
Abstract
Background:
Intrauterine growth restriction (IUGR) offspring with rapid catch-up growth are at increased risk for early obesity especially in males. Persistent insulin-like growth factor-1 (IGF-1) reduction is an important risk factor. Using a mouse model of maternal hypertension-induced IUGR, we examined IGF-1 levels, promoter DNA methylation, and histone H3 covalent modifications at birth (D1). We additionally investigated whether prenatal perturbations could reset at preadolescence (D21).
Methods:
IUGR was induced via maternal thromboxane A2-analog infusion in mice.
Results:
IUGR uniformly decreased D1 IGF-1 mRNA and protein levels with reduced promoter 1 (P1) transcription and increased P1 DNA methylation. IUGR males also had increased H3K4ac at exon 5 and 3′ distal UTR. At D21, IUGR males continued to have decreased IGF-1 levels, originating from both P1 and P2 with reduced 1A variant. IUGR males also had decreased activation mark of H3K4me3 at P1 compared with sham males. In contrast, D21 IUGR females normalized their IGF-1 levels, in association with an increased activation mark of H3K4me3 at P1 compared with sham females.
Conclusion:
IUGR uniformly affected D1 hepatic IGF-1 epigenetic modifications in both sexes. However, at preadolescence, IUGR males are unable to correct for the prenatal reduction possibly due to a more perturbed IGF-1 chromatin structure.
Similar content being viewed by others

Main
Intrauterine growth restriction (IUGR) predisposes offspring toward early-onset metabolic syndrome. This predisposition is particularly pertinent with rapid postnatal catch-up growth and affects males more than females (1). The molecular mechanisms underlying this sex-specific predisposition remain elusive. Insulin-like growth factor 1 (IGF-1), a major regulator of growth and metabolism, has generated significant interest in the field (2). IGF-1 disruption has been implicated in aberrant growth and development of metabolic syndrome in both IUGR humans and animal models (3,4,5,6). In particular, decreased IGF-1 have been observed in IUGR humans who ultimately develop metabolic syndrome as adults (7,8).
The IGF-1 gene is an ideal candidate to examine IUGR’s effects not only because of its growth and metabolic properties but because of its complex gene structure allowing for developmental- and tissue-specific expression. The majority of serum IGF-1 is synthesized from the liver (9). The IGF-1 gene is regulated by two alternative promoters, promoter P1 initiates transcription from exon 1 while promoter P2 from exon 2. P1 is active in fetal life, whereas P2 becomes upregulated at ~3 wk of life when growth hormone exerts its effects on the rodent IGF-1 gene (10). The rodent IGF-1 gene also has an alternatively spliced exon, with the A variant excluding exon 5 and the B variant includes exon 5. Both alternative promoter selection and alternative exon splicing require specific epigenetic modifications to direct transcriptional machinery for specific mRNA transcript generation (11,12).
Given that prenatal insults are known to affect gene-specific DNA methylation and histone covalent modifications (13), we examined whether IUGR would disrupt hepatic IGF-1 epigenetics to result in aberrant IGF-1 expression in embryonic and postnatal life. We used our laboratory’s IUGR mouse model induced via maternal thromboxane A2-analog infusion from embryonic day (E) 12.5 to term to mimic pregnancy-induced hypertension (14), a common etiology of human IUGR. TXA2-analog, a potent vasoconstrictor, elicits maternal hypertension culminating in a dysfunctional oxygen delivery system to the fetus. E19.5 IUGR fetuses are hypoinsulinemic and have reduced hepatic IGF-1 mRNA levels. Sham and IUGR fetuses have similar levels of 11-dehydrothromboxane B2 (a TXA2 metabolite), tumor necrosis factor α (which could be induced by TXA2), and corticosterone levels indicating that these do not contribute to IUGR. Relative to other IUGR animal models in which fetuses are nutrient deficient, fetuses in this model have normal glucose and increased amino acids. This fetal metabolic profile duplicates that of human infants of pregnancy-induced hypertension mothers who also have increased serum nutrients including glucose and amino acids (15,16). Whether such increased fetal nutrients represent increased transplacental transport, decreased fetal utilization, and/or increased fetal catabolism is being investigated. In our model, pups are born ~15% symmetrically growth restricted. IUGR males catch up to sham males’ weight at postnatal day (D) 28 and surpass them at 8½ mo. IUGR females remain growth restricted until D77 after which they weigh similarly to sham females through the first year. Given this sex-specific growth pattern, we hypothesized that IUGR males and females would have a different pattern of hepatic IGF-1 expression and a different trajectory of promoter DNA methylation and histone H3 covalent modifications from birth to preadolescence.
