
Abstract
Nicotinic acetylcholine receptor (nAChR) blockers potentiate the effects of selective serotonin reuptake inhibitors (SSRIs) in some treatment-resistant patients; however, it is not known whether these effects are independent, or whether the two neurotransmitter systems act synergistically. We first determined that the SSRI fluoxetine and the nicotinic partial agonist cytisine have synergistic effects in a mouse model of antidepressant efficacy, whereas serotonin depletion blocked the effects of cytisine. Using a pharmacological approach, we found that the 5-HT1A agonist 8-OH-DPAT also potentiated the antidepressant-like effects of cytisine, suggesting that this subtype might mediate the interaction between the serotonergic and cholinergic systems. The 5-HT1A receptors are located both presynaptically and postsynaptically. We therefore knocked down 5-HT1A receptors in either the dorsal raphe (presynaptic autoreceptors) or the hippocampus (a brain area with high expression of 5-HT1A heteroreceptors sensitive to cholinergic effects on affective behaviors). Knockdown of 5-HT1A receptors in hippocampus, but not dorsal raphe, significantly decreased the antidepressant-like effect of cytisine. This study suggests that serotonin signaling through postsynaptic 5-HT1A receptors in the hippocampus is critical for the antidepressant-like effects of a cholinergic drug and begins to elucidate the molecular mechanisms underlying interactions between the serotonergic and cholinergic systems related to mood disorders.
Similar content being viewed by others


INTRODUCTION
Major depressive disorder (MDD) is one of the most common psychiatric illnesses, with a lifetime prevalence of ∼15%. The most widely prescribed class of antidepressant medications is the selective serotonin reuptake inhibitors (SSRIs) that increase levels of serotonin at the synapse. Although current antidepressant therapies are effective in ∼60% of depressed individuals, remission is only observed in approximately a third of subjects (Han et al, 2013; Preston and Shelton, 2013). The incomplete response of patients, combined with side effects of current treatments (Ginsberg, 2009), has led to the hope that investigation of other targets or the development of an augmentation strategy to potentiate the effects of commonly used pharmacotherapies (for a review, see Duric and Duman, 2013) could result in remission for a greater proportion of patients. For example, the NMDA receptor antagonist ketamine induces rapid-onset antidepressant effects, opening new possibilities for alternative treatment (Duman et al, 2012). Similarly, targeting the cholinergic system by altering signaling through its muscarinic and nicotinic receptor families (muscarinic acetylcholine receptor (mAChR) and nicotinic acetylcholine receptor (nAChR), respectively) has shown some promising results (Drevets et al, 2012; Furey and Drevets, 2006; Shytle et al, 2002b). Despite the recent failure of one clinical trial of a nicotinic antagonist, several studies have demonstrated that SSRI-resistant patients show improvement of depressive symptoms when a nicotinic antagonist or partial agonist is added (George et al, 2008; Philip et al, 2009). The idea that limiting cholinergic signaling could be antidepressant is in line with human studies indicating that blocking the breakdown of acetylcholine (ACh), thereby increasing ACh levels, can induce symptoms of depression. More recently, human imaging studies have suggested that ACh levels are high throughout the brain in patients who are actively depressed (Saricicek et al, 2012; Hannestad et al, 2013). Thus, although targeting the monoamine system is effective for treating depression in a significant subset of patients, emerging data suggest that abnormalities in the cholinergic system could be one of the etiological factors underlying the development of depression.
Although the nicotinic antagonist mecamylamine has been effective in relieving the symptoms of some human depressed subjects, full blockade of nAChRs can result in side effects, limiting the tolerability of the drug. Therefore, decreasing ACh signaling through these receptors while preserving some function, as can be achieved by treating with a nicotinic partial agonist such as cytisine or varenicline, may be a more effective strategy for targeting the nicotinic cholinergic system in depressed individuals. The combination of a nicotinic partial agonist with an SSRI antidepressant is therefore one augmentation strategy that could be effective in patients refractory to current antidepressant treatments (Rollema et al, 2009); however, the mechanisms underlying the interaction between cholinergic and serotonergic medications are unknown. Most antidepressant drugs can also antagonize nAChRs in the nM μM range (Shytle et al, 2002a). It is therefore possible that cholinergic compounds and current antidepressant medications could converge to decrease activity of nAChRs. Alternatively, these drugs could affect serotoninergic and cholinergic pathways independently, or changes in cholinergic signaling could alter serotonin neurotransmission directly. We therefore investigated the nature and site of interaction between the cholinergic and monoaminergic neurotransmitter systems in the regulation of behavior in rodent tests of antidepressant efficacy using a combined pharmacological and molecular genetic approach.
MATERIALS AND METHODS
Animals
Male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME), allowed to acclimate to the animal facility for at least 14 days before testing, and housed under standard laboratory conditions (temperature 21±2 °C, 12:12 light/dark cycle, lights on at 0700 h). Food and water were available ad libitum throughout the studies. All procedures were approved by the Yale University Animal Care and Use Committee and conformed to the standards of the NIH Guide for the Care and Use of Laboratory Animals.
