home cages. On day 11, the conditioning session, 12 h after the termination of chronic treatment, rats were re-injected with saline, 10 mg/kg of morphine or 1.0 mg/kg of heroin and were confined randomly to one of the compartments in CPP test box for 1 h. At least 6 h later on the same day, all the animals received an additional injection of saline and were confined to the opposite side of the test box. On day 13, the test day, animals were all given a saline injection and allowed to roam freely between the two sides. The time spent on each side was recorded, and data are expressed as time spent on drug-paired side minus time spent on saline-paired side. A positive number indicates a preference for the drug-paired side, whereas a negative number indicates an aversion to the drug-paired side. A number at or near zero indicates no preference for either side.
Statistical Analysis
The data presented in the figures are mean±SEM. Statistical analysis of the data was carried out by using the Student's t-test or the one-way analysis of variance.
RESULTS
Differential Modulation of LTP by Subcutaneous Re-administration of Opiates
Chronic morphine or heroin treatment results in a reduction of LTP at CA1 synapses during drug withdrawal and re-exposure of the same opiates can restore this LTP reduction (Pu et al, 2002). In this study, we examined the effect of different opiates re-administration on the reduced LTP in morphine or heroin-dependent rats. For creating morphine or heroin dependence in rats, 10 mg/kg morphine or 1 mg/kg heroin was subcutaneously injected twice per day for 10 days, a procedure known to produce significant tolerance and dependence to drugs (Trujillo and Akil, 1991; Fan et al, 1999). Twelve hours after the termination of opiates treatment, morphine, heroin, or NS was subcutaneously re-injected, respectively. Then 30 min later, LTP was induced by high-frequency stimulation (HFS) of the Schaffer collateral–commissural inputs to CA1 pyramidal cells.
The results showed that during heroin withdrawal, normal LTP (∼190% of baseline) could be induced (Figure 1d) after the re-injection of morphine (10 mg/kg, s.c.), but during morphine withdrawal, LTP failed to be induced (Figure 1c) after the re-injection of heroin (1 mg/kg, s.c.), suggesting a differential effect between morphine and heroin on restoration of LTP in opiate-dependent rats. For control experiments, after re-injection of 1 mg/kg heroin (s.c.) in heroin-dependent rats, the LTP restoration was observed as compared with NS (Figure 1b), and the LTP reduction in morphine-dependent rats could be restored if morphine (10 mg/kg, s.c.) was re-injected (Figure 1a).

Differential restoration of the suppressed hippocampal LTP by subcutaneous re-administration of morphine and heroin. Twelve hours after the termination of chronic morphine (a, c) or herion (b, d) treatment, the opiate-dependent rats were s.c. re-injected with morphine (Mor; 10 mg/kg), heroin (Her; 1 mg/kg), or NS. In vivo hippocampal LTP was induced 30 min after the drug re-injection and expressed by the increase in fEPSP slopes (% of baseline). Insets, Sample fEPSPs (average of six consecutive sweeps) at the time indicated by the numbers. Calibration: 10 ms, 1 mV. Arrows indicate HFS and triangles indicate drug injection. (e) Statistic summary of LTP restoration by opiate re-injection in various conditions in morphine (Mor rat) or heroin (Her Rat)-dependent rats. LTP induction was analyzed 30 min after HFS using Student’s t-test, *p<0.01 compared with NS in each group. (f) Dose–response curve of chronic morphine or heroin treatment. Morphine, in doses of 0.1, 0.3, 1, 3, 10, and 30 mg/kg, or heroin, in doses of 0.03, 0.1, 0.3, 0.5, 1, and 3 mg/kg, was used b.i.d. for 10 days as LTP induction was measured 12 h after the termination of the last injection.
As heroin is rapidly hydrolyzed in the body and takes effect quickly (Inturrisi et al, 1984; see Supplementary material), a shorter period of time between heroin re-injection and LTP induction was chosen. As shown in Figure 1e, in morphine-dependent rats, 15 min after re-injection of heroin (1 mg/kg, s.c.), LTP still could not be restored; moreover, re-injection of a higher dose of heroin (10 mg/kg, s.c.; Figure 1e) also failed to restore the reduced LTP. However, both of those two conditions could significantly restore the LTP reduction in heroin-dependent rats (Figure 1e). These results indicate that morphine and heroin can differentially modulate hippocampal LTP in opiate-dependent rats.
To further evaluate the differential effects on LTP reduction by chronic morphine or heroin treatment, full dose–response curves of both opiates were generated. As shown in Figure 1f, the dose–response curve of heroin was approximately 10-fold left shift from that of morphine. When using lower dose of morphine (1 mg/kg) or heroin (0.1 mg/kg) for chronic treatment, LTP seemed normal during withdrawal, and when using 10 mg/kg morphine or 1 mg/kg heroin for chronic treatment, LTP declined to nearly the same level close to the plateau (Figure 1f). Thus, 10 mg/kg morphine and 1 mg/kg heroin were chosen for the chronic treatment.