Results
Body Weights of D1 and D21 Sham and IUGR Mice
IUGR significantly decreased birth weights in males by 13% and in females by 17% (sham males 1.43 ± 0.05 g vs. IUGR males 1.24 ± 0.11 g and sham females 1.35 ± 0.05 g vs. IUGR females 1.12 ± 0.07 g; P < 0.05). At D21, IUGR males remained 13%, while IUGR females remained 17% growth restricted (sham males 8.97 ± 0.42 g vs. IUGR males 7.83 ± 0.29 g and sham females 8.91 ± 0.32 g vs. IUGR females 7.33 ± 0.46 g; P < 0.05).
Fasting Blood Glucose, Serum Insulin, and IGF-1 Levels in D1 and D21 Sham and IUGR Mice
D1 sham and IUGR mice had similar fasting blood glucose (sham males 65 ± 5 mg/dl vs. IUGR males 65 ± 2 mg/dl and sham females 63 ± 5 mg/dl vs. IUGR females 70 ± 5 mg/dl) and serum insulin (sham males 0.81 ± 0.29 ng/ml vs. IUGR males 1.08 ± 0.22 ng/ml and sham females 0.83 ± 0.29 ng/ml vs. IUGR females 1.06 ± 0.20 ng/ml) levels. D1 IUGR mice had decreased fasting serum IGF-1 levels in both sexes compared with shams (sham males 34.3 ± 3.1 ng/ml vs. IUGR males 24.3 ± 2.9 ng/ml and sham females 35.2 ± 4.9 ng/ml vs. IUGR females 25.2 ± 1.7 ng/ml; P < 0.05). D21 sham and IUGR mice had similar fasting blood glucose (sham males 153 ± 21 mg/dl vs. IUGR males 157 ± 16 mg/dl and sham females 160 ± 24 mg/dl vs. IUGR females 142 ± 7 mg/dl) and serum insulin (sham males 0.119 ± 0.052 ng/ml vs. IUGR males 0.103 ± 0.023 ng/ml and sham females 0.124 ± 0.064 ng/ml vs. IUGR females 0.200 ± 0.075 ng/ml) levels. D21 IUGR males had decreased fasting serum IGF-1 levels compared with sham males (268 ± 23 ng/ml vs. 475 ± 72 ng/ml; P < 0.05). D21 IUGR females had similar serum IGF-1 levels as sham females (338 ± 38 ng/ml vs. 348 ± 45 ng/ml).
IUGR’s Effect on D1 and D21 Hepatic IGF-1 mRNA Levels
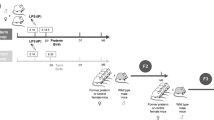
In D1 IUGR males, total IGF-1 mRNA level was decreased by 2.5-fold primarily due to reduced promoter P1 transcription ( Figure 1b ). Alternative splicing was unaffected since both variants were decreased equivalently. In D1 IUGR females, total IGF-1 mRNA level was also decreased, but the degree of reduction was less than IUGR males. Again, reduced transcription from P1 accounted for the total decrease, which was coupled to alternative splicing with the B variant ( Figure 1c ).

Mouse IGF-1 gene structure and mRNA transcript expression. (a) Genomic cartoon of the mouse IGF-1 gene with its alternative promoters and alternatively spliced exons. Transcription start sites (+1) are indicated as arrows above each promoter. ChIP primers are shown as black bars under exons. 1A variant lacks exon 5, while 1B variant contains exon 5. Graphs depicting total hepatic IGF-1 and its mRNA variants in (b) sham and IUGR males at D1, in (c) sham and IUGR females at D1, in (d) sham and IUGR males at D21, and in (e) sham and IUGR females at D21. mRNA levels are expressed as mean% of sham ± SEM. *P < 0.05 compared with age- and sex-matched shams with ANOVA with Fisher’s PLSD post-hoc test. White bars depict sham offspring, black bars depict IUGR offspring. n = 8/group from eight separate sham and IUGR litters. ChIP, chromatin immunoprecipitation; IGF-1, insulin-like growth factor-1; IUGR, intrauterine growth restriction; PLSD, protected least-significant difference.
At D21, IUGR males continued to have decreased total IGF-1 mRNA level. Unlike at D1, IUGR impacted transcripts originating from both promoters P1 and P2 in males. Moreover, the decrease in IGF-1 was predominately in the alternatively spliced A variant ( Figure 1d ). D21 IUGR females normalized their total hepatic IGF-1 mRNA levels ( Figure 1e ).
IUGR’s Effect on D1 and D21 Hepatic IGF-1 DNA Methylation of Promoters 1 and 2
At D1, IUGR increased promoter P1 DNA methylation at CG site located at -78 in males ( Figure 2b ) and at -142 in females ( Figure 2c ) compared with sex-matched shams. This 64 base-pair distance between IUGR males and females would be predicted to fall within the distance of the same nucleosome. IUGR also decreased P1 methylation at CG sites -110 and -328 in females at birth ( Figure 2c ). At D21, sham and IUGR males showed similar patterns of methylation ( Figure 2d ), while IUGR females had increased methylation at CG site -328 compared with sham females ( Figure 2e ).