Drugs
All drugs (8-OH-DPAT, cytisine, p-chlorophenylalanine (PCPA)) were purchased from Sigma Aldrich and were diluted in phosphate-buffered saline at 10 mg/ml (pH=7.4) and injected i.p. Serotonin depletion was performed as described in Lucki and O'Leary (2004) by injecting mice i.p. with PCPA (300 mg/kg) twice daily for 3 days and testing ∼18 h after the last treatment.
Design of Small Hairpin RNAs (shRNAs)
Two shRNAs directed against the htr1A mRNA encoding the 5-HT1A receptor were constructed by selecting unique 24 base sequences (5′ to 3′: 1. GCCAGTTGGACAGCGACAAAGTGA; 2. GCTAGACAGGTACTGGGCAATCAC; GenBank accession NM_008308.4) as previously described (Mineur et al, 2011). As a control, a random sequence of 24 bases without similarities to any known transcripts in mice was selected as a scrambled shRNA. Synthetic oligonucleotide duplexes were designed by adding antisense sequences directed against the selected mRNA region, followed by a miR23 loop of 10 nucleotides (CTTCCTGTCA) to the 5′ end of the sequences above, and by adding overhanging ends identical to those created by SapI and XbaI restriction enzyme digestion. Annealed oligonucleotides were ligated into a pAAV-EGFP-shRNA vector as described previously (Benavides et al, 2007; Hommel et al, 2003; Mineur et al, 2011) and positive clones were confirmed by sequencing.
Viral Production
Viruses were produced by transfecting HEK 293 cells with 135 μg each of pAAV2-shRNA, pHelper, and pAAV-RC plasmids using calcium phosphate (Hommel et al, 2003; Zolotukhin et al, 1999). Cells were harvested 96 to 120 h later, suspended in freezing buffer (0.15 M NaCl and 50 mM Tris, pH 8.0), freeze/thawed several times, and treated with 50 U/ml of benzonase for 30 min at 37 °C. The lysate was then centrifuged (3700 g for 20 min) and the supernatant was added to a centrifuge tube containing a 15, 25, 40, and 60% iodixanol step gradient. The gradient with lysate was then centrifuged (50 000 g for 200 min, 10 °C) before the 4/5 bottom of the 40% fraction was recovered. This extract was then diluted in PBS-MK (1 × PBS, 1 mM MgCl2, 2.5 mM KCl), and concentrated and purified with a Centricon Plus-20 (100K) filter device. Purified viruses were stored at 4 °C.
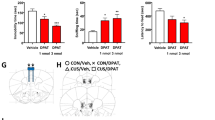
Efficacy of infectivity was first evaluated by qualitative assessment of GFP-positive cells (Figure 1a) following infection and observation of 5-HT1A-R immunostaining in mice that received AAV-sh5-HT1A compared with AAV-Scr in the hippocampus (Figure 1b). The efficacy of shRNA-mediated 5-HT1A knockdown in vivo was further confirmed by quantitative RT-PCR that identified >30% decrease in htr1A mRNA in hippocampal tissue (see ‘Quantitative real-time (q)-PCR and mRNA quantitation’ below). Because of the potential mismatch between mRNA levels and protein function (Vogel and Marcotte, 2012), functional evaluation of 5-HT1A KD was determined by measuring the reduction of 8-OH-DPAT-induced hypothermia in mice with knockdown of the 5-HT1A receptor in the dorsal raphe (Figure 1c). This assay is highly dependent on function of 5-HT1A autoreceptors (Ginefri-Gayet and Gayet, 1993).

(a) GFP expression in hippocampus and dorsal raphe after AAV infusion and shRNA expression. (b) Qualitative evaluation of 5-HT1A receptor expression following infusion of AAV-scrambled-shRNA or AAV-5-HT1A-shRNA into the hippocampus. (c) Change in body temperature following injection of 8-OH-DPAT (1 mg/kg) in mice with AAV-scrambled-shRNA or AAV-5-HT1A-shRNA infusion into the dorsal raphe. N=10 per group. (d) Time spent immobile in the tail suspension test following treatment with cytisine at subthreshold (1 mg/kg) or suprathreshold (1.5 mg/kg) doses in mice with AAV-scrambled-shRNA or AAV-5-HT1A-shRNA infusion into the dorsal raphe. N=7–9 per group after checking placements. (e) Time spent immobile in the tail suspension test following treatment with cytisine (1.5 mg/kg) or 8-OH-DPAT (1 mg/kg) in mice with AAV-scrambled-shRNA or AAV-5-HT1A-shRNA infusion into the hippocampus. N=9–10 per group after checking placements. (f) Social interaction following treatment with cytisine (1.5 mg/kg) in mice with AAV-scrambled-shRNA or AAV-5-HT1A-shRNA infusion into the hippocampus. N=10 per group after checking placements. Data are presented as means±SEM; (*)p<0.06; *p<0.05; **p<0.01; ***p<0.001.
