
Abstract
The mechanisms underlying neuronal pathology and death in the spinal cord (SC) during inflammation remain elusive. We previously showed the important role of plasma membrane calcium ATPases (PMCAs) in the survival of SC neurons, in vitro. We also postulated that a decrease in PMCA2 expression could cause neuronal death during experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis. The current studies were undertaken to define the specific contribution of PMCA2 to degeneration of SC neurons, the effectors downstream to PMCA2 mediating neuronal death and the triggers that reduce PMCA2 expression. We report that knockdown of PMCA2 in SC neurons decreases collapsin response mediator protein 1 (CRMP1) levels. This is followed by cell death. Silencing of CRMP1 expression also leads to neuronal loss. Kainic acid reduces both PMCA2 and CRMP1 levels and induces neuronal death. Administration of an α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate receptor antagonist, at onset or peak of EAE, restores the decreased PMCA2 and CRMP1 levels to control values and ameliorates clinical deficits. Thus, our data link the reduction in PMCA2 expression with perturbations in the expression of CRMP1 and the ensuing death of SC neurons. This represents an additional mechanism underlying AMPA/kainate receptor-mediated excitotoxicity with relevance to neurodegeneration in EAE.
Similar content being viewed by others

Main
Multiple sclerosis is a demyelinating, inflammatory and neurodegenerative disorder of the central nervous system (CNS). Axonal damage and neuronal degeneration are the pathological hallmarks of multiple sclerosis,1, 2 occur early in the disease,3 are determinants of permanent disability4 and correlate with neurological deficits.5 Despite increasing information about gray and white matter pathology6 and the onset, progression and extent of neuronal injury in multiple sclerosis,7 the molecular mechanisms underlying neuronal degeneration remain elusive.
Our earlier studies on experimental autoimmune encephalomyelitis (EAE) suggested that plasma membrane calcium ATPase (PMCA)2 contributes to neuronal pathology in the inflamed spinal cord (SC). PMCA2 mRNA and protein levels are decreased at the onset of symptoms in myelin basic protein-induced acute EAE in the Lewis rat and myelin oligodendrocyte glycoprotein (MOG35–55)-mediated non-remitting EAE in the C57Bl/6 mouse.8, 9 We also reported that a pharmacological inhibitor of all PMCA isoforms delays the clearance of depolarization-induced calcium transients, which is followed by neuritic beading and cell loss in pure SC neuronal cultures.10 However, this latter study did not determine whether suppression of PMCA2 expression alone is sufficient to induce neuronal death, an issue that is addressed in the current report.
PMCA2 is a member of P-type ATPases, which mediate calcium extrusion from cells. The four PMCA isoforms have a highly restricted cellular and tissue distribution.11 In contrast to PMCA1 and 4, which are ubiquitously expressed, PMCA2 and 3 are primarily localized to specific regions of the CNS and are found predominantly in neurons.12 The unique role of PMCA2 is indicated by the phenotype of the PMCA2-null mice and the deafwaddler2J (dfw2J), a mouse with a mutation in the gene encoding PMCA2 that affects its activity.13, 14 These mice exhibit hearing and vestibular deficits, cerebellar abnormalities and loss of motor neurons.10, 13, 14, 15 Motor dysfunction and delayed motor neuron loss are observed even in the PMCA2-heterozygous mice, which express half the levels of PMCA2 as compared with the wild-type littermates.16
Little is known about the mechanisms downstream to PMCA2 that mediate death of SC neurons and the triggers that downregulate PMCA2 expression. The current investigations were undertaken in order to elucidate these issues.
Results
Silencing of PMCA2 in SC neurons leads to cell death, in vitro
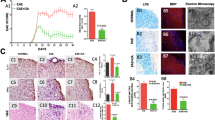
Our previous studies have shown that inhibition of PMCA activity by carboxyeosin (CE) in SC neuronal cultures results in neuritic damage followed by neuronal loss.10 CE is an inhibitor of all PMCA isoforms. To determine the specific contribution of PMCA2 to neuronal degeneration, we silenced PMCA2 expression and evaluated the viability of neurons by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. SC neurons were transfected with PMCA2 small interfering ribonucleic acid (siRNA) for 6 h and protein was collected at 24, 36, 48 and 72 h after initiation of treatment. Western blots indicated a 37% decrease in PMCA2 protein levels at 24 h. PMCA2 protein levels were further reduced (56%) at 72 h post transfection, the latest time examined (Figure 1a and b). We further corroborated the reduction in PMCA2 expression at the cellular level by performing immunocytochemistry on pure neuronal cultures 24 h after transfection with PMCA2 siRNA. Strong staining was detected on the membranes and cytoplasm of neurons in non-manipulated controls or negative controls transfected with scrambled siRNA (Figure 1c and d). In contrast, a reduction in the staining intensity was observed in neurons treated with PMCA2 siRNA, confirming the results obtained by western analysis (Figure 1e).

Viability of SC neurons after selective silencing of PMCA2 expression, in vitro. (a) Representative western blots probed with an anti-PMCA2 antibody showing a decrease in the intensity of a band corresponding to the molecular weight of PMCA2 (∼129–133 kDa), at different times post-transfection. Total protein visualized by BLOT-FastStain was used to account for experimental variations. Lanes 1–2: untreated control neurons; 3–4: neurons transfected with scrambled siRNA (negative control siRNA); 5–6: neurons transfected with PMCA2-specific siRNA. (b) Graphic representation of the data obtained by western blot analysis. Results from two to three experiments were combined (n=12 for each group except for 36 h n=6). (c–e) PMCA2 immunoreactivity in pure neuronal cultures after transfection with PMCA2 siRNA. (c) A composite showing PMCA2-labeled cells in a control, untreated culture. (d) A composite showing PMCA2 immunoreactive cells in a culture transfected with scrambled siRNA (negative control) (e) A composite showing reduced PMCA2 staining in a culture transfected with PMCA2 siRNA. Arrows point at examples of labeled cells. Bar represents 50 μm. (f) PMCA1 levels, 24 and 72 h after transfection of neurons with PMCA2 siRNA (n=3 for each group). (g) MTT-positive viable cell number at various times after transfection of neurons with PMCA2 siRNA (results are obtained from two experiments combined, n=6). Values in the graph represent mean±S.E.M. *P<0.001, significantly different from control and negative control siRNA by two-way ANOVA, Bonferroni post hoc test
To further ensure that PMCA2 siRNA did not have a generic effect, we quantified PMCA1 levels. We did not observe significant changes in PMCA1 levels at any time (Figure 1f). Knockdown of PMCA2 expression caused a significant loss of SC neurons at 48 h post transfection. Viable cell number in siRNA-treated neuronal cultures was 58% of controls (Figure 1g) and remained at the same level until 72 h. Our results indicate that silencing of PMCA2 alone is sufficient to induce death of SC neurons.
