
Abstract
Although TRAIL (tumor necrosis factor (TNF)-related apoptosis inducing ligand) is a well-known apoptosis inducer, we have previously demonstrated that acidic extracellular pH (pHe) switches TRAIL-induced apoptosis to regulated necrosis (or necroptosis) in human HT29 colon and HepG2 liver cancer cells. Here, we investigated the role of RIPK1 (receptor interacting protein kinase 1), RIPK3 and PARP-1 (poly (ADP-ribose) polymerase-1) in TRAIL-induced necroptosis in vitro and in concanavalin A (Con A)-induced murine hepatitis. Pretreatment of HT29 or HepG2 with pharmacological inhibitors of RIPK1 or PARP-1 (Nec-1 or PJ-34, respectively), or transient transfection with siRNAs against RIPK1 or RIPK3, inhibited both TRAIL-induced necroptosis and PARP-1-dependent intracellular ATP depletion demonstrating that RIPK1 and RIPK3 were involved upstream of PARP-1 activation and ATP depletion. In the mouse model of Con A-induced hepatitis, where death of mouse hepatocytes is dependent on TRAIL and NKT (Natural Killer T) cells, PARP-1 activity was positively correlated with liver injury and hepatitis was prevented both by Nec-1 or PJ-34. These data provide new insights into TRAIL-induced necroptosis with PARP-1 being active effector downstream of RIPK1/RIPK3 initiators and suggest that pharmacological inhibitors of RIPKs and PARP-1 could be new treatment options for immune-mediated hepatitis.
Similar content being viewed by others


Main
Tumor necrosis factor (TNF)-related apoptosis inducing ligand (TRAIL) is a key inducer of apoptosis in tumor cells1 and a mediator of tumor immune surveillance.2 TRAIL has also a crucial role in Con A-induced hepatitis.3
TRAIL activates apoptosis by binding to death receptors DR4 (TRAIL-R1) and DR5 (TRAIL-R2)4 in humans and only to mDR5 (mTRAIL-R2/mKILLER) in mice.5 Besides apoptosis induction, TRAIL induces necrosis in Jurkat cells6 or in murine prostate adenocarcinoma TRAMP-C2 cells.7 More recently, we have shown that acidic pHe switches TRAIL-induced apoptosis to RIPK1 (receptor interacting protein kinase 1)-dependent regulated necrosis in human HT29 colon adenocarcinoma and human HepG2 hepatocarcinoma cell lines.8, 9 Such an acidic extracellular environment could occur under some pathological conditions including cancer and inflammation.10, 11
Necrosis has long been considered as an accidental cell death process until it has been shown that necrosis is also a well-orchestrated programmed cell death12, 13 like apoptosis. However, there was no consensus on the specific biochemical changes, as many initiators, modulators or effectors of necrosis have been described.14 Nonetheless, some specific molecular mechanisms have been pointed out in PARP-1 (poly (ADP-ribose) polymerase-1)-mediated necrotic cell death induced by DNA-damaging agents, called parthanatos,15 and in RIPK1-dependent programmed necrosis induced by TNF-like cytokines.16 Recently, RIPK3, another RIP family kinase, has been identified as an essential mediator for TNF-, FasL- and TRAIL-mediated necroptosis.17, 18, 19 This emerging necrotic pathway in vitro has been shown to be relevant in vivo in vaccinia virus infection17 and cerulein-induced pancreatitis.18
The mouse model of immune-mediated liver injury, after i.v. administration of the T-cell mitogen Con A, results in fulminant hepatitis.20 Several lines of evidence suggest that NKT cells are critically involved in Con A-induced hepatitis,21, 22, 23 and that TRAIL expressed by these cells in the liver is directly responsible for mediating hepatic cell death.3
Here, we studied the molecular mechanisms involved in TRAIL-induced necroptosis at acidic pHe and found that TRAIL induced an early PARP-1-dependent intracellular ATP depletion, which was dependent on both RIPK1 and RIPK3 demonstrating for the first time an interrelationship between RIPK1/RIPK3 and PARP-1. We also provided evidence that Con A-induced hepatitis was a relevant model for TRAIL-induced necroptosis in vivo, as Nec-1 and PJ-34 protected mice from this hepatitis.
Results
TRAIL-induced necroptosis at acidic pHe is dependent on RIPK1 and RIPK3
TRAIL induced apoptosis at pHe 7.4 (∼25–30% of apoptotic cells) (Figure 1a, upper panel) with very low ATP depletion Figure 1b) whereas it induced regulated necrosis at pHe 6.5 (∼70% of necrotic permeabilized cells) (Figure 1a, lower panel) with high ATP depletion (∼90%/NT (non-treated cells)) (Figure 1b). As TRAIL-induced necrosis was significantly inhibited by Nec-1 (Figure 1a, lower panel), a specific inhibitor of RIPK1 kinase activity, this type of cell death has been designated as necroptosis in the following text. Moreover, Nec-1 or geldanamycin (GA) pretreatment led to a switch from necroptosis to apoptosis at acidic pHe (Figure 1a) with partial intracellular ATP repletion (Figure 1b). Then, we studied the role of RIPK1 and RIPK3 in TRAIL-induced necroptosis at acidic pHe and observed that transient transfections with siRNA targeting RIPK1 or RIPK3 almost totally reduced the expression of RIPK1 and RIPK3 in HT29 cells (Figure 1d and Supplementary Figure S1a) and significantly inhibited both TRAIL-induced necroptosis (Figure 1c, lower panel) and ATP depletion (Figure 1e). However, gene knockdown of RIPK1 and RIPK3 did not achieve complete inhibition of TRAIL-induced necroptosis at acidic pHe (Figure 1c, lower panel) probably due to increased RIPK1 and RIPK3 expression upon TRAIL treatment (data not shown). Moreover, gene knockdown of RIPK1 and/or RIPK3 led to a switch from TRAIL-induced necroptosis to apoptosis at acidic pHe with partial restoration of intracellular ATP level (Figures 1c and e). We then tested mouse embryonic fibroblasts (MEFs) from Ripk1−/− and Ripk3−/− mice with respective littermate WT controls. Both WT MEFs (MEFs RIPK1 and MEFS RIPK3) were resistant to TRAIL-induced apoptosis at physiological pHe (Figures 2a and b, left panels) but were sensitized to TRAIL-induced necroptosis at acidic pHe (Figures 2a and b, right panels). As expected, TRAIL-induced necroptosis at acidic pHe was almost completely inhibited in MEFs RIPK1 KO and MEFs RIPK3 KO, which do not express RIPK1 or RIPK3, respectively, (Figure 2c and Supplementary Figure S1b), confirming a role for both RIPK1 and RIPK3 in TRAIL-induced necroptosis at acidic pHe.