DNA methylation patterns of sham and IUGR mouse hepatic IGF-1 P1 at D1 and D21. (a) Mouse IGF-1 promoter 1 (P1) CG sites based on cDNA clone accession #Ensmust0122386. Smaller vertical bars indicate the eight CG sites upstream of +1 of exon 1. Hepatic IGF-1 P1 DNA methylation in sham and IUGR males at D1 (b), in sham and IUGR females at D1 (c), in sham and IUGR males at D21 (d), in sham and IUGR females at D21 (e). Results are expressed as mean% ± SEM. *P < 0.05 compared with age- and sex-matched shams with Mann–Whitney U-test. White bars depict sham offspring, black bars depict IUGR offspring. n = 6/group from six separate sham and IUGR litters. IGF-1, insulin-like growth factor-1; IUGR, intrauterine growth restriction.
IUGR did not affect promoter P2 DNA methylation in either sex at birth consistent with its reduced usage in fetal life ( Figures 3b , 3c ). At D21, IUGR males showed no change in DNA methylation compared with sham males ( Figure 3d ), whereas IUGR females showed increased P2 methylation at -93 and -347 compared with sham females ( Figure 3e ).

DNA methylation patterns of sham and IUGR mouse hepatic IGF-1 P2 at D1 and D21. (a) Mouse IGF-1 promoter 2 (P2) CG sites based on cDNA clone accession #Ensmust062862. Smaller vertical bars indicate the eight CG sites upstream of +1 of exon 2. Hepatic IGF-1 P2 DNA methylation in (b) sham and IUGR males at D1, in (c) sham and IUGR females at D1, in (d) sham and IUGR males at D21, and in (e) sham and IUGR females at D21. Results are expressed as mean% ± SEM. *P < 0.05 compared with age- and sex-matched shams with Mann–Whitney U-test. White bars depict sham offspring, black bars depict IUGR offspring. n = 6/group from six separate sham and IUGR litters. IGF-1, insulin-like growth factor-1; IUGR, intrauterine growth restriction.
Determining Histone H3 Covalent Modifications Along the Mouse Hepatic IGF-1 Gene
We have previously shown that histone covalent modifications vary across the IGF-1 locus for the gene to define its 5′ and 3′ ends (17). Therefore, before we can determine the effects of IUGR, we established the baseline histone code for the C57BL/6J mouse. For this set of data, we have chosen to express each modification as a percent of that at promoter P1 as a reference point.
In general, D1 and D21 sham males and females had similar baseline hepatic IGF-1 histone H3 covalent modifications showing that the gene structure appeared to be set up correctly. These findings reassured us that sham surgery did not perturb the IGF-1 chromatin. Specifically at D1, P2 had decreased H3K4me2 but increased H3K9me3 and H3K36me3 compared with P1 ( Figure 4a , b ). The directionality of change in these histone modifications is consistent with decreased activation of P2 at D1 (ref. 18). Exon 5 had increased H3K14ac, H3K9me3, and H3K36me3 ( Figure 4a , b ), showing that 1B transcripts with this exon retained is the predominant transcript at D1 compared with 1A transcripts that have this exon alternatively spliced out (12). The proximal and distal 3′ UTRs which encode for the first and fourth polyadenylation signals, respectively, in the liver had decreased H3K9ac, H3K4me2, and H3K4me3 and increased H3K36me3 ( Figure 4a , b ). This histone signature is consistent with the definition of the 3′ end of a gene (19). By D21, P2 had a dramatic increase in the promoter activation mark of H3K4me3 in both sexes ( Figure 4c , d ), ascertaining that P2 is active in preadolescence. The remaining histone code for exon 5 and proximal and distal 3′ UTRs is similar to that of D1.

Baseline histone (H) 3 covalent modifications along the hepatic IGF-1 gene in C57BL/6J (a) D1 males, (b) D1 females, (c) D21 males, and (d) D21 females. Values for P2, exon 5, proximal and distal 3′ UTR are presented as mean% of P1 ± SEM where P1 is normalized to 100% to denote relative changes in H3 modifications within the gene itself. *P < 0.05 compared with P1 values with Mann–Whitney U-test. White bars depict K9ac, black bars depict K14ac, gray bars depict K4me2, white hatched bars depict K4me3, black hatched bars depict K9me3, and gray hatched bars depict K36me3. n = 6/group from six separate sham and IUGR litters. IGF-1, insulin-like growth factor-1; IUGR, intrauterine growth restriction.