Differential Modulation of LTP by i.c.v. Re-administration of Opiates
To further confirm the differential effect by morphine and heroin on LTP, the central actions of both opiates were tested directly in CNS by specific microinjection into the brain. Rats were chronic treated with morphine or heroin 10 days as above. Twelve hours after the termination of opiates treatment, a second injection was given i.c.v. 10 nmol per rat of morphine or 1 nmol per rat of heroin was used, a dose being able to induce analgesia in rodents. As shown in Figure 2a and b, after re-injection of morphine (10 nmol, i.c.v.) in morphine-dependent rats (Figure 2a) or heroin (1 nmol, i.c.v.) in heroin-dependent rats (Figure 2b), normal LTP (∼190% of baseline) could be induced by HFS as compared with NS control respectively, indicating a restoration of LTP by i.c.v. opiate readministration. Dissimilarly, LTP was apparently restored in heroin-dependent rats with i.c.v. re-injection of morphine (Figure 2d), but was not restored in morphine-dependent rats with i.c.v. re-injection of heroin (Figure 2c). Additionally, re-injection of heroin at a higher dose (10 nmol/rat, i.c.v.) was able to restore the reduced LTP in heroin-dependent rats as shown in Figure 2e, but still failed to restore the reduced LTP in morphine-dependent rats.

Differential restoration of the suppressed hippocampal LTP by i.c.v. re-administration of morphine and heroin. The morphine (a, c) or herion (b, d)-dependent rats were i.c.v. injected with morphine (Mor; 10 nmol), heroin (Her; 1 nmol), or NS. Hippocampal LTP was induced 30 min after the drug application. Insets, sample fEPSPs (average of six consecutive sweeps) at the time indicated by the numbers. Calibration: 10 ms, 1 mV. Arrows indicate HFS and triangles indicate drug injection. (e) LTP restoration by i.c.v. administration of opiates under various conditions in NS, morphine Mor or Her dependent rats. Data of LTP induction were analyzed 30 min after HFS using Student’s t-test; *p<0.01 compared with NS in each group.
As drugs took effect faster by i.c.v. route than s.c. injection, a shorter period of time between i.c.v. injection and LTP induction was also tested. In heroin-dependent rats, 15 min after the re-injection of heroin (1 nmol/rat, i.c.v.), LTP could be restored to the normal level, but in morphine-dependent rats, LTP remained uninducable (Figure 2e).
The above results further illustrate the differential effect of morphine and heroin in CNS on the modulation of LTP.
Involvement of MOR in LTP Restoration by Morphine and Heroin
A MOR-specific agonist, DAMGO, and antagonist, CTOP, were also used here to further investigate whether the differential effect of morphine and heroin on LTP restoration was mainly mediated by MOR. Rats were chronically treated with morphine or heroin as above. And during drug withdrawal DAMGO or CTOP was i.c.v. administrated before LTP induction. As shown in Figure 3a and b, 12 h after the termination of drug treatment, i.c.v. injection of DAMGO (1.5 nmol/rat) could significantly restore the reduced LTP to apparent normal level (∼190% of baseline), in both morphine-(Figure 3a) and heroin (Figure 3b)-dependent rats, which mimicked the effect of morphine re-injection on LTP restoration.

DAMGO restoration of the suppressed LTP. Twelve hours after the termination of chronic morphine (a) or heroin (b) treatment, the opiate-dependent rats were injected with DAMGO (1.5 nmol/rat, i.c.v.) or vehicle, respectively. Hippocampal LTP was induced 30 min after the drug application. Arrows indicateHFS; triangles indicate drug injection.
As shown in Figure 4a, i.c.v. injection of CTOP in morphine-dependent rats could significantly block the restoration of LTP by morphine re-injection (10 mg/kg, s.c.) to approximately the baseline as compared with that of vehicle (Figure 4a). Actually, the blockage effect by CTOP was observed if LTP was restored by re-injection of morphine in morphine-dependent rats, re-injection of heroin in heroin-dependent rats, or i.c.v. injection of DAMGO in rats dependent on both opiates (Figure 4b). Taken together, these results of DAMGO and CTOP indicate that the restoration of LTP during withdrawal by re-application of opiates may be mediated by MOR.

The restoration of LTP can be blocked by CTOP. Twelve hours after the termination of chronic opiates treatment, rats were injected with CTOP (1.5 nmol/rat, i.c.v.) or vehicle and 2 h later re-injected with NS, morphine (10 mg/kg, s.c.), or heroin (1 mg/kg, s.c.). Hippocampal LTP was then induced 30 min later. (a) As compared with vehicle, CTOP could block the LTP restoration by morphine re-injection in morphine-dependent rats. Arrows indicateHFS; triangles indicate drug injection. (b) Statistic summary on the effects of CTOP in Mor- or Her dependent rats. LTP induction was analyzed 30 min after high-frequency stimulation using Student’s t-test, *p<0.01 compared with vehicle in each group.
Involvement of Delta-Opioid Receptor in LTP Restoration by Morphine and Heroin
As is known, most of opiate-induced antinociception and dependence was mediated by MOR; still, there was evidence indicating an involvement of delta-opioid receptor (DOR) system in response to morphine analgesia and tolerance (Zhu et al, 1999; Nitsche et al, 2002). Therefore, the effects of DOR-specific agonist and antagonist on LTP restoration following chronic morphine or heroin treatment were also studied in the paper. Rats were chronically treated with morphine or heroin as above, and 12 h after the termination of drug treatment, DOR-specific agonists, DPDPE or deltorphin, and antagonist NTI were i.c.v. administrated before LTP induction. As shown in Figure 5a and b, both DPDPE (10 nmol/rat, i.c.v.) and deltorphin I (1 nmol/rat, i.c.v.) could restore LTP to approximately 150% (Figure 5a), which was near half of the normal LTP induction level. Moreover, this LTP restoration could be completely blocked by DOR-specific antagonist, NTI (2 nmol/rat, i.c.v. 30 min before agonist application; Figure 5b), indicating a specific DOR-mediated reaction in the current condition.

