
Abstract
Viral infections of the central nervous system (CNS) are associated with an increased risk for seizures during the acute infection period and the subsequent development of chronic epilepsy that is often difficult to treat. In previous work, we have shown that mice of the C57BL/6 strain infected with Theiler's murine encephalomyelitis virus (TMEV) exhibit a similar sequence, thereby providing a potential useful model of virus-induced epilepsy. The present study examines spontaneous and miniature excitatory postsynaptic currents in CA3 pyramidal cells recorded from brain slices prepared during both the acute phase during encephalitis and 2 months following TMEV infection. Animals that develop chronic epilepsy following TMEV infection exhibit considerable hippocampal sclerosis, directly implicating this brain region in the process of epileptogenesis. There are significant increases in amplitude and frequency of spontaneous and miniature excitatory currents in CA3 cells recorded in brain slices prepared during the acute infection period and 2 months after infection. However, the patterns of changes observed are markedly different during these two periods, suggesting that there are underlying changes in the network over time. These differences have implications for the treatment used during the acute infection and after chronic seizures develop.
Similar content being viewed by others


Background
Viral infections of the central nervous system (CNS) are associated with an increased risk for seizures and the subsequent development of chronic epilepsy (Eeg-Olofsson 2003; Eisenschenk 2000; Getts et al. 2007; Labar and Harden 1998; Misra et al. 2008; Schmutzhard 2001). Viral infections that have been associated with clinical seizures such as human immunodeficiency virus (HIV), West Nile virus, and human herpes simplex virus-6 have a tropism for the hippocampus (Beers et al. 1993; Hunsperger and Roehrig 2006; Wu et al. 2003; Yamashita and Morishima 2005), the limbic region implicated in temporal lobe epilepsy, a common form of epilepsy often resistant to treatment. However, epileptic encephalitis has been difficult to address experimentally because, despite the fact that acute viral infection of rabbits, rats, and mice is often associated with seizures (Beers et al. 1993; Griffith et al. 1967; Lehrmann et al. 2008; Wu et al. 2003), infected animals either die from the acute viral encephalitis, or if they do not die, they do not go on to develop spontaneous recurrent seizures. We have determined that C57BL/6 mice infected with Theiler's murine encephalomyelitis virus (TMEV) display acute spontaneous seizures several days postinfection (Libbey et al. 2008) that correlate with infection and subsequent cell death of pyramidal neurons in the hippocampus (Stewart et al. 2010b). During the acute infection, a variety of cytokines are elevated including TNF-α, which has been shown to be important in the development of seizures in this animal model (Kirkman et al. 2010). TMEV-infected mice survive the initial infection, the virus is cleared from the CNS, and the seizures resolve within a few days. However, the hippocampus and limbic system exhibit neuronal death and astrogliosis, there are long-lasting reductions in seizure thresholds (Stewart et al. 2010b), and a significant number of TMEV-injected mice go on to develop epilepsy following an undefined latent period (Stewart et al. 2010a). This pattern of infection: tropism for the limbic system, acute seizures, hippocampal sclerosis, and the development of epilepsy is consistent with the temporal pattern of epileptogenesis seen in patients (Annegers et al. 1988) and therefore has the potential of being a useful animal model for studying epileptogenesis in viral-induced epilepsies.
The existence of a wide range of viruses that can cause acute seizures during infection, including Flaviviridae (West Nile virus), lentiviruses (HIV), Orthomyxoviridae (influenza), and DNA viruses like HSV-1 in the herpes family, has been taken to suggest a potential common mechanism for seizure generation (Getts et al. 2008). Additionally, it was hypothesized by Getts et al.(2008) that the mechanisms involved in the seizures during the acute infection are different from those causing the seizures after the virus is cleared. This hypothesis is consistent with a variety of non-viral animal models of epilepsy where the seizures associated with the initial insult show different pharmacological sensitivities than the spontaneous seizures that develop after a latent period (Anderson et al. 1987; Hellier et al. 2009; Stasheff et al. 1989). Because TMEV-infected mice survive, clear the initial infection, and have resolution of acute seizures prior to developing epilepsy, this model allows us to address the hypothesis of different etiologies for the acute and later spontaneous seizures. The present work is the first electrophysiological study examining neural network excitability at these two time points in the disease progression of a viral-induced epilepsy exhibiting acute seizures, a distinct latent period, and later spontaneous seizures. In particular, we examine spontaneous and miniature excitatory postsynaptic currents (EPSCs) in CA3 pyramidal neurons both during the period of acute seizures while the infection is active and 2 months following infection, when the virus is cleared and spontaneous seizures can occur (Stewart et al. 2010a, b). We found that the frequency number and amplitude of spontaneous EPSCs were increased both during the acute infection period and after 2 months. Additionally, there was a higher frequency and changes in the amplitude distributions of miniature EPSCs both acutely and after 2 months. However, the pattern of the changes in miniature EPSCs (mEPSCs) following TMEV infection was different in slices prepared acutely during infection and 2 months after virus clearance, suggesting different states for CA3 networks during these periods. These differences might have implications for the treatment of seizures in patients that occur during different periods following CNS infection.
Methods
Animals
Male C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME), 4 to 5 weeks old, were used for all experiments. Animals were kept on a 12-h light/dark cycle and allowed free access to food and water. All animal care and experimental manipulations were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Utah Institutional Animal Care and Use Committee.
TMEV infection and selection of mice
Under isoflurane anesthesia, mice were injected intracerebrally (in the posterior parietal cortex) with 2 × 104 plaque forming units of the DA strain of TMEV in a total volume of 20 μl sterile phosphate-buffered saline (PBS) as previously described (Stewart et al. 2010b) and similar to previous animal models of viral infections of the CNS (Solbrig 2010). Sham animals were injected with 20 μl PBS only. Injections were performed with an insulin syringe and a 28-gauge needle. The needle was fitted with a plastic William's collar to limit needle penetration through the skin, skull, and cortex to a total depth of 2 mm. After the injection, all animals were observed 2 h daily over the course of 0–8 days postinfection (d.p.i.) as described previously (Libbey et al. 2008) to detect signs of illness and the presence of behavioral seizures. TMEV-inoculated mice that displayed at least one behavioral seizure during the acute observation period were used in the present study.
Selection of the CA3 region for study
TMEV exhibits a strong tropism for the limbic region in C57BL/6 mice (Libbey et al. 2008; Stewart et al. 2010a, b). TMEV-infected mice exhibit neuronal death in CA1 of the hippocampus during the acute infection while CA3 is spared (Stewart et al. 2010a). These brain regions have been implicated as a potential source of seizures in temporal lobe epilepsies (Lothman 1994; McNamara 1994). The CA3 region in particular can initiate substantial synchronous behavior under normal physiological conditions (Buzsaki 1986; Ylinen et al. 1995) and robust synchronous bursts in pathological situations both in vivo and in vitro (Hablitz 1984; Schwartzkroin and Prince 1977; Wong and Traub 1983). Different patterns of activity between subregions of CA3 are involved in both interictal and ictal epileptiform activity (Dzhala and Staley 2003) suggesting that the interictal activity commonly used as a diagnostic tool and the synchronous activity generating the behavioral seizures could both be originating from CA3 in temporal lobe epilepsies. Additionally, different animal models of virus-induced seizures have observed changes in the CA3 region suggestive of hyperexcitability (Chen et al. 2004; Wu et al. 2003). For these reasons, we have chosen to initially examine the CA3 region for electrophysiological changes following TMEV infection.