Silencing of PMCA2 causes downregulation of collapsin response mediator protein (CRMP)1 expression
To identify cellular changes that occur following the knockdown of PMCA2, we compared the proteome profiles of SC neurons transfected with PMCA2 siRNA versus controls by use of two-dimensional (2DE) gel electrophoresis followed by identification of proteins by matrix-assisted laser desorption/ionization mass spectrometry (time-of-flight (MALDI-TOF MS); Figure 2a–c). We found that CRMP1 is one of the proteins showing the most prominent downregulation after knockdown of PMCA2 for 24 h. This finding was corroborated by western blots, which detected two bands of ∼70 and ∼62 kDa molecular weight (Figure 2d). Although 70 kDa is the molecular weight observed by the manufacturer (Abcam), 62 kDa has been reported as the expected molecular weight.17 It is possible that the two bands represent splice variants of CRMP1. The intensity of the 70 kDa band was significantly reduced by 48% in SC neurons treated with PMCA2 siRNA, as compared with negative control siRNA or untreated neurons (Figure 2d and e). Although there was a 20% decrease in the intensity of the 62 kDa band, the difference was not statistically significant (P>0.06 by one-way ANOVA). The decrease in CRMP1 was further supported by immunocytochemical studies showing reduced CRMP1 staining 24 h after transfection of neurons with PMCA2 siRNA, in vitro (Figure 2f–h).

Alterations in CRMP1 levels after silencing of PMCA2 expression. (a) 2DE gel patterns of lysates obtained from the control (top) and PMCA2 siRNA transfected (bottom) SC neuronal cultures. Arrows point at the spot representing CRMP1. (b) Quantification of the signal intensity shows a decrease in the CRMP1 level. (c) A representative tandem mass spectrum of a rat CRMP1 peptide. Peptide sequences of CRMP1 were deduced from the observation of continuous series of either N-terminal (b-series) or C-terminal (y-series) ions. (d) A representative western blot showing CRMP1 levels 24 h after transfection of SC neurons with PMCA2 siRNA. Two bands with molecular weight of ∼70 and ∼62 kDa were detected. Lanes 1–2: untreated control neurons; 3–4: neurons transfected with scrambled siRNA (negative control siRNA); 5–6: neurons transfected with PMCA2 specific siRNA. (e) The quantitative analysis of the bands at ∼70 and ∼62 kDa from two independent experiments (n=6). *P<0.0336, significantly different from control and negative control siRNA by one-way ANOVA, Tukey's post hoc test. (f) Composite showing CRMP1 positive cells and neurites in control, non-manipulated pure neuronal cultures. (g) Composite showing CRMP1 immunoreactive cells and processes in cultures transfected with scrambled siRNA (negative control) (h) Composite showing decreased CRMP1 staining in cultures transfected with PMCA2 siRNA. Arrows point at examples of labeled cells and arrowheads illustrate the examples of immunoreactive neurites. Bar represents 50 μm
These results raised the possibility that a reduction in CRMP1 could mediate neurodegeneration when PMCA2 expression is decreased. In fact, CRMPs have been implicated in neurodegenerative disorders.18 To establish a direct link between a decrease in CRMP1 expression and neurodegeneration, we transfected SC neurons with CRMP1-specific siRNA. We observed a 58 and 27% decrease in the ∼70 and ∼62 kDa CRMP1 bands, respectively, at 24 h (Figure 3a and b) and a 54 and 30% decline in the ∼70 and ∼62 kDa CRMP1 bands, respectively, after 48 h (Figure 3a and c). This was also confirmed by immunocytochemistry showing a reduction in the CRMP1 staining in cultures transfected with the CRMP1 siRNA as compared with non-manipulated controls or negative controls transfected with scrambled siRNA (Figure 3d–f). The decrease in CRMP1 level was paralleled by a 40% reduction in MTT-positive cell number after 48 h (Figure 3g). Thus, a decline in CRMP1 protein expression causes loss of SC neurons, in vitro.

Effects of selective silencing of CRMP1 expression on SC neurons. (a) Representative western blots showing a decrease in the intensity of the bands corresponding to the molecular weight of CRMP1 after transfection of neurons with a CRMP1 specific siRNA. Lanes 1–2: two individual cultures of untreated neurons; 3–4 neurons transfected with scrambled siRNA (negative control siRNA; 5–6 neurons transfected with CRMP1 siRNA. (b) Graphic representation of the data obtained by the western blots at 24 h (n=6/group). (c) Graphic representation of the data obtained by western blots at 48 h (n=6/group). (d) A composite showing CRMP1 labeling in neuronal cultures. (e) CRMP1-stained cells in neuronal cultures transfected with scrambled siRNA. (f) A composite showing decreased CRMP1 levels in cultures transfected with CRMP1 siRNA. Arrows point at examples of labeled cells and arrowheads illustrate examples of labeled neurites. Bar represents 50 μm. (g) MTT-positive cell number at 48 h following transfection of neurons with CRMP1 siRNA (n=8). (h) Representative western blots showing unaltered PMCA2 levels 24 or 48 h after knockdown of CRMP1 in neuronal cultures. Total protein shows similar loading. (i) Graphic representation of the results obtained by the western blots (n=6 for 24 h and n=3 for 48 h). Values in the graph represent mean±S.E.M. *P<0.0091, **P<0.0001 significantly different from control and negative control siRNA by one-way ANOVA, Tukey's post hoc test. Results from two experiments were combined
Subsequently, we assessed whether silencing of CRMP1 expression leads to decreased PMCA2 levels. We did not observe any significant changes in PMCA2 protein levels 24 or 48 h after knockdown of CRMP1 (Figure 3h and i). The lack of reciprocity supports the notion that changes in CRMP1 occur downstream to PMCA2.