The effects of DPDPE, deltorphine I and NTI on LTP restoration. (a) The effects of DPDPE and DELT on restoration of the suppressed LTP. Twelve hours after the termination of chronic morphine treatment, the rats were injected with DPDPE (10 nmol/rat, i.c.v.), deltorphin I (DELT, 1 nmol/rat, i.c.v.), or vehicle, respectively. Hippocampal LTP was induced 30 min after the drug application. Arrow indicates HFS; triangle indicates drug injection. (b) Statistic summary of LTP restoration by drug injection in various conditions in Mor or Her- dependent rats. DOR antagonist, NTI (2 nmol/rat, i.c.v.), was administrated 30 min before morphine, heroin, DPDPE, or DELT administration. LTP induction was analyzed 30 min after HFS.
Unfortunately, pre-application of NTI (2 nmol/rat, i.c.v.) could not block the restoration of LTP by re-administration of morphine or heroin in opiate-dependent rats as shown in Figure 5b, suggesting that response induced by re-administration of morphine or heroin in opiates dependent rats was not mediated by DOR.
These results indicate that DOR system may be partially involved in the signaling pathways related to LTP restoration and, on the other hand, DOR itself may not mediate the reaction induced by re-administration of morphine or heroin in opiate-dependent rats.
Differential Effect of Morphine and Heroin on PKA Activity and CPP Formation in Opiate-Dependent Rats
Stimulation of opioid receptors, which are coupled with inhibitory G-proteins, is known to inhibit both the cAMP formation and the PKA activation (Childers, 1991; Chalecka-Franaszek et al, 2000). On the other hand, the rebound of the cAMP system is a well-known biochemical adaptation that correlates with chronic opiate exposure (Nestler and Aghajanian, 1997; Koob et al, 1998; Bohn et al, 2000). Our previous result showed that PKA activity was elevated after the termination of chronic morphine treatment and re-administration of morphine at that moment could reduce this overactivated PKA to normal level (Pu et al, 2002). However, differential modulation of PKA activity by morphine or heroin re-administration could also be found during morphine withdrawal. As shown in Figure 6a and b, 12 h after the termination of chronic morphine or NS treatment, rats were subcutaneously re-injected with morphine, heroin, and NS, respectively, and, 30 min later, PKA activity was tested. In chronic NS-treated group (NS group), PKA activity did not show any difference among the re-injection of morphine, heroin, or NS. On the contrary, in chronic morphine-treated group, PKA activity was significantly increased when NS was re-injected during withdrawal as compared with NS group, which was consistent with our previous results. However, the overactivated PKA activity was apparently suppressed to the control level by re-exposure of rats to morphine (10 mg/kg, s.c.), conversely, no apparent alteration was observed and PKA activity remained in its enhanced state by re-exposure of rats to heroin (1 mg/kg, s.c.); Thus, these results indicate a differential regulation of hippocampal PKA activity by morphine and heroin in morphine-dependent rats.

Differential suppression of the upregulated hippocampal PKA activity. In opiate-dependent or NS rat, PKA activity of the hippocampal extracts was measured 30 min after re-injection of morphine (10 mg/kg; s.c.), heroin (1 mg/kg; s.c.), or NS. (a) The upper band indicates the unphosphorylated substrates, and the lower band shows the phosphorylated peptides that reflect the PKA activity. (b) Statistical analysis of the optical density of fluorescence using Student's t-test. *p<0.01 vs NS re-injection; #p<0.01 vs corresponding drugs in the NS group.
The hippocampus plays a key role in learning and memory, and it was reported to be involved in a test of CPP (Ferbinteanu and McDonald, 2001; Zarrindast et al, 2005), a learning paradigm requiring formation of associations between reward and particular locations (Bardo and Bevins, 2000). Hence, CPP assay was carried out to see if the behavior consequence followed the pattern of differential modulation of hippocampal function by opiates. Rats, 12 h after chronic morphine treatment (10 mg/kg, s.c., b.i.d. 10 days), were re-exposed to morphine (10 mg/kg, s.c.), heroin (1 mg/kg, s.c.), or saline, respectively, and then trained 30 min later for 1 h. On the test day, time of occupancy in each compartment was recorded. Figure 7a was the timeline of the whole procedure. The results in Figure 7b showed that, after chronic morphine treatment, the rats with re-injection of morphine spent longer time in or significant preference to drug-paired side, whereas the rats with re-injection of heroin displayed no significant preference in either side, indicating that, in morphine-dependent rats, morphine re-administration could significantly induce CPP, but heroin could not. CPP experiment with single opiate injection was also performed in control rats (10 days NS treatment) as a comparison study. Twelve hours after single injection of morphine (10 mg/kg, s.c.), heroin (1 mg/kg, s.c.), or NS, the rats were exposed to CPP training and, on the test day, the time spent in each compartment was recorded. As shown in Figure 7c, when compared with control group, the group treated with either morphine or heroin could form CPP. These results illustrate that a differential influence on animal behavior exists between morphine and heroin, which matches the previous patterns of LTP restoration by re-injection of those opiates during drug withdrawal.

