
Abstract
Alcoholism is a pervasive disorder perpetuated in part to relieve negative mood states like anxiety experienced during alcohol withdrawal. Emerging evidence demonstrates a role for the serotonin-rich dorsal raphe (DR) in anxiety following ethanol withdrawal. The current study examined the effects of chronic ethanol vapor exposure on the DR using slice electrophysiology in male DBA2/J mice. We found that chronic ethanol exposure resulted in deficits in social approach indicative of increased anxiety-like behavior at both 24 h and 7 days post-ethanol exposure. At 24 h post-ethanol exposure, we observed increased excitability and decreased spontaneous inhibitory transmission (inhibitory postsynaptic currents, IPSCs) in the DR. At 7 days post-ethanol exposure, we observed increased spontaneous and miniature excitatory transmission (excitatory postsynaptic currents, EPSCs). Because acute ethanol alters GABA transmission in other brain regions, we assessed the effects of ex vivo ethanol (50 mM) on miniature IPSCs (mIPSCs) in the DR 24-h post-ethanol exposure. Bath application of ethanol enhanced the amplitude of mIPSCs in cells from ethanol-naive and chronic intermittent ethanol-exposed (CIE) mice, but significantly enhanced the frequency of mIPSCs only in cells from CIE mice, suggesting that DR neurons are more sensitive to the inhibitory effects of acute ethanol following CIE. On the basis of these findings, we hypothesize that net excitation of DR neurons following chronic ethanol exposure contributes to enhanced anxiety during ethanol withdrawal, and that increased sensitivity of DR neurons to subsequent ethanol exposure may mediate acute ethanol’s ability to relieve anxiety during ethanol withdrawal.
Similar content being viewed by others


INTRODUCTION
Alcoholism is a pervasive chronic, relapsing disorder that is characterized by increased prevalence of negative mood states such as anxiety during abstinence (Hamilton et al, 2013). Alcohol’s ability to relieve such negative mood states is positively associated with craving for alcohol (Fox et al, 2012) and is a common motivation to resume alcohol consumption (Glöckner-Rist et al, 2013).
Serotonin is a monoamine neurotransmitter that is widely expressed throughout the central nervous system, where it has a principle role in stress responses, mood regulation, and, a growing body of evidence suggests, alcohol use disorders. Dysfunction in the serotonin system contributes significantly to the symptoms of many mood disorders (Miller et al, 2013; Arango et al, 2001; López-Figueroa et al, 2004) that commonly present with alcohol use disorders (Hasin et al, 2007), and alterations in serotonin markers are well-documented in human alcoholics (Underwood et al, 2007; Pivac et al, 2004; Beck et al, 1983). In preclinical models, ethanol drinking and enhanced anxiety-like behavior stemming from chronic ethanol exposure are attenuated by serotonergic compounds (Yoshimoto et al, 2012; Overstreet et al, 2006), whereas feelings of nervousness, anxiety, and craving in alcoholics are exacerbated by the serotonin agonist meta-chlorophenylpiperazine (Umhau et al, 2011; Krystal et al, 1996). Moreover, a series of studies from the Kranzler group demonstrate that in a subpopulation of alcohol-dependent individuals, treatment with selective serotonin reuptake inhibitors (SSRIs) can increase both alcohol consumption and anxiety, and worsen treatment outcomes relative to placebo (Kranzler et al, 2013; Kranzler et al, 2012; Kranzler et al, 1996; Dundon et al, 2004). Taken together, these data suggest that dysregulation in the serotonin system underlies negative mood states associated with alcohol use disorders.
The majority of serotonin in the brain arises from the dorsal raphe nucleus (DR), a midbrain region that is known to modulate neurophysiological and behavioral responses to stress. Neurons containing serotonin are found throughout this structure and are especially enriched in the dorsomedial and ventromedial aspects of the DR (Fu et al, 2010). This portion of the DR is innervated by limbic inputs of particular interest to the study of alcoholism and negative affect, such as the central nucleus of the amygdala and bed nucleus of the stria terminalis (Hale et al, 2012). Emerging preclinical evidence suggests that the DR is involved in behavioral phenotypes associated with ethanol (Tomkins et al, 1994; Hwa et al, 2013; Takahashi et al, 2010) including enhanced anxiety following chronic ethanol exposure (Knapp et al, 2011; Huang et al, 2010; Overstreet et al, 2006). However, to date, little is known about the effects of chronic ethanol exposure on the functional activity of the DR.
In the current study, we examined the effects of chronic ethanol exposure on DR activity in DBA2/J mice. As a high anxiety and stress-vulnerable strain (Millstein and Holmes 2007), DBA2/J mice resemble human alcoholics, for whom stress and anxiety disorders typically precede the development of alcoholism (Marquenie et al, 2007). In addition, DBA2/J mice have high-functioning serotonin transporters (Carneiro et al, 2009), a trait that predicts depression, anxiety, and neuroticism among individuals that have a positive family history of alcoholism (Lovallo et al, 2014). Here, we demonstrate that DBA2/J mice display robustly enhanced anxiety during ethanol withdrawal much like human alcoholics. Following a regimen of chronic ethanol vapor exposure and withdrawal to establish an ethanol history, changes in DR activity were assessed using electrophysiology.
MATERIALS AND METHODS
Animals
Male DBA2/J mice (Jackson Laboratories, Bar Harbor, ME) arrived at 6 weeks of age and were immediately housed with littermates (2–4 per cage) in a vivarium maintained on a 12-h/12-h light/dark cycle with lights off at 0700 hours. Mice had access to standard rodent chow and water ad libitum for the duration of experiments.
Chronic Intermittent Ethanol Exposure
Five to eleven days after arrival, mice began a 5-day regimen of exposure to chronic intermittent ethanol vapors (chronic intermittent ethanol-exposed; CIE). This protocol has been used previously to induce behavioral indices of dependence in rodents (Becker and Lopez, 2004). Briefly, daily beginning at 1630 h, mice were removed from their home cages, injected with the alcohol dehydrogenase inhibitor pyrazole (1 mmol/kg) and placed with cagemates in vapor chambers, where they were continuously exposed to ethanol vapor and oxygen (15 l/min) for 16 consecutive hours. The relative flow rate of ethanol vapor and oxygen was carefully maintained to yield target blood ethanol concentrations (BECs) of 150–200 mg/dl. At 0900 hours, mice were removed from the vapor chambers and returned to their home cages. This cycle of ethanol vapor exposure and withdrawal was repeated over the next 4 days, resulting in 5 × 16-h exposures to ethanol vapors and 5 × 8-h periods of ethanol withdrawal. Following the final ethanol vapor exposure, mice remained in their home cages without disturbance for the duration of withdrawal (24 h or 7 days). A control group underwent identical procedures, including injections of pyrazole (1 mmol/kg) but was exposed to forced air (air-exposed; AE) rather than ethanol vapors. BECs were assessed in two additional cohorts of mice: one cohort of AE and ethanol-exposed mice was used to assess BECs immediately following removal from ethanol vapor exposure and a second cohort of AE and ethanol-exposed mice was used to assess BECs 24 h following removal from ethanol vapor exposure. Briefly, mice were restrained, and ∼20 μl of blood was collected from a small incision made in the tail. Plasma extracted from each blood sample was analyzed using the Analox Alcohol Analyzer (Analox Instruments).
