
Abstract
Actin filaments (F-actin) are the major structural component of excitatory synapses, being present in presynaptic terminals and in postsynaptic dendritic spines. In the last decade, it has been appreciated that actin dynamics, the assembly and disassembly of F-actin, is crucial not only for the structure of excitatory synapses, but also for pre- and postsynaptic physiology. Hence, regulators of actin dynamics take a central role in mediating neurotransmitter release, synaptic plasticity, and ultimately behavior. Actin depolymerizing proteins of the ADF/cofilin family are essential regulators of actin dynamics, and a number of recent studies highlighted their crucial functions in excitatory synapses. In dendritic spines, ADF/cofilin activity is required for spine enlargement during initial long-term potentiation (LTP), but needs to be switched off during spine stabilization and LTP consolidation. Conversely, active ADF/cofilin is needed for spine pruning during long-term depression (LTD). Moreover, ADF/cofilin controls activity-induced synaptic availability of glutamate receptors, and exocytosis of synaptic vesicles. These data show that the activity of ADF/cofilin in synapses needs to be spatially and temporally tightly controlled through several upstream regulatory pathways, which have been identified recently. Hence, ADF/cofilin-controlled actin dynamics emerged as a critical and central regulator of synapse physiology. In this review, I will summarize and discuss our current knowledge on the roles of ADF/cofilin in synapse physiology and behavior, by focusing on excitatory synapses of the mammalian central nervous system.
Similar content being viewed by others

Introduction
Actin is highly abundant at excitatory synapses, and actin filaments (F-actin) are present in presynaptic terminals and in postsynaptic dendritic spines (for review: [1]). As the major structural postsynaptic component, F-actin controls spine morphology, and assembly and disassembly of F-actin (termed actin dynamics) drive spine morphological changes that occur in response to synaptic activity: spine enlargement is associated with enhanced synaptic activity and depends on F-actin assembly and stabilization, while spine shrinkage and elimination induced by low-frequency stimulation requires F-actin disassembly [2–5]. Although less well-understood, the sub-synaptic distribution of F-actin emphasizes a role for actin dynamics in presynaptic processes such as synaptic vesicle recruitment and exocytosis, or endocytosis (for review: [6]). Hence, actin-binding proteins (ABP) that control F-actin assembly and/or disassembly have moved into the focus as critical regulators of neurotransmitter release, synaptic plasticity, and ultimately behavior.
ADF/cofilin has been recognized as a key regulator of actin dynamics that controls F-actin assembly and disassembly in a complex, concentration-dependent manner (for review: [7]), and studies on gene-targeted mice linked ADF/cofilin to neuronal actin dynamics relevant for central nervous system development [8–10]. At low concentrations, ADF/cofilin promotes F-actin disassembly by accelerating the dissociation of monomeric actin (G-actin) from the filaments’ minus ends and by severing F-actin [11, 12]. Filament severing can either result in a net assembly or disassembly of F-actin, depending on the local G-actin concentration and on the activity of actin polymerizing proteins [13]. Conversely, at high concentrations, ADF/cofilin can promote F-actin assembly by nucleating new and by stabilizing preexisting filaments [14]. Actin binding of ADF/cofilin is controlled via phosphorylation (inactivation) and dephosphorylation (activation) of a conserved serine residue at position 3 (Ser3) (for review: [15]), and LIM kinases (LIMK) and slingshot phosphatases have been recognized as potent regulators [16, 17]. In mammals, the ADF/cofilin family comprises three proteins: cofilin1 (non-muscle cofilin, n-cofilin), cofilin2 (muscle cofilin, m-cofilin), and ADF (actin depolymerizing factor, destrin). Cofilin1 and ADF are broadly expressed in the adult brain [9, 18], they are both present at excitatory synapses [18, 19], and their synaptic function has been investigated in numerous recent studies. These studies established that (1) ADF/cofilin is critical for spine morphology [20–23], (2) ADF/cofilin controls functional and structural aspects of synaptic plasticity [4, 21–23], (3) cofilin1 and ADF have redundant and overlapping functions in neurotransmitter release [24, 25], and (4) genetic inactivation of cofilin1 and ADF impairs learning and causes severe behavioral abnormalities [21, 25, 26]. Moreover, a number of regulatory pathways upstream of ADF/cofilin in excitatory synapses have been identified. Together, these studies highlight the relevance of ADF/cofilin for synapse physiology and behavior, and identified ADF/cofilin as a key regulator of synaptic plasticity.