Differential effect of morphine and heroin on the induction of CPP. Rats were chronically treated as above. Twelve hours after the termination of chronic morphine or heroin treatment, CPP training was carried out using single injection of morphine (5 mg/kg, s.c.) or heroin (1 mg/kg, s.c.). (a) Timeline of the CPP procedure. (b) In morphine-dependent rats, re-exposure of morphine could significantly activate CPP but re-exposure of heroin could not. (c) In control rats, both opiates could induce CPP. Data were analyzed using Student’s t-test; *p<0.01 compared with NS re-injection in each group.
DISCUSSION
Morphine and heroin share many similarities (see Introduction). However, heroin, as a morphine derivative, still has its own properties that exhibit more potent analgesia and lower LD50 than morphine. It has been reported that heroin and morphine could produce dose-dependent antinociception, and heroin was 10- to 20-fold more potent than morphine in rhesus monkeys (Dykstra et al, 1987; Negus et al, 1998a, 1998b). In addition, heroin was found to be approximately 40 times more potent than morphine in lowering the reward threshold and approximately 6.5 times more potent in raising the escape threshold in rat (Hubner and Kornetsky, 1992). In the present study, heroin (1 mg/kg), at one-tenth of the morphine dose, could produce the same inhibition in hippocampal LTP as morphine, which was 10 mg/kg (Figure 1a, b and f). Nevertheless, when 1 mg/kg morphine was applied for all the treatment, it could hardly influence the LTP (Figure 1f). Such findings on the modulation of hippocampal LTP further confirmed the previous reports that heroin shows much more potency than morphine. On the other hand, the acute analgesic effect of both morphine and heroin on rats were also tested (see Supplementary), in order to compare the effects of opiate drugs themselves. Under the current experimental conditions, the antinociceptive response of 10 mg/kg morphine (s.c.) and 1 mg/kg heroin (s.c.) reached approximately the same level of the maximum possible effect , and the time to peak analgesic effect of heroin, which was near 15 min, was much shorter (maybe owing to its higher lipophility) than that of morphine, which was about 45 min. These results are also consistent with the previous reports.
As the findings demonstrated, LTP in both morphine-and heroin-dependent rats declined during withdrawal and the reduced LTP could be restored to a normal level by the re-exposure of animals to the corresponding opiates. However, the differential patterns of LTP modulation responding to morphine and heroin following long-term exposure of opiates still could be observed in the present study. Here, when drug-dependent rats were exposed to a different opiate (distinguished from the drug used for chronic treatment) during withdrawal, the reduction of LTP could be differentially regulated. Morphine applied either systemically or locally could recover the LTP reduction in heroin-dependent rats, whereas heroin could not restore this reduction in morphine-dependent rats. Such differences suggest that chronic treatment of morphine and heroin differentially alter the neuronal plasticity in rat hippocampus, and this may indicate some different molecular mechanisms involved in the circuits of signal transduction pathways including the opioid receptors and the downstream reaction cascades.
So far plentiful evidence has illustrated that heroin itself has low affinity for opioid receptors, it is rapidly metabolized to 6-MAM, morphine, and further M6G, all of which have relatively high affinity for MOR and are almost considered as potent analgesics. It has also been reported that one of heroin metabolites, 6-MAM, possesses higher efficacy than other heroin metabolites at mu-opioid receptors, which may contribute to the higher efficacy of heroin compared with morphine in certain behavioral paradigms in vivo (Selley et al, 2001). And systemically administrated heroin and its active metabolite, 6-MAM, were significantly more potent than all the other derivatives, including morphine in analgesic test (Gilbert et al, 2004). These above reports seem to give us the hint that heroin metabolites, especially the 6-MAM, might be mainly responsible for the differential effect between morphine and heroin. And they also bring forward the question how heroin metabolites and opioid receptors are differentially involved in hippocampal LTP.
As we know, of these three families of opioid receptors, mu, delta, and kappa (Pasternak, 1993; Snyder and Pasternak, 2003), the MOR plays a crucial role in mediating the actions of morphine and morphine-like drugs such as heroin (Matthes et al, 1996; Kitanaka et al, 1998; Waldhoer et al, 2004). As opioid receptors belong to the large family of seven transmembrane domains G-protein-coupled receptors, they share a very classical signal-transduction pathway, in which the binding of agonist to receptor initiates the release of G-protein alpha, and beta–gamma subunits then trigger (activates or inhibits) many different kinds of targets, and, in turn, relays the signals from outside to inside of the cells. Reports show that only a single MOR gene, Oprm1, has been identified (Min et al, 1994; Giros et al, 1995; Wendel and Hoehe 1998). Owing to the differential performance of morphine and heroin, as well as M6G on CXBK mice (Rossi et al, 1996), exon1 KO mice (Schuller et al, 1999) and exon2 KO mice (Matthes et al, 1996; Contarino et al, 2002), it is likely suggested that the different effect between morphine and heroin may be through an opioid receptor mechanism distinct from the classical theory of pharmacokinetics (Inturrisi et al, 1983, 1984). They probably act on different subtypes of MOR populations, the alternative splicing variants (Schuller et al, 1999). In rat, MOR is alternatively spliced into at least six isoforms (Pasternak et al, 2004). And mu-opioid agonists or antagonists show different binding affinities to those different splicing variants (Pasternak et al, 2004; Pasternak, 2004). Koch et al (1998) also discover that different c-terminal splicing variants of rat MOR, MOR1 and MOR1b, may modulate different receptor-internalization and resensitization (Koch et al, 1998). Given the above backgrounds, one of our hypotheses is that different MOR groups of the spliced variants may be activated by different agonists like morphine or heroin, and the different fates of those receptors may occur during the chronic opiates treatment and result in a different pattern of receptor variants in cell surface, which could then differentially alter the signal transduction pathway in that cell. Therefore, it is worth further studying, whether differential changes of the mu-receptor variants really exist during the chronic morphine or heroin treatment and whether heroin metabolites, 6-MAM or further M6G, may share the different signaling pathways other than morphine owing to the different receptor populations. From another point of view, mechanisms involved in phosphorylation, ubiquitination, and glycosylation of opioid receptors may also respond for such difference (Wei et al, 2004).