Social Approach
Mice from CIE and AE groups were injected with buspirone (1 mg/kg, i.p.) or 0.9% saline 30 min before behavioral testing in the social approach apparatus, a three-chambered Plexiglass container divided by two panels with a small opening at the base to allow movement between the chambers as described previously (Moy et al, 2009). Following a 10-min habituation to the test chamber, during which crossings between chambers were observed, the time spent sniffing a novel mouse vs an empty cage was evaluated over 10 min. Two cohorts of mice were used to evaluate CIE effects on social approach, following either 24 h of withdrawal or 7 days of withdrawal.
Electrophysiology
Twenty-four hours or 7 days following the final ethanol vapor exposure, mice were killed via deep isoflurane anesthesia, and brain slices containing the DR were prepared as previously described (Li et al, 2012). Slice electrophysiology was conducted to assess the effects of CIE on basal functioning of neurons in the medial aspect of the DR using previously published methods (Li et al, 2012). In light of emerging data suggesting that the dorsomedial and ventromedial portions of the DR are especially relevant to anxiety-like phenotypes (Hale et al, 2012), cellular recordings were focused in these subregions. The effects of CIE on excitability parameters were first assessed in current clamp using a potassium gluconate-based intracellular solution (in mM: 135 K+ gluconate, 5 NaCl, 2 MgCl2, 10 HEPES, 0.6 EGTA, 4 Na2ATP, 0.4 Na2GTP, pH 7.3, 285–290 mOsmol). The resting membrane potential of the cell was noted before an increasing amount of current was injected (beginning at 0 pA) to assess the amount of current required to fire an action potential (termed the rheobase) and the membrane potential at which an action potential first fired. The number of action potentials fired at a given amount of current (0–200 pA; increased 10 pA per step) was also assessed. The effects of CIE on basal synaptic transmission was assessed in voltage clamp by adjusting the membrane potential and using a cesium methanesulfonate-based intracellular solution (in mM: 135 cesium methanesulfonate, 10 KCl, 1 MgCl2, 0.2 EGTA, 2 QX-314, 4 MgATP, 0.3 GTP, 20 phosphocreatine, pH 7.3, 285–290 mOsmol). Excitatory transmission was recorded at −55 mV, the reversal potential for GABAA-mediated events and inhibitory transmission was recorded at +10 mV, the reversal potential for AMPAR-mediated events. These experiments were repeated in the presence of the sodium channel blocker tetrodotoxin (0.5 μM) to assess the effects of CIE on network-independent synaptic transmission. To examine acute ethanol effects on inhibitory transmission, and to determine whether it is altered by CIE, inhibitory events were recorded in voltage clamp at −70 mV using a cesium chloride-based intracellular solution (in mM: 130 CsCl, 10 EGTA, 10 HEPES, 2 MgATP, 0.2 NaGTP, pH 7.3, 285–290 mOsmol) in the presence of tetrodotoxin (0.5 μM) and the glutamate receptor antagonist kynurenic acid (3 mM). After a stable baseline was recorded, acute ethanol (50 mM) was bath applied at a rate of 2 ml/min for 10 min. The GABAA receptor antagonist picrotoxin (200 μM) was then applied in addition to ethanol to confirm that these events were GABA mediated (Fleming et al, 2007). Parallel experiments were conducted in age-matched DBA mice maintained under identical colony housing conditions but not subjected to AE or CIE protocols (naive) to control for potential effects of stress on electrophysiological parameters. Naive mice did not differ from AE mice in any of the assessed parameters (see Supplementary Materials).
Statistics
BECs were analyzed using a repeated measures analysis of variance (ANOVA) to assess between-group (CIE vs AE) differences immediately following removal from ethanol exposure and an unpaired t-test to assess between-group (CIE vs AE) differences at 24 h post ethanol exposure. Behavioral data were analyzed using two-way ANOVAs to assess between-group (CIE vs AE) and between-treatment (buspirone vs vehicle) differences. All significant ANOVA terms were further analyzed for specific differences using Bonferroni post hoc tests. For all between-group comparisons that violated homogeneity of variance assumptions (deduced using F tests to compare variance), Welch’s t-tests were performed and, where appropriate, then corrected for multiple comparisons with a Bonferonni correction. A priori hypotheses about between-group differences in behavior were assessed using unpaired t-tests. Electrophysiologcal data were analyzed using unpaired t-tests or Welch’s t-tests, where appropriate, to assess the differences between groups (CIE vs AE, AE vs naive). Grubb’s test was used to identify statistical outliers and only one outlier was removed per group. Repeated measures two-way ANOVAs with Bonferonni-adjusted multiple comparisons as post hocs were used to assess between-group differences in the responses to ascending steps of current injection and bath application of acute ethanol. All values presented are means±SEM and significance was accepted when p<0.05.