Function of cofilin1 and ADF in structural plasticity
A role of cofilin1 in dendritic spine morphology and structural plasticity has been postulated from its accumulation in sub-spinous regions that contain a dynamic F-actin network responsible for spine morphological changes during synaptic plasticity [19, 27]. Indeed, by exploiting short synthetic peptides mimicking active or inactive ADF/cofilin, early studies implicated ADF/cofilin in spine shrinkage associated with long-term depression (LTD), and ADF/cofilin inactivation in spine enlargement associated with long-term potentiation (LTP) [4, 28, 29]. Moreover, enhanced ADF/cofilin activity upon genetic ablation of its negative regulator LIMK1 resulted in an abnormal thin spine profile [30], while spine enlargement upon LTP induction or during learning was associated with elevated synaptic levels of phosphorylated (inactive) ADF/cofilin [31, 32]. More direct proofs for the role of ADF/cofilin in synaptic plasticity have been provided by genetic approaches: while overexpression of constitutively inactive cofilin1 in hippocampal cultures resulted in more mature spines and elevated spine densities [22, 33]; overexpression of constitutive active cofilin1 induced an immature spine profile, similar to low-frequency stimulation that induce N-methyl-d-aspartic acid receptor (NMDAR)-mediated LTD [34, 35]. In line with these studies, acute, small interfering RNA (siRNA)-mediated knockdown of cofilin1 in hippocampal neurons caused longer dendritic protrusions and a reduced number of thin spines [20], and spine length and spine head width were increased in primary hippocampal cultures derived from cofilin1 mutant mice [21]. The latter study also revealed spine enlargement and an increased spine density in hippocampal slices from cofilin1 mutant mice, thereby providing evidence for a role of cofilin1 in spine morphology in vivo [21]. Notably, these two studies reported impaired actin dynamics in dendritic spines (reduced actin turnover by fluorescent recovery after photobleaching) and an increase in the F/G-actin ratio in synaptosomes (isolated nerve terminals generated from fresh brain tissue [36]) from cofilin1 mutants, suggesting that cofilin1 controls spine morphology via an actin-dependent mechanism [20, 21]. In summary, these studies are consistent with a model in which cofilin1 inactivation is required for spine enlargement and stabilization during LTP, while cofilin1 activation can drive F-actin disassembly and spine shrinkage during LTD.
This model has been expanded recently by demonstrating that dephosphorylated (active) cofilin1 rapidly moves into dendritic spines during the initial phase of structural LTP, resulting in a strong and persistent synaptic enrichment which is required for spine enlargement [23]. Hence, this study revealed a novel and crucial role for cofilin1 in promoting spine enlargement, and it paints a more detailed picture on its function during structural plasticity: on the one hand, cofilin1 promotes F-actin assembly during LTP, and on the other hand, it is required for F-actin disassembly and spine shrinkage during LTD. As described in the introduction, such a dual function of cofilin1 in F-actin assembly/disassembly is in good agreement with its biochemical properties, which depends on the local ratio of cofilin1 and actin [7, 14]. Compared to actin, but also to other important regulators of synaptic actin dynamics such as Arp2/3, profilin2 or drebrin [37–42], the accumulation of cofilin1 in dendritic spines during initial LTP was the strongest [23]. Hence, the cofilin1-actin ratio increased, and this increase could be sufficient to promote cofilin1-dependent stabilization of preexisting F-actin and/or F-actin assembly, while under basal conditions or during LTD, a lower cofilin1-actin ratio would rather promote F-actin disassembly. Indeed, Förster resonance energy transfer-based experiments revealed a peak in cofilin1’s interaction with F-actin during initial LTP [23]. Further findings in this study suggest that cofilin1, in concert with Arp2/3, promotes F-actin assembly and spine enlargement, and that these structural changes become consolidated by actin stabilizers thereafter. Interestingly, consolidation of spine enlargement required cofilin1 inactivation, which was paralleled by its gradual migration towards sub-spinous regions with a more stable actin cytoskeleton [23]. Such a temporal sequence of cofilin1 activation and inactivation during LTP is in good agreement with a previous study which reported that phosphorylation (inactivation) of ADF/cofilin in dendritic spines of rat hippocampal slices was not detectable until at least 2 min after LTP induction [31]. Together, cofilin1 acts as a bidirectional regulator of spine structural plasticity that is crucial for both spine enlargement and shrinkage (Fig. 1).