Stereotaxic Surgery
C57BL/6J mice, 10–12 weeks old, were placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA) under isoflurane anesthesia. Purified high-titer AAV2 constructs were injected bilaterally into the dorsal raphe nucleus (from bregma: −2.5 mm anteroposterior, ±1.5 mm lateral, 2.7 mm dorsoventral) or the hippocampus (from bregma: −2 mm anteroposterior, ±1.5 mm lateral, 2.8 mm dorsoventral) over 15 min using a 26-s beveled-tipped Hamilton syringe (Hamilton, Reno, NE). For the raphe experiments, injection volume was 0.5 μl of AAV (knockdown or scrambled); for hippocampus, the volume was 1.5 μl per side. At the end of the infusion, the needle was kept in place for an additional 5 min before it was slowly withdrawn. Following stereotaxic surgery, mice were allowed to recover for at least 2–4 weeks before behavioral assays to allow for decrease in 5-HT1A expression.
Validation of Injection and Infection
Mice were anesthetized and perfused intracardially with PBS (∼50 ml) followed by 4% PFA in PBS (∼75 ml) for 5 min each. Brains were removed, postfixed for 24 h in 4% PFA at 4 °C, and then placed in 30% sucrose in PBS for cryoprotection and stored at 4 °C. Then, 40 μm brain sections were cut with a sliding microtome and placed in PBS before injection sites were examined by visual confirmation of EGFP expression by direct visualization with an actinic flashlight or using a Nikon fluorescence microscope after mounting the tissue on slides. For some of the animals used to determine knockdown efficacy by qPCR, we also ran experiments to detect GFP expression to confirm viral infection.
Quantitative Real-Time (q)-PCR and mRNA Quantitation
Mice infused with AAV-sh5-HT1A or AAV-Scr in the hippocampus were killed 3 weeks after viral infusion. Following rapid decapitation, the hippocampus was removed, and RNA was extracted using a RNeasy Lipid Tissue Mini Kit (Qiagen). Reverse transcription was performed and cDNA was quantified by quantitative PCR with SYBR Green using a StepOnePlus Thermal Cycler (Applied Biosystems). Primer efficiency was determined to be optimal and sequences were as follows: forward, 5′-TACTCCACTTTCGGCGCTTT-3′; reverse, 5′-GGCTGACCATTCAGGCTCTT-3′. Loading control was the TATAbox bing protein 1 (TBP) gene: forward, 5′-AAAGGGAGAATCATGGACCAGAACAA-3′; reverse, 5′-TGGACTAAAGATGGGAATTCCAGGAG-3′.
5-HT1A Immunohistochemistry
Free-floating brain slices obtained as described above were washed in PBS 0.1 M (pH=7.4), incubated for 1 h with Triton-X100 0.3%/Normal Goat Serum/PBS, and incubated overnight at 4 °C with a 5-HT1A primary antibody in PBS (1 : 1000; Abcam ab101914, Cambridge, MA), followed by a fluorophore-conjugated anti-goat secondary antibody (1 : 2000; Alexafluor 647) for 2 h at room temperature. One of every six slices of hippocampus was then mounted onto gelatin-coated slides, covered with mounting media (Vectashield, Vector Laboratories, Burlingame, CA), sealed with a coverslip, and observed under a Zeiss ApoTome fluorescence microscope.
Behavioral Assays
Following AAV infusions, mice were allowed to recover for 21–28 days to achieve viral infection and 5-HT1A receptor knockdown. Mice were then tested in the tail suspension test (TST) and subsequently evaluated for any changes in locomotor activity 48 h later. Another cohort of animals was tested in the social defeat paradigm, following the same timeline.
Mice were habituated to testing rooms for at least 30 min before behavioral testing, and testing took place between 1000 and 1800 h.
Tail suspension test
The TST is used to screen antidepressant compounds or assess depression-like phenotypes (Cryan et al, 2005; Steru et al, 1985). This test was chosen particularly because it is not confounded by changes in body temperature that could result from manipulations of the 5-HT system (Abdel-Fattah et al, 1997; Sheard and Aghajanian, 1967). It is also not affected by changes in appetite that can result from treatment with cholinergic compounds (Mineur et al, 2011).
Mice were gently suspended by the tail, filmed and subsequently scored, or observed live, depending on the number of animals tested. The time spent immobile over a 6-min period was determined as described previously (Mineur et al, 2007a). Immobility was defined as absence of movement except for respiration. Subjects were returned to their home cage at the end of the test.
Social interaction test following social defeat
We screened CD1 mice (retired breeders) for aggressive behavior, and only the CD1 mice with attack latencies 1 min were used in defeat episodes (Wilkinson et al, 2011). Tested animals were subjected to two defeat episodes per day for 4 days and then tested for social interaction. On day 5, experimental mice were placed in a novel open field (39 × 39 cm) in dim light for two consecutive sessions of 2.5 min. During the first session (no target), the open field contained an empty metal grid cage (10 × 6 cm) at one end of the arena. In the second session (target present), an unfamiliar CD1 mouse was introduced in the metal grid. Interaction score was defined as the ratio of time spent in the arena around the grid cage with or without the CD1 mouse.