IUGR’s Effect on D1 and D21 Hepatic IGF-1 Histone H3 Covalent Modifications
For these data, we have expressed each IUGR modification as a percent of the sham modification to denote IUGR-specific changes. In D1 IUGR males, the histone covalent modifications were similar to D1 sham males except for increased H3K9ac in exon 5 and distal 3′UTR ( Figure 5a ). Similar to D1 IUGR males, D21 IUGR males showed a similar histone code as D21 sham males except for decreased H3K9me3 in proximal 5′ UTR ( Figure 5c ). D1 IUGR females had increased H3K9me3 at promoter P2 and exon 5. In addition, the polyadenylation signals for proximal and distal 3′ UTRs showed increased H3K14ac and H3K9me3 but decreased H3K9ac and H3K4me3 ( Figure 5b ). The histone code continued to show changes in D21 IUGR females where promoter P1 had increased H3K4- and K9-me3, exon 5 had decreased H3K14ac but increased H3K4me2, and proximal and distal 3′ UTR had increased H3K9me3 ( Figure 5d ).

Histone (H) 3 covalent modifications along the hepatic IGF-1 gene in C57BL/6J (a) D1 IUGR males, (b) D1 IUGR females, (c) D21 IUGR males, and (d) D21 IUGR females. IUGR values are presented as mean% of sham ± SEM where sham values are normalized to 100% to denote IUGR-induced differences. *P < 0.05 compared with age- and sex-matched shams with Mann–Whitney U-test. White bars depict K9ac, black bars depict K14ac, gray bars depict K4me2, white hatched bars depict K4 me3, black hatched bars depict K9me3, and gray hatched bars depict K36me3. n = 6/group from six separate sham and IUGR litters. IGF-1, insulin-like growth factor-1; IUGR, intrauterine growth restriction.
Discussion
The most important finding of this study is that in a mouse model of maternal hypertension-induced IUGR, juvenile males have persistently decreased IGF-1 mRNA and protein levels, whereas juvenile females have normal IGF-1 mRNA and protein levels. In association with these IGF-1 levels, juvenile IUGR males and females have a different pattern of promoter DNA methylation and histone H3 covalent modifications. These epigenetic modifications may provide a molecular explanation for the varying IUGR male and female IGF-1 expression.
IUGR at D1 significantly affected transcription originating from promoter P1 compared with promoter P2. This finding is consistent with promoter P1 being more active in early embryonic life (20). In association with promoter P1 transcript decrease, IUGR increased DNA methylation in promoter P1 at CG site -78 in males and -142 in females. These sites lie within the projected nucleosome-free region (NFR). NFR occurs during active transcription when the region 200–300 base pairs upstream of the transcription start site lacks a nucleosome and is typically unmethylated. Methylation within the NFR, in contrast, leads to nucleosome formation and gene repression. Our results therefore suggest that DNA methylation in IUGR offspring may have contributed to NFR loss on some of the DNA templates which would result in reduced IGF-1 transcripts. In addition to DNA methylation, D1 IUGR males had an abnormal H3K9ac pattern. H3K9ac, along with H3K4me3, are usually localized to the 5′ end of genes that have a first exon that is <1 kb (like the IGF-1 gene) to attract general transcription factors for gene activation (21). These two modifications then disappear within 1 kb downstream of the transcription start site. In our study, H3K9ac is aberrantly localized in exon 5 and 3′ UTR in IUGR males ( Figure 6 ). DNA methylation within the NFR coupled with aberrant H3K9ac in the body of the IGF-1 gene indicate that IUGR males have more disruptions to their epigenome at birth compared with IUGR females, potentially setting them up for a reduced ability to normalize IGF-1 expression over time. Another interesting finding is that IUGR females preferentially decreased their 1B splice variant. Increased H3K9me3 is normally associated with gene repression (12), but this mark also correlates with increased inclusion of alternative exons (22); therefore, increased inclusion of exon 5 may have led to 1B splice variant decrease in IUGR females.

An illustrative figure depicting H3 covalent modifications along the IGF-1 gene that may underlie IUGR-induced sex-specific gene expression. K9ac, K4me3, and K9me3 peaks illustrate relative qualitative differences between sham and IUGR. IUGR-induced covalent modifications that predict transcriptional outcomes are compared with sham modifications. ♂ = males; ♀ = females. IGF-1, insulin-like growth factor-1; IUGR, intrauterine growth restriction.