Blockade of α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate receptors at the peak of clinical symptoms restores PMCA2 and CRMP1 levels and reverses neurological deficits during EAE
The triggers that induce a downregulation of PMCA2 in EAE have not been identified. As we have previously shown that kainic acid (KA) suppresses neuronal PMCA2 mRNA expression in SC slice cultures,8 we assessed whether blockade of AMPA/kainate receptors with 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) during MOG35–55-induced EAE in C57Bl/6 mice restores PMCA2 levels. We also chose to study this paradigm because reports have indicated that NBQX prevents the progression of EAE symptoms19 and reverses the neurological deficits.20 We initiated the NBQX treatment either at 24 h after onset of symptoms (clinical score 1; early intervention) or at the peak of the disease (clinical score 2.5–3; late intervention). In agreement with previous findings, administration of NBQX at the onset of symptoms prevented the progression of EAE as indicated by the clinical scores (Figure 4a). In accordance with our earlier findings, PMCA2 levels in the lumbar SC were significantly decreased by ∼40% at onset of the symptoms9 and were restored to control values after early intervention with NBQX (Figure 4b). α-Tubulin, a housekeeping protein, was used to account for the experimental variations and did not show any significant alterations (Figure 4c).

Restoration of PMCA2 and CRMP1 levels after blockade of AMPA/kainate receptors in EAE. (a) Clinical scores of mice with EAE following administration of NBQX or vehicle 24 h after onset of symptoms (early intervention). Bar represents the duration of the treatment. (b) Representative western blot (left panel) showing PMCA2 levels in the lumbar SC of individual control and EAE mice with and without NBQX administration at 24 h after onset of symptoms. Lanes 1–2: samples from two individual control mice; 3–4: samples from EAE mice which received vehicle and lanes 5–6: samples from EAE mice treated with NBQX. Graphic representation of the results obtained by western blot analysis (right panel). (c) Western blot probed with an anti-α-tubulin (∼50 kDa) antibody to account for experimental variations. (d) Clinical scores of mice with EAE following administration of NBQX or vehicle at peak of symptoms (late intervention). Bar represents the duration of the treatment. (e) Representative western blot (left panel) showing PMCA2 levels in the lumbar SC of individual control and EAE mice with and without NBQX administration at peak of symptoms. Graphic representation of the results obtained by western blot analysis (right panel). (f) Western blot probed with an anti-α-tubulin antibody. (g) Western blot showing no changes in the level of PMCA1 (∼129 kDa). The western blot shown in e was stripped and re-probed with the PMCA1 antibody. (h) Modulation of CRMP1 levels. The western blot shown in E was stripped and probed with an anti-CRMP1 antibody (∼70 kDa, left panel). Graphic representation of the data (right panel). Values in graph represent mean±S.E.M. *P<0.0056, **P<0.0001, +P<0.0004 significantly different from other groups by one-way ANOVA, Tukey's post hoc test. (n=3–4 for a–c and g; n=6 for d–f and h). The experiment was repeated twice and yielded similar results
Subsequently, we assessed whether the decrease in PMCA2 is reversed when NBQX treatment is initiated at the peak of the disease. NBQX reversed neurological decline even when administered at the more advanced stage of EAE (Figure 4d). In mice treated with vehicle alone, PMCA2 levels were low throughout the course of EAE, as observed before.9 NBQX reversed the decrease in PMCA2 (Figure 4e). In contrast to PMCA2, α-tubulin and PMCA1 levels were not altered in the lumbar SC of EAE-afflicted mice, with and without NBQX treatment (Figure 4f and g).
As we found a link between PMCA2 and CRMP1 expression, in vitro, we investigated whether there is a correlation between the expression of the two proteins during EAE. CRMP1 protein level was reduced by 45% at the peak of the disease but returned to control values after late NBQX intervention (Figure 4h).
Activation of neuronal AMPA/kainate receptors reduces PMCA2 and CRMP1 protein levels, in vitro
We previously reported that neuronal PMCA2 mRNA levels are decreased in slice cultures continuously exposed to very low concentrations (4 μM) of KA.8 In addition, the current investigations indicate that blockade of AMPA/kainate receptors restores PMCA2 protein levels in EAE. It is not yet known whether the modulation of PMCA2 expression is due to the direct activation of AMPA/kainate receptors on neurons or is mediated indirectly, through factors released by other cells such as glia, which also express AMPA/kainate receptors. To address this issue, we treated pure SC neuronal cultures with a low (4 μM) or a higher (20 μM) concentration of KA. These concentrations were chosen because 4 μM is the lowest KA concentration that induces a decrease in PMCA2 mRNA expression in slice cultures8 and 20 μM causes a more robust delay in the clearance of neuronal calcium transients leading to cell death.10
KA (4 μM) caused a 36% decrease in PMCA2 protein levels at 36 h (Figure 5a and b). In cells exposed to 20 μM KA, PMCA2 protein levels were significantly reduced by 34% already at 12 h and progressively decreased by 44% at 24 h and by 69% at 36 h (Figure 5c).