Fos and NeuN immunohistochemistry
For all acute histology studies, animals were sacrificed 3–7 d.p.i. Animals were sacrificed 2 h postseizure. Mice were anesthetized with sodium pentobarbital (60 mg/kg, intraperitoneal) and perfused with phosphate-buffered saline followed by buffered 4% paraformaldehyde (PFA). Following perfusion, brains were dissected out and post-fixed overnight in 4% PFA at 4°C. Forty-micrometer-thick coronal sections were then cut and processed accordingly.
In order to reveal Fos expression levels in the brain after TMEV-induced seizures, sections were processed for Fos immunohistochemistry with avidin–biotin peroxidase methods (Klein et al. 2004). Specifically, free-floating brain sections were pre-treated for 30 min with 0.5% H2O2 to reduce endogenous peroxidase activity. They were then rinsed thoroughly with PBS and mounted on glass slides. Sections were incubated in 4% normal goat serum (Vector Laboratories, Burlingame, CA) for 1 h in PBT (PBS containing 0.3% Triton X-100) to reduce nonspecific binding. They were then incubated with Fos antiserum (Ab-5; Calbiochem, Gibbstown, NJ) diluted 1:1,000 in PBT and 1% bovine serum albumin at room temperature. After rinsing with PBS, sections were incubated with biotinylated goat anti-rabbit IgG secondary antiserum (1:400 dilution in PBT; Vector Laboratories, Burlingame, CA) for 2 h. After a thorough rinse, sections were further incubated in avidin–biotin peroxidase complex (Vectastain Elite ABC kit, Vector Laboratories, Burlingame, CA) for 1 h. Labeling was revealed by exposure to 3,3′-diaminobenzidine (DAB) substrate (Peroxidase Substrate kit, Vector Laboratories, Burlingame, CA) for 5–10 min. Fos immunoreactivity was visualized as a blue–gray staining in the cell nuclei.
Digital images were obtained with a Zeiss Axioskop microscope equipped with an AxioCam digital camera system and AxioVision 3.0 software (Carl Zeiss Inc., Jena, DE). In order to quantify the relative difference in Fos staining, ImageJ (US National Institutes of Health, Bethesda, MD) was used for automated counts of Fos-labeled cells. Images of the dorsal CA3 pyramidal layer from the beginning of the pyramidal cell layer in the hilar region to the lateral bend of CA3 were cropped and used for cell counting. At least three slides from the dorsal hippocampus were used for cell counts. The length of the CA3 layer in each image was measured using ImageJ and used to normalize cell counts per 100 μm of CA3. This procedure resulted in an average count of cells with positive Fos labeling per 100 μm of CA3 using at least three slides containing both hemispheres for each animal. While this procedure is not appropriate for estimating an absolute number of cells labeled for each dorsal hippocampus, it is adequate for determining relative differences of Fos-related activity between control and TMEV-infected animals.
Sections were also stained with an antibody against NeuN, a neuronal marker, to confirm previously reported structural damage within the hippocampus as a consequence of TMEV infection. Slices were stored in TRIS buffer until processing for the neuronal marker NeuN. Sections were mounted onto slides and air-dried for 15 min. Sections were blocked in 4.5% normal goat serum and then incubated in the primary antibody, mouse anti-NeuN (1:1000; MAB377, Chemicon, Billerica, MA). Sections were rinsed in PBS and incubated with the secondary antibody, Alexa 546 goat anti-mouse (1:2000; A11003, Invitrogen, Carlsbad, CA). After a final rinsing in PBS, the sections were mounted on slides in DPX mounting medium (Fluka, St. Louis, MO) for visualization.
Brain slice preparation and electrophysiology
Mice were anesthetized with pentobarbital (Nembutal) (25 mg/kg) and decapitated. The brains were rapidly removed and placed in ice-cold sucrose Ringer's solution bubbled with 95% O2/5% CO2 and containing (in millimolar): 124 sucrose, 3 KCl, 1.2 NaPO4, 2 MgSO4, 6 NaHCO3, 10 glucose, and 2 CaCl2. For all external solutions, the pH was between 7.34 and 7.36, and the osmolality was 295–305 mOsm. Brains were blocked and glued ventral side down on a chuck, and horizontal slices 300 μm thick were cut using a Vibratome slicer (Vibratome, St. Louis, MO). Brain slices were placed in an incubation chamber at room temperature containing artificial cerebrospinal fluid (ACSF) consisting of (in millimolar) 124 NaCl, 3 KCl, 1.4 Na2PO4, 10 glucose, 6 NaHCO3, 3.0 MgCl2, and 3.0 CaCl2. Values for KCl, MgCl2, and CaCl2, were selected to suppress the spontaneous bursting in CA3 that often occurs following blocking of the inhibitory network (Henze et al. 1997; Oliver et al. 1980).
For recording, slices were placed in a submersion chamber and constantly superfused at room temperature (2.5–3.0 ml/min) with oxygenated Ringer's solution. The whole-cell patch–clamp technique was used to record from pyramidal neurons in the CA3b and CA3c regions. A microscope equipped with Nomarski optics and infrared illumination was used to visualize the slice and position the electrode above the CA3 pyramidal cell layer. Borosilicate patch electrodes (World Precision Instruments, Sarasota, FL) were pulled to 2.5–5-MΩ resistances when measured in Ringer's solution using a micropipette electrode puller (Sutter Instruments, Novato, CA). Electrodes were lowered past the surface of the slice into the pyramidal cell layer avoiding the superficial damaged tissue; therefore, while the CA3 layer could be visualized, the individual pyramidal cell was patched using the blind technique (Blanton et al. 1989). Spontaneous and miniature EPSCs were recorded with a cesium-based internal solution composed of (in millimolar): 140 CsCl, 10 Hepes, 1 EGTA/CsOH, 0.1 CaCl2, 4 Na2–ATP, 0.5 Na–GTP, 10 glucose, and 10 lidocaine N-ethyl bromide (QX-314; Sigma). Whole-cell recordings were obtained using a MultiClamp 700 A amplifier (Molecular Devices, Sunnyvale, CA). Signals in voltage-clamp mode were acquired at 20 kHz and filtered at 10 kHz for off-line analysis using MATLAB (Mathworks, Natick, MA) and Prism (GraphPad Software, San Diego, CA). Series resistance values of <15 MΩ were used as selection criteria for accepting recordings and were not corrected for. Additionally, only recordings that did not exhibit substantial changes (<20%) in either holding current or input resistance after 20 min of time allowing for the internal pipette solution to equilibrate with the neuron were included in the study. Positive and negative current injections of 500 ms length were used to assess blockage of action potentials by QX314 and the diffusion of cesium into the neuron and subsequent changes in membrane resistance. Spontaneous and miniature EPSCs were recorded in voltage clamp with a holding potential of −70 mV. Spontaneous EPSCs, postsynaptic currents resulting from both spontaneous release of glutamate at the synapse and action potential-mediated release of glutamate due to network activity, were recorded first. Following the recording of spontaneous EPSC (sEPSCs), network activity was blocked with tetrodotoxin, and mEPSCS were recorded representing only the spontaneous release of glutamate, an indicator of the function of the molecular machinery at the synapse. Pipette offset was zeroed in the bath solution. After whole-cell patching and diffusion of pipette solution into the cell, this technique of pipette offset zeroing results in an uncompensated liquid junction potential of approximately −4 mV resulting in a holding potential of ~−74 mV. Recordings were made for blocks of 3 min, a length of time found to smooth out time-dependent variations in excitatory postsynaptic potential (EPSP) frequency and thus reliably assess EPSP frequency (Chamberlin et al. 1990). Spontaneous EPSCs were isolated by the bath application of 50 μM picrotoxin (Sigma, St. Louis, MO) for a minimum of 20 min. Recordings of mEPSCs were done after 15 min of perfusion with ACSF containing 1 μM tetrodotoxin (TTX, Sigma, St. Louis, MO). Identification and measurement of EPSCs were done using the MiniAnalysis 6.0.1 program (Synaptosoft, Decatur, GA). The threshold for event detection was set at three times baseline root-mean-square noise. This threshold setting biased EPSC classification errors to false negatives, and visual inspection of analyzed traces determined the false-negative rate to be less than 3% of all EPSCs.