The most striking finding in our study is the sex specificity at D21, 3 wk after the prenatal insult. Whether the prenatal IUGR-induced epigenetic modifications could be reset during a relatively normal postnatal environment is important to address. Juvenile IUGR males decreased IGF-1 transcripts originating from both promoters P1 and P2 with now a predominant decrease in the alternatively spliced 1A variant. A surprising finding here however is that promoters P1 and P2 DNA methylation patterns and histone H3 covalent modifications in IUGR males are similar to those of sham males. This implies that IUGR males adjusted their specific epigenetic modifications detected as aberrant at birth back to sham levels, but these alterations were insufficient to normalize IGF-1 transcript levels. The explanation for this is unclear. We do know that growth hormone (GH) becomes the primary growth determinant in mice and humans at preadolescence. Plausible explanations for a persistent IGF-1 decrease may be due to either insufficient GH level or the cis elements where GH exerts its action on the IGF-1 gene are inaccessible. Indeed, IUGR children born small for gestational age have been described to have GH insufficiency, and current management of these small-for-gestational-age children who present with growth failure during childhood includes exogenous GH treatment (23). Alternatively, GH acts via the Jak/Stat pathway in which dimerized Stat proteins bind to GH responsive elements located in multiple sites on the IGF-1 gene to activate transcription (24). Perhaps these GH responsive elements are rendered inaccessible to Stat proteins due to a prenatal change in IGF-1 chromatin. Juvenile IUGR females, in contrast, had a new histone code that is different from sham females along the entire IGF-1 gene. We postulate that IUGR females may be able to normalize IGF-1 levels due to elevated H3K4me3 and H3K9me3 at P1 ( Figure 6 ). H3K9me3 was initially shown to be associated with transcriptional repression if the associated DNA was also methylated. In the absence of DNA methylation, H3K9me3 is shown to be associated with transcriptional activation and RNA polymerase elongation (25).
The epigenetic modifications detected in this study bear certain resemblance to the rat model of IUGR via bilateral uterine artery ligation. IUGR rat pups are 25% asymmetrically growth restricted with reduced oxygen, glucose, and branched-chain amino acids. IUGR also modified P1 and P2 DNA methylation patterns and histone code along the length of the hepatic IGF-1 gene (17). Unlike our mouse model, however, juvenile rat pups showed persistent decreases in IGF-1 levels in both sexes with their sex-specific epigenetic modifications similar at birth and preadolescence. The authors noted that less variability was found among IUGR rats compared with control rats, speculating that the response to IUGR results in a more uniform histone code alteration in the rat model.
The translational relevance of our D21 findings is that IUGR males are known to be at a disadvantage compared with IUGR females in many aspects of cardiovascular health especially in early adult life. Male offspring develop vascular dysfunction and hypertension, whereas female offspring seems to be protected (26,27). Literature over the past two decades has shown that IGF-1 not only controls growth properties but also has important effects on carbohydrate, lipid, and protein metabolism (2). IGF-1 shares structural homology and downstream signaling pathways with insulin and has insulin-like effects in lowering glucose levels by stimulating glucose uptake in skeletal muscle via glucose transporter 4 membrane translocation and by decreasing hepatic glucose production via inhibition of gluconeogenesis (28,29). IGF-1 also induces differentiation of preadipocytes into mature adipocytes for lipid storage as well as stimulates protein synthesis and decreases proteolysis (2). Therefore, a persistent decrease in IGF-1 in IUGR males from birth via altered IGF-1 chromatin may provide one mechanistic insight into the male sex vulnerability. Other explanations may lie in the effects of gonadal steroid hormones and sex chromosome (30). Gonadal steroid hormones act on nongonadal tissues to cause sex-specific phenotypes. Studies in rat and mouse liver models have identified >1,000 genes whose expression is sex dependent, imparting substantial sexual dimorphism to liver metabolic function and pathophysiology (31). Interestingly, the sexually dimorphic expression of liver genes is regulated by the temporal release of GH by the pituitary gland, which itself shows significant sex differences. In rats and mice, GH profiles are highly pulsatile (intermittent) in males vs. more frequent (continuous) in females, suggesting a level of IGF-1 gene regulation exists outside of the liver. The effects of sex chromosome genes acting outside of the gonads is also an important contributor to sex differences. Both X and Y genes, which are differentially present in each XX vs. XY cell, act in a sex-biased manner to cause sex differences in nongonadal phenotypes (32,33). In fact, the genetic sex difference leads to clear sex bias in the genome: Y genes act only in XY cells, X genes escaping X-inactivation may be expressed constitutively higher in XX than XY cells, XX cells receive both a paternal and maternal imprint on X chromosomes whereas XY cells receive only a maternal imprint, and X or Y chromosomes harbor segments of sex-specific heterochromatin that may alter the epigenetic status of autosomes in a sex-specific manner.
We recognize certain limitations exist in our study. One is the establishment of the causal relationship between epigenetic modifications and gene expression changes. Unfortunately, current technology is still unable to assign such cause-and-effect relationships; however, we present evidence that a developmentally regulated gene such as IGF-1 must undergo sequential epigenetic modifications prenatally and postnatally to maintain normal gene transcription. Any significant prenatal disruption in chromatin remodeling may prevent future modifications to take place for proper postnatal gene expression. A second limitation is that we have only examined a few IGF-1 epigenetic modifications to attempt to understand its role in later-life disease. Indeed, IGF-1 acts in concert with GH, GH receptors, IGF-receptors, and IGF-binding proteins. Therefore, perturbations in any of these growth regulators could have additional impact. A third limitation is the assurance of equal dietary intake during nursing between sham and IUGR groups. Measurement of actual milk intake would be ideal but pup rejection by constant handling is a genuine risk. We have tried to obviate this limitation by using cross-foster dams with no prior surgical manipulation and to maintain equal litter sizes.