Activation of AMPA/kainate receptors reduces neuronal PMCA2 and CRMP1 levels, in vitro. (a) A representative western blot probed with anti- PMCA2 and anti-CRMP1 antibodies showing a reduction in the levels of both proteins after treatment of neuronal cultures with 4 μM KA or vehicle (Control: C) for 36 h. Lowest panel (total protein) shows the BLOT-FastStain stained membrane, which was used as a control for the experimental variations. (b) PMCA2 levels following the exposure of neurons to 4 μM KA for 12 (n=8), 24 (n=11) or 36 (n=10) hours. (c) PMCA2 levels following exposure of neurons to 20 μM KA for 12 (n=7), 24 (n=5) or 36 (n=5) hours. (d) CRMP1 levels after the treatment of neuronal cultures with 4 μM KA for 36 h. Data from two independent experiments were combined (n=6). (e) MTT-positive cell number following treatment of neurons with 4 μM KA for 36 or 48 h. (f) MTT-positive cell number following the treatment of neurons with 20 μM KA. Data from two experiments were combined (n=6–8). (g) PMCA2 levels after co-administration of 10 μM NBQX with 20 μM KA (n=6). (h) A representative western blot showing CRMP1 levels after co-administration of 10 μM NBQX with 20 μM KA. Lanes 1 and 2: controls; 3 and 4: NBQX; 5 and 6: KA; 7 and 8: KA+NBQX. Values in graph represent mean±S.E.M. *P<0.05, **P<0.01, ***P<0.001 significantly different from the control and 12 h by two-way ANOVA, Bonferroni posttest; &P<0.001 significantly different from control and all the other groups; +P<0.02, ++P<0.0008 significantly different from control by Student's t-test; #P<0.0026 significantly different from all the other groups by one-way ANOVA, Tukey's post hoc test
To determine whether changes in PMCA2 are paralleled by alterations in CRMP1 levels, western blots were probed with an antibody against CRMP1. In cells treated with 4 μM KA for 36 h, there was a 50 and 26% drop in the intensity of the 70 and 62 kDa CRMP1 bands, respectively (Figure 5a and d), showing a correlation between the levels of the two proteins. The decrease in PMCA2 and CRMP1 was followed by a fall in cell viability. A 26% reduction in MTT-positive cell number was observed in neurons exposed to 4 μM KA for 48 h (Figure 5e). In cultures treated with 20 μM KA, MTT-positive cell number decreased by 30% at 24 and 36 h and by 52% by 48 h (Figure 5f). The specificity of the effect observed was confirmed by the simultaneous addition of 10 μM NBQX to cultures treated with 20 μM KA for 12 h. NBQX prevented the decrease in PMCA2 and CRMP1 protein levels (Figure 5g and h). These results show that direct activation of AMPA/kainate receptors causes reductions in PMCA2 and CRMP1 levels, which are followed by neuronal death.
Inhibition of calpain prevents the KA-induced decrease in PMCA2 levels, in vitro
To determine whether the KA-induced decrease in PMCA2 is due to alterations in transcription, we measured PMCA2 transcript levels by semi-quantitative RT-PCR. We exposed neurons to 4 μM KA for 24 and 36 h, time points before or concurrent to the observed changes in protein levels. Similarly, we evaluated PMCA2 transcript levels at 6 and 12 h after treatment of neurons with 20 μM KA. We did not find significant alterations in the transcript level (Figure 6a). Therefore, we assessed whether the reduction in PMCA2 protein level could be due to degradation by proteases. We focused on calpain as it is a calcium-activated protease implicated in multiple sclerosis, EAE and excitotoxicity.21, 22 We determined whether the calpain inhibitor, calpastatin, prevents the decrease in PMCA2 protein levels when SC neurons are exposed to KA. Simultaneous application of 4 μM KA and 1 μM calpastatin on SC neuronal cultures prevented the decrease in PMCA2 protein levels (Figure 6b and c). In contrast, 1 μM calpastatin was not able to inhibit the reduction in PMCA2 levels when the cells were exposed to 20 μM KA for 12 (results not shown) or 36 h (Figure 6b and c). However, a higher calpastatin concentration (10 μM) prevented the decrease of PMCA2 at both 24 and 36 h (Figure 6d). These results indicate that degradation of PMCA2 by calpain may be responsible for the KA-mediated drop in PMCA2 expression.

The effects of calpastatin on PMCA2 and CRMP1 levels in KA-treated SC neuronal cultures. (a) Top panel: RT-PCR showing PMCA2 transcript levels (1014 bp) in KA-treated cultures. Control: C. Bottom panel: 18S RNA (489 bp) was used to standardize against experimental variation. (b) A representative western blot probed with an anti-PMCA2 antibody showing the effects of KA in the presence and absence of 1 μM calpastatin. Cultures were treated for 36 h. Total protein was used as a control for experimental variations. Lanes 1–2: control, 3–4: 1 μM CP, 5–6: 4 μM KA, 7–8: 20 μM KA; 9–10: 4 μM KA+1 μM CP, 11–12: 20 μM KA+1 μM CP. (c) Graphic representation of the data obtained by use of western blotting. Data from two independent experiments were combined (n=6). Values in the graph represent mean±S.E.M. *P<0.0006 significantly different from the control, 1 μM CP and 1 μM CP+4 μM KA by one-way ANOVA, Tukey's post hoc test. (d) Representative western blots probed with an anti-PMCA2 antibody showing the effects of 20 μM KA in the presence and absence of 10 μM calpastatin at 24 and 36 h. Lane 1: control; lane 2: 10 μM CP, lane 3: 20 μM KA, lane 4: 20 μM KA+10 μM CP
Discussion
The current study indicates that downregulation of PMCA2 during EAE is the consequence of AMPA/kainate receptor-mediated excitotoxicity. The pivotal role of PMCA2 in SC neuronal survival is shown by the knockdown experiments confirming that a drop in the level of PMCA2 is sufficient to cause the death of SC neurons, in vitro. In addition, our findings establish, for the first time, a correlation between PMCA2 and CRMP1 levels in neuronal cultures and during EAE and show that restoration of the two proteins coincides with amelioration of neurological deficits. These findings raise the possibility that excitotoxicity-induced neuronal loss in EAE could be, at least partially, ascribed to a decrease in PMCA2 and CRMP1 levels after activation of AMPA/kainate receptors. A scheme representing a potential mechanism for PMCA2-mediated neuronal death is shown in Figure 7.