Locomotor activity
Locomotion was evaluated in an open field to control for any changes resulting from manipulation of the serotonergic or cholinergic system. Mice were placed in a clean Plexiglas cage (48 × 22 × 18 cm) for 30 min and locomotor activity was recorded using the OptiMax system (Columbus Instruments, Columbus, OH). Subjects were returned to their home cage at the end of the test.
Statistical Analysis
Results of behavioral assays following drug treatments were analyzed by Fisher’s protected least square deviation (PLSD). Results of behavioral assays following 5-HT1A knockdown were analyzed by ANOVA with ‘drug’ and/or ‘virus’ when relevant as between factors. Post hoc analyses were then performed by t-test with Bonferroni corrections. Significance was set at p<0.05.
RESULTS
A Low Dose of the SSRI Fluoxetine Potentiates the Antidepressant-Like Effects of Cytisine
Serotonin is one of the primary neurotransmitters targeted by commonly used antidepressants. To determine whether increasing serotonin signaling could potentiate the effects of a cholinergic drug in the TST, a test of antidepressant efficacy, we first identified a subthreshold dose of fluoxetine (7.5 mg/kg) that had no behavioral effects on its own (Figure 2a). Combining the subthreshold dose of fluoxetine (7.5 mg/kg) with a subthreshold dose of the nicotinic partial agonist cytisine (1 mg/kg; Mineur et al, 2007b) induced a significant decrease in immobility in the TST (Figure 2b; PLSD, p<0.001), and increased interaction with a novel mouse following social defeat (Figure 2c; PLSD, p<0.001). The combination of the two compounds did not alter locomotor activity significantly (F<1).

(a) Time spent immobile in the tail suspension test following treatment with different doses of fluoxetine. N=10–11 per group. (b) Time spent immobile in the tail suspension test following treatment with subthreshold doses of cytisine (1 mg/kg) and fluoxetine (7.5 mg/kg). N=10–12 per group. (c) Social interaction following social defeat. Treatment with subthreshold doses of cytisine (1 mg/kg) and fluoxetine (7.5 mg/kg). N=10 per group. Data are means±SEM; *p<0.05; **p<0.01; ***p<0.001.
Serotonin Depletion Prevents the Antidepressant-Like Effects Induced by the Nicotinic Partial Agonist Cytisine
In order to determine whether serotonin signaling is critical for the ability of a cholinergic drug to induce an antidepressant-like effect, we evaluated the effect of cytisine after serotonin depletion with PCPA. Treatment with cytisine (1.5 mg/kg) resulted in an antidepressant-like effect in C57BL/6J male mice (Figure 3; PLSD p<0.05), as has been published previously (Mineur et al, 2009; Mineur et al, 2007b). Pretreatment with PCPA did not induce an increase in time spent immobile on its own, suggesting that 5-HT depletion did not induce a change in depression-like phenotype (PLSD, p<0.5). However, PCPA-mediated serotonin depletion abolished the response to cytisine (1.5 mg/kg) in the TST, suggesting that serotonin signaling is required for its antidepressant-like effects.

Time spent immobile in the tail suspension test following serotonin depletion by PCPA without or with treatment with cytisine (1.5 mg/kg). N=7–8 per group (some animals had to be removed because of wounding). Data are means±SEM; *p<0.05.
The 5-HT1A Receptor Agonist 8-OH-DPAT Potentiates the Effects of Cytisine in the TST
To identify the receptor subtypes involved in the interaction between serotonin and cholinergic signaling, we determined whether stimulation of 5-HT1A receptors could augment the effects of cytisine in the TST by using 8-OH-DPAT, a 5-HT1A agonist that also has effects at the 5-HT7 subtype. The 8-OH-DPAT has antidepressant-like effects and is highly selective for 5-HT1A and 5-HT7 receptors (Lucki and Wieland, 1990; Wieland and Lucki, 1990). Using a subthreshold dose identified previously (Lucki and O'Leary, 2004; Wieland and Lucki, 1990), mice were administered a low dose of 8-OH-DPAT (0.1 mg/kg) combined with a subthreshold dose of cytisine (1 mg/kg). As expected, the subthreshold doses of each drug had no effect on their own, but the combination of 8-OH-DPAT (0.1 mg/kg) and cytisine (1 mg/kg) resulted in a significant decrease in the time spent immobile in the TST (p<0.01; Figure 4). No significant differences were observed in locomotor activity (F<1).

Time spent immobile in the tail suspension test following treatment with subthreshold doses of cytisine (1 mg/kg) and 8-OH-DPAT (0.1 mg/kg). N=10–11 per group. Data are means±SEM; **p<0.01.