RESULTS
Buspirone Attenuates Chronic Ethanol-Induced Deficits in Social Approach
The DR is implicated in behavioral stress and negative affective responses, and emerging evidence demonstrates its role in anxiety following chronic ethanol exposure (Overstreet et al, 2006). To confirm that our model of ethanol exposure elicits anxiety that involves the serotonin system, we assessed chronic ethanol effects on social approach. First, we characterized BECs immediately following CIE and 24 h post CIE to validate this method of ethanol exposure in DBA2/J mice (see Figure 1). To verify that CIE mice achieved pharmacologically relevant levels of intoxication (ie, >100 mg/dl), BECs were assessed immediately upon removal from ethanol exposure on days 1 and 3. The results of a two-way ANOVA showed a significant main effect of ethanol treatment (F(1, 12)=76.79, p<0.0001). The main effect of time (F(1, 12)=0.3541, p>0.05) and the interaction between time and ethanol treatment (F(1, 12)=0.4195, p>0.05) were not significant. Bonferonni post hoc analyses revealed that BECs in CIE mice were significantly higher than AE mice immediately following ethanol exposure. To verify that ethanol was no longer present at 24 h of withdrawal, BECs were assessed in an additional cohort of mice 24 h following removal from ethanol exposure. The results of an unpaired t-test showed that CIE mice did not significantly differ from AE mice in BECs (t(8)=1.243, p>0.05), demonstrating that ethanol has been fully metabolized at this time point. We then examined chronic ethanol effects on social approach during early (24 h post ethanol exposure) and late (7 days post ethanol exposure) withdrawal and the potential role of serotonin in this behavior using systemic administration of the 5HT1A-R agonist buspirone in two separate cohorts. In this model, reductions in social approach with a novel mouse are interpreted as an indication of increased anxiety (Knapp et al, 2004; Moy et al, 2007). To assess CIE or drug effects on overall exploration, we assessed the number of crossings between chambers during the 10-min habituation period (see Figure 2a and b). The results of a two-way ANOVA revealed a significant main effect of ethanol treatment at both 24 h and 7 days post ethanol exposure (F(1, 33)=13.47, p=0.0008 for 24 h and F(1, 33)=6.147, p=0.0184 for 7 days). The main effect of drug treatment as well as the ethanol × drug treatment interaction were not significant at either time point (F(1, 33)=0.06653, p>0.05 for drug treatment at 24 h; F(1, 33)=0.05978, p>0.05 for drug × ethanol treatment interaction at 24 h; F(1, 33)=0.6537, p>0.05 for drug treatment at 7 days; F(1, 33)=0.009, p>0.05 for drug × ethanol treatment interaction at 7 days). These results show that chronic ethanol exposure induces lasting deficits in exploratory behavior. For social approach, the results of a two-way ANOVA revealed significant main effects of ethanol treatment (F(1, 34)=7.131, p=0.0115), drug treatment (F(1, 34)=5.182, p=0.0292), and a significant ethanol × drug treatment interaction (F(1, 34)=5.905, p=0.0205) at 24 h post ethanol exposure (see Figure 2c). Bonferroni post hoc analyses revealed that at 24 h post ethanol exposure, vehicle-treated CIE mice spent significantly less time interacting with the novel mouse than vehicle-treated AE mice (p<0.05). Pretreatment with buspirone significantly attenuated deficits in social approach in CIE mice (p<0.05), but did not alter social approach in AE mice (p>0.05). At 7 days post-ethanol exposure, the results of a two-way ANOVA revealed significant main effects of ethanol treatment (F(1, 35)=4.692, p=0.0372) and drug treatment (F(1, 35)=9.763, p=0.0036), but the interaction was not significant (F(1, 35)=0.8817, p>0.05; see Figure 2d). Differences between vehicle-treated AE and CIE mice were assessed on the basis of the a priori hypothesis that CIE would reduce social approach at 7 days, an effect that was confirmed by an unpaired t-test (t(18)=4.149, p=0.0006). Likewise, an unpaired t-test confirmed our a priori hypothesis that buspirone would attenuate CIE-induced reductions in social approach at 7 days post-ethanol exposure (t(17)=3.232, p=0.0049) without effects in AE mice (Welch’s t(10.40)=1.420, p>0.05). Together, these results indicate that CIE induces both short-term and lasting deficits in both exploratory behaviors and social approach. However, it appears that only deficits in social approach involve the serotonin system, demonstrating a role for serotonin in anxiety following chronic ethanol exposure.

Chronic ethanol exposure (CIE) induces pharmacologically relevant blood ethanol concentrations (BECs) on days 1 and 3 of exposure. At 24 h post-CIE (Post), BECs of CIE mice do not significantly differ from those of air-exposed mice. * denotes significant between-group differences (p<0.05), ns denotes the absence of significant between-group differences; n=5–9 mice per group; error bars indicate SEM.

Chronic ethanol exposure (CIE) results in deficits in exploration at 24 h (a) and 7 days (b) post-ethanol exposure that are not altered by pretreatment with the 5HT1A agonist buspirone. Deficits in social approach are observed following CIE are rescued by pretreatment with buspirone at both 24 h (c) and 7 days (d) post-ethanol exposure relative to air exposure (AE). * denotes significant between-group differences (p<0.05); n=9–10 mice per group; error bars indicate SEM.
Chronic Ethanol Exposure Enhances the Activity and Excitability of DR Neurons
On the basis of our results with buspirone, we hypothesized that enhanced anxiety may result from increased DR activity induced by chronic ethanol exposure. We therefore used whole-cell-slice electrophysiology to assess chronic ethanol-induced alterations in excitability at 24 h and 7 days post-ethanol exposure. Unpaired t-tests revealed no between-group differences in resting membrane potential, the membrane potential at which the first action potential was fired, or the rheobase at 24 h post-ethanol exposure. Similar null results were observed at 7 days post-ethanol exposure. However, CIE-dependent between-group differences emerged at 24 h post-ethanol exposure in the number of action potentials fired over a range of stimulus intensities, as revealed by a significant repeated measures ANOVA (see Figure 3a and b). Specifically, significant main effects of stimulus intensity (F(20, 180)=29.53, p<0.001) and ethanol treatment (F(1, 9)=5.672, p=0.0411) and a significant interaction (F(20, 180)=4.858, p<0.001) were observed at 24 h post-ethanol exposure. This effect was transient, as at 7 days post-ethanol exposure, a repeated measures ANOVA indicated no differences between CIE and AE mice (Figure 3c and d). Together, these results indicate that chronic ethanol exposure induces a short-term hyperexcitability of DR neurons.

Chronic ethanol exposure enhances the excitability of DR neurons during early withdrawal. Representative traces showing action potentials fired in response to a 200 pA current injection in an air-exposed (AE) and chronic ethanol-exposed (CIE) mouse at 24 h post-ethanol exposure (a) and 7 days post-ethanol exposure (c). Chronic ethanol exposure significantly enhanced the number of action potentials fired in response to an increasing step of current at 24 h post-ethanol exposure (b) but not 7 days post-ethanol exposure (d) relative to AE. * denotes significant Bonferonni between-group differences (p<0.05); n=5–6 cells per group from during early withdrawal and 7–8 cells per group during late withdrawal; error bars indicate SEM.