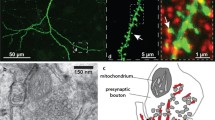
A schematic diagram summarizing the functions of cofilin1 in dendritic spines. Under basal conditions (b), before induction of either LTP or LTD, cofilin1 is present in dendritic spines [23]. Electron microscopy revealed an enrichment of cofiin1 in the spine head periphery [19], are sub-spinous region with a more dynamic actin network compared to the spine head core or the spine neck [27]. Under non-stimulated conditions, cofilin1 controls lateral diffusion of AMPAR, presumably via an actin-dependent mechanism [21]. Upon LTP induction (c), more cofilin1 moves into dendritic spines and this translocation is required for actin polymerization and spine enlargement [23]. Moreover, cofilin1-dependent actin dynamics control the synaptic accumulation of AMPAR during LTP [22]. Thereafter, during consolidation of structural changes (d), cofilin1 becomes phosphorylated (inactivated) and accumulates at the center to base sub-region of the spine head [22, 23]. a Upon induction of LTD, cofilin1 moves into dendritic spines [35], and cofilin1-dependent disassembly of F-actin mediates spine shrinkage [4]. Arrows indicate lateral diffusion of AMPAR in the extra-synaptic membrane
Apart from cofilin1, ADF has been located at dendritic spines [18], but, unlike cofilin1 [21], inactivation of ADF in mice did not change the morphology or density of dendritic spines [18]. Contradictory to this, a role of ADF in spine morphology has been observed upon acute, siRNA-mediated knockdown in hippocampal cultures, but has been mentioned just parenthetically in this study without showing experimental data [20]. The most obvious explanation for this discrepancy is compensation of ADF inactivation in mice, which has been corroborated by elevated synaptic cofilin1 levels [18]. Indeed, compared to cofilin1 mutants, spine size was increased in the hippocampus and the striatum of double mutants lacking both ADF and cofilin1 [24, 25], thereby demonstrating a role of ADF in spine morphology and overlapping, and redundant function for ADF and cofilin1 in dendritic spines. In line with these data, compound short hairpin RNA-mediated knockdown of cofilin1 and ADF, but not of cofilin1 alone, compromised spine enlargement during initial LTP in hippocampal slices, and this effect was rescued by the re-expression of cofilin1 [23]. Together, these studies suggest that cofilin1 is the predominant ADF/cofilin form in dendritic spine morphology and structural plasticity. ADF is functionally relevant for spine morphology as well, but its systemic inactivation in mice is rescued by compensational up-regulation of cofilin1 [18].
Regulation of ADF/cofilin activity in structural plasticity
As introduced above, the interaction of ADF/cofilin with actin is inhibited upon Ser3 phosphorylation, while Ser3 dephosphorylation enables actin binding [15]. Several tools are established to determine ADF/cofilin phosphorylation levels (pan-antibodies that specifically recognize all phosphorylated ADF/cofilin) [30–32, 43, 44], to interfere with the phosphorylation state of endogenous ADF/cofilin proteins (short synthetic peptides identical to the N-terminal sequence of ADF/cofilin that contain either phosphorylated or dephosphorylated Ser3 and act as competitive inhibitors) [4, 22, 28], or to overexpress either constitutively active or inactive full-length protein, in which Ser3 is mutated to alanine or glutamic acid, respectively [22, 23]. These tools have been extensively used in the past years to identify signaling cascades that act upstream of ADF/cofilin in structural plasticity.
Mechanisms that control ADF/cofilin activity during LTP
NMDAR stimulations that increase synaptic F-actin content, stabilize and enlarge dendritic spines and ultimately induce LTP have been associated with enhanced levels of ADF/cofilin phosphorylation (inactivation) [28, 31]. There is good evidence that p21-activated kinases (PAK) and their downstream effector LIMK1 promote ADF/cofilin inactivation during LTP [31]. Consequently, genetic ablation of either PAK or LIMK1 increased ADF/cofilin activity, altered spine morphology and density, and impaired synaptic plasticity [30, 43, 44]. PAK is a well-known effector of the Rho GTPases Cdc42 and Rac [45], which both have been implicated in spine morphology and synaptic plasticity [46, 47]. It is conceivable that deregulation of ADF/cofilin activity contributes to the synaptic defects found in Rac and Cdc42 mutant mice [48, 49]. Two other Rho GTPases, namely RhoA and RhoB, are relevant for spine morphology and synaptic plasticity, and both have been shown to mediate ADF/cofilin inactivation in dendritic spines [34, 50]. Stabilization of mature spines depends on signaling through the receptor tyrosine kinase EphB2 and focal adhesion kinase (FAK) which mediate ADF/cofilin inactivation via a pathway that includes RhoA, RhoA kinases (ROCK) and LIMK1 [34]. Consistent with a role of ROCK in controlling ADF/cofilin activity in dendritic spines, inactivation of ROCK2 resulted in elevated ADF/cofilin activity, reduced synaptic F-actin content, an immature spine profile, and impaired LTP [51]. Moreover, the releasable factor adenosine blocks LTP-induced ADF/cofilin inactivation and actin polymerization within dendritic spines via suppressing RhoA activity [52]. Interestingly, the RhoA-ROCK-LIMK-ADF/cofilin pathway is activated upon estrogen treatment, and it has been speculated that the acute effects of female sex hormones on synaptic function, LTP and learning are mediated via this pathway [53]. Additionally, RhoB- and LIMK1-mediated inactivation of ADF/cofilin has been shown recently, and this pathway is relevant specifically for the early, but not the late phase of LTP [50]. In RhoB-deficient mice, basal levels of ADF/cofilin phosphorylation were unchanged, but the LTP-induced phosphorylation (inactivation) of ADF/cofilin did not exist, resulting in larger spines [50]. Together, several Rho GTPase-dependent signaling pathways have been identified to control ADF/cofilin activity during LTP. Whether these pathways are involved in ADF/cofilin inactivation downstream of the extracellular matrix metalloproteinase-9 (MMP-9), which becomes activated during LTP [54] and controls activity-induced spine enlargement by inactivating ADF/cofilin via a mechanism that includes β1-containing integrin receptors [55], remains unknown. However, a functional link between integrins and Rho GTPases is well-established [56]. Rho GTPases have been also recognized as downstream effectors of the tumor suppressors, TSC1 and TSC2 [57]. Loss of either TSC1 or TSC2 resulted in enlarged spines, and functional data revealed that these morphological changes require ADF/cofilin inactivation [58].