Postsynaptic 5-HT1A in the Hippocampus Is Required for the Antidepressant-Like Effects of Cytisine
The 8-OH-DPAT is highly selective for 5-HT1A and 5-HT7 receptors, but cannot distinguish between the receptor subtypes. In addition, 5-HT1A receptors serve as inhibitory autoreceptors that decrease 5-HT release in the raphe, and also as postsynaptic 5-HT receptors, with high expression in the hippocampus. To determine whether the effect of cytisine in the TST is because of a decrease in 5-HT neuron activation due to 5-HT1A autoreceptors or postsynaptic effects in hippocampus, we designed AAV2 vectors carrying shRNAs targeting the 5-HT1A receptor. The AAV-5-HT1A-shRNA constructs are efficiently expressed in the hippocampus and raphe nuclei (Figure 1a) and decrease 5-HT1A receptor expression in vivo (Figure 1b). Note that sections were taken from perfused tissue for the hippocampus vs fresh frozen tissue for the dorsal raphe, and hence there is a difference in the intensity of the amplified immunocytochemistry signal as compared with the intrinsic fluorescence of GFP. In addition, functional knockdown of 5-HT1A was confirmed by measuring the hypothermic response to 8-OH-DPAT (1 mg/kg) challenge following infusion of the AAV-5-HT1A-shRNA construct into the dorsal raphe (Figure 1c).
Cytisine induced a dose-dependent decrease in immobility in the TST following infusion of the AAV-5-HT1A-shRNA construct into the dorsal raphe. ANOVA revealed that there was no interaction between knockdown in the dorsal raphe and the time spent immobile (F<1), suggesting that the 5-HT1A autoreceptors in the raphe do not contribute significantly to the antidepressant-like properties of cytisine (Figure 1d). The hippocampus receives a significant serotoninergic projection, and has one of the highest levels of 5-HT1A receptor expression in the brain (Hensler et al, 2007). In addition, recent studies have suggested that increased ACh signaling in the hippocampus exacerbates anxiety- and depression-like behaviors in mice (Mineur et al, 2013). We therefore infused the AAV-5-HT1A-shRNA construct into the hippocampus (Figure 1a) and measured immobility in the TST following cytisine treatment. Cytisine decreased immobility significantly in control mice (p<0.05), but had no significant effect in animals with 5-HT1A knockdown in the hippocampus (Figure 1e). The 8-OH-DPAT is an agonist at both 5-HT1A and 5-HT7 receptors, and hence the lack of behavioral effects of the compound in 5-HT1A knockdown mice confirms the specificity of the receptor subtype mediating the nicotinic interaction, and validates the efficacy of 5-HT1A knockdown. A similar pattern was observed in the social defeat paradigm: whereas cytisine increased social interaction in control animals, this effect was completely reversed in animals with the knockdown (Figure 1f; F(1, 36)=9.48, p<0.004). None of the treatment groups exhibited significant changes in locomotion (F<1).
DISCUSSION
Although there are several avenues for treatment of MDD, a significant subset of patients do not respond to current treatments, and many others receive some relief of symptoms, but not full remission. There is clear evidence demonstrating that serotonin signaling can be dysregulated in depressed subjects (for a review, see Araragi and Lesch, 2013) and SSRIs are currently the treatment of choice for mood disorders. Accumulating evidence also suggests that alteration of the cholinergic system may contribute to depression and be a target for antidepressant therapies. For example, human imaging studies of individuals with MDD or bipolar disorder show that ACh levels are high when individuals are actively depressed (Hannestad et al, 2013; Saricicek et al, 2012). These recent imaging studies are consistent with experiments showing that increasing ACh levels in human subjects by challenging with physostigmine, a drug that prevents breakdown of ACh, results in symptoms of anxiety and depression (Risch et al, 1981). Furthermore, recent rodent studies have shown that decreasing breakdown of ACh in the hippocampus is sufficient to cause anxiety- and depression-like behaviors in mice (Mineur et al, 2013).
Preclinical studies and clinical trials have demonstrated the possibility that decreasing cholinergic signaling through nAChRs using nicotinic antagonists or partial agonists can have antidepressant-like effects on their own and can also augment the efficacy of SSRIs (Andreasen et al, 2012; George et al, 2008; Philip et al, 2009; Rollema et al, 2009). A number of questions remain to be answered, however. A recent large clinical trial failed to show a significant effect of a nicotinic antagonist as an antidepressant in patients who were nonresponsive to SSRI administration (Vieta et al, 2013). The results of this trial contradict other smaller clinical trials that showed a significant improvement in depressive symptoms following the addition of nicotinic antagonist mecamylamine to patients taking an SSRI (Bacher et al, 2009; George et al, 2008). It is possible that complete blockade of nAChRs is not well tolerated, and hence the dose of the drug could not be increased sufficiently to achieve clinical efficacy. Thus, partial agonists of nAChRs that decrease ACh signaling, but do not have broad effects on their own, may be a better avenue for therapeutic development as antidepressant medications. Along this line, an intriguing study showed that the nicotinic partial agonist varenicline could also be antidepressant in subjects resistant to an SSRI in a similar add-on design (Philip et al, 2009).