Chronic Ethanol Exposure Alters Synaptic Transmission in the DR
We next evaluated alterations in excitatory and inhibitory synaptic transmission at 24 h and 7 days post ethanol exposure, by recording spontaneous excitatory postsynaptic currents (sEPSCs) and inhibitory postsynaptic currents (sIPSCs) in the same cell with temporal proximity and computed a ratio of excitation/inhibition (E/I) to obtain a relative measurement of net excitation or inhibition for each cell. A significant increase in the spontaneous E/I frequency (sE/I) of cells from CIE mice relative to AE mice was observed at 24 h post-ethanol exposure (Welch’s t(7.482)=2.699, p=0.0288; see Figure 4a). Further examination revealed that this effect was driven by reductions in the frequency of sIPSCs in cells from CIE mice, although this effect did not achieve statistical significance (Welch’s t(7.385)=1.983, p=0.0856; see Figure 4b). No between-group differences in sEPSC amplitude, sEPSC frequency, or sIPSC amplitude were observed at this time point (see Figure 4b and c). Although between-group differences in sE/I frequency were not observed at 7 days post-ethanol exposure (Figure 4d), a significant increase in sEPSC amplitude was observed in cells from CIE relative to AE mice (Welch’s t(8.682)=3.587, p=0.0062; Figure 4f). No between-group differences in sEPSC frequency, sIPSC amplitude, or sIPSC frequency were observed at this time point (see Figure 4e and f). Network-independent alterations in synaptic transmission following CIE were assessed in the presence of the sodium channel blocker, TTX. At 24 h post-ethanol exposure, no between-group differences in miniature excitatory postsynaptic (mEPSC) or miniature inhibitory postsynaptic (mIPSC) synaptic transmission were observed (see Figure 4g–i). However, at 7 days post CIE, a significant increase in miniature E/I frequency was observed in cells from CIE mice relative to AE mice (Welch’s t(10.91)=2.639, p=0.0232; see Figure 4j). Further examination revealed that this effect was driven by a significant increase in mEPSC frequency in these cells (t(16)=2.626; p=0.0183; see Figure 4k). No other between-group differences in network-independent synaptic transmission were observed at this time point (Figure 4l). Together, these results show that chronic ethanol exposure induces short-term reductions in inhibitory transmission and long-term enhancements in excitatory transmission in the DR.

Chronic ethanol alters synaptic transmission in the DR at 24 h post-ethanol exposure (a–c; g–i) and 7 days post-ethanol exposure (d–f; j–l). The balance between spontaneous excitatory and inhibitory transmission (sE/I frequency) was shifted towards net excitation at 24 h post-ethanol exposure (CIE 24 h) relative to air exposure (AE 24 h; a). Alterations in spontaneous inhibitory (sIPSC frequency) and excitatory transmission (sEPSC frequency; b), and sIPSC or sEPSC amplitude (c) were not observed. sE/I frequency (d), as well as sIPSC frequency and sEPSC frequency (e) at 7 days post- ethanol exposure (CIE 7 days) did not significantly differ from air exposure (AE 7 days). A significant increase in sEPSC amplitude was observed relative to AE at this time point (f). No differences in the balance of miniature inhibitory and excitatory transmission (mE/I frequency), miniature inhibitory (mIPSC) or miniature excitatory (mEPSC) transmission or amplitude were observed at 24 h post-ethanol exposure (CIE 24 h) relative to air exposure (AE 24 h; g–i). At 7 days post-ethanol exposure (CIE 7 days), the mE/I frequency was shifted toward net excitation relative to AE (AE 7 days; j), an effect that was driven by a significant enhancement of mEPSC frequency (k). No differences in mIPSC or mEPSC amplitude were observed at this time point (l). * denotes significant between-group differences (p<0.05); n=6–10 cells per group for 24 h post-ethanol exposure and 7–10 cells per group for 7 days post-ethanol exposure; error bars indicate SEM.
Acute Ethanol Exposure Enhances Inhibitory Transmission in the DR Following Chronic Ethanol Exposure
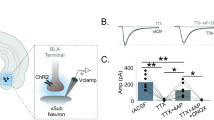
A previous investigation demonstrated that acute ethanol application to slices prepared from ethanol-naive rats enhanced GABAergic transmission in the DR (Lemos et al, 2011). We hypothesized that a history of chronic ethanol exposure would alter acute ethanol’s effects in the DR. To test this hypothesis, we examined the effects of acute bath application of ethanol (50 mM) on mIPSC frequency and amplitude in cells from CIE and AE mice 24 h post-ethanol exposure (see Figure 5). As shown in Figure 5a and b, acute ethanol significantly enhanced mIPSC amplitude in cells from both CIE and AE mice, revealed by a significant main effect of time in a repeated measures ANOVA (F(6, 102)=3.551, p=0.0031). However, the main effect of ethanol treatment (CIE vs AE) and the interaction between time and ethanol treatment were not significant (F(1, 17)=0.1590, p>0.05 for the main effect; F(6, 102)=1.066, p>0.05 for the interaction; see Figure 5b). The results of paired t-tests did not reveal significant differences in normalized mIPSC amplitude at baseline vs during acute ethanol bath application (minute 14 of the experiment) for either CIE or AE mice (t(8)=0.9890, p>0.05 for CIE mice; t(9)=1.839, p>0.05 for AE mice); between-group differences in normalized mIPSC amplitude during acute ethanol bath application (minute 14) assessed using unpaired t-tests were also not significant (t(17)=0.7030, p>0.05; see Figure 5c). In contrast to effects on mIPSC amplitude, acute ethanol effects on mIPSC frequency were significantly different between CIE and AE mice, as revealed in a repeated measures ANOVA by significant main effects of time (F(6, 102)=5.162, p=0.001) and ethanol treatment (F(1, 17)=4.607, p=0.0466) and a significant interaction (F(6, 102)=3.635, p=0.0026; see Figure 5d). Bonferonni-adjusted post hoc analyses indicated that normalized mIPSC frequency was significantly enhanced by acute ethanol following CIE, but not AE, at minutes 12 and 14. Likewise, unpaired t-tests revealed a significant increase in normalized mIPSC frequency during acute ethanol bath application (minute 14) relative to baseline in cells from CIE mice (t(8)=2.773, p=0.0242) but not in cells from AE mice (t(9)=0.6046, p>0.05). Additionally, normalized mIPSC frequency during acute ethanol (minute 14) was significantly greater in CIE relative to AE mice (t(17)=2.177, p=0.0438; Figure 5e). The decay kinetics of mIPSCs did not differ between groups at baseline (t(16)=0.7478, p>0.05) or during acute ethanol perfusion (analyzed at minute 14; t(16)=0.1853, p>0.05), nor did acute ethanol alter decay kinetics in AE mice (t(9)=0.6168, p>0.05; Figure 5f). In CIE mice, acute ethanol significantly increased the weighted decay tau (t(7)=2.608, p=0.0350; Figure 5g). A recent report in ethanol-naive C57BL/6J mice shows that acute ethanol induces a large tonic inward current mediated by glycine receptors (Maguire et al, 2014). Thus, we assessed whether acute ethanol produces a similar tonic current in AE or CIE mice, but found that it did not (the mean difference in holding current (pA) during baseline vs acute ethanol was −6.86±2.96 pA for AE and −1.86±2.6 pA for CIE and was not different between treatment groups, t(15)=1.196, p>0.05).