Mechanisms that control ADF/cofilin activity during LTD
While all aforementioned studies unraveled molecular mechanisms that inactivate ADF/cofilin in dendritic spines during LTP, calcineurin (protein phosphatase 2B) has been recognized as an important activator of ADF/cofilin during LTD [4, 33, 35, 59]. In these studies, calcineurin has been activated by low-frequency stimulation that activates NMDAR [4, 35], by physiological concentrations of naturally secreted dimers and trimers of amyloid-β-protein (Aβ) [33], or by the EphA4 receptor tyrosine kinase [59]. Notably, Aβ-induced synapse loss in hippocampal slices was rescued by overexpressing constitutively inactive cofilin1 [33], thereby suggesting a role for ADF/cofilin in the pathology of Alzheimer’s disease. Calcineurin has been identified as an activator of the ADF/cofilin phosphatase slingshot [60], and this pathway activates ADF/cofilin downstream of EphA4-mediated spine retraction [59]. Interestingly, signaling through EphA4 and its downstream effector phospholipase Cγ1 has been implicated in the release of ADF/cofilin from the plasma membrane of hippocampal neurons [61], suggesting a complex, EphA4-mediated regulation of ADF/cofilin that affects both its localization and its phosphorylation state. Apart from EphA4 and Cγ1, the scaffolding protein β-arrestin-2 has been unraveled as a regulator of cofilin1 localization [35], since cofilin1’s recruitment into dendritic spines during LTD was blocked in β-arrestin-2-deficient neurons and, as a consequence, spine shrinkage was impaired. Together, a multitude of signaling cascades that act upstream of activity-induced ADF/cofilin phosphorylation/dephosphorylation and/or localization has been identified in the past years. These studies identified ADF/cofilin as a central hub for structural plasticity where multiple signaling pathways merge and are translated into changes of the synaptic actin cytoskeleton.
Function of ADF/cofilin in synaptic AMPA receptor accumulation
Apart from its role in structural plasticity, several recent studies established a novel postsynaptic function for cofilin1 in trafficking and synaptic accumulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) [21, 22, 62]. Initially, lateral diffusion of the quantum dot-labeled AMPAR subunit GluR2 was shown to be compromised in the extrasynaptic, but not in the synaptic compartment of hippocampal neurons from cofilin1 mutants, and, presumably as a consequence of impaired lateral diffusion, spatial confinement of GluR2 was increased [21]. Similarly, F-actin stabilization by jasplakinolide reduced the mobility of extrasynaptic, but not of synaptic GluR2, while F-actin destabilization by latrunculin A increased GluR2 mobility independently of its location (same study). These findings led to the suggestions that cofilin1 controls AMPAR mobility via an actin-dependent mechanism and that cofilin1 controls synaptic strength via regulating AMPAR mobility, independently of its function in structural plasticity [21]. Indeed, this has been demonstrated in an independent study shortly after: during chemical LTP, cofilin1-dependent synaptic accumulation of the AMPAR subunit GluR1 precedes spine enlargement which, on the other hand, depends on cofilin1 inactivation [22]. A role of cofilin1 in AMPAR accumulation during synaptic plasticity is in very good agreement with the reduced LTP in cofilin1 mutant mice [21], and has been corroborated recently by the finding that manipulation of ADF/cofilin activity in the rat infralimbic cortex altered synaptic accumulation of GluR1 and GluR2 in a bidirectional manner, independently of spine morphological changes [62]. These studies, together with those discussed above (see section: Function of cofilin1 and ADF in structural plasticity), suggest a temporal sequence of ADF/cofilin activation and inactivation during LTP, relevant for initial spine enlargement and synaptic AMPAR accumulation (both dependent on ADF/cofilin activation), followed by consolidation of structural changes (dependent on ADF/cofilin inactivation). Intriguingly, ADF/cofilin could function as a molecular switch during synaptic plasticity, which allows the decoupling between synaptic strength and spine morphological changes. This model would explain previous findings of reduced spine size, but enhanced synaptic potentiation upon increased ADF/cofilin activity [4, 30], which contrasts that LTP is associated with spine enlargement under non-pathological conditions [2, 3, 28]. Together, ADF/cofilin controls functional aspects of synaptic plasticity and this activity is independent of its role in structural plasticity.