The current data support the idea that decreasing ACh signaling while simultaneously increasing serotonin signaling could be a useful approach for treating symptoms of depression and appears to increase resilience to social stress, a critical factor in the development of depression symptoms (Caspi et al, 2003). We show that these neurotransmitter systems can act synergistically to induce an antidepressant-like effect, even when they do not induce any behavioral change individually. The current studies also show that serotonin signaling is necessary for the antidepressant-like effects of cytisine, and indicate that signaling through postsynaptic 5-HT1A receptors of the hippocampus is critical for the antidepressant-like effect of this nicotinic partial agonist. Clinically, this suggests that even when SSRIs do not lead to a full antidepressant effect, potentiation of serotonin signaling may be sufficient to allow a decrease in cholinergic signaling to result in an antidepressant response. Furthermore, the current studies also emphasize that a deficit in serotonergic signaling in patients with depression could also decrease the response to augmentation therapy with cholinergic drugs. Interestingly, a recent study suggests that chronic treatment with nicotine can partially restore alterations induced by monoamine depletion (Khadrawy et al, 2011). As nicotine treatment can desensitize high-affinity nAChRs (Fenster et al, 1997; Grady et al, 1994), this provides some evidence that decreasing nAChR signaling could facilitate 5-HT signaling. These results are consistent with studies showing that treatment with either chronic nicotine or the nicotinic antagonist mecamylamine can increase serotonin release and signaling in the hippocampus (File et al, 2000; Kenny et al, 2000). Antagonism of muscarinic AChRs can also result in increased 5-HT release (Kenny et al, 2000), providing further evidence that decreasing ACh signaling in hippocampus can synergize with serotonin signaling. It should be noted that AAV2 is nonselective and targets many, if not all, neuronal types in the brain, and thus knockdown of 5-HT1A likely occurred in non-5-HT cells of the dorsal raphe as well. However, given the high rate of infectivity of the virus and the significant blunting of 8-OH-DPAT-induced hypothermia after knockdown, the data suggest that among the cell types affected by the 5-HT1A manipulation were ascending 5-HT neurons expressing 5-HT1A autoreceptors.
The precise cellular targets of 5-HT1A activation and nAChR blockade in the hippocampus remain to be elucidated but it seems possible that this interaction leads to decreased signaling in common pathways. 5-HT1A receptors are Gi/o-coupled receptors and will therefore decrease neuronal activity through inhibition of adenylyl cyclase and activation of potassium channels (Colino and Halliwell, 1987; De Vivo and Maayani, 1986). Postsynaptic effects of 5-HT1A stimulation in the hippocampus appear to be dependent on hyperpolarization mediated through GIRK2 potassium channels (Luscher et al, 1997). Blockade of nAChRs also decreases neuronal activation by decreasing ACh-mediated depolarization. Mechanistically, we propose the following model: acute stress leads to an increase in 5-HT and ACh release in different brain regions, including the hippocampus (Gartside et al, 2000; Grahn et al, 1999). Conversely, chronic stress, one of the main factors that can induce depression (Heim et al, 1997; Pace et al, 2006), decreases 5-HT release and sensitivity in the hippocampus that leads to depression-like symptoms (Kang et al, 2005; Luo et al, 2008) along with a decrease in 5-HT1A activation (for a broad review, see Mahar et al, 2014), but maintains higher ACh levels. SSRI treatment can restore the balance in serotonin signaling and lead to an increase in neurotrophic factor expression and neurogenesis, critical factors underlying antidepressant response (Brezun and Daszuta, 2000; Santarelli et al, 2003), mainly through brain 5-HT1A receptor stimulation of the hippocampus (Banasr et al, 2004).
This study shows that 5-HT1A signaling in the hippocampus is required for the antidepressant-like effects of the nAChR partial agonist cytisine. These data provide support for the idea that nAChR partial agonists could augment antidepressant effects of serotonergic medications, and highlight the neurobiological mechanisms that could underlie an augmentation strategy.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
Accession codes
References
Abdel-Fattah AF, Matsumoto K, Murakami Y, Adel-Khalek Gammaz H, Mohamed MF, Watanabe H (1997). Central serotonin level-dependent changes in body temperature following administration of tryptophan to pargyline- and harmaline-pretreated rats. Gen Pharmacol 28: 405–409.
Andreasen JT, Redrobe JP, Nielsen EO (2012). Combined alpha7 nicotinic acetylcholine receptor agonism and partial serotonin transporter inhibition produce antidepressant-like effects in the mouse forced swim and tail suspension tests: a comparison of SSR180711 and PNU-282987. Pharmacol Biochem Behav 100: 624–629.
Araragi N, Lesch KP (2013). Serotonin (5-HT) in the regulation of depression-related emotionality: insight from 5-HT transporter and tryptophan hydroxylase-2 knockout mouse models. Curr Drug Targets 14: 549–570.
Bacher I, Wu B, Shytle DR, George TP (2009). Mecamylamine - a nicotinic acetylcholine receptor antagonist with potential for the treatment of neuropsychiatric disorders. Expert Opin Pharmacother 10: 2709–2721.
Banasr M, Hery M, Printemps R, Daszuta A (2004). Serotonin-induced increases in adult cell proliferation and neurogenesis are mediated through different and common 5-HT receptor subtypes in the dentate gyrus and the subventricular zone. Neuropsychopharmacology 29: 450–460.
Benavides DR, Quinn JJ, Zhong P, Hawasli AH, DiLeone RJ, Kansy JW et al (2007). Cdk5 modulates cocaine reward, motivation, and striatal neuron excitability. J Neurosci 27: 12967–12976.