Scheme representing the potential mechanism of PMCA2-mediated neurodegeneration in the SC during EAE. Inflammation of the CNS during EAE leads to excess glutamate from non-neuronal cells and injured neurons (1). Glutamate binds to AMPA/kainate receptors on neurons (2a) and non-neuronal cells (2b). Over-activation of neuronal AMPA/kainate receptors results in excess calcium influx (3a), while non-neuronal cells release effectors (3b) such as IL-1β in response to glutamate. Effectors released by non-neuronal cells could affect PMCA2 at the transcriptional level, whereas excessive intracellular calcium can activate calcium-dependent proteases such as calpain (4), which, in turn, degrade PMCA2. The combined outcome is a decrease in the PMCA2 levels (5). The reduction in PMCA2 can lead to a decrease in CRMP1 levels by mechanisms that are not yet determined (6). This can cause perturbations in microtubule assembly (7). On the other hand, excess intracellular calcium activates apoptotic mechanisms including caspase-3 (8). These changes lead to neuronal pathology and death (9)
Glutamate excitotoxicity has been implicated in multiple sclerosis and EAE pathology. Glutamate levels are higher in active lesions and normal-appearing white matter of multiple sclerosis brains23 and in the cerebrospinal fluid of multiple sclerosis patients during relapse.24 In agreement with these findings, Werner et al.25 reported altered glutamate homeostasis in multiple sclerosis lesions and EAE. Expression of glutaminase, an enzyme involved in the synthesis of glutamate, was increased in the microglia and macrophages of active multiple sclerosis lesions. In contrast, the glutamate-metabolizing enzymes, glutamine synthetase and glutamate dehydrogenase, were absent in active and chronic lesions. The levels of the glutamate transporter (GLT)-1 were also decreased. In agreement with these results, excitatory amino acid transporter EAAT-1 (GLAST-1) and EAAT-2 (GLT1), which are expressed in astrocytes and are crucial for the maintenance of the extracellular glutamate concentration, were decreased in multiple sclerosis lesions.26 Such changes in glutamate homeostasis could lead to axonal injury as well as neuronal and oligodendrocyte death. In fact, administration of NBQX to mice with non-remitting EAE and rats with acute EAE, at the onset of symptoms, prevented oligodendrocyte and neuronal loss and improved neurological deficits.19, 27
We found that blockade of AMPA/kainate receptors at the onset or peak of EAE restores the PMCA2 protein levels. This correlated with the prevention of disease progression or reversal of clinical deficits. Thus, before very late disease stages are reached, there appears to be a phase during which the decrease in PMCA2 levels can be reversed. The reversible change in PMCA2 and its correlation with amelioration of EAE symptoms are in agreement with our previous report showing restoration of PMCA2 levels before spontaneous remission from acute EAE.9
Activation of AMPA/kainate receptors could modulate PMCA2 levels in at least two different ways: regulation of transcript level and degradation by proteases. This is indicated by our past and current results, combined. We previously reported a decrease in PMCA2 transcript levels after treatment of SC slice cultures with KA.8 We now found that in pure neuronal cultures, KA induces alterations only in protein levels. Because slice cultures contain both neuronal and non-neuronal cells, we postulate that the reduction in PMCA2 transcript level might be due to the actions of effectors secreted by non-neuronal cells in response to KA. In fact, glia also express AMPA/kainate receptors28 and microglia respond to kainate stimulation by upregulating interleukin 1β (IL-1β) expression.29 In agreement with this concept, we found that IL-1β suppresses neuronal PMCA2 expression, in vitro (unpublished results). Degradation by calpain could be another mechanism leading to a decrease in PMCA2 after activation of neuronal AMPA/kainate receptors, as indicated by our studies on pure neuronal cultures. Calpain activity is induced after AMPA/kainate receptor-mediated increases in intracellular calcium levels.30 PMCAs are calpain substrates31 and glutamate-induced loss of PMCA2 activity is prevented by inhibition of calpain in hippocampal neurons.32 Irrespective of the mechanism, decreased PMCA2 expression disrupts neuronal calcium homeostasis, which leads to cell death.
Abnormal increases in calpain expression or activity have been implicated in multiple sclerosis and EAE pathology.33 Inhibition of calpain attenuates axonal injury and reduces the clinical signs in EAE.34 Calpain activation induces apoptosis of SC motor neurons35 and inhibition of calpain activity prevents glutamate-induced apoptosis of SC motor neurons.36
We identified CRMP1 as one of the effectors whose expression is reduced when PMCA2 levels are decreased. Some CRMPs modulate microtubule assembly,37 reorganization of actin filaments and axonal protein trafficking.38 CRMP1 mediates neurotrophin-3-induced neurite extension.39
The decrease in CRMP1 levels during EAE and the amelioration of symptoms at the time when CRMP1 returns to control values suggest a role for this protein in the disease. A reduction in CRMP1 could affect microtubule assembly, resulting in the disruption of axonal stability and transport. This could lead to neuronal dysfunction and degeneration. In fact, neuronal death is observed when CRMP1 expression is silenced. This is a novel and significant finding in view of the importance of axonal pathology and neurodegeneration in multiple sclerosis. It is worth noting that such mechanisms might also be relevant to other pathological conditions of the SC, as PMCA2 mRNA levels are also decreased after SC injury.40
The interactions between PMCA2 and CRMP1 and the mechanisms underlying the decrease in CRMP1 when PMCA2 levels are reduced have not been defined and will be the focus of future investigations.
Materials and Methods
SC neuronal cultures
SCs, dissected from 15-day-old rat embryos (Charles River, Wilmington, MA, USA), were dissociated by trituration. The cells were plated at a density of 6 × 105 cells/35 mm dish in L-15 medium containing supplements as described before.10 The day after plating, the media were replaced with neurobasal medium (NBM) containing B-27 supplement, 6.3 mg/ml NaCl and 10 U/ml penicillin/streptomycin. Cultures were maintained for 7 days at 37 °C in a 95% air/5% CO2 incubator until neuronal differentiation and maturation. Media and B-27 were obtained from Invitrogen (Carlsbad, CA, USA) and all other chemicals were purchased from Sigma (St Louis, MO, USA).