Although MOR mediates most of morphine-or heroin-induced analgesia and dependence, evidence suggests that other opioid receptors and endogenous ligands, particularly those of the DOR system and preproenkephalin, the precursor of both MOR and DOR endogenous peptide agonists, are also intimately involved (Zhu et al, 1999; Nitsche et al, 2002). Moreover, the discovery of cell surface MOR–DOR heterodimerization and the existence of two subtypes of DOR encoded by only single gene even makes the regulation between those two kinds of receptors more complex (Gomes et al, 2004; Gomes et al, 2000; Mcnally and Akil, 2002). However, our results associated with DOR (see Figure 5a and b) show that NTI, a highly selective DOR antagonist, could not block LTP restoration induced by either re-administration of morphine or heroin in both opioid-dependent rats, suggesting that signaling through DOR pathway may contribute little to LTP restoration by morphine or heroin in those opiates dependent rats. Whereas, on the other hand, the results also indicate that activation of DOR alone by its specific agonists, DPDPE or deltorphin (DELT), can at least partially modulate LTP in opioid-dependent rats. Although the mechanism involved here is not clear, the hypotheses could be that DOR may share some part of down–stream pathway of MOR and/or there is regulation occurring between DOR-MOR dimer itself. Moreover, the using of DOR agonists might also be an implication of a new therapeutic treatment for opiate addiction.
CPP is a learning paradigm requiring formation of associations between reward and particular locations (Bardo and Bevins, 2000). The hippocampus is one of the most important sites involved in learning and memory (see Introduction). It is also thought to be necessary for complex reward-related learning, particularly when animals must remember the place and cues associated with drug administration (White, 1996; Rolls and Xiang, 2005). In our CPP experiment, the induction of place preference seems to follow a similar pattern of hippocampal LTP. During withdrawal, when morphine was re-injected, LTP was normal and CPP could also be formed. On the contrary, re-injection of heroin could neither restore LTP nor induce CPP (Figure 7). These results hint a correlation between LTP in rat hippocampus and CPP during opiates addiction. However, whether or not the lesion of hippocampal function of LTP being the reason for the failure to induce CPP and the involved mechanism is still not clear and needs further study.
PKA, a critical molecule involved in hippocampal CA1 LTP (Blitzer et al, 1995), modulates activity of AMPA receptors and CaMKII (Roche et al, 1996; Blitzer et al, 1998), the key mediator of the molecular machinery of CA1 LTP (Silva et al, 1992; Malenka and Nicoll, 1999; Lee et al, 2000). Our previous results show that upregulation of cAMP pathway and overactivation of PKA in hippocampus appear to be responsible for the reduction of LTP after the chronic opiate treatment, and suppression of high PKA activity by opiates may account for the restoration of LTP (Pu et al, 2002). The present study shows that, in morphine-dependent rats, the overactivated PKA can be suppressed by re-exposure of animals to morphine but not to heroin, whereas in heroin-dependent rats, this PKA overactivation could be suppressed by both morphine and heroin. Such patterns of PKA modulation in the hippocampus show similarities to those of LTP restoration by morphine and heroin following the chronic opioid treatment. And this casual relationship may indicate a likely underlying mechanism for the differential modulation of hippocampal LTP by two opiates. It has been reported that the activity of catalytic subunit of PKA requires the phosphate on Thr-197, which enhances the phosphoryl transfer rate and increases the affinity for ATP (Adams et al, 1995). The two factors, the protein levels of the catalytic subunits released by cAMP binding and the phosphorylation levels of Thr-197 on those released catalytic subunits, mainly determine the PKA activity. But how those two factors are involved in the differential modulation of LTP by morphine and heroin is still unclear. A differential alteration of mu receptor may be involved in camp-mediated PKA signal pathway.
In summary, the present study offers for the first time the evidence that morphine and heroin could differentially regulate the neuronal plasticity of the hippocampus and may provide a new insight into opiates and their derivatives on the differential regulation of the signal transduction pathway during long-term usage of the drugs.
References
Adams JA, McGlone ML, Gibson R, Taylor SS (1995). Phosphorylation modulates catalytic function and regulation in the cAMP-dependent protein kinase. Biochemistry 34: 2447–2454.
Bardo MT, Bevins RA (2000). Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berlin) 153: 31–43.