Presynaptic functions of ADF/cofilin
While studies on neuronal cultures established the postsynaptic functions of ADF/cofilin [20, 22, 23], its role in presynaptic mechanisms has just recently been unraveled by the analyses of gene-targeted mice. In double mutants lacking ADF and cofilin1, the number of synaptic vesicles attached to the active zone was increased and consequently synaptic vesicle exocytosis was enhanced [24, 25]. In line with a role of ADF/cofilin in vesicle exocytosis, inactivation of upstream regulators such as RhoB, ROCK2, PAK, LIMK1 or slingshot impaired vesicle exocytosis [30, 44, 50, 51, 63]. Interestingly, very similar to ADF/cofilin mutants, the number of docked vesicles and synaptic vesicle exocytosis was enhanced upon inactivation of profilin2 [39, 64], an actin-binding protein that promotes actin polymerization by accelerating the ATP-ADP exchange on G-actin and by funneling G-actin to F-actin’s plus ends [65]. This was an unexpected finding, since inactivation of ADF/cofilin and profilin2 resulted in strikingly different, almost contra-directional changes in synaptic actin dynamics: a shift towards F-actin under basal conditions and upon depolarization in ADF/cofilin mutants versus a block in depolarization-induced F-actin assembly in profilin2 mutants [24, 39]. Together, these findings led to the suggestion that the presynaptic actin cytoskeleton needs to be highly dynamic to fulfill its function in synaptic vesicle exocytosis. Hence, a dynamic actin cycle, rather than simple net F-actin assembly or disassembly, is the critical determinant of synaptic vesicle exocytosis, which extends the previous view of a barrier function of F-actin [1, 39].
Apart from the elevated numbers of docked vesicles, the vesicle distribution within presynaptic terminals was altered in double mutants lacking ADF and cofilin1: while their relative number adjacent to the active zone was reduced, it was increased in more distant regions [24]. Such changes could be caused by defective vesicle recruitment, which indeed was shown by reduced depolarization-induced glutamate release from synaptosomes of double mutants and by stronger synaptic depletion during 10 Hz stimulation for 2 min [24]. Interestingly, the defects in the distribution, recruitment and exocytosis of synaptic vesicles were evident only in double mutants, but not in ADF or cofilin1 single mutants [18, 21, 24, 25]. Hence, ADF and cofilin1 have the capacity to compensate the others’ inactivation in presynaptic terminals, which is in line with elevated synaptic cofilin1 levels in ADF mutants and, vice versa, elevated synaptic ADF levels in cofilin1 mutants [18, 21]. While cofilin1 emerged as the predominant ADF/cofilin form in dendritic spines, ADF and cofilin1 seem to be equally important for presynaptic physiology (Fig. 2).

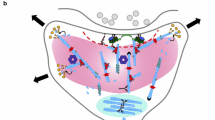
A schematic diagram showing the presynaptic defects in ADF/cofilin mutant mice. Compared to controls (a), the distribution of synaptic vesicles within the presynaptic bouton is changed in double mutants lacking cofilin1 and ADF (b) [24]. Moreover, the number of docked vesicles is increased. Electrophysiological experiments revealed an impairment in the recruitment of synaptic vesicles (indicated by a thinner arrow in the presynaptic terminal of double mutants when compared to controls) and an increase in synaptic vesicle exocytosis [24, 25]. No such defects were noted in cofilin1 or ADF single mutants [18, 21], thereby demonstrating that cofilin1 and ADF have redundant and overlapping functions in presynaptic physiology
Relevance of ADF/cofilin for learning, memory and complex behavior
The functions of ADF/cofilin in neurotransmitter release and synaptic plasticity suggest a crucial role also for behavior. Indeed, as expected from the altered spine morphology and the defects in synaptic plasticity (reduced LTP, absence of LTD, reduced AMPAR trafficking) upon cofilin1 inactivation, mutant mice lacking cofilin1 in principal neurons of the adult telencephalon performed substantially weaker in paradigms of associative learning [21]. Similarly, impaired learning has been reported for mutant mice lacking upstream regulators such as LIMK1, PAK1 and 3, or β-arrestin-2 [30, 35, 44]. A role of ADF/cofilin in spatial learning has been further corroborated by elevated spine numbers with high levels of phosphorylated (inactive) ADF/cofilin in the rat hippocampal CA1 region upon unsupervised learning in an enriched environment [32]. Notably, while long-term spatial memory was impaired in cofilin1 mutants, short-term spatial working memory was fully intact [21, 25]. Thus, cofilin1 mutant mice provide a genetic proof for the dissociation of short- and long-term spatial memory processes, thereby confirming the previously postulated existence of two distinct mechanisms for spatial memory [66]. Recently, increased ADF/cofilin activity in the rat infralimbic cortex has been shown to accelerate extinction of conditioned taste aversive memory via controlling synaptic AMPAR concentrations [62]. Intriguingly, ADF/cofilin-mediated AMPAR trafficking emerged as a central determinant for memory processes, controlling both memory acquisition and extinction [21, 62]. Moreover, apart from the defects in memory acquisition, cofilin1 mutants were less anxious when tested in mildly aversive environments, thereby confirming a role in anxiety that has been postulated from quantitative trait loci of heterogeneous stock mice [26].