Statistical analysis
Cumulative distributions were constructed from the total number of mEPSCs recorded across all experiments for the various groups. The Kolmogorov–Smirnoff (KS) test was used to assess significant differences between cumulative distributions. Distributions in the form of binned histograms were also constructed for some of the data sets. The differences in proportions in the data bins making up the distributions were tested using the two-proportion z test. Comparisons of averages from each experiment were done using the t test. For comparisons of averages between both injection type and time following infection, 2-way ANOVAs were used. Statistical tests were performed using Prism version 5, MATLAB, and Minianalysis.
Results
Fos is increased in the hippocampus following acute seizures
Fos immunoreactivity was used as a marker of acute spontaneous seizure-induced neuronal activation in TMEV-infected mice (Klein et al. 2004). Animals were sacrificed, and brains removed 2 h (see methods) after an observed seizure between 3–7 d.p.i., the time when seizures start and end. The 2-h time point represents the peak postseizure Fos immunoreactivity and was based on a pilot time-course experiment (data not shown). Diffuse Fos labeling was observed throughout most regions of the dorsal hippocampal formation of control mice (n = 4, Fig. 1a). In contrast, the granule cells of the dentate gyrus and the pyramidal cells of the CA fields of infected mice showed abundant Fos expression postseizure (n = 7, Fig. 1a). Automated cell counts of the CA3 region (see “Methods”) resulted in counts of 3.8 ± 0.9 cells per 100 μm of dorsal CA3 from control animals and 19.9 ± 3.9 cells per 100 μm of CA3 from TMEV-infected mice (p = 0.015, Student's t test; Fig. 1b). Qualitatively, similar results were seen for the ventral hippocampus. For the initial electrophysiological characterization of this animal model of viral-induced epilepsy, we focused on the CA3 region due to its sensitivity for hyperexcitability and implication in other animal models of epilepsy. Additionally, neurons in the CA1 region often die due to direct viral infection or by bystander killing and are either not present or heavily damaged at the 2-month time point as indicated by immmunohistochemistry for the neuronal marker, NeuN (Fig. 1c), rendering comparisons between time points impossible for the CA1 brain region (Stewart et al. 2010b).

Enhanced Fos immunoreactivity in the dorsal hippocampus following TMEV-induced seizure activity. a (top) Bright field image of dorsal hippocampus from a control animal showing minimal Fos labeling. (Bottom) Bright field image of a TMEV-infected dorsal hippocampus with a number of Fos-labeled neurons. Bar = 500 μm. b Bar graph showing the difference (p = 0.015) between control (N = 4) and TMEV-infected mice (N = 7) in average number of Fos-labeled cells per 100 μm of CA3 in the dorsal hippocampus. c Images of the dorsal hippocampus stained for the neuronal marker, NeuN, demonstrating the damage to CA1 in TMEV-infected mice (bottom) versus saline-injected control mice (top)
TMEV infection increases sEPSC frequency both acutely and 2 months after infection
In order to test if increased Fos labeling correlated with increased electrophysiological activity, sEPSC activity was recorded in CA3 pyramidal neurons in brain slices prepared during the acute infection. Mice usually begin to exhibit partial seizures that secondarily generalize on days 3–4 following TMEV infection (Stewart et al. 2010b). Seizure number and severity then crest during the next few days and rapidly resolve 5–6 days following the initial seizure. Brain slices prepared for study of the acute infection period were prepared from mice 4–7 d.p.i., a period when the virus has not been cleared in the animals (Stewart et al. 2010b). Of the animals infected with TMEV, 75% exhibit acute spontaneous seizures (Stewart et al. 2010a). Only mice observed to have stage 4–5 behavioral seizures on the Racine scale in the 4–7-day period following infection were used in the present study. A group of animals observed to have seizures during the acute period was set aside for experiments occurring at least 2 months p.i. While video EEG was not performed in the animals used for experiments at the 2-month time point, it is known that seizure threshold is persistently lowered in mice exhibiting acute behavioral seizures following TMEV infection (Stewart et al. 2010b) and that at least 65% of mice exhibiting acute seizures go on to exhibit spontaneous behavioral seizures by 2 months following a latent period (Stewart et al. 2010a).
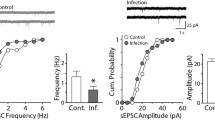
sEPSCs were isolated by bath application of 50 μM picrotoxin for a minimum of 20 min. Following the initiation of a whole-cell patch, an additional 20 min was allowed for the cesium-based internal solution to equilibrate with the neuron. Figure 2a and b shows examples of isolated sEPSCs on a longer and shorter time scale, respectively. The frequency of sEPSCs was increased in animals infected with TMEV relative to mock saline-infected controls. This increase was indicated by a significant leftward shift of the cumulative distribution of inter-event intervals (IEI) (Fig. 2c; Z = 12.31, p < 0.0001, 2-sample KS test) and a significant increase in the average frequency (Fig. 2c inset; control, 2.82 ± 0.51 Hz; TMEV, 4.92 ± 0.72 Hz; p = 0.039, t test). While the average frequency of sEPSCs increased during TMEV infection, the coefficients of variation (CV) of the IEIs were not significantly different (control, 0.87 ± 0.038, N = 8; TMEV, 0.80 ± 0.030, N = 10; t test, p = 0.168), suggesting no difference in the statistics of network activity at least by this measure. Average CVs for both control and TMEV groups were significantly different than 1, the theoretical IEI CV for a Poisson distribution (control, p = 0.012; TMEV, p = 0.0001; one-sample t test) so the network activity was not purely random. There was a significant increase of sEPSC amplitude indicated by a rightward shift of the cumulative distribution of amplitudes (Fig. 2d; Z = 3.28, p < 0.0001, KS test), but this difference was not observed in the comparison of averages (control, 20.63 ± 1.388 pA, N = 8; TMEV, 23.01 ± 3.875 pA, N = 10; t test, p = 0.61).