Transfection of SC neurons with siRNA
SC neurons, maintained 7 days in culture, were transfected with RNAi duplexes designed to silence PMCA2 (Invitrogen; RSS302023) or CRMP1 (RSS303593) expression or a non-targeted control siRNA (12935-300). In all, 7.5 μl of PMCA2 or CRMP1 siRNA was diluted in 180 μl NBM containing B-27 supplement and 6.3 mg/ml NaCl, but without antibiotics (NBM-abx). In addition, 5 μl of Lipofectamine LTX transfection reagent (Invitrogen) was diluted in 182.5 μl NBM-abx. The diluted siRNA and Lipofectamine LTX were combined, incubated at room temperature for 25–30 min and then added to the cultures. The final concentration of siRNA was 200 nM. Six hours later, the media were replaced with fresh NBM-containing antibiotics. Protein was collected after 24, 36, 48 and 72 h.
MTT assay
Viability of neurons was assessed by use of the MTT assay (Sigma). MTT was added to the cultures at a final concentration of 300 μg/ml and incubated at 37°C for 1 h. The number of MTT positive cells was quantified in 25 fields with a × 40 objective using a Nikon eclipse TS100 microscope.
2DE gel electrophoresis and protein quantification
SC neurons were washed with cold phosphate buffered saline and lysed by sonication in an isoelectric focusing rehydration buffer (7 M urea, 2 M thiourea, 4% CHAPS, 100 mM dithiothreitol (DTT), 0.2% biolytes (pH 5–8), 0.01% bromophenol blue and protease inhibitor). Fifty micrograms of protein in a total of 185 μl of the rehydration buffer was applied to 11 cm BioRad ReadyStrip IPG strips (pH 5–8) for overnight rehydration. First-dimension isoelectric focusing was performed on a BioRad (Hercules, CA, USA) PROTEAN IEF system at the University of Medicine and Dentistry of New Jersey Center for Advanced Proteomics Research (http://njms.umdnj.edu/proweb), as described by the manufacturer for a total focusing time of 75 000 V h. The strips were equilibrated with equilibration solution I (6 M urea, 0.375 M Tris-HCl, pH 8.8, 2% sodium dodecyl sulfate (SDS), 20% glycerol, 2% (w/v) DTT) for 15 min. The strips were further equilibrated with equilibration solution II containing 6 M urea, 0.375 M Tris-HCl, pH 8.8, 2% SDS, 20% glycerol, 2.5% (w/v) iodoacetamide, for 15 min and directly applied to a 12.5% isocratic SDS-polyacrylamide gel for the second-dimension electrophoresis. The gel was fixed in a solution consisting of 10% acetic acid and 40% ethanol for 30 min and stained overnight with SYPRO Ruby (Molecular Probes, OR, USA). Gels were destained with a solution containing 10% methanol and 7.5% acetic acid, for 2 h at room temperature and scanned on a 9400 typhoon variable mode imager (GE Healthcare, Piscataway, NJ, USA) using a green laser (532 nm) and a 610BP30 emission filter. Protein quantitation on SYPRO Ruby-stained gels was performed by PDQuest 2-D analysis software (BioRad) and protein spots which expression levels appreciably changed were isolated for mass spectrometric identification.
Mass spectrometry and protein identification
Protein spots from SYPRO Ruby-stained 2-D gels were picked for protein identification. The gel bands were diced into 1 mm3 pieces and washed with 30% acetonitrile (ACN) in 50 mM ammonium bicarbonate before DTT reduction and iodoacetamide alkylation. Trypsin was used for digestion at 37°C overnight. The resulting peptides were extracted with 30 μl of 1% trifluoroacetic acid followed by C18 ziptip desalting. For the MS analysis, the peptides were mixed with 7 mg/ml α-cyano-4-hydroxy-cinnamic acid matrix (in 60% ACN) in a 1 : 1 ratio and spotted onto a MALDI plate. The peptides were analyzed on a 4800 MALDI TOF/TOF mass analyzer (Applied Biosystem, Framingham, MA, USA). Mass spectra (m/z 880–3200) were acquired in positive ion reflector mode. The 15 most intense ions were selected for the subsequent MS/MS sequencing analysis in 1 kV mode. Protein identification was performed by searching the combined tandem mass spectra against the NCBI (National Center for Biotechnology Information) rat sequence database, using a local MASCOT search engine (V. 1.9) on a GPS (V. 3.5, ABI) server. A protein containing at least two peptides with confidence interval values not less than 95% was considered as having been identified.
Induction of EAE
C57BL/6 mice, 9 to 10-week-old (Taconic, Hudson, NY, USA) were immunized subcutaneously (s.c.) in the flank with an emulsion consisting of 200 μg myelin oligodendrocyte glycoprotein35–55 (MOG35–55, MEVGWYRSPFSRVVHLYRNGK; Stanford PAN Facility, Stanford, CA, USA) in saline and an equal volume of complete Freund's adjuvant containing 7 mg/ml Mycobacterium tuberculosis H37RA (MT; Difco Laboratories, Detroit, MI, USA). Control mice received an emulsion containing only saline and adjuvant/MT. All mice received an intraperitoneal injection of 350 ng of pertussis toxin (List Biological, Campbell, CA, USA) at 0 and 48 h post-immunization. Animals were given food and water ad libitum and were monitored daily for clinical symptoms. The neurological impairment was scored as no symptoms (0), tail weakness (0.5), tail paralysis (1), weakness in one hindlimb (1.5), weakness in both hindlimbs or paralysis of one hindlimb (2), weakness in one hindlimb and paralysis of one hindlimb (2.5), complete paralysis in both hindlimbs (3.0), tetraplegia (4) and moribund (5). Mice with EAE were treated daily with either NBQX (30 mg/kg; Tocris, Ellisville, MO, USA) or saline by s.c. injections. Control mice were treated with saline. All animal protocols were approved by institutional IACUC.