TRAIL-induced necrosis at acidic pHe is dependent on both RIPK1 and RIPK3. (a) HT29 cells were treated or not (NT) with 100 ng/ml TRAIL-Flag and 2 μg/ml anti-Flag M2 for 24 h, at physiological pHe 7.4 or acidic pHe 6.5, after a 16 h pretreatment with 1 μM GA or a 1 h pretreatment with 100 μM Necrostatin-1 (Nec-1). Percentages of apoptosis or necrosis were estimated as described in Materials and Methods. (b) Intracellular ATP concentration was measured as described in Materials and Methods. (c) HT29 cells were transiently transfected with the following siRNAs: si RIPK1, si RIPK3, si RIPK1/si RIPK3 (si RIPK1/3) or with si NT1 (non-targeting siRNA used as negative control). At 72 h after transfection, HT29 cells were treated with 100 ng/ml TRAIL-Flag and 2 μg/ml anti-Flag M2 for 24 h, at pHe 7.4 or 6.5. Percentages of apoptosis or necrosis were estimated as described in Materials and Methods. (d) Western blot analysis of RIPK1 and RIPK3 expression was carried out 72 h after transfection. Anti-human HSC70 was used as a control of protein loading (one representative of three independent experiments). (e) Intracellular ATP concentration was measured as described in Materials and Methods. Mean±S.D. #P<0.05, **, ##P<0.01 and ***, ###P<0.001

MEFs RIPK1 KO and MEFs RIPK3 KO are resistant to TRAIL-induced necroptosis at acidic pHe. (a) MEFs RIPK1 and MEFs RIPK1 KO (b) MEFs RIPK3 and MEFs RIPK3 KO were treated with the indicated TRAIL-Flag concentrations cross-linked with 2 μg/ml anti-Flag M2 for 24 h, at pHe 7.4 or 6.5. Percentages of apoptosis and necrosis were estimated as described in Materials and Methods. (c) Western blot analysis of RIPK1 and RIPK3 expression was performed. Anti-human HSC70 was used as a control of protein loading (one representative of three independent experiments). Mean±S.D. *, $P<0.05, $$P<0.01 and ***, $$$P<0.001
Interestingly, an acidic pHe sensitized HT29 cells only to TRAIL-induced cell death but not to FasL or TNF (Supplementary Figure S2a, right panel), although the pro-death activities of FasL or TNF in Jurkat or L929 cells were not impaired, respectively (data not shown). TRAIL-induced cell death in HT29 cells was inhibited by the use of antagonistic antibodies directed against DR4 or DR5, but not by an antagonistic antibody directed against Fas or by a TNF inhibitor (Supplementary Figure S2b). However, anti-Fas or TNF inhibitor inhibited FasL-induced cell death in Jurkat cells or TNF-induced cell death in L929 cells, respectively (Supplementary Figures S2c and d). Moreover, transient transfection of HT29 cells with siRNA targeting DR4, DR5, Fas or TNF-R1 led to a decreased expression of DR4, DR5, Fas or TNF-R1, respectively (Supplementary Figures S3b and c), but only reduced expression of DR4 and DR5 inhibited TRAIL-induced necroptosis at acidic pHe (Supplementary Figure S3a, right panel). Besides, transient transfection of Jurkat with siRNA targeting Fas or L929 cells with siRNA targeting TNF-R1, significantly inhibited FasL-induced cell death and TNF-induced cell death, respectively (Supplementary Figures S4a–d). Finally, upon TRAIL treatment at physiological or acidic pHe, very low concentrations of TNF were secreted by HT29 cells (Supplementary Figure S4e). All these data suggested that TRAIL-induced necroptosis at acidic pHe was only triggered via TRAIL death receptors (DR4 or DR5) independently of TNF secretion.
RIPK1/RIPK3-dependent PARP-1 activation initiates TRAIL-induced necroptosis at acidic pHe
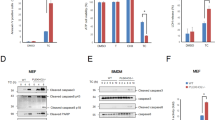
PARP-1 activity was not detected in HT29 cells treated with TRAIL at pHe 7.4 but was increased between 4 and 16 h after TRAIL treatment at acidic pHe (Figure 3a, upper panel). Protein poly ADP-ribosylation (PAR), analyzed by western blot, increased accordingly (Figure 3a, lower panel and Supplementary Figure S5a). To confirm the role of PARP-1, transient transfection with siRNA targeting PARP-1 almost totally reduced PARP-1 expression in HT29 cells (inset Figure 3b and Supplementary Figure S5b), inhibited TRAIL-induced ATP depletion (Figure 3b) and necroptosis at acidic pHe (Figure 3c, lower panel) and switched to TRAIL-induced apoptosis (Figure 3c, upper panel). Moreover, pretreatment with PJ-34, a pharmacological inhibitor of PARP-1, switched from TRAIL-induced necroptosis to apoptosis and partially restored intracellular ATP levels (Figures 3c and d). Pretreatment with PJ-34 or gene knockdown of PARP-1 by RNA interference completely inhibited TRAIL-induced PARP-1 activation at acidic pHe (Figures 3e and f). Moreover, pretreatment with GA or Nec-1, or gene knockdown of RIPK1 or RIPK3 by RNA interference inhibited TRAIL-induced PARP-1 activation at acidic pHe (Figures 3e and f), suggesting that RIPK1/RIPK3-dependent signaling is required for PARP-1 activation. In accordance with the above results obtained with HT29 cells, MEFs PARP-1 KO cells were more resistant than WT MEFs (MEFs PARP-1) to TRAIL-induced necroptosis at acidic pHe (Figure 4a, lower panel). Moreover, TRAIL-induced ATP depletion at acidic pHe was partially inhibited by pretreatment of MEFs PARP-1 with GA, Nec-1 or PJ-34 (Figure 4b), suggesting a role for RIPK1 and PARP-1 in TRAIL-induced necroptosis at acidic pHe. Transient transfection of MEFs PARP-1 with siRNA targeting PARP-1 decreased PARP-1 expression (Figure 4c and Supplementary Figure S5c) and rendered these cells more resistant to TRAIL-induced cell death at acidic pHe (Figure 4d). Finally, gene knockdown of RIPK1, RIPK3 or PARP-1 by RNA interference in human HepG2 hepatocarcinoma cells inhibited TRAIL-induced necroptosis at acidic pHe and led to a switch from necroptosis to apoptosis with restoration of intracellular ATP level (Figures 5a and b). All these data confirmed a role for PARP-1, RIPK1 and RIPK3 in TRAIL-induced necroptosis at acidic pHe in HT29, MEFs and HepG2 cells.