Black YD, Green-Jordan K, Eichenbaum HB, Kantak KM (2004). Hippocampal memory system function and the regulation of cocaine self-administration behavior in rats. Behav Brain Res 151: 225–238.
Bliss TV, Collingridge GL (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39.
Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R et al (1998). Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 280: 1940–1942.
Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM (1995). Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron 15: 1403–1414.
Bohn LM, Gainetdinov RR, Lin F-T, Lefkowitz RJ, Caron MG (2000). opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408: 720–723.
Chalecka-Franaszek E, Weems HB, Crowder AT, Cox BM, Côt TE (2000). Immunoprecipitation of high-affinity, guanine nucleotide-sensitive, solubilized μ-opioid receptors from rat brain: coimmunoprecipitation of the G proteins Galpha o, Galpha i1, Galpha i3. J Neurochem 74: 1068–1078.
Childers SR (1991). Opioid receptor-coupled second messenger systems. Life Sci 48: 1991–2003.
Contarino A, Picetti R, Matthes HW, Koob GF, Kieffer BL, Gold LH (2002). Lack of reward and locomotor stimulation induced by heroin in mu-opioid receptor-deficient mice. Eur J Pharmacol 446: 103–109.
Corrigall WA, Coen KM (1990). Selective D1 and D2 dopamine antagonists decrease response rates of food-maintained behavior and reduce the discriminative stimulus effects of heroin. Pharmacol Biochem Behav 35: 351–355.
Dykstra LA, Gmerek DE, Winger G, Woods JH (1987). Kappa opioids in rhesus monkeys. II. Analysis of the antagonistic actions of quadazocine and beta-funaltrexamine. J Pharmacol Exp Ther 242: 421–427.
Eisch AJ, Barrot M, Schad CA, Self DW, Nestler EJ (2000). Opiates inhibit neurogenesis in the adult rat hippocampus. Proc Natl Acad Sci USA 97: 7579–7584.
Fan GH, Wang LZ, Qiu HC, Ma L, Pei G (1999). Inhibition of calcium/calmodulin-dependent protein kinase II in rat hippocampus attenuates morphine tolerance and dependence. Mol Pharmacol 56: 39–45.
Ferbinteanu J, McDonald RJ (2001). Dorsal/ventral hippocampus, fornix, and conditioned place preference. Hippocampus 11: 187–200.
Ghodse AH, Reed JL (1984). Gastric emptying, glucose tolerance and associated hormonal changes in heroin addiction. Psychol Med 14: 521–525.
Gilbert AK, Hosztafi S, Mahurter L, Pasternak GW (2004). Pharmacological characterization of dihydromorphine, 6-acetyldihydromorphine and dihydroheroin analgesia and their differentiation from morphine. Eur J Pharmacol 492: 123–130.
Giros B, Pohl M, Rochelle JM, Seldin MF (1995). Chromosomal localization of opioid peptide and receptor genes in the mouse. Life Sci 56: PL369–PL375.
Glare PA, Walsh TD (1991). Clinical pharmacokinetics of morphine. Ther Drug Monit 13: 1–23.
Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA (2004). A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci USA 101: 5135–5139.
Gomes I, Jordan BA, Gupta A, Trapaidze N, Nagy V, Devi LA (2000). Heterodimerization of mu and delta opioid receptors: a role in opiate synergy. J Neurosci 20: RC110.
Gutstein HB, Akil H (2001). Opioid analgesics. In: Hardman JG, Limbard LE and Gilman AG (eds). Goodman & Gilman's The Pharmacological Basis of Therapeutics. McGraw-Hill Book Company: New York. pp 569–619.
Hubner CB, Kornetsky C (1992). Heroin, 6-acetylmorphine and morphine effects on threshold for rewarding and aversive brain stimulation. J Pharmacol Exp Ther 260: 562–567.
Inturrisi CE, Max MB, Foley KM, Schultz M, Shin SU, Houde RW (1984). The pharmacokinetics of heroin in patients with chronic pain. N Engl J Med 310: 1213–1217.
Inturrisi CE, Schultz M, Shin S, Umans JG, Angel L, Simon EJ (1983). Evidence from opiate binding studies that heroin acts through its metabolites. Life Sci 33 (Suppl 1): 773–776.
Kamendulis LM, Brzezinski MR, Pindel EV, Bosron WF, Dean RA (1996). Metabolism of cocaine and heroin is catalyzed by the same human liver carboxylesterases. J Pharmacol Exp Ther 279: 713–717.
Kitanaka N, Sora I, Kinsey S, Zeng Z, Uhl GR (1998). No heroin or morphine 6beta-glucuronide analgesia in mu-opioid receptor knockout mice. Eur J Pharmacol 355: R1–R3.
Koch T, Schulz S, Schroder H, Wolf R, Raulf E, Hollt V (1998). Carboxyl-terminal splicing of the rat mu opioid receptor modulates agonist-mediated internalization and receptor resensitization. J Biol Chem 273: 13652–13657.
Koob GF, Sanna PP, Bloom FE (1998). Neuroscience of addiction. Neuron 21: 467–476.
Lange DG, Roerig SC, Fujimoto JM (1980). Absence of cross-tolerance to heroin in morphine-tolerant mice. Science 206: 72–74.
Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405: 955–959.