Acute TMEV infection results in increased number and amplitude of sEPSCs in CA3 neurons. a Recordings of sEPSCS from CA3 neurons from control (PBS injected) and TMEV-injected animals. b Example traces on a faster time scale collected from the traces in a. c Cumulative distributions of IEIs showing a leftward shift of the experimental distribution indicating a higher frequency of events (KS test, p < 0.0001) and the inset bar graph shows the significant increase in average frequency (control, 2.82 ± 0.51 Hz, N = 8; TMEV, 4.92 ± 0.72 Hz, N = 10; p = 0.039, t test). d Cumulative distributions of sEPSC amplitudes with a shift toward larger amplitudes in the TMEV-infected mouse (KS test, p < 0.0001), but there is no increase (inset) in average sEPSC amplitudes (p = 0.61; control, N = 8; TMEV, N = 10)
Previous work in this model has found that animals infected with TMEV have a persistent decrease in seizure threshold and frequent interictal spikes on EEG recordings, and that a significant proportion of animals develop spontaneous seizures by 2 months p.i. after a latent period (Stewart et al. 2010a, b). Therefore, spontaneous EPSCs were also examined in brain slices prepared at least 2 months following infection. TMEV-infected mice were only kept for later examination if they were observed to have stage 4–5 behavioral seizures on the Racine scale during the acute infection. Example traces of sEPSC recorded from CA3 neurons prepared from saline-injected and TMEV-infected mice are shown in Fig. 3a. As during the acute stage of disease, there was a significant leftward shift of the IEI cumulative distribution indicating an increase in sEPSC (Fig. 3b; Z = 15.91, p < 0.0001, 2-sample KS test). This change in IEI distribution was accompanied by a significant increase in average frequency (control, 3.199 ± 0.4410 Hz, N = 8; TMEV, 6.299 ± 1.028 Hz, N = 9; t test, p = 0.018). The CVs of the IEIs after 2 months were not significantly different (control, 0.82 ± 0.021, N = 9; TMEV, 0.83 ± 0.085, N = 10; t test, p = 0.908) as seen during the acute stage. Similar to the acute time period, sEPSC amplitudes exhibited a significant increase in amplitude as indicated by cumulative probability curves (Fig. 2c; Z = 6.22, p < 0.0001, 2-sample KS test) that was not observed in comparisons of averages (control, 12.24 ± 1.649 pA, N = 8; TMEV, 14.98 ± 1.234 pA, N = 9; t test, p = 0.197).

Frequency and amplitude of sEPSCs remain enhanced in CA3 neurons 2 months following TMEV clearance. a Recordings of sEPSCs from CA3 neurons from control and TMEV-injected animals. b Cumulative distributions of IEIs showing a leftward shift of the experimental distribution indicating a higher frequency of events (KS test, p < 0.0001), which is accompanied by an increase in average frequency (inset; control, 3.20 ± 0.44 Hz, N = 8; TMEV, 6.30 ± 1.02 Hz, N = 9; t test, p = 0.018). c Cumulative distributions of sEPSC amplitudes with a shift toward larger sEPSC amplitudes in the TMEV-infected mouse (KS test, p < 0.0001), which is not accompanied by a significant increase in average amplitude (inset; control, 12.24 ± 1.65 pA, N = 8; TMEV, 14.98 ± 1.23 pA, N = 9; t test, p = 0.197). d Results of a 2-way ANOVA revealing an overall effect of TMEV injection (p = 0.0015) but not time (p = 0.248) for sEPSC frequency. e Results of a 2-way ANOVA revealing no overall effect on sEPSC amplitude of TMEV injection (p = 0.319). An overall effect for time was observed (p = 0.0028)
To examine the evolution of changes in sEPSC frequency and amplitude over time, 2-way ANOVAs were applied to the distribution of averages derived from the individual experiments with injection (saline versus TMEV) and time (acute versus 2 months) forming the two groups. There was a significant overall effect of infection on frequency of sEPSCs (p = 0.0015) shown in Fig. 3d while there was not an overall effect of time (p = 0.248), nor was there an interaction (p = 0.503). The situation was different for the amplitude of the sEPSCs, with no overall effect of infection (p = 0.319), but an overall effect for time (Fig. 3e, p = 0.0028). The irregular shape of the cumulative distributions of sEPSC amplitudes (compare Fig. 3c with Fig. 2d) and the fact that distributions were significantly different while averages were not suggested that changes might be occurring preferentially in specific amplitude ranges due to TMEV infection. Indeed, the difference between the distributions of sEPSC amplitudes following infection for each time point (comparing TMEV groups from both time points) is confirmed using a Kolmogorov–Smirnoff test (acute TMEV vs. 2-month TMEV, Z = 72.13, p < 0.0001, 2-sample KS test). Additionally, the distribution of averages sampled from any distribution of data is Gaussian, while the distribution of the data making up the cumulative distributions is not Gaussian (D'Agostino and Pearson normality test, p < 0.0001 for both control and TMEV mEPSC amplitude data sets at 2 months). Examination of the cumulative distributions for the control and TMEV groups (Fig. 3c) reveals that the separations between the two distributions tend to happen away from the median; therefore, a comparison of averages may miss some relevant differences. The potential for changes that occur in specific amplitude ranges is explored further in the analysis of mEPSCs where there is evidence linking amplitude ranges with specific anatomy (Henze et al. 1997). The sEPSC frequency data suggest that CA3 network activity is increased at both stages of disease, while sEPSC amplitude distributions suggest that the synaptic locations of that activity are different between the stages of disease.
TMEV infection increases number and size of mEPSCs both acutely and 2 months after infection
To investigate further potential changes at the synapse, mEPSCs were isolated by the bath application of TTX for 10 min in the continued presence of picrotoxin following the initial recording of sEPSCs. The examination of the amplitude and frequency of mEPSCs suggests changes at the synapse postsynaptically and presynaptically, respectively, resulting from TMEV infection. As for the sEPSCs, 3-min epochs of mEPSCs were recorded for analysis. Examples of average mEPSCs from a control and experimental animal during the acute period are shown in Fig. 4a. An increase in frequency was indicated by a significant leftward shift of the cumulative distribution of inter-event intervals (Fig. 4b; Z = 5.89, p < 0.0001, 2-sample KS test). The CVs of the IEIs were not significantly different (control, 1.08 ± 0.056, N = 8; TMEV, 1.14 ± 0.11, N = 10; t test, p = 0.690). Average CVs for both control and TMEV groups were not significantly different than 1, consistent with random release of synaptic vesicles (control, p = 0.165; TMEV, p = 0.231; one-sample t test). While IEI cumulative distributions were significantly different, mean mEPSC frequency was not significantly different in the TMEV-infected mice relative to controls (control, 1.05 ± 0.32 Hz, N = 8; TMEV, 1.63 ± 0.44 Hz, N = 10; t test, p = 0.349). Cumulative distributions also indicated significant rightward shifts in mEPSC amplitude (Fig. 4c; Z = 3.11, p < 0.0001, 2-sample KS test) and charge transfer (Fig. 4d; Z = 5.10, p < 0.0001, 2-sample KS test) in the TMEV-infected mouse. The increase in charge transfer was consistent with a lengthening of rise times (Fig. 4e; Z = 4.08, p < 0.0001, 2-sample KS test) and decay times (Fig. 4f; Z = 6.31, p < 0.0001, 2-sample KS test) in the TMEV mouse relative to controls. The shifts in the IEI and amplitude cumulative distributions indicate changes at the synapse both presynaptically and postsynaptically, respectively, during the acute stage of infection.