Cells of the DR are more sensitive to the inhibitory effects of acute ethanol (50 mM) 24 h following chronic ethanol exposure. (a) Representative traces showing miniature inhibitory transmission (mIPSCs) at baseline and during the final minute of 50 mM acute ethanol perfusion (minute 14). (b) Acute ethanol modestly enhanced normalized mIPSC amplitude irrespective of chronic ethanol treatment. (c) Single subject plots show no between-group differences in the effect of acute ethanol on mIPSC amplitude during the final minute of 50 mM acute ethanol perfusion and the normalized mIPSC amplitude during acute ethanol did not significantly differ from baseline following chronic ethanol exposure (CIE 24 h) or air exposure (AE 24 h). Horizontal bars denote the group means and standard error at this time point. (d) Acute ethanol significantly enhanced normalized mIPSC frequency following chronic ethanol exposure (CIE 24 h) but not air exposure (AE 24 h). (e) Single subject plots show a significant increase in normalized mIPSC frequency during the final minute of acute ethanol perfusion following CIE relative to AE. Normalized mIPSC frequency was significantly enhanced from baseline following CIE but not following AE. Horizontal bars denote the group means at this time point. (f) Representative traces of an averaged mIPSC events at baseline (black line) and during the final minute of acute ethanol perfusion (50 mM; gray line) following AE or CIE. (g) A significant increase in the weighted tau was observed during the final minute of acute ethanol perfusion relative to baseline following CIE, but not following AE (data not shown). * denotes significant between-group differences (p<0.05); n=9–10 cells per group; error bars indicate SEM.
DISCUSSION
Here, we provide an initial and novel characterization of chronic ethanol effects on neurons of the DR, revealing a potentially significant role for this circuit in the pathophysiology of alcoholism. First, we show that chronic ethanol exposure enhances anxiety-like behavior during both early and late withdrawal, an effect that involves the serotonin system. At parallel time points, we show increases in the net activity of DR neurons, manifested as hyperexcitability and reduced inhibitory transmission during early withdrawal and increased excitatory transmission during late withdrawal. Further, we show that DR neurons are more sensitive to the inhibitory effects of acute ethanol following chronic ethanol exposure. On the basis of these findings, we propose that increases in the net activity of DR neurons induced by chronic ethanol contribute to enhanced anxiety during ethanol withdrawal, and that by dampening this activity, reintroduction of ethanol may provide relief from this negative mood state. Taken together, these results demonstrate that the DR is a critical site of neuroadaptations contributing to the pathology of alcoholism.
Chronic Ethanol Exposure Enhances Anxiety-Like Behavior in DBA2/J Mice
Impairments in sociability are a common feature of alcoholism. Social anxiety is highly comorbid with alcoholism (Schneier et al, 2010), and is reported as a precipitating factor in excessive alcohol use (Buckner et al, 2008). Individuals with alcoholism experience greater interpersonal conflict and less social support (Dutton et al, 2014; Booth et al, 1992), which may contribute to mood disorders that are often comorbid in this population (Booth et al, 1992). A lack of social engagement may contribute to poor treatment outcomes, as treatment-seeking alcoholics who relapse report more severe social difficulties than those who remain abstinent (Charney et al, 2010). On the other hand, improving social ties may promote treatment efforts (Booth et al, 1992; Richter et al, 1991; Satre et al, 2012; Kelly et al, 2012), including pharmacological interventions (Pettinati et al, 2009).
In agreement with these clinical observations and previous studies in rats (Overstreet et al, 2002), we found that chronic ethanol exposure impairs social approach in DBA2/J mice. Previous preclinical characterization of this behavior, relates this impairment to an increase in anxiety (Moy et al, 2009; Meeker et al, 2013; Samaco et al, 2012; Overstreet et al, 2002). The observed deficits in social approach were lasting and could be normalized by pretreatment with the 5HT1A agonist buspirone during both early and late withdrawal. Previous work has shown a similar effect of buspirone on anxiety during withdrawal from chronic ethanol in rats when administered peripherally or into the DR (Overstreet et al, 2006; Overstreet et al, 2003). Interestingly, site-specific administration of busprione into the amygdala, nucleus accumbens, or paraventricular nucleus of the hypothalamus did not alter this phenotype, suggesting that 5HT1A receptors within the DR are particularly important for anxiety-like behavior during withdrawal from ethanol (Overstreet et al, 2006). Taken together, the results of the current study and the work of Overstreet et al, (2006) demonstrate roles for both the DR and serotonin in chronic ethanol-induced anxiety.
We also observed reduced exploration during the habituation period of the test among mice that underwent chronic ethanol exposure, although this phenotype does not appear to involve 5HT1A receptors. This effect of chronic ethanol exposure was persistent, as it was observed at both early and late withdrawal time points, therefore suggesting that reduced exploration may be due to a lasting aversion to novelty rather than feelings of physical illness that are typically observed during acute withdrawal (ie, up to 8 h post ethanol exposure). Interestingly, unlike chronic ethanol’s effect on social approach, deficits in exploratory behavior were not rescued by buspirone, suggesting that divergent neurochemical systems or neural circuits may govern these behaviors. In addition, this phenotype is consistent with findings from the Breese group (Knapp et al, 2005). Importantly, these data also demonstrate that deficits in exploratory behavior do not impede the ability to engage in social approach behaviors.
Chronic Ethanol Effects on the DR
Although serotonin has long been implicated in alcoholism, few studies to date have assessed ethanol’s effects on serotonin neurons or the larger circuit of the DR in which they are contained. To our knowledge, the current study provides the first evidence that synaptic transmission and excitability within the DR are altered by chronic ethanol exposure. With respect to synaptic transmission, we observed a shift towards enhanced excitation during early withdrawal that was driven by reductions in spontaneous inhibitory transmission on the presynaptic side, perhaps due to reductions in GABA release. Because changes in network-independent inhibitory transmission were not observed at this time point, these findings suggest that chronic ethanol exposure may enhance the net activity of DR neurons via adaptations that reduce the net activity of DR interneurons, which are located primarily in the lateral wings of the structure (Fu et al, 2010), thus resulting in reduced GABA release. Although further investigation is required to characterize alterations in release probability and conclusively determine the source of GABA inputs that are altered following chronic ethanol exposure, recent work by Challis et al (2013) suggests that synapses between interneurons and serotonin neurons are a significant site of adaptations that confer behavioral pathology induced by chronic stressors. Thus, it is reasonable to hypothesize that this synapse is also a potential site of ethanol-induced adaptations.