Western blot analysis
Lumbar SCs were homogenized on ice in 200 μl lysis buffer containing 0.2 mM PMSF, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 2 μg/ml pepstatin. Protein was collected from neurons cultured in 35 mm Petri dishes, in a total volume of 200 μl lysis buffer. The lysates were vortexed five times for 15 s, incubated on ice for 15 s, sonicated for 15 s in a low-frequency ultrasonic bath and placed on ice for 30 min. The samples were centrifuged at 11 000 r.p.m. for 20 min at 4°C. The supernatant was collected and stored at −80°C. Protein concentrations were determined using the BCA protein quantification kit according to the manufacturer's instructions (Pierce Biotechnology, Inc., Rockford, IL, USA). A total protein of 10 μg was loaded in each lane of a 10-well 3–8% NuPage Tris-acetate gel (Invitrogen, Carlsbad, CA, USA) or 18-well 3–8% Tris-acetate gel (BioRad). Electrophoresis was performed for 60 min at 150 V. The protein was then electrotransferred onto a polyvinylidene difluoride (PVDF, Invitrogen) membrane for 60 min at 30–100 V, followed by staining with BLOT-FastStain (Millipore, Temecula, CA, USA) according to the manufacturer's instructions. Immunodetection was performed using the following primary antibodies: rabbit anti-PMCA1 (1 : 2000; Abcam, Cambridge, MA, USA), rabbit anti-PMCA2 (1 : 5000, Abcam), rabbit anti-CRMP1 (1 : 5000, Abcam) and mouse anti-α-tubulin (1 : 20 000, Sigma). Signal was visualized by exposure of blots to hyperfilm ECL (GE Healthcare). Bands were quantified using the Un-Scan-It software (Silk Scientific, Orem, UT, USA). Between each immunoblot, membranes were stripped with Re-Blot recycling kit (Millipore), according to the manufacturer's instructions.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde/10 mM phosphate buffer pH 7.5/0.15 M NaCl. They were then blocked in 10% horse serum in phosphate buffered saline (PBS, PMCA2 staining) or 10% horse serum in PBS/0.1% triton X-100 (CRMP1 staining), for 1 h at room temperature. The cells were incubated with anti-PMCA2 (Abcam; 1 : 500) or anti-CRMP1 (Abcam; 1 : 500) antibodies, overnight at 4°C. The cells were then washed, and incubated with biotinylated anti-rabbit IgG (1 : 500) for 1 h at room temperature. Signal was visualized by the use of avidin-biotin complex (ABC, Vector, Burlingame, CA, USA) and 3,3′-diaminobenzidine (DAB; Sigma). In the case of PMCA2, signal was amplified by the use of Nickel/DAB.
Abbreviations
- AMPA:
-
α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate
- CE:
-
5(6)carboxyeosin
- CNS:
-
central nervous system
- CRMP1:
-
collapsin response mediator protein 1
- DTT:
-
dithiothreitol
- EAAT:
-
excitatory amino acid transporter
- EAE:
-
experimental autoimmune encephalomyelitis
- GLT-1:
-
glutamate transporter 1
- GLAST-1:
-
glutamate aspartate transporter
- IL-1β:
-
interleukin 1β
- KA:
-
kainic acid
- MALDI-TOF MS:
-
matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- MOG:
-
myelin oligodendrocyte glycoprotein
- MS:
-
mass spectrometry
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NCBI:
-
National Center for Biotechnology Information
- NBQX:
-
2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione
- PMCA:
-
plasma membrane calcium ATPase
- PVDF:
-
polyvinylidene
- SC:
-
spinal cord
- SDS:
-
sodium dodecyl sulfate
- siRNA:
-
small interfering ribonucleic acid
References
Ferguson B, Matyszak MK, Esiri MM, Perry VH . Axonal damage in acute multiple sclerosis lesions. Brain 1997; 120: 393–399.
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L . Axonal transaction in the lesions of multiple sclerosis. N Engl J Med 1998; 338: 278–285.
Filippi M, Bozzali M, Rovaris M, Gonen O, Kesavadas C, Ghezzi A et al. Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis. Brain 2003; 126: 433–437.
Arnold DL . Magnetic resonance spectroscopy: imaging axonal damage in MS. J Neuroimmunol 1999; 98: 2–6.
De Stefano N, Matthews PM, Fu L, Narayanan S, Stanley J, Francis GS et al. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain 1998; 121: 1469–1477.
Filippi M, Rocca MA . MRI evidence for multiple sclerosis as a diffuse disease of the central nervous system. J Neurol 2005; 252: 16–24.
Kuhlmann T, Lingfield G, Bitsch A, Schurchardt J, Bruck W . Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain 2002; 125: 2202–2212.
Nicot A, Ratnakar PV, Ron Y, Chen CC, Elkabes S . Regulation of gene expression in experimental autoimmune encephalomyelitis indicates early neuronal dysfunction. Brain 2003; 126: 398–412.
Nicot A, Kurnellas M, Elkabes S . Temporal pattern of plasma membrane calcium ATPase 2 expression in the spinal correlates with the course of clinical symptoms in two rodent models of autoimmune encephalomyelitis. Eur J Neurol 2005; 21: 2660–2670.
Kurnellas MP, Nicot A, Shull GE, Elkabes S . Plasma membrane calcium ATPase deficiency causes neuronal pathology in the spinal cord: a potential mechanism for neurodegeneration in multiple sclerosis and spinal cord injury. FASEB J 2005; 19: 298–300.
Strehler EE, Zacharias DA . Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol Rev 2001; 81: 21–50.
Garcia ML, Strehler EE . Plasma membrane calcium ATPases as critical regulators of calcium homeostasis during neuronal cell function. Front Biosci 1999; 4: D869–D882.
Kozel PJ, Friedman RA, Erway LC, Yamoah EN, Liu LH, Riddle T et al. Balance and hearing deficits in mice with a null mutation in the gene encoding plasma membrane Ca2+-ATPase isoform 2. J Biol Chem 1998; 273: 18693–18696.
Street VA, McKee-Johnson JW, Fonseca RC, Tempel BL, Noben-Trauth K . Mutations in a plasma membrane Ca2+-ATPase gene cause deafness in deafwaddler mice. Nat Genet 1998; 19: 390–394.
Kurnellas MP, Lee AK, Li H, Deng L, Ehrlich DJ, Elkabes S . Molecular alterations in the cerebellum of the plasma membrane calcium ATPase 2 (PMCA2)-null mouse indicate abnormalities in Purkinje neurons. Mol Cell Neurosci 2007; 34: 178–188.