Acute TMEV infection results in increased mEPSC frequency and amplitude coupled with slower kinetics. a Example average mEPSC recordings from PBS-injected and TMEV-injected mice. b Cumulative IEI distributions showing shortening of mEPSC intervals (KS test, p < 0.0001). c The amplitude cumulative distribution is shifted to the right in the TMEV-infected mouse (KS test, p < 0.0001). d Consistent with the change in amplitude, mEPSC charge transfer is also increased (KS test, p < 0.0001). e A rightward shift in the cumulative distribution of rise times shows a slowing of kinetics (KS test, p < 0.0001). f Decay times also have slower kinetics (KS test, p < 0.0001)
Miniature EPSCs were also analyzed at the 2-month time point. Examples of mEPSCs from this period are shown in Fig. 5a. The pattern of changes of the cumulative distributions is similar to that of the acute period. A leftward shift in the cumulative distribution for mEPSC IEIs is significant (Fig. 5b; Z = 8.83, p < 0.0001, 2-sample KS test) indicating an increase in frequency, which was not associated with an increase in mean mEPSC frequency (control, 1.63 ± 0.44 Hz, N = 9; TMEV, 3.22 ± 0.77 Hz, N = 10; t test, p = 0.093) suggesting a more subtle change compared to spontaneous EPSC data. The CVs of IEIs from control and TMEV animals are not different from each other (control, 1.01 ± 3.84, N = 9; TMEV, 0.945 ± 5.17, N = 10; t test, p = 0.891) or 1 (control, p = 0.868; TMEV, p = 0.150; one-sample t test). As in the case of the acute infection, cumulative distributions also indicate significant rightward shifts in mEPSC amplitude (Fig. 5c; Z = 16.2, p < 0.0001, 2-sample KS test) and charge transfer (Fig. 5d; Z = 9.52, p < 0.0001, 2-sample KS test) coupled with a lengthening of rise times (Fig. 5e; Z = 7.76, p < 0.0001, 2-sample KS test) and decay times (Fig. 5f; Z = 6.25, p < 0.0001, 2-sample KS test). As for the results during the acute phase, the shifts in the IEI and amplitude cumulative distributions indicate changes at the synapse both presynaptically and postsynaptically.

mEPSC frequency and amplitude remain elevated 2 months following TMEV infection and clearance. a Example average mEPSC recordings from PBS-injected and TMEV-injected mice. b Cumulative IEI distributions remain shifted to shorted intervals at 2 months (KS test, p < 0.0001). c The amplitude cumulative distribution is still shifted to the right 2 months following clearance of the virus (KS test, p < 0.0001). d Consistent with the change in amplitude, mEPSC charge transfer is also increased (KS test, p < 0.0001). e A rightward shift in the cumulative distribution of rise times shows a slowing of kinetics (KS test, p < 0.0001). f Decay times also have slower kinetics (KS test, p < 0.0001)
Similar to the results for sEPSCs, the evolution of changes in average mEPSC frequency and amplitude over time was examined using 2-way ANOVAs with injection (control versus TMEV) and time (acute versus 2 months) comprising the two groups. There was a significant overall effect of infection on frequency of mEPSCs (p = 0.037) shown in Fig. 6a while there was not an overall effect of time (p = 0.089), nor was there an interaction (p = 0.487). Consistent with the sEPSC results, 2-way ANOVAs revealed no overall effect on mEPSC amplitude (p = 0.138, Fig. 6b); however, an overall decrease in mEPSC amplitude was observed as animals aged from 1 month to approximately 3 months (p = 0.044, Fig. 6b). The shifts in the cumulative distributions for mEPSC amplitude, rise time, and decay time for both the acute and 2-month time points shown in Figs. 4 and 5 are consistent with more charge transfer following TMEV infection. However, these significant shifts as detected by the KS test were not generally borne out in the statistical comparison of averages. Cumulative distributions and the KS test are a very common method of presentation for this type of data, but this method is particularly sensitive at detecting small changes due to the large number of samples (many hundreds) (Goodman 1999). Therefore, we examined the distribution of mEPSC amplitudes in more detail to determine their potential biological relevance.

Patterns of change in mEPSC characteristics differ at the acute and 2-month time points. a There is an overall effect of TMEV injection on frequency (2-way ANOVA, p = 0.037), but not an overall effect of time (2-way ANOVA, p = 0.089). b Results of a 2-way ANOVA revealing no overall effect on mEPSC amplitude of TMEV injection (p = 0.138). An overall effect for time was observed (p = 0.044). c Distributions of mEPSC amplitude with 10-pA bin size during the acute period show TMEV-induced changes vary depending on the mEPSC amplitude range examined (*p < 0.01, **p < 0.0001). d Distribution of mEPSC amplitudes showing regions of change for the 2-month time point (*p < 0.01, **p < 0.0001). e Bar graph showing the change in total proportion for each of the 10-pA amplitude bins examined in c and d. f Plot of the mEPSC amplitude cumulative distributions for both acute and 2-month time points
The cumulative distributions for mEPSC amplitudes for the control and TMEV-infected conditions for both the acute and 2-month time points are shown in Fig. 6f. The maximal differences between the control and TMEV cumulative distributions derived from the acute and epilepsy phase occur at different ranges of mEPSC amplitudes. Because different ranges of mEPSC amplitudes have been associated with different locations of synapses of the dendritic arbors of CA3 neurons (Henze et al. 1997), this result suggests different synapses are affected on the CA3 neuron between the two phases. To examine the meaning of the detected differences between the cumulative distributions in more detail, we constructed binned distributions with bin sizes of 10 pA elaborating on the method of Henze et al. (1997). The binned distributions for the acute and 2-month time points are shown in Fig. 6c and d, respectively. The proportion of total events falling within each bin range is shown; using proportions also normalizes the different number of total mEPSCs across conditions. With the data presented in this way, we can use the two-proportion z test to determine significant shifts in the proportions falling within each bin between the control and experimental groups. Significant differences computed from the two-proportion z test are shown on Fig. 6c and d (*p < 0.01 and **p < 0.0001). Because the two-proportion z test also is sensitive to detecting differences due to the large number of events, we focused on p values less than 0.0001 (**). During the acute period of active infection (Fig. 6c), there was no change in the 0–10-pA bin and a significant decrease in the 10–20-pA bin; these are mEPSC amplitudes shown to be dominated by the CA3 collaterals (Henze et al. 1997). A significant increase occurred (p < 0.0001) in the 30–40 bin, which is an amplitude range dominated by the mossy fiber input (Henze et al. 1997). Two months following the infection, changes in proportions were more dramatic (Fig. 6d). There was a large decrease (~25% of total recorded events, p < 0.0001) in the 0–10-pA bin that was almost mirrored by a corresponding increase in the 10–20-pA bin. As both these mEPSC amplitude ranges are dominated by CA3 collaterals, this suggests that there is an increase in amplitudes among the collaterals at the 2-month time point. There are also significant increases (p < 0.0001) in the 20–30- and 30–40-pA bins suggesting the mossy fibers are still affected at this late time point, but the changes occurred relative to a lower level of control activity in these amplitude ranges. Figure 6e summarizes the changes. The bar graph shows the change in total proportion recorded for each amplitude range; therefore, a decrease of 25% means that a quarter of all recorded events shifted out of that size range and into other ranges. The most dramatic changes occurred in the 0–20 suggesting no increase in mEPSC amplitude of the collaterals during the acute period but a large increase after 2 months. Amplitude ranges associated with mossy fiber input are increased at both time points, but at 2 months, there are fewer overall mEPSCs in these ranges. The changes 2 months after infection are more extensive, covering a wider range of amplitudes, and this change might result from the increased involvement of the CA3 region at this stage of the disease. The coefficients of variation of mEPSC amplitude were not significantly different at the acute time point (control, 0.41 ± .06; TMEV, 0.53 ± 0.07, p = 0.25) nor at the 2-month time point (control, 0.37 ± 0.03; TMEV, 0.42 ± 0.05, p = 0.43) suggesting that these shifts in amplitude are postsynaptic. Therefore, the data suggest that after the virus is cleared and epilepsy begins to develop at the late stage, the CA3 collaterals are preferentially strengthened. These collaterals have been shown via positive feedback to destabilize the CA3 region in non-infection models of epilepsy (Hablitz 1984; Schwartzkroin and Prince 1977; Wong and Traub 1983).