ADF is present at excitatory synapses and it shares very similar actin dynamizing activities with cofilin1 [15, 18]. However, gene-targeted systemic ADF mutants performed normal in paradigms of associative learning, and they showed no obvious behavioral abnormalities [18, 25]. Normal learning skills and unaltered behavior was in accordance with unchanged synapse morphology or physiology in these mice [18, 24, 25]. However, by analyzing a mouse strain carrying a spontaneous ADF mutation [67], ADF has been implicated in the development of methamphetamine-induced place preference [68]. Unfortunately, the ADF-dependent mechanism in methamphetamine-conditioning has not been elucidated, and it remains unknown whether synaptic function is altered in these mutants.
Interestingly, compound inactivation of ADF (systemically) and cofilin1 (adult telencephalon) in mice resulted in a deregulation of dopaminergic transmission causative for behavioral abnormalities reminiscent of attention-deficit/hyperactivity disorder (ADHD) [25]. These behavioral abnormalities comprised strongly increased locomotion in accustomed and novel environment, a paradox calming effect of psycho-stimulants including the ADHD-medication methylphenidate, impaired working memory, impulsive traits, and a lack of nesting behavior [25]. Since these abnormalities were not present in ADF or cofilin1 single mutants, they provide compelling evidence for an overlapping function of ADF and cofilin1 in vivo. Hence, double mutants lacking cofilin1 and ADF are a representative example for gene–gene interactions in the patho-physiology of ADHD [69]. In summary, the analyses of mutant mice revealed that cofilin1, and potentially also ADF, is critical for learning and memory. Moreover, ADF/cofilin appears to be relevant for the control of complex behavior. Hence, deregulation of ADF/cofilin activity could cause or contribute to the pathology of human neuropathies, as it has been speculated for Williams’ syndrome [70, 71], a particular form of mental retardation and autism spectrum disorder, or for tuberous sclerosis complex, a dominant hamartomatous disorder that often presents with mental retardation, epilepsy and autism [58, 72].
Concluding remarks and future perspectives
Since its first implication in dendritic spine morphology and structural plasticity in the early 2000 [4, 30], the regulation and function of the ABP ADF/cofilin in dendritic spines has been investigated in a multitude of studies. These studies revealed that the ADF/cofilin family member cofilin1 acts as a bidirectional regulator of spine structural plasticity that is crucial for F-actin assembly during initial LTP which is relevant for spine enlargement [23], and for F-actin disassembly during LTD which is relevant for spine shrinkage and elimination [4, 20–22]. Notably, consolidation of spine enlargement during LTP requires inactivation of cofilin1 [23], thereby suggesting a temporal sequence of cofilin1 activation and inactivation during LTP which is required for initial spine enlargement and consolidation of structural changes, respectively. Hence, cofilin1 activity in dendritic spines needs to be tightly regulated, and several regulatory pathways acting upstream of ADF/cofilin in dendritic spines during LTP and LTD have been identified to date. However, based on the complexity of the signaling cascades that control spine morphological changes during synaptic plasticity, it is very likely that many more molecular pathways involved in the control of ADF/cofilin activity in dendritic spines will be discovered in the near future. Apart from regulating activity-induced spine morphological changes, cofilin1-dependent actin dynamics has been implicated in the synaptic accumulation of the AMPAR subunits GluR1 and GluR2 [21, 22, 62], and, hence, these studies unraveled a novel function of cofilin1 in functional plasticity that is independent from its role in structural plasticity. However, the mechanisms that control cofilin1-dependent AMPAR trafficking largely remained unknown and these mechanisms likely will be identified in future studies. Moreover, future studies will show whether cofilin1 specifically controls the synaptic accumulation of AMPAR subunits during LTP or whether cofilin1-dependent actin dynamics provides a general mechanism that controls the concentration of postsynaptic transmembrane proteins.
Apart from cofilin1, ADF has been located in dendritic spines [18]. However, unlike cofilin1, its synaptic function remains unknown, mainly because ADF has been neglected in most studies on neuronal cultures and because ADF mutant mice do not display any obvious synaptic defect [18]. Nevertheless, compared to cofilin1 single mutants, spines were severely enlarged in double mutants lacking cofilin1 and ADF [24, 25], thereby demonstrating that (1) ADF is functionally relevant for spine morphology, (2) cofilin1 and ADF share overlapping functions in dendritic spines, and (3) cofilin1 and ADF can (partially) compensate for the others inactivation. Although cofilin1 and ADF share similar actin dynamizing activities, in vitro studies on human cofilin1 and ADF revealed quantitative differences between both proteins and demonstrated that cofilin1 is more efficient in actin nucleation and F-actin severing, while ADF is more efficient in G-actin sequestering [73]. Moreover, while phospho-regulation of Ser3 is quite similar for cofilin1 and ADF [15], differences on the transcriptional and post-transcriptional levels have been worked out for ADF and cofilin1 in non-neuronal cells [74, 75]. Although experimental evidences are missing yet, it is conceivable that ADF fulfills a specific function in dendritic spine actin dynamics that, compared to cofilin1, is less obvious and more difficult to decipher.