In addition to alterations in synaptic modulation, we observed a significant enhancement of excitability during early withdrawal from chronic ethanol exposure. Because resting membrane potential, the threshold for excitation, and the rheobase were not altered by chronic ethanol, this finding reflects enhancements in neuronal activity that are stimulation-dependent rather than an alteration in intrinsic neuronal properties at rest. Such increases in excitability could have functional consequences at downstream targets of the DR, such as the nucleus accumbens, where serotonin release is increased following chronic ethanol exposure (Yoshimoto et al, 2012), or the central nucleus of the amygdala, where serotonin receptor compounds normalize chronic ethanol-induced anxiety-like behavior (Overstreet et al, 2006). Indeed, enhanced serotonin activity during periods of abstinence have documented effects on AUD-related behavioral pathology, such as serotonin agonist-induced exacerbation of anxiety and craving (Umhau et al, 2011) and SSRI-induced worsening of long-term drinking outcomes in human alcoholics (Dundon et al, 2004). Although further investigation is required to confirm that chronic ethanol enhancement of net activity in the DR confers increased serotonin output, our initial findings provide insight into a potential mechanism by which the serotonin system is dysregulated by chronic ethanol.
Although alterations in neuronal excitability and inhibitory transmission were resolved during late withdrawal, a shift toward excitation was still evident at this time point. In contrast to early withdrawal, this shift was driven primarily by enhancements in excitatory transmission. Specifically, we observed a significant increase in spontaneous postsynaptic excitatory transmission as well as a significant increase in network-independent presynaptic excitatory transmission, results that suggest changes in postsynaptic glutamate receptor availability or types as well as increases in presynaptic glutamate release. Enhanced presynaptic excitatory transmission has also been observed in the DR following exposure to forced swim stress (Kirby et al, 2007). Interestingly, this effect was specific to serotonin neurons, suggesting that this neuronal population may be particularly sensitive to stressors and perhaps chronic ethanol exposure. Considering that glutamatergic activity in the DR has been implicated in stress-induced behavioral changes related to negative mood states (Grahn et al, 2000), enhanced excitatory transmission in the DR induced by chronic ethanol may contribute to increases in anxiety-like behavior during withdrawal although further examination is required to confirm this possibility. The observed alterations in synaptic transmission during both early and late withdrawal are particularly interesting in light of emerging data showing that GABA bidirectionally modulates glutamate release onto DR neurons (Soiza-Reilly et al, 2013). Thus, reductions in GABA transmission during early withdrawal may facilitate adaptations resulting in enhanced glutamate release during late withdrawal.
Acute Ethanol Effects in the DR
We found that following chronic ethanol exposure, cells from the DR are more sensitive to the inhibitory effects of a moderate concentration of ethanol applied acutely to slices. In contrast to ethanol-naive (both AE and naive) mice, in which postsynaptic inhibition is enhanced by acute ethanol without an appreciable effect on presynaptic inhibition, both pre- and postsynaptic inhibition is enhanced by acute ethanol following chronic ethanol treatment. Indeed, while acute ethanol enhances postsynaptic GABA transmission in the DR of both ethanol-naive and chronically exposed mice, the decay kinetics of inhibitory events were slower during acute ethanol perfusion only following chronic ethanol exposure. Thus, the postsynaptic effect of acute ethanol is augmented by chronic ethanol exposure. Further, we found that although basal GABA transmission is not altered by chronic ethanol, presynaptic mechanisms are engaged by acute ethanol exclusively following chronic ethanol exposure, conferring increased GABA release. This effect contrasts with previous observations in the central nucleus of the amygdala, where chronic ethanol exposure results in enhanced basal levels of GABA that are augmented further by reintroduction of acute ethanol (Roberto et al, 2010).
Acute ethanol facilitation of inhibitory transmission in the DR has been reported previously in ethanol-naive rodents. In contrast with our findings, an initial investigation found that moderate concentrations of acute ethanol enhances both pre- and postsynaptic GABAA receptor mediated transmission in rats (Lemos et al, 2011), perhaps reflecting a species difference in acute ethanol responses. In addition, recent findings suggest that strain may also be a factor in differential effects of ethanol as a recent report shows enhancement of only presynaptic GABAA receptor mediated transmission in ethanol-naive C57BL/6J mice (Maguire et al, 2014). In addition to effects on GABA transmission, acute ethanol activated a large tonic inward current mediated by extrasynaptic glycine receptors in the DR of C57BL/6J mice. In the current study conducted in DBA2/J mice, we found no evidence of an ethanol-induced tonic current under naive or chronic ethanol conditions. Thus, DBA2/J mice and C57BL/6J mice appear to fundamentally differ in aspects of inhibitory modulation of the DR. In support of this possibility, there is evidence showing basal differences in GABA transmission in the basolateral amygdala of these mouse strains (Dubois et al, 2006). It is of note that these strains also differ behaviorally, with DBA2/J mice showing higher levels of basal anxiety than C57BL/6J mice (Dubois et al, 2006). Likewise, C57BL/6J mice appear to be resistant to chronic ethanol-induced anxiety (Cox et al, 2013), whereas the current results show that DBA2/J mice are vulnerable. It will be interesting to ascertain whether such behavioral differences arise from strain differences in DR function and/or serotonin activity.
CONCLUSIONS
The present findings demonstrate that chronic ethanol exposure enhances the net activity of DR neurons at time points during withdrawal when anxiety-like behavior is increased. Because the inhibitory effects of acute ethanol are augmented following chronic ethanol treatment, reintroduction of acute ethanol appears to functionally oppose withdrawal effects on GABA transmission in the DR. As such, the DR may have a critical role in the negative reinforcing properties of acute ethanol that have been linked with relapse in alcoholic populations. Overall, these results provide the first compelling evidence implicating chronic ethanol-induced hyperactivity in the DR as a potential mechanism underlying ethanol-related behavioral pathologies related to serotonin that have been extensively documented in the literature.
FUNDING AND DISCLOSURE
This work was funded by NIH-NIAAA grants U01-AA020911, R01-AA019454, and P60-AA011605 to TLK, NIH-NIAAA grant T32-AA007573 and F32-AA022549 to EGLG and NIH-NIAAA grant F32-AA021319 to CAM. The authors declare no conflict of interest.
References
Arango V, Underwood MD, Boldrini M, Tamir H, Kassir SA, Hsiung S et al (2001). Serotonin 1A receptors, serotonin transporter binding and serotonin transporter mRNA expression in the brainstem of depressed suicide victims. Neuropsychopharmacology 25: 892–903.
Beck O, Borg S, Sedvall G (1983). Tryptophan levels in human cerebrospinal acute and chronic ethanol consumption fluid after. DrugAlcohol Depend 12: 217–222.
Becker HC, Lopez MF (2004). Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in c57bl/6 mice. Alcohol Clin Exp Res 28: 1829–1838.
Booth B, Russell DW, Yates WR, Laughlin PR, Brown K, Reed D (1992). Social support and depression in men during alcoholism treatment. J Subst Abuse 4: 57–67.
Buckner JD, Timpano KR, Zvolensky MJ, Sachs-Ericsson N, Schmidt NB (2008). Implications of comorbid alcohol dependence among individuals with social anxiety disorder. Depress Anxiety 25: 1028–1037.