Leshner AI (1997). Addiction is a brain disease, and it matters. Science 278: 45–47.
Lou LG, Pei G (1997). Modulation of protein kinase C and cAMP-dependent protein kinase by delta-opioid. Biochem Biophys Res Commun 236: 626–629.
Malenka RC, Nicoll RA (1999). Long-term potentiation—a decade of progress? Science 285: 1870–1874.
Manzanares J, Corchero J, Romero J, Fernandez-Ruiz JJ, Ramos A, Fuentes JA (1999). Pharmacological and biochemical interactions between opioids and cannabinoids. Trends Pharmacol Sci 20: 287–294.
Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I et al (1996). Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383: 819–823.
Mcnally GP, Akil H (2002). Opioid peptides and their receptors: overview and function in pain modulation. In: Davis KL, Charney D, Coyle JT, Nemeroff C (eds). Neuropsychopharmacology: The Fifth Generation of Progress. Lippincott Williams & Wilkins: Philadelphia. pp 35–46.
Milne RW, Nation RL, Somogyi AA (1996). The disposition of morphine and its 3- and 6-glucuronide metabolites in humans and animals, and the importance of the metabolites to the pharmacological effects of morphine. Drug Metab Rev 28: 345–472.
Min BH, Augustin LB, Felsheim RF, Fuchs JA, Loh HH (1994). Genomic structure and analysis of promoter sequence of a mouse μ opioid receptor gene. Proc Natl Acad Sci USA 91: 9081–9085.
Morris RG, Moser EI, Riedel G, Martin SJ, Sandin J, Day M et al (2003). Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Philos Trans R Soc Lond B Biol Sci 358: 773–786.
Moser EI, Krobert KA, Moser MB, Morris RGM (1998). Impaired spatial learning after saturation of long-term potentiation. Science 281: 2038–2042.
Negus SS, Gatch MB, Mello NK (1998a). Discriminative stimulus effects of a cocaine/heroin ‘speedball’ combination in rhesus monkeys. J Pharmacol Exp Ther 285: 1123–1136.
Negus SS, Gatch MB, Mello NK (1998b). Effects of mu opioid agonists alone and in combination with cocaine and D-amphetamine in rhesus monkeys trained to discriminate cocaine. Neuropsychopharmacology 18: 325–338.
Nestler EJ (2001a). Neurobiology. Total recall—the memory of addiction. Science 292: 2266–2267.
Nestler EJ (2001b). Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2: 119–128.
Nestler EJ (2004). Historical review: molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci 25: 210–218.
Nestler EJ, Aghajanian GK (1997). Molecular and cellular basis of addiction. Science 278: 58–63.
Nitsche JF, Schuller AG, King MA, Zengh M, Pasternak GW, Pintar JE (2002). Genetic dissociation of opiate tolerance and physical dependence in delta-opioid receptor-1 and preproenkephalin knock-out mice. J Neurosci 22: 10906–10913.
O’Brien CP (1997). A range of research-based pharmacotherapies for addiction. Science 278: 66–70.
O’Mahony S, Coyle N, Payne R (2001). Current management of opioid-related side effects. Oncology (Williston Park). 15: 61–73, 77; discussion 77–78, 80–82.
Pasternak DA, Pan L, Xu J, Yu R, Xu MM, Pasternak GW et al (2004). Identification of three new alternatively spliced variants of the rat mu opioid receptor gene: dissociation of affinity and efficacy. J Neurochem 91: 881–890.
Petersen DW, Fujimoto JM (1983). Differential tolerance to the intestinal inhibitory effect of opiates in mice. Eur J Pharmacol 95: 225–230.
Pasternak GW (1993). Pharmacological mechanisms of opioid analgesics. Clin Neuropharmacol 16: 1–18.
Pasternak GW (2004). Multiple opiate receptors: deja vu all over again. Neuropharmacology 47 (Suppl 1): 312–323.
Pouletty P (2002). Drug addictions: towards socially accepted and medically treatable diseases. Nat Rev Drug Discov 1: 731–736.
Pu L, Bao GB, Xu NJ, Ma L, Pei G (2002). Hippocampal long-term potentiation is reduced by chronic opiate treatment and can be restored by re-exposure to opiates. J Neurosci 22: 1914–1921.
Rezayof A, Zarrindast MR, Sahraei H, Haeri-Rohani A (2003). Involvement of dopamine receptors of the dorsal hippocampus on the acquisition and expression of morphine-induced place preference in rats. J Psychopharmacol 17: 415–423.
Riedel G, Micheau J, Lam AGM, Roloff E, Martin SJ, Bridge H et al (1999). Reversible neural inactivation reveals hippocampal participation in several memory processes. Nat Neurosci 2: 898–905.
Robbins TW, Everitt BJ (1999). Drug addiction: bad habits add up. Nature 398: 567–570.
Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL (1996). Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16: 1179–1188.
Rolls ET, Xiang JZ (2005). Reward-spatial view representations and learning in the primate hippocampus. J Neurosci 25: 6167–6174.
Rossi GC, Brown GP, Leventhal L, Yang K, Pasternak GW (1996). Novel receptor mechanisms for heroin and morphine-6 beta-glucuronide analgesia. Neurosci Lett 216: 1–4.
Rossi GC, Leventhal L, Pan YX, Cole J, Su W, Bodnar RJ et al (1997). Antisense mapping of MOR-1 in rats: distinguishing between morphine and morphine-6beta-glucuronide antinociception. J Pharmacol Exp Ther 281: 109–114.