Discussion
We have recently described a novel model of virus-induced epilepsy in C57BL/6 mice (Libbey et al. 2008; Stewart et al. 2010a, b). These mice, when infected with Theiler's virus, develop acute seizures and a large increase in proinflammatory cytokines (including TNF-α coincident with the viral infection) that subside as the virus is cleared (Kirkman et al. 2010; Libbey et al. 2008; Stewart et al. 2010b). Following the clearance of the virus, TMEV-infected mice have a chronically lowered seizure threshold (Stewart et al. 2010b), and by 2 months, the majority of mice develop spontaneous seizures (Stewart et al. 2010a), the primary symptom defining epilepsy. TMEV-infected mice have damage to the CA1 region and exhibit indicators of gliosis as do other non-viral animal models of epilepsy, but have very little mossy fiber sprouting, a common feature of many animal models of epilepsy (Stewart et al. 2010b). While a previous animal model examining Borna disease does exhibit spontaneous seizures during disease progression (Solbrig 2010; Solbrig et al. 2006), the virus is not cleared, and acute seizures are not observed making it a fundamentally different disease progression compared to the TMEV infection model. Previous models of viral-induced CNS disease that do exhibit acute seizures result in high mortality rates making study of the chronic disease difficult (Chen et al. 2004; Stringer 2006; Wu et al. 2003). Additionally, TMEV is not a human pathogen; the mouse is the natural host for the virus, which makes it easier to work with than other viral models exhibiting seizures during infection (Chen et al. 2004; Stringer 2006; Wu et al. 2003). Using this model, we have begun to study the disease process resulting in epilepsy following acute CNS infection and seizures by examining the excitatory activity in the CA3 network in control and TMEV-infected mice both during the acute and 2-month time points, time points both associated with spontaneous seizures but separated by a latent period.
Fos immunohistochemisty was used to examine neural excitability in the hippocampal region during the acute viral infection. Quantitative cell counts show that Fos labeling is greatly enhanced in the dorsal CA3 region of the hippocampus as a consequence of seizures in the TMEV-infected mice. The ventral CA3 region has qualitatively similar Fos staining. The ventral CA3 region is known to exhibit more excitability in slices (Gilbert et al. 1985), and subsequent patch clamp recordings were done from this region. Electrophysiological recordings confirm that activity in the ventral CA3 excitatory network is enhanced both acutely during the infection and at the 2-month time point as evidenced by enhanced frequency and amplitude of sEPSCs as quantified by cumulative distributions. ANOVA performed on the averages of sEPSC frequency from each experiment during the two time points revealed an overall effect of TMEV infection. Interestingly, ANOVA performed on average amplitude of sEPSCs did not reveal an overall effect of infection but did show an overall effect of time with the amplitude of sEPSCs decreasing as the animal aged.
To better isolate changes at the level of the synapse from network level changes, miniature EPSCs were examined at both time points by blocking the network activity with tetrodotoxin. Analogous to the changes observed for sEPSCs, mEPSCs also exhibited changes in both frequency and amplitude at both time points as measured using cumulative distributions. ANOVA performed on average frequencies from each experiment revealed an overall effect of infection, and similar analysis of mEPSC amplitudes only revealed an overall effect of time, with amplitudes decreasing as the animal aged in both control and experimental conditions, again consistent with sEPSCs. Because the Kolmogorov–Smirnoff test of cumulative distributions can detect small differences that may not be biologically relevant, we constructed distributions of amplitudes (10-pA bin size) on which we could perform proportion tests. Focusing only on changes in the bins detected with very low p values (p < 0.0001), it was found that while there were increases in amplitude ranges thought to correspond to the large mossy fiber synapses (Henze et al. 1997), the dramatic differences between the acute and 2-month time points occurred in the mEPSC ranges associated with CA3 collaterals (Henze et al. 1997). Specifically, during the acute infection, the proportion of mEPSCs occurring in the upper range of amplitudes associated with CA3 collaterals decreased, while at 2 months, this range showed a large increase. That the increase at 2 months of the 10–20-pA bin was mirrored by a decrease in the 0–10-pA bin suggests that these synapses at 2 months were potentiated. Cell capacitance values calculated from resistance and time constant measurements from hyperpolarizing current injections assessing cesium diffusion into the cell at the beginning of experiments were not significantly different over time or following infection. These capacitance measurements suggest that at least changes in cell size are not responsible for observed changes. The lack of any changes in the coefficient of variation of mEPSC amplitudes suggests the changes in amplitude are postsynaptic. These observations are consistent with studies showing that synchronized activity in the CA3 network results in changes specifically affecting the CA3 collateral synapses (Bains et al. 1999). These changes in the CA3 collateral network cause the CA3 region to be more excitable, a result that can be blocked if NMDA receptor blockers are used during early synchronized activity (Bains et al. 1999). If the TMEV infection in mice and other viral infections in general result in a similar sequence of events, then early intervention during the acute seizures would potentially be beneficial.
It has been observed for many years that the CA3 region of the hippocampus is a source of bursts of synchronized activity both in vivo (Buzsaki 1986) and in vitro (Hablitz 1984; Wong and Traub 1983). The extensive recurrent excitatory connections in this brain region (MacVicar and Dudek 1980; Traub and Wong 1982) are an important factor in this excitability. The damage to neurons of the CA1 region and the increase in excitatory activity in the CA3 region in the current model are similar to models of ischemia-induced epilepsy (Congar et al. 2000; Epsztein et al. 2008, 2006). The authors of these ischemia studies suggested the increase in CA3 excitability following injury could be the result of intrinsic excitability of the CA3 neurons (Congar et al. 2000) and sprouting new CA3 recurrent collaterals (Epsztein et al. 2006). In the current study, we observed an increase in sEPSC frequency at both the acute and 2-month time point. An increase in sEPSCs in CA3 has been observed months after an ischemic insult and was associated with CA3 sprouting (Epsztein et al. 2006). In these ischemia studies (Congar et al. 2000; Epsztein et al. 2006), the CA3 network was not studied during the acute insult. Increases in sEPSC frequency have also been observed in CA1 weeks after insult in the kainate-induced seizure model (Esclapez et al. 1999; Shao and Dudek 2004) and pilcarpine models (Esclapez et al. 1999). We did not study the intrinsic excitability of the CA3 neurons, which could result in increased excitatory network activity, due to the use of an internal solution for enhancing the electrotonic compactness of the neurons. Although previous studies of chronic kainate insults in slices did not exhibit changes in CA3 intrinsic excitability (Ben-Ari and Gho 1988), a change in resting membrane potential of CA3 neurons has been observed months after ischemic insult (Congar et al. 2000); therefore, future excitability studies are warranted.