RIPK1/RIPK3-dependent PARP-1 activation initiates TRAIL-induced necroptosis at acidic pHe. (a) HT29 cells were treated with 100 ng/ml TRAIL-Flag and 2 μg/ml anti-Flag M2 for the indicated times, at pHe 7.4 or 6.5. PARP-1 activity was determined as described in Materials and Methods. Western blot analysis of protein poly ADP-ribosylation (PAR) was carried out. Anti-human HSC70 was used as a control of protein loading (one representative of three independent experiments). (b) HT29 cells were transiently transfected with a siRNA directed against PARP-1 or with si NT1 (non-targeting siRNA used as negative control). At 72 h after transfection, cells were treated as above. Intracellular ATP concentration was measured as described in Materials and Methods. Western blot analysis of PARP-1 expression was carried out 72 h after transfection. Anti-human HSC70 was used as a control of protein loading (one representative of three independent experiments). (c) HT29 cells were transiently transfected with si NT1 or si PARP-1 for 72 h, or were pretreated for 1 h with DMSO or 10 μM PJ-34. Then, HT29 cells were treated or not (NT) with 100 ng/ml TRAIL-Flag and 2 μg/ml anti-Flag M2 for 24 h, at pHe 7.4 or 6.5. Percentages of apoptosis and necrosis were estimated as described in Materials and Methods. (d) Intracellular ATP concentration was measured as described in Materials and Methods. (e) HT29 cells were treated or not (NT) with 100 ng/ml TRAIL-Flag and 2 μg/ml anti-Flag M2 for 4 h, at pHe 7.4 or 6.5, after a pretreatment or not (DMSO) with 1 μM GA for 16 h, 100 μM Nec-1 or 10 μM PJ-34 for 1 h. PARP-1 activity was determined as described in Materials and Methods. (f) HT29 cells were transiently transfected with the following siRNAs: si RIPK1, si RIPK3, si PARP-1 or with si NT1 for 72 h, and then treated with TRAIL as above. PARP-1 activity was determined as described in Materials and Methods. Mean±S.D. *, #P<0.05, **, ##P<0.01 and ***, ###P<0.001

Additional evidence for the role of PARP-1 in TRAIL-induced necroptosis at acidic pHe. (a) MEF PARP-1 and MEF PARP-1 KO cells were treated with the indicated TRAIL-Flag concentrations cross-linked with 2 μg/ml anti-Flag M2 for 24 h, at pHe 7.4 or 6.5. Percentages of apoptosis and necrosis were estimated as described in Materials and Methods. (b) MEF PARP-1 cells were treated with 100 ng/ml TRAIL-Flag and 2 μg/ml anti-Flag M2 for 24 h, at pHe 7.4 or 6.5, after a pretreatment or not (DMSO) with 1 μM GA for 16 h, 100 μM Nec-1 or 10 μM PJ-34 for 1 h. Intracellular ATP concentration was measured as described in Materials and Methods. (c) MEF PARP-1 KO and MEF PARP-1 cells were transiently transfected for 72 h with si NT1 (non-targeting siRNA used as negative control) or si PARP-1. Western blot analysis of PARP-1 expression was performed. Anti-human HSC70 was used as a control of protein loading (one representative of three independent experiments). (d) MEF PARP-1 cells were transiently transfected with si NT1 or si PARP-1 for 72 h, and then treated with the indicated TRAIL-Flag concentrations cross-linked with 2 μg/ml anti-Flag M2 for 24 h, at pHe 7.4 or 6.5. Percentage of cell death was estimated by a methylene blue viability assay as described in Materials and Methods. Mean±S.D. *, #, $P<0.05, **, ##P<0.01 and ***P<0.001

Role of PARP-1, RIPK1 and RIPK3 in TRAIL-induced necroptosis at acidic pHe, in HepG2 cells. (a) HepG2 cells were transiently transfected for 72 h with si NT1 (non-targeting siRNA used as negative control) or with siRNAs directed against RIPK1, RIPK3 or PARP-1, and then treated with 100 ng/ml TRAIL-Flag and 2 μg/ml anti-Flag M2 for 24 h, at pHe 7.4 or 6.5. Percentages of apoptosis and necrosis were estimated as described in Materials and Methods. (b) Intracellular ATP concentration was measured as described in Materials and Methods. Mean±S.D. **, ##P<0.01 and ***, ###P<0.001
ROS production is not involved in TRAIL-induced necroptosis at acidic pHe
As reactive oxygen species (ROS) generation has been involved in TNF-induced necrosis24, 25 and as ROS-mediated DNA damage might contribute to activation of PARP-1, we looked for such a role. We first measured ROS generation by flow cytometry using dihydroethidium probe. We observed an important ROS production (superoxide anion, O2.−) in HT29 cells, from 12 to 24 h after TRAIL treatment at acidic pHe (Supplementary Figure S6a). ROS generation 24 h after TRAIL treatment at acidic pHe or after menadione treatment was inhibited by several antioxydants (Thiourea, N-acetyl-cysteine (NAC) and TEMPOL) (Supplementary Figures S6b and c). However, TRAIL-induced necroptosis (Supplementary Figure S7a), PARP-1 activation (Supplementary Figure S7b) or intracellular ATP depletion (Supplementary Figure S7c) at pHe 6.5 were not inhibited by NAC or TEMPOL, which were able to inhibit ROS-dependent cell death induced by propionate/acetate in HT29 cells26 (Supplementary Figures S7d and e). Altogether, these results suggested that TRAIL-induced necroptosis at acidic pHe involved a RIPK1/RIPK3-dependent PARP-1 activation pathway independent of ROS production leading to high ATP depletion.
Con A-induced hepatitis is inhibited by Nec-1 or PJ-34 pretreatment
As CD1d-deficient (CD1d KO) mice have been shown to be protected from Con A-induced hepatitis demonstrating a role for NKT cells21, 22, 23 and TRAIL has been shown to have a crucial role in Con A-induced hepatitis,3 we questioned whether TRAIL-induced necroptosis has a causal role in this model. Pathological examination of liver tissues stained with Hematoxylin/Eosin showed that a single injection of Con A led to a clear liver damage as presented by large diffused perivascular and parenchymal zones of necrosis observed at 6 h or 10 h in WT (data not shown) but not in CD1d KO mice, confirming a role for NKT cells in Con A-induced hepatitis. These histological findings were corroborated with biochemical findings showing that levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were elevated in serum of WT mice but were very low in serum of CD1d KO mice at 6 and 10 h after Con A injection (Figure 6a). As TRAIL has been shown to have a crucial role in Con A-induced hepatitis, we performed RT-qPCR to determine mRNA levels of TRAIL and its death receptor DR5 in mouse liver tissues. TRAIL and DR5 mRNA levels were increased at 6 and/or 10 h after Con A injection in WT but not in CD1d KO mice (Figure 6b, left and right panels, respectively).

Con A-induced hepatitis requires NKT cells and is associated with TRAIL-induced necrotic cell death. WT and CD1d KO C57Bl/6 mice were treated with 20 mg/kg Con A for 6 h (WT, n=6; CD1d KO, n=7) or 10 h (WT, n=7; CD1d KO, n=6). Control mice were treated with PBS only (NT, WT, n=7; CD1d KO, n=6). (a) Serum levels of ALT and AST were determined as described in Materials and Methods. (b) Relative mTRAIL and mDR5 mRNA levels were determined by RT-qPCR in liver tissues as described in Materials and Methods. (c) Intracellular LDH levels were measured in lysates of liver tissues as described in Materials and Methods. (d) Caspase-3 activity was measured in lysates of liver tissues as described in Materials and Methods. (e) Western blot analysis of HMGB1 or cyclophilin A was carried out in lysates of liver tissues. Anti-human HSC70 was used as a control of protein loading (one representative of three experiments). Mean±S.D. *, #P<0.05, **P<0.01 and ***, ###P<0.001
Besides, the mode of hepatic cell death induced by Con A was rather necrotic than apoptotic, as a time-dependent decrease of intracellular LDH level and very low caspase-3 activity were measured in lysates of liver tissues from Con A-treated WT mice (Figures 6c and d). We also observed an increased expression of necrotic markers such as HMGB1 and cyclophilin A in lysates of liver tissues from Con A-treated mice at 6 and 10 h after ConA injection (Figure 6e and Supplementary Figure S8a). Interestingly, intracellular LDH levels were preserved in lysates of liver tissues from Con A-treated CD1d KO mice confirming protection of these mice against Con A-induced hepatitis (Figure 6c).