Souayah N, Sharovetskaya A, Kurnellas MP, Myerson M, Deitch JS, Elkabes S . Reductions in motor unit number estimates (MUNE) precede motor neuron loss in the plasma membrane calcium ATPase 2 (PMCA2)-heterozygous mice. Exp Neurol 2008; 214: 341–346.
Wang LH, Strittmatter SM . Brain CRMP forms heterotetramers similar to liver dihydropyrimidinase. J Neurochem 1997; 69: 2261–2269.
Charrier E, Reibel S, Rogemond V, Aguera M, Thomasset N, Honnorat J . Collapsin response mediator proteins (CRMPs): involvement in nervous system development and adult neurodegenerative disorders. Mol Neurobiol 2003; 28: 51–64.
Pitt D, Werner P, Raine CS . Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 2000; 6: 67–70.
Kanwar JR, Kanwar RK, Krissansen GW . Simultaneous neuroprotection and blockade of inflammation reverses autoimmune encephalomyelitis. Brain 2004; 127: 1313–1331.
Shields DC, Schaecher KE, Saido TC, Banik NL . A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc Natl Acad Sci USA 1999; 96: 11486–11491.
Rami A, Ferger D, Krieglstein J . Blockade of calpain proteolytic activity rescues neurons from glutamate excitotoxicity. Neurosci Res 1997; 27: 93–97.
Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D . Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain 2005; 128: 1016–1025.
Sarchielli P, Greco L, Floridi A, Floridi A, Gallai V . Excitatory amino acids and multiple sclerosis. Arch Neurol 2003; 60: 1082–1088.
Werner P, Pitt D, Raine CS . Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol 2001; 50: 169–180.
Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GV et al. Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol 2007; 66: 732–739.
Smith T, Groom A, Zhu B, Turski L . Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med 2000; 6: 62–66.
Martin LJ, Blackstone CD, Levey AI, Huganir RL, Price DL . AMPA glutamate receptor subunits are differentially distributed in rat brain. Neurosci 1993; 53: 327–358.
Eriksson C, Tehranian R, Iverfeldt K, Winblad B, Schultzberg M . Increased expression of mRNA encoding interleukin-1beta and caspase-1, and the secreted isoform of interleukin-1 receptor antagonist in the rat brain following systemic kainic acid administration. J Neurosci Res 2000; 60: 266–279.
Corona JC, Tapia R . Calpain inhibition protects spinal motoneurons from the excitotoxic effects of AMPA in vivo. Neurochem Res 2008; 33: 1428–1434.
Guerini D, Pan B, Carafoli E . Expression, purification, and characterization of isoform 1 of the plasma membrane Ca2+ pump: focus on calpain sensitivity. J Biol Chem 2003; 278: 38141–38148.
Pottorf II WJ, Johanns TM, Derrington SM, Strehler EE, Enyedi A, Thayer SA . Glutamate-induced protease-mediated loss of plasma membrane Ca2+ pump activity in rat hippocampal neurons. J Neurochem 2006; 98: 1646–1656.
Shields DC, Banik NL . Upregulation of calpain activity and expression in experimental allergic encephalomyelitis: a putative role for calpain in demyelination. Brain Res 1998; 794: 68–74.
Hassen GW, Feliberti J, Kesner L, Stracher A, Mokhtarian F . Prevention of axonal injury using calpain inhibitor in chronic progressive experimental autoimmune encephalomyelitis. Brain Res 2008; 1236: 206–215.
Momeni HR, Azadi S, Kanje M . Calpain activation and apoptosis in motor neurons of cultured adult mouse spinal cord. Funct Neurol 2007; 22: 105–110.
Kieran D, Greensmith L . Inhibition of calpains, by treatment with leupeptin, improves motoneuron survival and muscle function in models of motoneuron degeneration. Neuroscience 2004; 125: 427–439.
Fukuta Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T et al. CMRP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol 2002; 4: 583–591.
Yuasa-Kawada J, Suzuki R, Kano F, Ohkawara T, Murata M, Noda M . Axonal morphogenesis controlled by antagonistic roles of two CRMP subtypes in microtubule organization. Eur J Neurosci 2003; 17: 2329–2343.
Quach TT, Duchemin AM, Rogemond V, Aguera M, Honnorat J, Belin MF et al. Involvement of collapsin response mediator proteins in the neurite extension induced by neurotrophins in dorsal root ganglion neurons. Mol Cell Neurosci 2004; 25: 433–443.
Tachibana T, Noguchi K, Ruda MA . Analysis of gene expression following spinal cord injury in rat using complementary DNA microarray. Neurosci Lett 2002; 327: 133–137.
Acknowledgements
This study was supported by grant NS046363 from the NIH-NINDS and grant 08-3073-SCR-E-0 from NJCSCR to SE, NIH-NINDS fellowship F31NS054336 to MPK and NIH grant NS046593 to HL. We are grateful to the Tim Reynolds family for their support of spinal cord research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by N Bazan
Rights and permissions
About this article
Cite this article
Kurnellas, M., Li, H., Jain, M. et al. Reduced expression of plasma membrane calcium ATPase 2 and collapsin response mediator protein 1 promotes death of spinal cord neurons. Cell Death Differ 17, 1501–1510 (2010). https://doi.org/10.1038/cdd.2010.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2010.54
Keywords
This article is cited by
-
Pathological pain processing in mouse models of multiple sclerosis and spinal cord injury: contribution of plasma membrane calcium ATPase 2 (PMCA2)
Journal of Neuroinflammation (2019)
-
Maternal autoantibody related autism: mechanisms and pathways
Molecular Psychiatry (2019)
-
CRMPs Function in Neurons and Glial Cells: Potential Therapeutic Targets for Neurodegenerative Diseases and CNS Injury
Molecular Neurobiology (2017)
-
Apolipoprotein D Overexpression Protects Against Kainate-Induced Neurotoxicity in Mice
Molecular Neurobiology (2017)
-
Regulation of GAP43/calmodulin complex formation via calcineurin-dependent mechanism in differentiated PC12 cells with altered PMCA isoforms composition
Molecular and Cellular Biochemistry (2015)