Examination of cumulative distributions of sEPSC amplitudes revealed increases both during the acute period and 2 months following TMEV infection. Interestingly, ANOVA revealed an overall decrease in amplitudes of sEPSCs in slices prepared from 5-week-old mice compared to slices prepared from 13-week-old mice. In rat CA3 mini slices, sEPSC amplitudes showed a decrease between early time points (postnatal days 9–14) and older animals (postnatal days 90–100) in normal animals (Shao and Dudek 2009), although they also observed decreases in frequency, which we did not observe. The present and previous results (Shao and Dudek 2009) showing a decrease in sEPSC amplitudes in CA3 neurons as the animal ages are consistent with anatomical observations of rapid growth and increasing complexity in rat CA3 neurons until 2 weeks followed by a decrease in branching complexity as the animal ages (Gomez-Di Cesare et al. 1997).
The increase of mEPSC amplitude and frequency at 2 months following infection is consistent with the hypothesis of sprouting of mossy fiber input to CA3 observed in other animal models of epilepsy (Congar et al. 2000; Epsztein et al. 2006; Esclapez et al. 1999; Shao and Dudek 2004; Wuarin and Dudek 2001), but was most pronounced in amplitude ranges associated with the CA3-CA3 collaterals. However, increases in mEPSC amplitude and frequency were also observed during the acute infection when it seems improbable that sprouting has had time to occur. We have shown that the cytokine TNF-α is greatly increased during TMEV infection (Kirkman et al. 2010). In addition to TNF-α's role in the immune response, it has also been shown to be involved in the synaptic scaling that strengthens synapses (Stellwagen et al. 2005; Stellwagen and Malenka 2006). This could provide a fast-acting mechanism for the acute synaptic changes that we have observed. The different pattern of changes occurring in mEPSC amplitude distributions at the two time points is consistent with two different mechanisms operating during the acute insult of infection and following months of subsequent changes finally leading to spontaneous seizures. The potential for different mechanisms for seizures arising from the acute insult and the chronic spontaneous seizures is also consistent with observed differences in pharmacological susceptibility of acute and the later-developing epileptiform activity (Anderson et al. 1987; Hellier et al. 2009; Stasheff et al. 1989). The study by Bain et al. suggests that a variety of insults resulting in aberrant network synchronization could result in a pathological change in CA3 collateral synapses resulting in spontaneous bursts of activity in the CA3 region (Bains et al. 1999), a sequence of events that might be occurring following the TMEV infection in mice. Their observation that early intervention with NMDA antagonists blocked this process (Bains et al. 1999) is consistent with previous in vitro models of network bursting where NMDA antagonists blocked early, but not late, bursts (Anderson et al. 1987; Stasheff et al. 1989). In total, the present and previous studies cited suggest seizures associated with the acute TMEV infection are distinct from the later seizures, which may be associated with pathological long-term changes in the network and should be treated in different ways. Additionally, these studies suggest that the early treatment is important for ameliorating the pathological changes that might result in developing epilepsy.
Conclusions
TMEV infection of C57BL/6 mice results in an acute infection associated with seizures and a greatly increased risk of chronic spontaneous seizures. The present study examines the excitatory network in the CA3 region of the hippocampus at these two time points. Examination of sEPSCs demonstrates an increase in CA3 network excitability at both time points. This increase in network activity is correlated with increases in mEPSC amplitude and frequency implicating changes at the synapse in shifts of network excitability. However, the patterns of these changes are different at these two time points suggesting that different mechanisms might underlie the acute and chronic seizures. If different mechanisms underlie acute and chronic seizures, different treatments might be warranted at various time points of the disease. Additionally, the present animal model of epilepsy provides a new research tool for examination of intervention during the early phase of viral infection.
References
Anderson WW, Swartzwelder HS, Wilson WA (1987) The NMDA receptor antagonist 2-amino-5-phosphonovalerate blocks stimulus train-induced epileptogenesis but not epileptiform bursting in the rat hippocampal slice. J Neurophysiol 57:1–21
Annegers JF, Hauser WA, Beghi E, Nicolosi A, Kurland LT (1988) The risk of unprovoked seizures after encephalitis and meningitis. Neurology 38:1407–1410
Bains JS, Longacher JM, Staley KJ (1999) Reciprocal interactions between CA3 network activity and strength of recurrent collateral synapses. Nat Neurosci 2:720–726
Beers DR, Henkel JS, Schaefer DC, Rose JW, Stroop WG (1993) Neuropathology of herpes simplex virus encephalitis in a rat seizure model. J Neuropathol Exp Neurol 52:241–252
Ben-Ari Y, Gho M (1988) Long-lasting modification of the synaptic properties of rat CA3 hippocampal neurones induced by kainic acid. J Physiol 404:365–384
Blanton MG, Lo Turco JJ, Kriegstein AR (1989) Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods 30:203–210
Buzsaki G (1986) Hippocampal sharp waves: their origin and significance. Brain Res 398:242–252
Chamberlin NL, Traub RD, Dingledine R (1990) Role of EPSPs in initiation of spontaneous synchronized burst firing in rat hippocampal neurons bathed in high potassium. J Neurophysiol 64:1000–1008
Chen SF, Huang CC, Wu HM, Chen SH, Liang YC, Hsu KS (2004) Seizure, neuron loss, and mossy fiber sprouting in herpes simplex virus type 1-infected organotypic hippocampal cultures. Epilepsia 45:322–332
Congar P, Gaiarsa JL, Popovici T, Ben-Ari Y, Crepel V (2000) Permanent reduction of seizure threshold in post-ischemic CA3 pyramidal neurons. J Neurophysiol 83:2040–2046
Dzhala VI, Staley KJ (2003) Transition from interictal to ictal activity in limbic networks in vitro. J Neurosci 23:7873–7880
Eeg-Olofsson O (2003) Virological and immunological aspects of seizure disorders. Brain Dev 25:9–13
Eisenschenk SGR (2000) Seizures associated with non- neurological medical conditions. In: Wyllie E (ed) The treatment of epilepsy: principles and practice. Lippincott Williams and Wilkins Co., New York, pp 657–669
Epsztein J, Milh M, Bihi RI, Jorquera I, Ben-Ari Y, Represa A, Crepel V (2006) Ongoing epileptiform activity in the post-ischemic hippocampus is associated with a permanent shift of the excitatory-inhibitory synaptic balance in CA3 pyramidal neurons. J Neurosci 26:7082–7092
Epsztein J, Ben-Ari Y, Represa A, Crepel V (2008) Late-onset epileptogenesis and seizure genesis: lessons from models of cerebral ischemia. Neuroscientist 14:78–90