Next, we looked for a role of PARP-1. PARP-1 activity was increased at 6 and 10 h after Con A injection in lysates of liver tissues from WT mice but not in those from CD1d KO mice (Figure 7a). Accordingly, PAR residues formation, analyzed by western blot in lysates of liver tissues, was increased at 6 and 10 h after Con A injection in WT but not in CD1d KO mice (Figure 7b). The Spearman Rank Correlation analyses to compare ALT or AST serum levels with PARP-1 activity after Con A injection in WT mice demonstrated a positive correlation between these biological markers of hepatic injury (Spearman r=0.8166, P<0.001 for AST/PARP-1 and Spearman r=0.7669, P<0.05 for ALT/PARP-1) (Figure 7c).

PARP-1 is activated in Con A-induced hepatitis. WT and CD1d KO C57Bl/6 mice were treated as in Figure 6. (a) PARP-1 activity was determined as described in Materials and Methods. (b) Western blot analysis of PAR expression was carried out in lysates of liver tissues. Anti-human HSC70 was used as a control of protein loading (one representative of three experiments). Densitometry analysis of PAR expression was carried out (three independent western blot experiments). Relative PAR expression was expressed in arbitrary units (AU) as percentage of absorbance measured in NT. (c) Graphic representation of the Spearman’s rank correlation analysis between AST/ALT and PARP-1 activity. Mean±S.D. *, #P<0.05 and ***, ###P<0.001
At last, to investigate the role of RIPK1 and PARP-1 in Con A-induced hepatitis, WT mice were pretreated with Nec-1, PJ-34 or both inhibitors before Con A injection. Pretreatment with either Nec-1 or PJ-34 significantly decreased Con A-induced hepatotoxicity (Figure 8a, see arrows), associated AST/ALT serum levels (Figure 8b) and PARP activation in lysates from affected liver tissue (Figure 8c). This suggests a potential role of RIPK1 and PARP-1 in this type of hepatic injury. Interestingly, Con A-induced PARP-1 activation (Figure 8c) and increased PAR residues formation (Figure 8d and Supplementary Figure S8b) were significantly attenuated in lysates of liver tissues from mice pretreated with Nec-1, suggesting that RIPK1 might be involved upstream of PARP-1 activity. In agreement with a hepatic necrotic death, no proteolytic fragments of caspase-3 (p32→p17 and p10) and PARP-1 (p116→p84) were detected in lysates of liver tissues from mice treated with Con A (Figure 8d and Supplementary Figure S8b). Moreover, the increase in PARP-1 expression in liver tissues after Con A treatment (Supplementary Figure S8b) was inhibited by pretreatment with Nec-1 or PJ-34 (Figure 8d). All these data pointed out that Con A-induced hepatitis is mediated by TRAIL-induced necroptosis driven by RIPK1. We did not observe any increased protective effect when both inhibitors were combined (Figures 8b and c). Moreover, Con A-induced increase in PARP-1 expression and protein poly ADP-ribosylation were inhibited to the same extent by either Nec-1 or PJ-34 or the combination (Figure 8d). Altogether, these data suggest that PARP-1 activation has a significant role in Con A-induced hepatitis and that RIPK1 and PARP-1 constitute a major cell death signaling pathway.

RIPK1 kinase and PARP-1 are involved in Con A-induced hepatitis. C57Bl/6 WT mice were pretreated or not (NT) with 10 mg/kg PJ-34 for 1 h, 125 μg Nec-1 for 15 min, or with both inhibitors before treatment or not with 12 mg/kg Con A for 10 h (PBS, n=7; PBS+Con A, n=10; Nec-1+Con A, n=6; PJ-34+Con A, n=6; PJ-34+Nec-1+Con A, n=7; Nec-1+PBS, n=7; PJ-34+PBS, n=7; PJ-34+Nec-1+PBS, n=7). (a) Hematoxylin- and Eosin-stained liver sections were shown. Scale bars represent 200 μM. (b) ALT and AST serum levels were determined as described in Materials and Methods. (c) PARP-1 activity was determined in lysates of liver tissues as described in Materials and Methods. (d) Western blot analysis of protein poly ADP-ribosylation (PAR), PARP-1 and caspase-3 expression was carried out. Anti-human HSC70 was used as a control of protein loading (one representative of three experiments). Mean±S.D. $P<0.05, **P<0.01 and ***, $$$P<0.001
Discussion
We reported earlier that TRAIL induces a RIPK1-dependent necrosis-like cell death pathway at acidic pHe.9 We further investigated here the underlying molecular mechanisms involved in this cell death pathway. By using Nec-1, an RIPK1 allosteric inhibitor,27 and RNA interference targeting RIPK1 or RIPK3, we demonstrated that TRAIL-induced cell death at acidic pHe can indeed be defined as necroptosis and depends on both RIPK1 and RIPK3. The role of RIPK1 and RIPK3 was clearly confirmed in MEFs derived from ripk1 and ripk3 knockout mice, as in both MEFs TRAIL-induced necroptosis at acidic pHe was completely inhibited in contrast to control MEFs. Moreover, an acidic pHe sensitized HT29 cells only to TRAIL-induced cell death but not to TNF or FasL. And only decreased expression of DR4 or DR5 by RNA interference, or use of antagonistic antibodies directed against DR4 and DR5, significantly inhibited TRAIL-induced necroptosis showing that TRAIL-induced necroptosis at acidic pHe was specific of the TRAIL death pathway. Although a TNF autocrine production is implicated in zVAD-fmk-induced necroptosis in L929 cells,28 it was not involved in TRAIL-induced necroptosis at acidic pHe, as we observed no inhibition of this cell death pathway by using a TNF inhibitor and low increased production of TNF by ELISA.