The analysis of double mutants lacking cofilin1 and ADF not only revealed a function of ADF in dendritic spine morphology, but also demonstrated an overlapping and redundant function for cofilin1 and ADF in presynaptic mechanisms [24, 25]. Electron microscopy and electrophysiology data from double mutants are in good agreement with a role of ADF/cofilin in vesicle recruitment and in vesicle exocytosis. However, more direct experimental evidences for this suggestion are missing, e.g. time lapse experiments to study activity-induced vesicle mobility and exocytosis in ADF/cofilin-deficient neurons. Such experiments should include analysis of the presynaptic actin cytoskeleton to test whether ADF/cofilin control synaptic vesicle recruitment and exocytosis via an actin-dependent mechanisms, as it has been suggested from the strongly increased synaptosomal F-actin levels in double mutant mice [24]. Compared to the ADF/cofilin regulation in dendritic spines, very little is known about the mechanisms upstream of ADF/cofilin in presynaptic boutons. Known ADF/cofilin regulators such as RhoB, ROCK2, PAK, LIMK1 or slingshot have been implicated in presynaptic physiology [30, 44, 50, 51, 63], and it is tempting to speculate that these molecules control ADF/cofilin activity also in presynaptic boutons, which needs to be tested in future studies.
References
Cingolani LA, Goda Y (2008) Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 9:344–356
Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766
Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7:1104–1112
Zhou Q, Homma KJ, Poo MM (2004) Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44:749–757
Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T (2004) Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44:759–767
Rust MB, Maritzen T (2015) Relevance of presynaptic actin dynamics for synapse function and mouse behavior. Exp Cell Res. doi:10.1016/j.yexcr.2014.12.020
Hild G, Kalmar L, Kardos R, Nyitrai M, Bugyi B (2014) The other side of the coin: functional and structural versatility of ADF/cofilins. Eur J Cell Biol 93:238–251
Gurniak CB, Perlas E, Witke W (2005) The actin depolymerizing factor n-cofilin is essential for neural tube morphogenesis and neural crest cell migration. Dev Biol 278:231–241
Bellenchi GC, Gurniak CB, Perlas E, Middei S, Ammassari-Teule M, Witke W (2007) N-cofilin is associated with neuronal migration disorders and cell cycle control in the cerebral cortex. Genes Dev 21:2347–2357
Flynn KC, Hellal F, Neukirchen D, Jacob S, Tahirovic S, Dupraz S, Stern S, Garvalov BK, Gurniak C, Shaw AE, Meyn L, Wedlich-Soldner R, Bamburg JR, Small JV, Witke W, Bradke F (2012) ADF/cofilin-mediated actin retrograde flow directs neurite formation in the developing brain. Neuron 76:1091–1107
Maciver SK, Zot HG, Pollard TD (1991) Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J Cell Biol 115:1611–1620
Blanchoin L, Pollard TD (1999) Mechanism of interaction of Acanthamoeba actophorin (ADF/Cofilin) with actin filaments. J Biol Chem 274:15538–15546
Van Troys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J, Ampe C (2008) Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol 87:649–667
Andrianantoandro E, Pollard TD (2006) Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell 24:13–23
Bernstein BW, Bamburg JR (2010) ADF/cofilin: a functional node in cell biology. Trends Cell Biol 20:187–195
Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K (1998) Cofilin phosphorylation by LIM-kinase 1 and its role in Rac mediated actin reorganization. Nature 393:809–812
Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T (2002) Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108:233–246
Görlich A, Wolf M, Zimmermann AM, Gurniak CB, Al Banchaabouchi M, Sassoe-Pognetto M, Witke W, Friauf E, Rust MB (2011) N-cofilin can compensate for the loss of adf in excitatory synapses. PLoS One 6:26789
Racz B, Weinberg RJ (2006) Spatial organization of cofilin in dendritic spines. Neuroscience 138:447–456
Hotulainen P, Llano O, Smirnov S, Tanhuanpaa K, Faix J, Rivera C, Lappalainen P (2009) Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol 185:323–339
Rust MB, Gurniak CB, Renner M, Vara H, Morando L, Gorlich A, Sassoe-Pognetto M, Banchaabouch MA, Giustetto M, Triller A, Choquet D, Witke W (2010) Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J 29:1889–1902
Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, Zheng JQ (2010) ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci 13:1208–1215
Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M, Hayashi Y (2014) Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82:444–459
Wolf M, Zimmermann AM, Görlich A, Gurniak CB, Sassoè-Pognetto M, Friauf E, Witke W, Rust MB (2014) ADF/cofilin controls synaptic actin dynamics and regulates synaptic vesicle mobilization and exocytosis. Cereb Cortex [Epub ahead of print]