Esclapez M, Hirsch JC, Ben-Ari Y, Bernard C (1999) Newly formed excitatory pathways provide a substrate for hyperexcitability in experimental temporal lobe epilepsy. J Comp Neurol 408:449–460
Getts DR, Matsumoto I, Muller M, Getts MT, Radford J, Shrestha B, Campbell IL, King NJ (2007) Role of IFN-gamma in an experimental murine model of West Nile virus-induced seizures. J Neurochem 103:1019–1030
Getts DR, Balcar VJ, Matsumoto I, Muller M, King NJ (2008) Viruses and the immune system: their roles in seizure cascade development. J Neurochem 104:1167–1176
Gilbert M, Racine RJ, Smith GK (1985) Epileptiform burst responses in ventral vs dorsal hippocampal slices. Brain Res 361:389–391
Gomez-Di Cesare CM, Smith KL, Rice FL, Swann JW (1997) Axonal remodeling during postnatal maturation of CA3 hippocampal pyramidal neurons. J Comp Neurol 384:165–180
Goodman SN (1999) Toward evidence-based medical statistics. 1: The P value fallacy. Ann Intern Med 130:995–1004
Griffith JF, Kibrick S, Dodge PR, Richardson EP (1967) Experimental herpes simplex encephalitis. Electroencephalographic, clinical, virologic, and pathologic observations in the rabbit. Electroencephalogr Clin Neurophysiol 23:263–269
Hablitz JJ (1984) Picrotoxin-induced epileptiform activity in hippocampus: role of endogenous versus synaptic factors. J Neurophysiol 51:1011–1027
Hellier JL, White A, Williams PA, Edward Dudek F, Staley KJ (2009) NMDA receptor-mediated long-term alterations in epileptiform activity in experimental chronic epilepsy. Neuropharmacology 56:414–421
Henze DA, Card JP, Barrionuevo G, Ben-Ari Y (1997) Large amplitude miniature excitatory postsynaptic currents in hippocampal CA3 pyramidal neurons are of mossy fiber origin. J Neurophysiol 77:1075–1086
Hunsperger EA, Roehrig JT (2006) Temporal analyses of the neuropathogenesis of a West Nile virus infection in mice. J Neurovirol 12:129–139
Kirkman NJ, Libbey JE, Wilcox KS, White HS, Fujinami RS (2010) Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia 51:454–464
Klein BD, Fu YH, Ptacek LJ, White HS (2004) c-Fos immunohistochemical mapping of the audiogenic seizure network and tonotopic neuronal hyperexcitability in the inferior colliculus of the Frings mouse. Epilepsy Res 62:13–25
Labar DR, Harden C (1998) Infection and inflammatory diseases. In: Engel J Jr, Pedley TA (eds) Epilepsy: a comprehensive textbook. Lippincott Williams & Wilkins, New York, pp 2587–2596
Lehrmann E, Guidetti P, Love A, Williamson J, Bertram EH, Schwarcz R (2008) Glial activation precedes seizures and hippocampal neurodegeneration in measles virus-infected mice. Epilepsia 49(Suppl 2):13–23
Libbey JE, Kirkman NJ, Smith MC, Tanaka T, Wilcox KS, White HS, Fujinami RS (2008) Seizures following picornavirus infection. Epilepsia 49:1066–1074
Lothman EW (1994) Seizure circuits in the hippocampus and associated structures. Hippocampus 4:286–290
MacVicar BA, Dudek FE (1980) Local synaptic circuits in rat hippocampus: interactions between pyramidal cells. Brain Res 184:220–223
McNamara JO (1994) Cellular and molecular basis of epilepsy. J Neurosci 14:3413–3425
Misra UK, Tan CT, Kalita J (2008) Viral encephalitis and epilepsy. Epilepsia 49(Suppl 6):13–18
Oliver AP, Carman JS, Hoffer BJ, Wyatt RJ (1980) Effect of altered calcium ion concentration on interictal spike generation in the hippocampal slice. Exp Neurol 68:489–499
Schmutzhard E (2001) Viral infections of the CNS with special emphasis on herpes simplex infections. J Neurol 248:469–477
Schwartzkroin PA, Prince DA (1977) Penicillin-induced epileptiform activity in the hippocampal in vitro prepatation. Ann Neurol 1:463–469
Shao LR, Dudek FE (2004) Increased excitatory synaptic activity and local connectivity of hippocampal CA1 pyramidal cells in rats with kainate-induced epilepsy. J Neurophysiol 92:1366–1373
Shao LR, Dudek FE (2009) Both synaptic and intrinsic mechanisms underlie the different properties of population bursts in the hippocampal CA3 area of immature versus adult rats. J Physiol 587:5907–5923
Solbrig MV (2010) Animal models of CNS viral disease: examples from borna disease virus models. Interdiscip Perspect Infect Dis 2010:709791
Solbrig MV, Adrian R, Baratta J, Lauterborn JC, Koob GF (2006) Kappa opioid control of seizures produced by a virus in an animal model. Brain 129:642–654
Stasheff SF, Anderson WW, Clark S, Wilson WA (1989) NMDA antagonists differentiate epileptogenesis from seizure expression in an in vitro model. Science 245:648–651
Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial TNF-alpha. Nature 440:1054–1059
Stellwagen D, Beattie EC, Seo JY, Malenka RC (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 25:3219–3228
Stewart KA, Wilcox KS, Fujinami RS, White HS (2010a) Development of postinfection epilepsy after Theiler's virus infection of C57BL/6 mice. J Neuropathol Exp Neurol 69:1210–1219
Stewart KA, Wilcox KS, Fujinami RS, White HS (2010b) Theiler's virus infection chronically alters seizure susceptibility. Epilepsia 51:1418–1428
Stringer JL (2006) Models available for infection-induced seizures. In: Pitkanen A, Schwartzkroin PA, Moshe SL (eds) Models of seizures and epilepsy. Elsevier Academic Press, New York, pp 521–526
Traub RD, Wong RK (1982) Cellular mechanism of neuronal synchronization in epilepsy. Science 216:745–747
Wong RK, Traub RD (1983) Synchronized burst discharge in disinhibited hippocampal slice. I. Initiation in CA2-CA3 region. J Neurophysiol 49:442–458
Wu HM, Huang CC, Chen SH, Liang YC, Tsai JJ, Hsieh CL, Hsu KS (2003) Herpes simplex virus type 1 inoculation enhances hippocampal excitability and seizure susceptibility in mice. Eur J Neurosci 18:3294–3304
Wuarin JP, Dudek FE (2001) Excitatory synaptic input to granule cells increases with time after kainate treatment. J Neurophysiol 85:1067–1077
Yamashita N, Morishima T (2005) HHV-6 and seizures. Herpes 12:46–49
Ylinen A, Bragin A, Nadasdy Z, Jando G, Szabo I, Sik A, Buzsaki G (1995) Sharp wave-associated high-frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms. J Neurosci 15:30–46
Acknowledgments
The authors would like to acknowledge the financial support of Robert and Joyce Rice (Salt Lake City, UT), the Margolis Foundation (Salt Lake City, UT), Citizens United for Research in Epilepsy (HSW and RSF), National Institutes of Health 1R01NS065714 (RSF), the Epilepsy Foundation (KAS), and the American Epilepsy Society (KAS), the Dumke Foundation (KSW) and National Institutes of Health R21 NS41673 (KSW). We would also like to thank Dr. Alla Borisyuk for advice about data analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Smeal, R.M., Stewart, KA., Iacob, E. et al. The activity within the CA3 excitatory network during Theiler's virus encephalitis is distinct from that observed during chronic epilepsy. J. Neurovirol. 18, 30–44 (2012). https://doi.org/10.1007/s13365-012-0082-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-012-0082-5