Although the effector mechanisms of apoptosis are well defined, little is known about those involved in regulated necrosis.29 The role of mitochondrial ROS in necrosis has been previously shown.24 And recent data have demonstrated that RIPK1 and RIPK3 act upstream mitochondria to regulate ROS production during regulated necrosis.17, 19 However, ROS are not required for regulated necrosis in all cell types, particularly in U937, HT29 or Jurkat cells.18, 30 Accordingly, we demonstrated here that TRAIL-induced necroptosis at acidic pHe was not inhibited in HT29 cells by Thiourea, NAC or TEMPOL, three antioxidants. However, we found a clear link between RIPK1/RIPK3 and PARP-1 activation in TRAIL-induced necroptosis at acidic pHe. PARP-1 has been critically implicated in the necroptosis induced by TNF in L929 cells31 and more recently in parthanatos.15, 32 Our data showed that PARP-1 activity increased with time during the course of TRAIL-induced necroptosis and concomitantly resulted in increased ATP depletion. Indeed, PJ-34, a pharmacological inhibitor of PARP-1, and RNA interference targeting PARP-1 inhibited TRAIL-induced ATP depletion leading to a switch from necroptosis to apoptosis at acidic pHe, thus further supporting an active role of PARP-1 in determining the mode of death receptor-induced cell death.31 Such a switch was also observed after Nec-1 or GA pretreatment (Meurette et al.9 and present data). Our previous and present data are in agreement with literature reporting that GA induced a switch from TNF-induced necroptosis to apoptosis.33 Moreover, the role of PARP-1 in TRAIL-induced necroptosis at acidic pHe was confirmed in parp-1 KO MEFs, in which cell death was completely inhibited. In addition, knockdown of PARP-1 mRNA in MEFs rendered these cells more resistant to TRAIL-induced cell death. Interestingly, Nec-1 and knockdown of RIPK1 or RIPK3 revealed that TRAIL-induced PARP-1 activation at acidic pHe was dependent on both RIPK1 and RIPK3, suggesting that these two kinases most likely act upstream of PARP-1 and ATP depletion during necroptosis. Until now, RIPK1 activation leading to mitochondrial dysfunction has been shown to occur downstream of PARP-1 activation in DNA alkylating agent-induced necrosis,34 and the relationship between this cell death process and necroptosis remains unclear. More studies are needed to understand the relationship between RIPK1/RIPK3 and PARP-1 activation in TRAIL-induced necroptosis at acidic pHe. It has been suggested that PARP-1 activity may be regulated by phosphorylation.35 Therefore, it is tempting to speculate that PARP-1 might be a direct or indirect substrate for RIPK1- and/or RIPK3-mediated kinase cascade.
Besides, NKT cells and TRAIL have been shown to have a crucial role in Con A-induced hepatitis and liver damage (large area of necrosis) was associated with high serum levels of AST and ALT.3, 22 Accordingly, we reported increased TRAIL mRNA expression levels in the liver of Con A-treated WT mice but not in Con A-treated CD1d KO mice suggesting that the increase might be confined to certain immune cells such as NKT cells. In line with this, we observed increased mDR5 mRNA levels in liver of Con A-treated animals, which could mediate hepatocyte cell death induced by TRAIL-expressing NKT cells. Increased expression of HMGB1 and cyclophilin A in the liver of Con A-treated mice are other evidence in favor of ongoing necrosis.36, 37 Moreover, we demonstrated that Con A injection activated PARP-1 in the liver of WT mice but not of CD1d KO mice. In agreement with our in vitro data showing high PARP-1 activation during TRAIL-induced necroptosis at acidic pHe, high PARP-1 activity was correlated with liver damage and high AST/ALT serum levels in WT mice treated with Con A. Moreover, we observed an increased PARP-1 expression in injured liver tissues suggesting that PARP-1 may serve as a biological marker of Con A-induced hepatitis. Interestingly, pretreatment of mice with Nec-1 significantly protected mice from Con A-induced hepatitis by decreasing serum AST/ALT levels, PARP-1 expression and PARP-1 activation in the liver suggesting a role for RIPK1 upstream of PARP-1 in necrosis of hepatocytes. This was confirmed by the protective effect of Nec-1 and PJ-34 on Con A-induced hepatitis. However, no increased protective effect was shown when combining both inhibitors.
All these data suggest that Con A-induced hepatitis may be a relevant in vivo model of TRAIL-induced necroptosis at acidic pHe, as it involves the TRAIL/mDR5 pathway and the RIPK1/PARP-1 axis. The role of RIPK3 in Con A-induced hepatitis remains to be determined. The required acidic conditions during Con A-induced hepatitis or TRAIL-mediated necroptosis could be generated by the inflamed liver environment,38 which could lead to acidification of the extracellular environment.10
Necrotic death is associated with diverse pathological conditions, including myocardial infarction, cerebral ischemia, acute organ failure and infection.16 The in vitro and in vivo models of TRAIL-induced necroptosis described in this work may be the basis to identify new modulators or inhibitors. These drugs would be particularly useful in the treatment of degenerative diseases where necrotic pathways are involved. In addition, programmed necrosis could be triggered to induce cell death in apoptotic/drug-resistant cancers against chemotherapy to counteract the ‘Achilles heel’ of cancers.39 In this context, in solid tumors with acidic pHe, TRAIL may be an efficient inducer of necroptosis and the release of damage-associated molecular pattern molecules and proinflammatory cytokines may favor the recruitment of immune cells leading to tumor regression. In conclusion, our findings provide evidence that TRAIL-induced necroptosis involves RIPK1/RIPK3-dependent PARP-1 activation.
Materials and Methods
Chemicals and antibodies
Recombinant human Flag-tagged TRAIL and Flag-tagged FasL, recombinant human TNF-α, Necrostatin-1 (Nec-1) and PJ-34 were obtained from Alexis Biochemicals (Enzo Life Sciences, Villeurbanne, France). Propidium iodide, Methylene blue, NAC, TEMPOL, Menadione, acetate and propionate sodium salts, concanavalin A (Con A) and anti-Flag M2 IgG1 antibody were obtained from Sigma-Aldrich (Lyon, France). Hoechst 33342 was obtained from Invitrogen (Life Technologies, Villebon sur Yvette, France). GA and pan-caspase inhibitor (z-VAD) were obtained from Calbiochem (VWR International, Fontenay-sous-Bois, France). Thiourea was obtained from Fluka (Sigma-Aldrich). Bradford reagent was obtained from Bio-Rad (Marnes-la-Coquette, France). Proteases inhibitors cocktail and PHOSstop were obtained from Roche (Roche Applied Science, Meylan, France).
Antagonistic mouse monoclonal antibodies directed against extracellular domain of human TRAIL-R1/DR4 and TRAIL-R2/DR5 were from Alexis Biochemicals. The antagonistic mouse monoclonal antibody directed against Fas receptor (ZB4) was from Immunotech (Beckman Coulter, Marseille, France). The TNFα inhibitor (Etanercept, TNFR2 (p75)-IgG1) was obtained from Amgen (Thousand Oaks, CA, USA).
Antibodies used for immunoblotting were mouse monoclonal antibodies to RIPK1 (clone 38), PARP-1 (clone 7D3–6) (Becton Dickinson, Le Pont de Claix, France), PAR (clone 10H) (Alexis Biochemicals), TNFR1 (clone H-5) and HSC70 (clone B-6) (Santa-Cruz, Tebu, Le Perray en Yvelines, France); rabbit polyclonal antibodies to TRAIL-R1/DR4, TRAIL-R2/DR5 (ProSci Incorporated, Interchim, Montluçon, France), PAR (Alexis Biochemicals), Fas/CD95 (clone C-20), Caspase-3 (clone H-277) (Santa-Cruz), HMGB1 (Sigma-Aldrich), Cyclophilin A, PARP-1 (clone 46D11) and RIPK1 (Cell Signaling Technology, St. Quentin, France) and goat polyclonal antibodies to RIPK3 (clone N-14) (Santa-Cruz). Horseradish-peroxydase-conjugated secondary antibodies were provided by Dako France (Trappes, France) and Upstate Technology Incorporated (Milton Keynes, UK).