Carneiro AMD, Airey DC, Thompson B, Zhu CB, Lu L, Chesler EJ et al (2009). Functional coding variation in recombinant inbred mouse lines reveals multiple serotonin transporter-associated phenotypes. Proc Natl Acad Sci USA 106: 2047–2052.
Challis C, Boulden J, Veerakumar A, Espallergues J, Vassoler FM, Pierce RC et al (2013). Raphe GABAergic neurons mediate the acquisition of avoidance after social defeat. J Neurosci 33: 13978–13988 13988a.
Charney DA, Zikos E, Gill KJ (2010). Early recovery from alcohol dependence : factors that promote or impede abstinence. J Subst Abuse Treat 38: 42–50.
Cox BR, Olney JJ, Lowery-Gionta EG, Sprow GM, Rinker JA, Navarro M et al (2013). Repeated cycles of binge-like ethanol (EtOH)-drinking in male C57BL/6J mice augments subsequent voluntary EtOH intake but not other dependence-like phenotypes. Alcohol Clin Exp Res 37: 1688–1695.
Dubois DW, Perlegas A, Floyd DW, Weiner JL, McCool BA (2006). Distinct functional characteristics of the lateral/basolateral amygdala GABAergic system in C57BL/6J and DBA/2J mice. J Pharmacol Exp Ther 318: 629–640.
Dundon W, Lynch KG, Pettinati HM, Lipkin C (2004). Treatment Outcomes in Type A and B Alcohol Dependence 6 Months After Serotonergic Pharmacotherapy. Alcohol Clin Exp Res 28: 1065–1073.
Dutton CE, Adams T, Bujarski S, Badour CL, Feldner MT (2014). Posttraumatic stress disorder and alcohol dependence: individual and combined associations with social network problems. J Anxiety Disord 28: 67–74.
Fleming RL, Wilson Wa, Swartzwelder HS (2007). Magnitude and ethanol sensitivity of tonic GABAA receptor-mediated inhibition in dentate gyrus changes from adolescence to adulthood. J Neurophysiol 97: 3806–3811.
Fox HC, Anderson GM, Tuit K, Hansen J, Kimmerling A, Siedlarz KM et al (2012). Prazosin effects on stress- and cue-induced craving and stress response in alcohol-dependent individuals: preliminary findings. Alcohol Clin Exp Res 36: 351–360.
Fu W, Le Maître E, Fabre V, Bernard JF, David Xu ZQ, Hökfelt T (2010). Chemical neuroanatomy of the dorsal raphe nucleus and adjacent structures of the mouse brain. J Comp Neurol 518: 3464–3494.
Glöckner-Rist A, Lémenager T, Mann K (2013). Reward and relief craving tendencies in patients with alcohol use disorders: results from the PREDICT study. Addict Behav 38: 1532–1540.
Grahn RE, Watkins LR, Maier SF (2000). Impaired escape performance and enhanced conditioned fear in rats following exposure to an uncontrollable stressor are mediated by glutamate and nitric oxide in the dorsal raphe nucleus. Behav Brain Res 112: 33–41.
Hale MW, Shekhar A, Lowry Ca (2012). Stress-related serotonergic systems: implications for symptomatology of anxiety and affective disorders. Cell Mol Neurobiol 32: 695–708.
Hamilton KR, Ansell EB, Reynolds B, Potenza MN, Sinha R (2013). Self-reported impulsivity, but not behavioral choice or response impulsivity, partially mediates the effect of stress on drinking behavior. Stress 16: 3–15.
Hasin DS, Stinson FS, Ogburn E, Grant BF (2007). Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry 64: 830–842.
Huang MM, Overstreet DH, Knapp DJ, Angel R, Wills TA, Navarro M et al (2010). Corticotropin-releasing factor (CRF) sensitization of ethanol withdrawal-induced anxiety-like behavior is brain site specific and mediated by CRF-1 receptors: Relation to stress-induced sensitization. J Pharmacol Exp Ther 332: 298–307.
Hwa LS, Debold JF, Miczek Ka (2013). Alcohol in excess: CRF1 receptors in the rat and mouse VTA and DRN. Psychopharmacology 225: 313–327.
Kelly JF, Hoeppner B, Stout RL, Pagano M (2012). Determining the relative importance of the mechanisms of behavior change within alcoholics anonymous: a multiple mediator analysis. Addiction 107: 289–299.
Kirby LG, Pan YZ, Freeman-Daniels E, Rani S, Nunan JD, Akanwa A et al (2007). Cellular effects of swim stress in the dorsal raphe nucleus. Psychoneuroendocrinology 32: 712–723.
Knapp DJ, Overstreet DH, Huang M, Wills TA, Whitman BA, Angel RA et al (2011). Effects of a stressor and corticotrophin releasing factor on ethanol deprivation-induced ethanol intake and anxiety-like behavior in alcohol-preferring P rats. Psychopharmacology 218: 179–189.
Knapp DJ, Overstreet DH, Moy SS, Breese GR (2004). SB242084, flumazenil, and CRA1000 block ethanol withdrawal-induced anxiety in rats. Alcohol 32: 101–111.
Knapp DJ, Overstreet DH, Breese GR (2005). Modulation of ethanol withdrawal-induced anxiety-like behavior during later withdrawals by treatment of early withdrawals with benzodiazepine/gamma-aminobutyric acid ligands. Alcohol Clin Exp Res 29: 553–563.
Kranzler HR, Armeli S, Tennen H, Covault J (2013). 5-HTTLPR genotype and daily negative mood moderate the effects of sertraline on drinking intensity. Addict Biol 18: 1024–1031.
Kranzler HR, Feinn R, Armeli S, Tennen H (2012). Comparison of alcoholism subtypes as moderators of the response to sertraline treatment. Alcohol Clin Exp Res 36: 509–516.
Kranzler HR, Burleson JA, Brown J, Babor TF (1996). Fluoxetine treatment seems to reduce the beneficial effects of cognitive-behavioral therapy in type B alcoholics. Alcohol Clin Exp Res 20: 1534–1541.
Krystal JH, Webb E, Cooney NL, Kranzler HR, Southwick SW, Heninger GR et al (1996). Serotonergic and noradrenergic dysregulation in alcoholism: m-chlorophenylpiperazine and yohimbine effects in recently detoxified alcoholics and healthy comparison subjects. Am J Psychiatry 153: 83–92.
Lemos JC, Zhang G, Walsh T, Kirby LG, Akanwa A, Brooks-Kayal A et al (2011). Stress-hyperresponsive WKY rats demonstrate depressed dorsal raphe neuronal excitability and dysregulated CRF-mediated responses. Neuropsychopharmacology 36: 721–734.