In summary, we have shown that maternal-induced hypertension in the mouse disrupts IGF-1 upregulation in preadolescent IUGR males. Intriguingly, in the next week, IUGR males in this model catch up to sham males in weight and even surpass them at 8½ mo of age. It remains to be determined whether IGF-1 levels stay suppressed or become accelerated as catch-up growth continues and whether weight gain is a function of gain in lean or fat mass. Further dissection into other aspects of the GH-mediated IGF-1 upregulation is in progress.
Methods
IUGR Mouse Model via Maternal Thromboxane A2-Analog Infusion
All procedures were approved by the University of Utah Animal Care committee and carried out in accordance with US National Institutes of Health Guide for the Care and Use of Laboratory Animals. IUGR offspring were induced according to Fung et al. (14). Briefly, timed matings of C57BL/6J mice were set up. At E12.5, pregnant females were anesthetized and micro-osmotic pumps infusing either vehicle (sham group) or 4,000 ng/μl U-46619 were implanted for the remainder of pregnancy (~20 d). Pups delivered spontaneously at term (D1). Unmanipulated dams cross-fostered all pups from D1 to D21. We measured D1 and D21 body weights and harvested sera and livers.
Fasting Whole Blood Glucose and Serum Insulin and IGF-1 Measurements
All mice were fasted for 1 h prior to decapitation and blood collection. The first drop of blood (~0.5 μl) was used to measure whole blood glucose using an Accu-Chek Aviva glucometer (Roche Diagnostics, Indianapolis, IN). Remaining blood was collected into serum separator microtubes and centrifuged for 20 min at 2,000g. Sera were separated, flash-frozen, and stored at −80 °C until analysis. We measured insulin levels with Ultrasensitive Mouse Insulin ELISA kit (90080; Crystal Chem, Downers Grove, IL) following manufacturer’s protocol using the Low Range Assay and IGF-1 levels with Mouse IGF-1 Immunoassay (MG100; R&D Systems, Minneapolis, MN) following manufacturer’s protocol (n = 6/group from three separate sham and IUGR litters for both time points).
RNA Isolation, cDNA Synthesis, and Real-Time RT-PCR Quantification
Total RNA was extracted from frozen livers using a Nucleospin RNA II kit (Macherey-Nagel, Bethlehem, PA). cDNA was synthesized from 2 μg total RNA using random hexamers (High-Capacity cDNA Reverse Transcription kit, Applied Biosystems, Foster City, CA). Total hepatic IGF-1 mRNA and variants ( Figure 1a ) were quantified by real-time RT-PCR using Taqman Assays-on-Demand ( Table 1 ) as previously described (n = 8/group from eight separate sham and IUGR litters) (34). Each sample was run in quadruplicate. Relative quantitation comparing 2-▵CT values was used to analyze gene expression changes between sham and IUGR pups with GAPDH as a loading control. To validate GAPDH as an appropriate loading control, parallel serial dilutions between sham and IUGR cDNA were quantified as previously described (35).
DNA Methylation Analysis
Genomic DNA extracted from livers (n = 6/group from six separate sham and IUGR litters) was converted with sodium bisulfite (EZ DNA Methylation-Gold kit, Zymo Research, Irvine, CA). IGF-1 P1 and P2 have multiple transcription start sites therefore we analyzed cytosine-guanine (CG) sites that were upstream of the longest known 5′ UTR transcripts for each promoter. Using cDNA clone accession #Ensmust0122386, the eight P1 CG sites analyzed were at -417, -363, -328, -255, -239, -142, -110, and -78 ( Figure 2a ). Bisulfite primers used were: forward 5′-TAAGTAGGTTTTTATTTATGGGGTAG and rev 5′-TAAGTAGGTTTTTATTTATGGGGTAG for -417 to -328, and forward 5′-TGGGAAAGTATATTTGGAGAGATATT and rev 5′- TTATAATATCATTCAAATCCCTCAACT for -255 to -78. Using cDNA clone accession #Ensmust062862, the eight P2 CG sites analyzed were at -372, -347, -311, -306, -285, -255, -185, and -93 ( Figure 3a ). Bisulfite primers used were: forward 5′-GGTAGAAATTATTGTAAGGTATTTTTGTT and rev 5′-ATCACACACATACATAAACACAAA for -372 to -255, and forward 5′ GTTTGTGTTTATGTATGTGTGTGA and rev 5′-CTTCCCCTAACTTTAACTTTCTAAAC for -185 to -93. PCR conditions were 95 °C for 10 min, 94 °C for 30 s, annealing at 55 °C (for P1) or 56 °C (for P2) for 30 s, 72 °C for 30 s, 35 cycles. PCR products from bisulfite-treated genomic DNA were cloned into pSC-A (Stratagene, Cedar Creek, TX). Plasmid DNA was prepared and sequenced using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems) with M13 forward or reverse primers.