Cell culture
Human HT29 colon carcinoma, HepG2 hepatocarcinoma and Jurkat lymphoma cell lines were obtained from ATCC (American Type Culture Collection, Rockville, MD, USA) and cultured in EMEM (Eurobio, les Ulis, France), DMEM high glucose (Invitrogen) or RPMI (Invitrogen), respectively, supplemented with 10% FBS (Invitrogen) and 2 mM L-glutamine (Invitrogen). Mouse L929 cell line was a gift from Vandenabeele lab (VIB, Ghent University, Belgium) and cultured in DMEM high glucose supplemented with 10% FCS and 2 mM L-glutamine. Immortalized A1.47 MEFs (MEFs PARP-1 KO) and C9 MEFs (MEFs PARP-1) were a gift of Girish Shah (University of Laval, Canada) and cultured as previously described. 40 Immortalized MEFs RIPK1 and MEFs RIPK1 KO (prepared by Linde Duprez, Vandenabeele lab, VIB, Ghent University, Belgium), primary MEFs RIPK3 and MEFs RIPK3 KO (prepared by Mathieu Bertrand, Vandenabeele lab, VIB, Ghent University, Belgium) were cultured in DMEM high glucose supplemented with 10% FCS and 2 mM L-glutamine. All cells were cultured under a 5% CO2 atmosphere at 37°C.
Cell treatments
To set the pHe of culture medium to either 7.4 or 6.5, we supplemented medium without sodium hydrogenocarbonate (Eurobio) with 23 mM or 5 mM sodium hydrogenocarbonate, respectively, in an atmosphere of 5% CO2. For all experiments, cells were treated in proliferative conditions.
Cell death assays
Cell viability was assessed by a methylene blue colorimetric assay. Briefly, cells were seeded in MW96 at a density of 30 000 cells/well. After treatment, cells were washed in PBS and fixed for 30 min in 95% ethanol. Following removal of ethanol, fixed cells were dried and colored for 40 min in methylene blue. After two washes in tap water, 100 μl of 0.1 N HCl per well were added. Plates were then analyzed with a spectrometer at 620 nm (VersaMax Microplate Reader, Molecular Devices, Wokingham Berkshire, UK) with SoftMax Pro Software. The percentage of viability was calculated by the following formula: 100 × (optical density values of treated cells/optical density values of non-treated cells).
Microscopic detection of apoptosis or necrosis was carried out in both floating and adherent cells recovered after TRAIL treatment using nuclear chromatin staining with 1 μg/ml Hoechst 33342 and 1 μg/ml propidium iodide for 15 min at 37°C. Apoptotic cells (i.e., with condensed blue chromatin or fragmented blue nuclei) or necrotic cells (i.e., with non-fragmented red nuclei) were counted in comparison with total population (n=300 cells).
Caspase-3 activity assay
Caspase-3 activity was measured by monitoring fluorescence continuously in a dual luminescence fluorimeter (Spectra max Gemini XS, Molecular Devices). Briefly, 50–80 μg of cell lysates obtained in a lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EGTA pH 7.4, 0.1% Tween, 10% glycerol, 1 mM DTT) were incubated at 37°C in a caspase assay buffer (20 mM PIPES pH 7.2, 100 mM NaCl, 10% sucrose, 1 mM EDTA, 0.1% CHAPS, 1 mM dithiothreitol) containing 80 μM of DEVD-AMC (Sigma-Aldrich). Enzyme activities were determined as initial velocities and expressed as relative intensity/hour.
LDH concentration measurement
LDH concentration was measured with the CytoTox-One homogenous membrane integrity assay (Promega, Charbonnières, France) according to the manufacturer’s instructions. LDH concentration was expressed as %/NT.
ATP concentration measurement
ATP concentration was measured using the CellTiter-Glo Luminescent Cell Viability Assay kit (Promega). Briefly, cells were seeded in MW96 at a density of 30 000 cells/well. Half an hour before the end of treatment, cells were allowed to equilibrate to room temperature. Then, medium was thrown, cells were lysed in the provided buffer and the luminescent substrate was added. Luminescence was measured in a dual luminescence fluorimeter (Spectra max Gemini XS). In parallel, cell viability was determined by a methylene blue assay. ATP concentration was next expressed as %/NT/cell number.
ELISA
Conditionned media were collected after TRAIL treatment for indicated times and hTNF (DuoSet ELISA, R&D Systems Europe, Lille, France) was measured according to the manufacturer’s instructions.
Western blot analysis
Immunoblotting analysis was performed as previously described.9 Revelation was performed by chemiluminescence using the LAS-3000 analyzer (Fujifilm, Bois-d’Arcy, France). Image processing and densitometry analyses were performed using Multi Gauge software (Fujifilm).
Cell transfections
HT29, HepG2, Jurkat, L929 and MEFs cells were transfected by reverse transfection with double-stranded siGenome SMARTpool siRNAs targeting mRNAs of hRIPK1 (ID=8737), hRIPK3 (ID=11 035), hFas/CD95 (ID=355), hTNFR1 (ID=7132), mTNFR1 (ID=21 937), hPARP-1 (ID=142), mPARP-1 (ID=11 545) (ThermoFisher Scientific, Illkirch, France), or Silencer Select siRNA targeting mRNAs of hDR4 (ID=8797) and hDR5 (ID=8795) (Ambion, Applied Biosystems, Courtaboeuf, France). A non-specific targeting siRNA (siNT1, control siRNA, ThermoFisher Scientific) was used as a negative control for all experiments.
Cells were seeded in MW96 at a concentration of 20 000 cells/well for cell death assays and ATP measurement experiments. Per well, 80 nM siRNA and 0,3 μl of DharmaFECT-4 transfection reagent (ThermoFisher Scientific) were applied in 25 μl Opti-MEMI (Invitrogen), for HT29, HepG2, L929 and MEFs cells. Then, 100 μl cells/well was added. For Jurkat cells, 0,3 μl of Lipofectamine LTX (Invitrogen) and 0.1 μl of PLUS Reagent (Invitrogen) were used instead of DharmaFECT-4. Following a 72-h transfection period, cells were treated with TRAIL at pHe 7.4 or 6.5, for indicated times.
Cells were seeded in MW6 at a concentration of 400 000 cells/well for immunoblotting experiments and PARP-1 activity measurements. Per well, 80 nM siRNA and 5 μl of DharmaFECT-4 transfection reagent (ThermoFisher Scientific) were applied in 1 ml Opti-MEMI (Invitrogen), for HT29, HepG2, L929 and MEFs cells. Then, 1 ml cells/well was added. For Jurkat cells, 5 μl of Lipofectamine LTX (Invitrogen) and 2.5 μl of PLUS Reagent (Invitrogen) were used instead of DharmaFECT-4. For immunoblotting, cells were harvested 72 h after transfection. For PARP-1 activity measurement, following a 72-h transfection period, cells were treated with TRAIL at pHe 7.4 or 6.5, for 4 h.