Li C, Pleil KE, Stamatakis AM, Busan S, Vong L, Lowell BB et al (2012). Presynaptic inhibition of gamma-aminobutyric acid release in the bed nucleus of the stria terminalis by kappa opioid receptor signaling. Biolo Psychiatry 71: 725–732.
López-Figueroa AL, Norton CS, López-Figueroa MO, Armellini-Dodel D, Burke S, Akil H et al (2004). Serotonin 5-HT1A, 5-HT1B, and 5-HT2A receptor mRNA expression in subjects with major depression, bipolar disorder, and schizophrenia. Biol Psychiatry 55: 225–233.
Lovallo WR, Enoch MA, Yechiam E, Glahn DC, Acheson A, Sorocco KH et al (2014). Differential impact of serotonin transporter activity on temperament and behavior in persons with a family history of alcoholism in the oklahoma family health patterns project. Alcohol Clin Exp Res 38: 1575–1581.
Maguire E, Mitchell EA, Greig SJ, Corteen N, Balfour DJ, Swinny JD et al (2014). Extrasynpatic glycine receptors of roden dorsal raphe serotonergic neurons: a sensitive target for ethanol. Neuropsychopharmacology 39: 1232–1244.
Marquenie LA, Schadé A, van Balkom AJ, Comijs HC, de Graaf R, Vollebergh W et al (2007). Origin of the comorbidity of anxiety disorders and alcohol dependence: findings of a general population study. Eur Addict Res 13: 39–49.
Meeker HC, Chadman KK, Heaney AT, Carp RI (2013). Assessment of social interaction and anxiety-like behavior in senescence-accelerated-prone and -resistant mice. Physiol Behav 118: 97–102.
Miller JM, Hesselgrave N, Ogden RT, Zanderigo F, Oquendo MA, Mann JJ et al (2013). Brain serotonin 1A receptor binding as a predictor of treatment outcome in major depressive disorder. Biol Psychiatry 74: 760–767.
Millstein Ra, Holmes A (2007). Effects of repeated maternal separation on anxiety- and depression-related phenotypes in different mouse strains. Neurosci Biobehav Rev 31: 3–17.
Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP et al (2007). Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav Brain Res 176: 4–20.
Moy SS, Nadler JJ, Young NB, Nonneman RJ, Grossman AW, Murphy DL et al (2009). Social approach in genetically-engineered mouse lines relevant to autism. Genes Brain Behav 8: 129–142.
Overstreet D, Knapp D, Breese G (2002). Accentuated decrease in social interaction in rats subjected to repeated ethanol withdrawals. Alcohol Clin Exp Res 26: 1259–1268.
Overstreet DH, Knapp DJ, Angel RA, Navarro M, Breese GR (2006). Reduction in repeated ethanol-withdrawal-induced anxiety-like behavior by site-selective injections of 5-HT1A and 5-HT2C ligands. Psychopharmacology 187: 1–12.
Overstreet DH, Moy SS, Breese GR (2003). A 5-HT1A agonist and 5-HT2C antagonist reduce social interaction deficit induced by multiple ethanol withdrawals in rats. Pyschopharmacology 167: 344–352.
Pettinati HM, Gastfriend DR, Dong Q, Kranzler HR, O'Malley SS (2009). Effect of extended-release naltrexone (XR-NTX) on quality of life in alcohol-dependent patients. Alcohol Clin Exp Res 33: 350–356.
Pivac N, Mück-Seler D, Mustapić M, Nenadić-Sviglin K, Kozarić-Kovacić D (2004). Platelet serotonin concentration in alcoholic subjects. Life Sci 76: 521–531.
Richter S, Brown S, Mott M (1991). The impact of social support and self-esteem on adolescent substance abuse treatment outcome. J Subst Abuse 3: 371–385.
Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M et al (2010). Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry 67: 831–839.
Samaco RC, Mandel-Brehm C, McGraw CM, Shaw CA, McGill BE, Zoghbi HY (2012). Crh and Oprm1 mediate anxiety-related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nat Genet 44: 206–211.
Satre DD, Chi FW, Mertens JR, Weisner CM (2012). Effects of age and life transitions on alcohol and drug treatment outcome over nine years. J Studies Alcohol Drugs 73: 459–468.
Schneier FR, Foose TE, Hasin DS, Heimberg RG, Liu SM, Grant BF et al (2010). Social anxiety disorder and alcohol use disorder co-morbidity in the national epidemiologic survey on alcohol and related conditions. Psychol Med 40: 977–988.
Soiza-Reilly M, Anderson WB, Vaughan CW, Commons KG (2013). Presynaptic gating of excitation in the dorsal raphe nucleus by GABA. Proc Natl Acad Sci USA 110: 15800–15805.
Takahashi A, Kwa C, Debold JF, Miczek KA (2010). GABA(A) receptors in the dorsal raphé nucleus of mice: escalation of aggression after alcohol consumption. Psychopharmacology 211: 467–477.
Tomkins D, Sellers E, Fletcher P (1994). Effect of dorsal raphe injections of the GABAA agonist, muscimol, on ethanol intake and measures of intoxication in Wistar rats. Alcohol Alcohol Suppl 2: 551–558.
Umhau JC, Schwandt ML, Usala J, Geyer C, Singley E, George DT et al (2011). Pharmacologically induced alcohol craving in treatment seeking alcoholics correlates with alcoholism severity, but is insensitive to acamprosate. Neuropsychopharmacology 36: 1178–1186.
Underwood MD, Mann JJ, Arango V (2007). Morphometry of dorsal raphe nucleus serotonergic neurons in alcoholism. Alcohol Clin Exp Res 31: 837–845.
Yoshimoto K, Watanabe Y, Tanaka M, Kimura M (2012). Serotonin2C receptors in the nucleus accumbens are involved in enhanced alcohol-drinking behavior. Eur J Neurosci 35: 1368–1380.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article

Cite this article
Lowery-Gionta, E., Marcinkiewcz, C. & Kash, T. Functional Alterations in the Dorsal Raphe Nucleus Following Acute and Chronic Ethanol Exposure. Neuropsychopharmacol 40, 590–600 (2015). https://doi.org/10.1038/npp.2014.205
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.205
This article is cited by
-
Estrogen signaling in the dorsal raphe regulates binge-like drinking in mice
Translational Psychiatry (2024)
-
Neural serotonergic circuits for controlling long-term voluntary alcohol consumption in mice
Molecular Psychiatry (2022)
-
Serotonin modulates an inhibitory input to the central amygdala from the ventral periaqueductal gray
Neuropsychopharmacology (2022)
-
5-HT1A receptor-dependent modulation of emotional and neurogenic deficits elicited by prolonged consumption of alcohol
Scientific Reports (2018)
-
Opposing actions of CRF-R1 and CB1 receptors on VTA-GABAergic plasticity following chronic exposure to ethanol
Neuropsychopharmacology (2018)