Chromatin Immunoprecipitation and Real-Time PCR Quantification
We followed the protocol developed by Fu et al. (17). with some modifications. Twenty micrograms of liver tissue was ground in liquid nitrogen and fixed in 1% formaldehyde for 10 min (n = 6/group from six separate sham and IUGR litters). Chromatin was sonicated 12× for 10 s on ice (Sonic Dismembrator model 100; Fisher Scientific, Pittsburgh, PA) to generate 500–2,000 base-pair chromatin fragments. DNA content was quantified spectrophotometrically at A260. Chromatin equivalent to 20 μg DNA was used for each immunoprecipitation. Five micrograms of anti-H3K14ac (Millipore Upstate, Charlottesville, VA) and anti-H3K36me3 (Abcam, Cambridge, MA) antibodies and 10 μg of anti-H3K9ac (Cell Signaling Technologies, Beverly, MA), anti-H3K4me2, anti-H3K4me3, and anti-H3K9me3 (Millipore Upstate) antibodies were used for immunoprecipitation. Purified immunoprecipitation DNA fragments containing IGF-1 site-specific sequences ( Figure 1a ) and an intergenic region were quantified by real-time PCR. Table 2 lists primers and probe sequences. Intergenic sequences are accepted internal controls in chromatin immunoprecipitation assays (36); therefore, we used an untranscribed region upstream of the IGF-1 gene and showed that this region contained low levels of all six histone covalent modifications ( Table 3 ). We used the comparative CT method to quantify value differences between target and intergenic control. To quantitate the five sites in each sample and to establish that all primers and probes annealed equivalently, we generated a synthetic template by cloning the five sites into a single plasmid of pBIIKS (Stratagene, Cedar Creek, TX; synthetic template cloning primers are shown in Table 4 ). The synthetic template was serially diluted, and each site was quantified by real-time PCR. Table 5 shows that the CT values for all five primer/probe sets as equivalent.
Statistics
IUGR-induced changes in mRNA and promoter DNA methylation were expressed as mean% of sham ± SEM. Changes in H3 covalent modifications in the mouse hepatic IGF-1 gene were expressed as mean% of P1 ± SEM where P1 is normalized to 100% to denote relative changes in H3 modifications within the gene itself, and as mean% of sham ± SEM where sham values are normalized to 100% to denote IUGR-induced differences. ANOVA with Fisher’s protected least-significant difference post-hoc analysis was used for body weight, glucose, insulin, and serum IGF-1 protein analyses. The Mann–Whitney U-test was used for IGF-1 mRNA, DNA methylation, and chromatin immuno precipitation analyses. Statistical significance was declared at P < 0.05.
Statement of Financial Support
This study was supported by divisional funds at the University of Utah.
Disclosure
All authors have nothing to disclose and no conflicts of interest to resolve.
References
Claris O, Beltrand J, Levy-Marchal C. Consequences of intrauterine growth and early neonatal catch-up growth. Semin Perinatol 2010;34:207–10.
LeRoith D, Yakar S. Mechanisms of disease: metabolic effects of growth hormone and insulin-like growth factor 1. Nat Clin Pract Endocrinol Metab 2007;3:302–10.
Remacle C, Bieswal F, Reusens B. Programming of obesity and cardiovascular disease. Int J Obes Relat Metab Disord 2004;28:Suppl 3:S46–53.
Dunger DB, Salgin B, Ong KK. Session 7: early nutrition and later health early developmental pathways of obesity and diabetes risk. Proc Nutr Soc 2007;66:451–7.
Guilloteau P, Zabielski R, Hammon HM, Metges CC. Adverse effects of nutritional programming during prenatal and early postnatal life, some aspects of regulation and potential prevention and treatments. J Physiol Pharmacol 2009;60:Suppl 3:17–35.
Kamei H, Ding Y, Kajimura S, Wells M, Chiang P, Duan C. Role of IGF signaling in catch-up growth and accelerated temporal development in zebrafish embryos in response to oxygen availability. Development 2011;138:777–86.
Akanji AO, Smith RJ. The insulin-like growth factor system, metabolic syndrome, and cardiovascular disease risk. Metab Syndr Relat Disord 2012;10:3–13.
Cohen DH, LeRoith D. Obesity, type 2 diabetes, and cancer: the insulin and IGF connection. Endocr Relat Cancer 2012;19:F27–45.
Barton ER. The ABCs of IGF-I isoforms: impact on muscle hypertrophy and implications for repair. Appl Physiol Nutr Metab 2006;31:791–7.
Kikuchi K, Bichell DP, Rotwein P. Chromatin changes accompany the developmental activation of insulin-like growth factor I gene transcription. J Biol Chem 1992;267:21505–11.