Brezun JM, Daszuta A (2000). Serotonin may stimulate granule cell proliferation in the adult hippocampus, as observed in rats grafted with foetal raphe neurons. Eur J Neurosci 12: 391–396.
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H et al (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science (New York, NY) 301: 386–389.
Colino A, Halliwell JV (1987). Differential modulation of three separate K-conductances in hippocampal CA1 neurons by serotonin. Nature 328: 73–77.
Cryan JF, Mombereau C, Vassout A (2005). The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev 29: 571–625.
De Vivo M, Maayani S (1986). Characterization of the 5-hydroxytryptamine1A receptor-mediated inhibition of forskolin-stimulated adenylate cyclase activity in guinea pig and rat hippocampal membranes. J Pharmacol Exp Ther 238: 248–253.
Drevets WC Jr., Zarate CA, Furey ML (2012). Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry 73: 1156–1163.
Duman RS, Li N, Liu RJ, Duric V, Aghajanian G (2012). Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 62: 35–41.
Duric V, Duman RS (2013). Depression and treatment response: dynamic interplay of signaling pathways and altered neural processes. Cell Mol Life Sci 70: 39–53.
Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA (1997). Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci 17: 5747–5759.
File SE, Kenny PJ, Cheeta S (2000). The role of the dorsal hippocampal serotonergic and cholinergic systems in the modulation of anxiety. Pharmacol Biochem Behav 66: 65–72.
Furey ML, Drevets WC (2006). Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry 63: 1121–1129.
Gartside SE, Hajos-Korcsok E, Bagdy E, Harsing LG Jr., Sharp T, Hajos M (2000). Neurochemical and electrophysiological studies on the functional significance of burst firing in serotonergic neurons. Neuroscience 98: 295–300.
George TP, Sacco KA, Vessicchio JC, Weinberger AH, Shytle RD (2008). Nicotinic antagonist augmentation of selective serotonin reuptake inhibitor-refractory major depressive disorder: a preliminary study. J Clin Psychopharmacol 28: 340–344.
Ginefri-Gayet M, Gayet J (1993). Hypothermia induced by infusion of methionine sulfoximine into the dorsal raphe nucleus of the rat: involvement of 5-HT1A and GABAB receptors. Eur J Pharmacol 235: 189–196.
Ginsberg LD (2009). Impact of drug tolerability on the selection of antidepressant treatment in patients with major depressive disorder. CNS Spectr 14: 8–14.
Grady SR, Marks MJ, Collins AC (1994). Desensitization of nicotine-stimulated [3H]dopamine release from mouse striatal synaptosomes. J Neurochem 62: 1390–1398.
Grahn RE, Will MJ, Hammack SE, Maswood S, McQueen MB, Watkins LR et al (1999). Activation of serotonin-immunoreactive cells in the dorsal raphe nucleus in rats exposed to an uncontrollable stressor. Brain Res 826: 35–43.
Han C, Yeh TL, Kato M, Sato S, Chang CM, Pae CU (2013). Management of chronic depressive patients with residual symptoms. CNS Drugs 27: S53–S57.
Hannestad JO, Cosgrove KP, DellaGioia NF, Perkins E, Bois F, Bhagwagar Z et al (2013). Changes in the cholinergic system between bipolar depression and euthymia as measured with [123I]5IA single photon emission computed tomography. Biol Psychiatry 74: 768–776.
Heim C, Owens MJ, Plotsky PM, Nemeroff CB (1997). Persistent changes in corticotropin-releasing factor systems due to early life stress: relationship to the pathophysiology of major depression and post-traumatic stress disorder. Psychopharmacol Bull 33: 185–192.
Hensler JG, Advani T, Monteggia LM (2007). Regulation of serotonin-1A receptor function in inducible brain-derived neurotrophic factor knockout mice after administration of corticosterone. Biol Psychiatry 62: 521–529.
Hommel JD, Sears RM, Georgescu D, Simmons DL, DiLeone RJ (2003). Local gene knockdown in the brain using viral-mediated RNA interference. Nat Med 9: 1539–1544.
Kang M, Pyun KH, Jang CG, Kim H, Bae H, Shim I (2005). Nelumbinis Semen reverses a decrease in hippocampal 5-HT release induced by chronic mild stress in rats. J Pharm Pharmacol 57: 651–656.
Kenny PJ, File SE, Neal MJ (2000). Evidence for a complex influence of nicotinic acetylcholine receptors on hippocampal serotonin release. J Neurochem 75: 2409–2414.
Khadrawy YA, El-Shamy KA, Mohamed SI (2011). Nicotine restores monoamine neurotransmitter changes in the cortex and hippocampus of reserpinized rats as a model of depression. Eur Rev Med Pharmacol Sci 15: 863–870.
Lucki I, O'Leary OF (2004). Distinguishing roles for norepinephrine and serotonin in the behavioral effects of antidepressant drugs. J Clin Psychiatry 65: 11–24.
Lucki I, Wieland S (1990). 5-Hydroxytryptamine1A receptors and behavioral responses. Neuropsychopharmacology 3: 481–493.