RNA extraction and RT-qPCR analysis
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. To measure the relative mRNA levels of mTRAIL and mDR5, quantitative real-time RT-PCR was performed using Power SYBRGreen PCR (Applied Biosystems, Life Technologies), in conjunction with an ABI7300 real-time PCR system (Applied Biosystems). Each sample was tested in duplicate in at least three independent experiments. rRNA 18S was used as the invariant control and primers efficiency was measured in each plate. The following primer sets were used. mTRAIL: 5′-CCCTGCTTGCAGGTTAAGAG-3′ (forward), 5′-GGCCTAAGGTCTTTCCATCC-3′ (reverse); mDR5: 5′-TGACGGGGAAGAGGAACTGA-3′ (forward), 5′-GGCTTTGACCATTTGGATCT-3′ (reverse).
PARP-1 activity assay
PARP-1 activity was measured with the PARP Universal Colorimetric Assay (Trevigen Inc., R&D Systems, Lille, France) according to the manufacturer’s instructions. Briefly, 5 × 106 cells were lysed in PARP buffer containing 0.4 M NaCl, 1% Triton X-100 and proteases inhibitors; protein concentration was determined by Bradford assay and at least 20 μg of protein were used in triplicate to measure, by a colorimetric assay, the incorporation of biotinylated PAR onto histone proteins coated in a 96-well plate. PARP-1 activity was expressed as arbitrary units.
Measurement of intracellular superoxide anion by flow cytometry
Dihydroethidium (DHE, Molecular Probes, Life Technologies) was used to detect intracellular superoxide anion (O2.−) production. After treatment, floating and adherent cells (1 × 106 cells) were recovered, washed and incubated in PBS containing 5 μM DHE for 30 min at 37°C. Dye oxidation (increase in FL-2 fluorescence) was measured using a FC-500 flow cytometer (Beckman Coulter). A positive control was obtained by incubating cells for 1 h with Menadione (100 μM). Each measurement was conducted on 10 000 cells and analyzed on CXP Analysis (Beckman Coulter). Superimposition of control and Menadione histograms allowed defining a gate for calculating the percentage of cells with accumulated superoxide anion.
Con A-induced hepatitis
Seven to nine-week-old female C57Bl/6 WT (Janvier, Le Genest St. Isle, France) or CD1d KO mice (provided by Dr. A Lehuen, Hôpital St. Vincent de Paul, INSERM U986, Paris) were treated with a retro-orbital administration of 20 mg/kg Con A (Sigma-Aldrich) diluted in PBS. Mice were killed at 6 h (WT, n=6; CD1d KO, n=7) or 10 h (WT, n=7; CD1d KO, n=6) following treatment. Control mice were treated with vehicle only (PBS) (WT, n=7; CD1d KO, n=6). All mice used were bred in our local house facilities.
For Nec-1 and PJ-34 experiment, C57Bl/6 WT mice were pretreated with i.p. administration of 10 mg/kg PJ-34 for 1 h, or i.v. administration of 125 μg Nec-1 for 15 min, or with both inhibitors before i.v. administration of 12 mg/kg Con A for 10 h. At the end of treatment, mice were killed (PBS+Con A, n=10; Nec-1+Con A, n=6; PJ-34+Con A, n=6; PJ-34+Nec-1+Con A, n=7). Control mice were treated with Nec-1+PBS (n=7), PJ-34+PBS (n=7) or PJ-34+Nec-1+PBS (n=7). All animals received human care and all study protocols comply with the institution’s guidelines.
Fragments of mouse livers were fixed in 4% paraformaldehyde and embedded in paraffin for Hematoxylin/Eosin staining. Serum AST and ALT were measured in serum according to the IFCC primary reference procedures and using the Olympus AU2700 Autoanalyser (Olympus France, Rungis, France).
For liver lysates preparation, 100 mg of liver was homogenized on ice in 650 μl PARP assay lysis buffer containing 0.4 M NaCl, 1% Triton X-100 and proteases inhibitors, with an ULTRA-TURRAX homogenizer. Lysates were incubated on ice with periodic vortexing for 40 min, and then centrifuged at 10 000 × g for 10 min at 4°C to remove insoluble material. Protein concentration was determined by Bradford assay before further analysis.
Statistical analyses
Data presented were acquired from a minimum of three independent experiments. They are expressed as means±S.D. Statistical analyses for in vitro studies were performed with a Student’s t-test considering the variances as unequal. For in vivo studies, Mann–Whitney U test was used for comparison of control group parameters with treatment group and multiple group analysis was carried out by one-way ANOVA with post Mann–Whitney U test. The correlation between continuous variables was examined using Spearman’s rank-order coefficients. All statistical analyses were performed with GraphPad Prism5 software (GraphPad Software, San Diego, CA, USA). The significance is shown as follows: *P<0.05, **P<0.01 and ***P<0.001, TRAIL versus NT. #P<0.05, ##P<0.01, ###P<0.001, TRAIL with inhibitors versus TRAIL with DMSO, or TRAIL with specific siRNAs versus TRAIL with siNT1. $P<0.05, $$P<0.01 and $$$P<0.001, MEF KO cells versus MEF WT cells, or TRAIL pHe 6.5 versus TRAIL pHe 7.4. And for the in vivo experiments: *P<0.05, **P<0.01 and ***P<0.001, mice treated with Con A versus mice treated with PBS. #P<0.05, ##P<0.01 and ###P<0.001, CD1d KO mice treated with Con A versus WT mice treated with Con A. $P<0.05, $$$P<0.001, mice treated with Nec-1+ConA, PJ-34+Con A or Nec-1+PJ-34+Con A versus mice treated with PBS+Con A.
Abbreviations
- ALT:
-
alanine aminotransferase
- AST:
-
aspartate aminotransferase
- Con A:
-
concanavalin A
- GA:
-
geldanamycin
- MEFs:
-
mouse embryonic fibroblasts
- NAC:
-
N-acetyl-cysteine
- Nec-1:
-
necrostatin-1
- NKT:
-
natural killer T
- PAR:
-
poly ADP ribosylation
- PARP-1:
-
poly (ADP-ribose) polymerase-1
- pHe:
-
extracellular pH
- RIPK1/3:
-
receptor interacting protein kinase 1/3
- ROS:
-
reactive oxygen species
- TNF:
-
tumor necrosis factor
- TRAIL:
-
TNF-related apoptosis inducing ligand
References
Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest 1999; 104: 155–162.
Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med 2001; 7: 94–100.
Zheng SJ, Wang P, Tsabary G, Chen YH . Critical roles of TRAIL in hepatic cell death and hepatic inflammation. J Clin Invest 2004; 113: 58–64.
Gonzalvez F, Ashkenazi A . New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 2010; 29: 4752–4765.
Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry WS . Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res 1999; 59: 2770–2775.
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000; 1: 489–495.
Kemp TJ, Kim JS, Crist SA, Griffith TS . Induction of necrotic tumor cell death by TRAIL/Apo-2L. Apoptosis 2003; 8: 587–599.
Meurette O, Huc L, Rebillard A, Le Moigne G, Lagadic-Gossmann D, Dimanche-Boitrel MT . TRAIL (TNF-related apoptosis-inducing ligand) induces necrosis-like cell death in tumor cells at acidic extracellular pH. Ann NY Acad Sci 2005; 1056: 379–387.
Meurette O, Rebillard A, Huc L, Le Moigne G, Merino D, Micheau O et al. TRAIL induces receptor-interacting protein 1-dependent and caspase-dependent necrosis-like cell death under acidic extracellular conditions. Cancer Res 2007; 67: 218–226.
Lardner A . The effects of extracellular pH on immune function. J Leukoc Biol 2001; 69: 522–530.
Wike-Hooley JL, van den Berg AP, van der Zee J, Reinhold HS . Human tumour pH and its variation. Eur J Cancer Clin Oncol 1985; 21: 785–791.
Boujrad H, Gubkina O, Robert N, Krantic S, Susin SA . AIF-mediated programmed necrosis: a highly regulated way to die. Cell Cycle 2007; 6: 2612–2619.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005; 1: 112–119.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G . Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11: 700–714.
Andrabi SA, Dawson TM, Dawson VL . Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann NY Acad Sci 2008; 1147: 233–241.
Degterev A, Yuan J . Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol 2008; 9: 378–390.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–1123.
He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325: 332–336.
Tiegs G, Gantner F . Immunotoxicology of T cell-dependent experimental liver injury. Exp Toxicol Pathol 1996; 48: 471–476.
Kaneko Y, Harada M, Kawano T, Yamashita M, Shibata Y, Gejyo F et al. Augmentation of Valpha14 NKT cell-mediated cytotoxicity by interleukin 4 in an autocrine mechanism resulting in the development of concanavalin A-induced hepatitis. J Exp Med 2000; 191: 105–114.
Takeda K, Hayakawa Y, Van Kaer L, Matsuda H, Yagita H, Okumura K . Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci USA 2000; 97: 5498–5503.
Toyabe S, Seki S, Iiai T, Takeda K, Shirai K, Watanabe H et al. Requirement of IL-4 and liver NK1+ T cells for concanavalin A-induced hepatic injury in mice. J Immunol 1997; 159: 1537–1542.
Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W . Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem 1992; 267: 5317–5323.
Goossens V, De Vos K, Vercammen D, Steemans M, Vancompernolle K, Fiers W et al. Redox regulation of TNF signaling. Biofactors 1999; 10: 145–156.
Jan G, Belzacq AS, Haouzi D, Rouault A, Métivier D, Kroemer G et al. Propionibacteria induce apoptosis of colorectal carcinoma cells via short-chain fatty acids acting on mitochondria. Cell Death Differ 2002; 9: 179–188.
Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008; 4: 313–321.
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008; 135: 1311–1323.
Festjens N, Vanden Berghe T, Vandenabeele P . Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta 2006; 1757: 1371–1387.
Temkin V, Huang Q, Liu H, Osada H, Pope RM . Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol Cell Biol 2006; 26: 2215–2225.
Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell 2002; 13: 978–988.
Wang Y, Dawson VL, Dawson TM . Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol 2009; 218: 193–202.
Vanden Berghe T, Kalai M, van Loo G, Declercq W, Vandenabeele P . Disruption of HSP90 function reverts tumor necrosis factor-induced necrosis to apoptosis. J Biol Chem 2003; 278: 5622–5629.
Xu Y, Huang S, Liu ZG, Han J . Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol Chem 2006; 281: 8788–8795.
Gagne JP, Moreel X, Gagne P, Labelle Y, Droit A, Chevalier-Paré M et al. Proteomic investigation of phosphorylation sites in poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase. J Proteome Res 2009; 8: 1014–1029.
Christofferson DE, Yuan J . Cyclophilin A release as a biomarker of necrotic cell death. Cell Death Differ 2010; 17: 1942–1943.
Scaffidi P, Misteli T, Bianchi ME . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418: 191–195.
Sass G, Heinlein S, Agli A, Bang R, Schümann J, Tiegs G . Cytokine expression in three mouse models of experimental hepatitis. Cytokine 2002; 19: 115–120.
Hu X, Han W, Li L . Targeting the weak point of cancer by induction of necroptosis. Autophagy 2007; 3: 490–492.
Lepretre C, Scovassi AI, Shah GM, Torriglia A . Regulation of poly(ADP-ribose) polymerase-1 functions by leukocyte elastase inhibitor/LEI-derived DNase II during caspase-independent apoptosis. Int J Biochem Cell Biol 2009; 41: 1046–1054.
Acknowledgements
We are grateful to Dr. Alicia Torriglia and Dr. Mathieu Bertrand for helpful discussions. We thank in particular Linde Duprez and Mathieu Bertrand for preparing and making available ripk1 and ripk3 knockout MEF cells. We also thank the animal house facilities, the flow cytometry platform and the immunohistology platform (SFR Biosit, Rennes). Research in the IRSET group was supported by grants from the Ligue Nationale Contre le Cancer (the Côte d’Armor, Ille et Vilaine, Morbihan, Vendée and Sarthe Comittees), INSERM, University of Rennes 1 and the Region Bretagne. S Jouan-Lanhouet was supported by the Association pour la Recherche sur le Cancer (doctoral fellowship). Research in the Vandenabeele group has been supported by Flanders Institute for Biotechnology (VIB), by European grants (FP6 ApopTrain, MRTN-CT-035624; FP7 EC RTD Integrated Project, Apo-Sys, FP7–200767; Euregional PACT II), Belgian grants (Interuniversity Attraction Poles, IAP 6/18), Flemish grants (Fonds Wetenschappelijk Onderzoek Vlaanderen, FWO G.0875.11 and FWO G.0973.11) and Ghent University grants (MRP, GROUP-ID). P Vandenabeele is also holder of a Methusalem grant (BOF09/01M00709) from the Flemish Government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by C Borner
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Jouan-Lanhouet, S., Arshad, M., Piquet-Pellorce, C. et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ 19, 2003–2014 (2012). https://doi.org/10.1038/cdd.2012.90
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.90
Keywords
This article is cited by
-
Immunogenic cell death in cancer: targeting necroptosis to induce antitumour immunity
Nature Reviews Cancer (2024)
-
Roles of RIPK3 in necroptosis, cell signaling, and disease
Experimental & Molecular Medicine (2022)
-
AATF Competitively Interacts with Nuclear AIF and Inhibits Parthanatos of Neurons in dMCAO/R and OGD/R Models
Journal of Molecular Neuroscience (2022)
-
The regulation of necroptosis by ubiquitylation
Apoptosis (2022)
-
The molecular mechanism of acute liver injury and inflammatory response induced by Concanavalin A
Molecular Biomedicine (2021)