Karlić R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci USA 2010;107:2926–31.
Zhou HL, Luo G, Wise JA, Lou H. Regulation of alternative splicing by local histone modifications: potential roles for RNA-guided mechanisms. Nucleic Acids Res 2014;42:701–13.
Sookoian S, Gianotti TF, Burgueño AL, Pirola CJ. Fetal metabolic programming and epigenetic modifications: a systems biology approach. Pediatr Res 2013;73(4 Pt 2):531–42.
Fung C, Brown A, Cox J, Callaway C, McKnight R, Lane R. Novel thromboxane A2 analog-induced IUGR mouse model. J Dev Orig Health Dis 2011;2:291–301.
von Versen-Hoeynck FM, Powers RW. Maternal-fetal metabolism in normal pregnancy and preeclampsia. Front Biosci 2007;12:2457–70.
Evans RW, Powers RW, Ness RB, et al. Maternal and fetal amino acid concentrations and fetal outcomes during pre-eclampsia. Reproduction 2003;125:785–90.
Fu Q, Yu X, Callaway CW, Lane RH, McKnight RA. Epigenetics: intrauterine growth retardation (IUGR) modifies the histone code along the rat hepatic IGF-1 gene. FASEB J 2009;23:2438–49.
Benevolenskaya EV. Histone H3K4 demethylases are essential in development and differentiation. Biochem Cell Biol 2007;85:435–43.
Wozniak GG, Strahl BD. Hitting the ‘mark’: interpreting lysine methylation in the context of active transcription. Biochim Biophys Acta 2014;1839:1353–61.
Lowe WL Jr, Roberts CT Jr, Lasky SR, LeRoith D. Differential expression of alternative 5’ untranslated regions in mRNAs encoding rat insulin-like growth factor I. Proc Natl Acad Sci USA 1987;84:8946–50.
Bieberstein NI, Carrillo Oesterreich F, Straube K, Neugebauer KM. First exon length controls active chromatin signatures and transcription. Cell Rep 2012;2:62–8.
Saint-André V, Batsché E, Rachez C, Muchardt C. Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat Struct Mol Biol 2011;18:337–44.
Ali O, Cohen P. Insulin-like growth factors and their binding proteins in children born small for gestational age: implication for growth hormone therapy. Horm Res 2003;60:Suppl 3:115–23.
Chia DJ, Ono M, Woelfle J, Schlesinger-Massart M, Jiang H, Rotwein P. Characterization of distinct Stat5b binding sites that mediate growth hormone-stimulated IGF-I gene transcription. J Biol Chem 2006;281:3190–7.
Wiencke JK, Zheng S, Morrison Z, Yeh RF. Differentially expressed genes are marked by histone 3 lysine 9 trimethylation in human cancer cells. Oncogene 2008;27:2412–21.
Hemmings DG, Williams SJ, Davidge ST. Increased myogenic tone in 7-month-old adult male but not female offspring from rat dams exposed to hypoxia during pregnancy. Am J Physiol Heart Circ Physiol 2005;289:H674–82.
Brawley L, Itoh S, Torrens C, et al. Dietary protein restriction in pregnancy induces hypertension and vascular defects in rat male offspring. Pediatr Res 2003;54:83–90.
Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. Insulin-like growth factors. N Engl J Med 1997;336:633–40.
Rinderknecht E, Humbel RE. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J Biol Chem 1978;253:2769–76.
Arnold AP. Mouse models for evaluating sex chromosome effects that cause sex differences in non-gonadal tissues. J Neuroendocrinol 2009;21:377–86.
Waxman DJ, Holloway MG. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol 2009;76:215–28.
Arnold AP. Sex chromosomes and brain gender. Nat Rev Neurosci 2004;5:701–8.
Turner ME, Ely D, Prokop J, Milsted A. Sry, more than testis determination? Am J Physiol Regul Integr Comp Physiol 2011;301:R561–71.
MacLennan NK, James SJ, Melnyk S, et al. Uteroplacental insufficiency alters DNA methylation, one-carbon metabolism, and histone acetylation in IUGR rats. Physiol Genomics 2004;18:43–50.
Pham TD, MacLennan NK, Chiu CT, Laksana GS, Hsu JL, Lane RH. Uteroplacental insufficiency increases apoptosis and alters p53 gene methylation in the full-term IUGR rat kidney. Am J Physiol Regul Integr Comp Physiol 2003;285:R962–70.
Lan F, Collins RE, De Cegli R, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 2007;448:718–22.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fung, C., Yang, Y., Fu, Q. et al. IUGR prevents IGF-1 upregulation in juvenile male mice by perturbing postnatal IGF-1 chromatin remodeling. Pediatr Res 78, 14–23 (2015). https://doi.org/10.1038/pr.2015.70
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.70