Luo DD, An SC, Zhang X (2008). Involvement of hippocampal serotonin and neuropeptide Y in depression induced by chronic unpredicted mild stress. Brain Res Bull 77: 8–12.
Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA (1997). G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19: 687–695.
Mahar I, Bambico FR, Mechawar N, Nobrega JN (2014). Stress, serotonin, and hippocampal neurogenesis in relation to depression and antidepressant effects. Neurosci Biobehav Rev 38: 173–192.
Mineur YS, Abizaid A, Rao Y, Salas R, DiLeone RJ, Gundisch D et al (2011). Nicotine decreases food intake through activation of POMC neurons. Science (New York, NY) 332: 1330–1332.
Mineur YS, Eibl C, Young G, Kochevar C, Papke RL, Gundisch D et al (2009). Cytisine-based nicotinic partial agonists as novel antidepressant compounds. J Pharmacol Exp Ther 329: 377–386.
Mineur YS, Obayemi A, Wigestrand MB, Fote GM, Calarco CA, Li AM et al (2013). Cholinergic signaling in the hippocampus regulates social stress resilience and anxiety- and depression-like behavior. Proc Natl Acad Sci USA 110: 3573–3578.
Mineur YS, Picciotto MR, Sanacora G (2007a). Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry 61: 250–252.
Mineur YS, Somenzi O, Picciotto MR (2007b). Cytisine, a partial agonist of high-affinity nicotinic acetylcholine receptors, has antidepressant-like properties in male C57BL/6J mice. Neuropharmacology 52: 1256–1262.
Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH et al (2006). Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry 163: 1630–1633.
Philip NS, Carpenter LL, Tyrka AR, Whiteley LB, Price LH (2009). Varenicline augmentation in depressed smokers: an 8-week, open-label study. J Clin Psychiatry 70: 1026–1031.
Preston TC, Shelton RC (2013). Treatment resistant depression: strategies for primary care. Curr Psychiatry Rep 15: 370.
Risch SC, Cohen RM, Janowsky DS, Kalin NH, Sitaram N, Gillin JC et al (1981). Physostigmine induction of depressive symptomatology in normal human subjects. Psychiatry Res 4: 89–94.
Rollema H, Guanowsky V, Mineur YS, Shrikhande A, Coe JW, Seymour PA et al (2009). Varenicline has antidepressant-like activity in the forced swim test and augments sertraline’s effect. Eur J Pharmacol 605: 114–116.
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S et al (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science (New York, NY) 301: 805–809.
Saricicek A, Esterlis I, Maloney KH, Mineur YS, Ruf BM, Muralidharan A et al (2012). Persistent beta2*-nicotinic acetylcholinergic receptor dysfunction in major depressive disorder. Am J Psychiatry 169: 851–859.
Sheard MH, Aghajanian GK (1967). Neural release of brain serotonin and body temperature. Nature 216: 495–496.
Shytle RD, Silver AA, Lukas RJ, Newman MB, Sheehan DV, Sanberg PR (2002a). Nicotinic acetylcholine receptors as targets for antidepressants. Mol Psychiatry 7: 525–535.
Shytle RD, Silver AA, Sheehan KH, Sheehan DV, Sanberg PR (2002b). Neuronal nicotinic receptor inhibition for treating mood disorders: preliminary controlled evidence with mecamylamine. Depress Anxiety 16: 89–92.
Steru L, Chermat R, Thierry B, Simon P (1985). The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85: 367–370.
Vieta E, Thase ME, Naber D, D'Souza B, Rancans E, Lepola U et al (2013). Efficacy and tolerability of flexibly-dosed adjunct TC-5214 (dexmecamylamine) in patients with major depressive disorder and inadequate response to prior antidepressant. Eur Neuropsychopharmacol 24: 564–574.
Vogel C, Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13: 227–232.
Wieland S, Lucki I (1990). Antidepressant-like activity of 5-HT1A agonists measured with the forced swim test. Psychopharmacology 101: 497–504.
Wilkinson MB, Dias C, Magida J, Mazei-Robison M, Lobo M, Kennedy P et al (2011). A novel role of the WNT-dishevelled-GSK3beta signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. J Neurosci 31: 9084–9092.
Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K et al (1999). Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Therapy 6: 973–985.
Acknowledgements
This work was supported by Grants MH077681, DA14241, and P50 DA033945 from the National Institute on Drug Abuse (NIDA), the Food and Drug Administration (FDA), and the Office of Research on Women’s Health (ORWH).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mineur, Y., Einstein, E., Bentham, M. et al. Expression of the 5-HT1A Serotonin Receptor in the Hippocampus Is Required for Social Stress Resilience and the Antidepressant-Like Effects Induced by the Nicotinic Partial Agonist Cytisine. Neuropsychopharmacol 40, 938–946 (2015). https://doi.org/10.1038/npp.2014.269
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.269
This article is cited by
-
Dipeptide tyrosyl-leucine exhibits antidepressant-like activity in mice
Scientific Reports (2020)
-
Multiple Nicotinic Acetylcholine Receptor Subtypes in the Mouse Amygdala Regulate Affective Behaviors and Response to Social Stress
Neuropsychopharmacology (2016)