Rossi GC, Pan YX, Brown GP, Pasternak GW (1995a). Antisense mapping the MOR-1 opioid receptor: evidence for alternative splicing and a novel morphine-6 beta-glucuronide receptor. FEBS Lett 369: 192–196.
Rossi GC, Standifer KM, Pasternak GW (1995b). Differential blockade of morphine and morphine-6 beta-glucuronide analgesia by antisense oligodeoxynucleotides directed against MOR-1 and G-protein alpha subunits in rats. Neurosci Lett 198: 99–102.
Schuller AG, King MA, Zhang J, Bolan E, Pan YX, Morgan DJ et al (1999). Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat Neurosci 2: 151–156.
Selley DE, Cao CC, Sexton T, Schwegel JA, Martin TJ, Childers SR (2001). mu Opioid receptor-mediated G-protein activation by heroin metabolites: evidence for greater efficacy of 6-monoacetylmorphine compared with morphine. Biochem Pharmacol 62: 447–455.
Silva AJ, Stevens CF, Tonegawa S, Wang Y (1992). Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science 257: 201–206.
Snyder SH, Pasternak GW (2003). Historical review: opioid receptors. Trends Pharmacol Sci 24: 198–205.
Thronhill JA, Hirst M, Gowdey CW (1976). Changes in diurnal temperature and feeding patterns of rats during repeated injections of heroin and withdrawal. Arch Int Pharmacodyn Ther 223: 120–131.
Trujillo KA, Akil H (1991). Opiate tolerance and dependence: recent findings and synthesis. New Biol 3: 915–923.
Vallejo R, de Leon-Casasola O, Benyamin R (2004). Opioid therapy and immunosuppression: a review. Am J Ther 11: 354–365.
Volkow ND, Li TK (2004). Drug addiction: the neurobiology of behaviour gone awry. Nat Rev Neurosci 5: 963–970.
Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL (2001). Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science 292: 1175–1178.
Waldhoer M, Bartlett SE, Whistler JL (2004). Opioid receptors. Annu Rev Biochem 73: 953–990.
Weber RJ, Gomez-Flores R, Smith JE, Martin TJ (2004). Immune, neuroendocrine, and somatic alterations in animal models of human heroin abuse. J Neuroimmunol 147: 134–137.
Wei LN, Law PY, Loh HH (2004). Post-transcriptional regulation of opioid receptors in the nervous system. Front Biosci 9: 1665–1679.
Wendel B, Hoehe MR (1998). The human mu opioid receptor gene: 5′ regulatory and intronic sequences. J Mol Med 76: 525–532.
White NM (1996). Addictive drugs as reinforcers: multiple partial actions on memory systems. Addiction 91: 921–949.
Yang Y, Zheng X, Wang Y, Cao J, Dong Z, Cai J et al (2004). Stress enables synaptic depression in CA1 synapses by acute and chronic morphine: possible mechanisms for corticosterone on opiate addiction. J Neurosci 24: 2412–2420.
Zarrindast MR, Nasehi M, Rostami P, Rezayof A, Fazli-Tabaei S (2005). Repeated administration of dopaminergic agents in the dorsal hippocampus and morphine-induced place preference. Behav Pharmacol 16: 85–92.
Zhang XF, Hu XT, White FJ (1998). Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J Neurosci 18: 488–498.
Zhu Y, King MA, Schuller AG, Nitsche JF, Reidl M, Elde RP et al (1999). Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron 24: 243–252.
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology (2003CB515405, 2005CB522406), the National Natural Science Foundation of China (30021003, 30400129, 30230130, 30625014, and 30400230), the Ministry of Education (PCSIRT), Chinese Academy of Sciences (KSCX2-SW) and Shanghai Municipal Commission for Science and technology (03DZ19213, 06JC14008 and 06ZR14098), and the German Max-Planck Society. We greatly thank Drs Xu Zhang and Lan Bao, as well as Johannes Richter, for their critical comments on this manuscript and Yalan Wu and Xiajing Tong for their technical help.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp)
Supplementary information
Rights and permissions
About this article
Cite this article
Bao, G., Kang, L., Li, H. et al. Morphine and Heroin Differentially Modulate In Vivo Hippocampal LTP in Opiate-Dependent Rat. Neuropsychopharmacol 32, 1738–1749 (2007). https://doi.org/10.1038/sj.npp.1301308
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301308
Keywords
This article is cited by
-
Alcohol exposure disrupts mu opioid receptor-mediated long-term depression at insular cortex inputs to dorsolateral striatum
Nature Communications (2018)
-
Glucocorticoid Homeostasis in the Dentate Gyrus Is Essential for Opiate Withdrawal-Associated Memories
Molecular Neurobiology (2017)
-
Formation of aversive memories associated with conditioned drug withdrawal requires BDNF expression in the amygdala in acute morphine-dependent rats
Acta Pharmacologica Sinica (2015)
-
Neonatal Morphine Administration Leads to Changes in Hippocampal BDNF Levels and Antioxidant Enzyme Activity in the Adult Life of Rats
Neurochemical Research (2013)
-
Impaired hippocampal synaptic plasticity in C6 glioma-bearing rats
Journal of Neuro-Oncology (2011)