Zimmermann AM, Jene T, Wolf M, Gorlich A, Gurniak CB, Sassoe-Pognetto M, Witke W, Friauf E, Rust MB (2014) Attention-deficit/hyperactivity disorder-like phenotype in a mouse model with impaired actin dynamics. Biol Psychiatry [Epub ahead of print]
Goodson M, Rust MB, Witke W, Bannerman D, Mott R, Ponting CP, Flint J (2012) Cofilin-1: a modulator of anxiety in mice. PLoS Genet 8:e1002970
Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GC, Kasai H (2008) The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 57:719–729
Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K (2003) Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38:447–460
Morishita W, Marie H, Malenka RC (2005) Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci 8:1043–1050
Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z (2002) Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35:121–133
Chen LY, Rex CS, Casale MS, Gall CM, Lynch G (2007) Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci 27:5363–5372
Fedulov V, Rex CS, Simmons DA, Palmer L, Gall CM, Lynch G (2007) Evidence that long-term potentiation occurs within individual hippocampal synapses during learning. J Neurosci 27:8031–8039
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27:2866–2875
Shi Y, Pontrello CG, DeFea KA, Reichardt LF, Ethell IM (2009) Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J Neurosci 29:8129–8142
Pontrello CG, Sun MY, Lin A, Fiacco TA, DeFea KA, Ethell IM (2012) Cofilin under control of beta-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci USA 109:E442–E451
Dunkley PR, Jarvie PE, Robinson PJ (2008) A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc 3:1718–1728
Hayashi K, Shirao T (1999) Change in the shape of dendritic spines caused by overexpression of drebrin in cultured cortical neurons. J Neurosci 19:3918–3925
Ackermann M, Matus A (2003) Activity-induced targeting of profilin and stabilization of dendritic spine morphology. Nat Neurosci 6:1194–1200
Pilo Boyl P, Di Nardo A, Mulle C, Sassoe-Pognetto M, Panzanelli P, Mele A, Kneussel M, Costantini V, Perlas E, Massimi M, Vara H, Giustetto M, Witke W (2007) Profilin2 contributes to synaptic vesicle exocytosis, neuronal excitability, and novelty-seeking behavior. EMBO J 26:2991–3002
Ivanov A, Esclapez M, Pellegrino C, Shirao T, Ferhat L (2009) Drebrin A regulates dendritic spine plasticity and synaptic function in mature cultured hippocampal neurons. J Cell Sci 122:524–534
Nakamura Y, Wood CL, Patton AP, Jaafari N, Henley JM, Mellor JR, Hanley JG (2011) PICK1 inhibition of the Arp2/3 complex controls dendritic spine size and synaptic plasticity. EMBO J 30:719–730
Kim IH, Racz B, Wang H, Burianek L, Weinberg R, Yasuda R, Wetsel WC, Soderling SH (2013) Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J Neurosci 33:6081–6092
Asrar A, Meng Y, Zhou Z, Todorovski Z, Huang WW, Jia Z (2009) Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 56:73–80
Huang W, Zhou Z, Asrar S, Henkelman M, Xie W, Jia Z (2011) p21-Activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Mol Cell Biol 31:388–403
Rane CK, Minden A (2014) P21 activated kinases: structure, regulation, and functions. Small GTPases. doi:10.4161/sgtp.28003
Irie F, Yamaguchi Z (2002) EphB receptors regulate dendritic spine development via intersection, Cdc42 and N-WASP. Nat Neurosci 5:1117–1118
Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME (2005) The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 45:525–538
Haditsch U, Leone DP, Farinelli M, Chrostek-Grashoff A, Brakebusch C, Mansuy IM, McConnell SK, Palmer TD (2009) A central role for the small GTPase Rac1 in hippocampal plasticity and spatial learning and memory. Mol Cell Neurosci 41:409–419
Kim IH, Wang H, Soderling SH, Yasuda R (2014) Loss of Cdc42 leads to defects in synaptic plasticity and remote memory recall. eLife 8:3
McNair K, Spike R, Guilding C, Prendergast GC, Stone TW, Cobb SR, Morris BJ (2010) A role for RhoB in synaptic plasticity and the regulation of neuronal morphology. J Neurosci 30:3508–3517
Zhou Z, Meng Y, Asrar S, Todorovski Z, Jia Z (2009) A critical role of Rho-kinase ROCK2 in the regulation of spine and synaptic function. Neuropharmacology 56:81–89
Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G (2009) Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol 186:85–97
Kramar EA, Chen LY, Brandon NJ, Rex CS, Liu F, Gall CM, Lynch G (2009) Cytoskeletal changes underlie estrogen’s acute effects on synaptic transmission and plasticity. J Neurosci 29:12982–12993
Nagy V, Bozdagi O, Matynia A, Balcerzyk M, Okulski P, Dzwonek J, Costa RM, Silva AJ, Kaczmarek L, Huntley GW (2006) Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J Neurosci 26:1923–1934
Wang XB, Bozdagi O, Nikitczuk JS, Zhai ZW, Zhou Q, Huntley GW (2008) Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc Natl Acad Sci USA 105:19520–19525
Legate KR, Fassler R (2009) Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J Cell Sci 122:187–198
Lamb RF, Roy C, Diefenbach TJ, Vinters HV, Johnson MM, Jay DG, Hall A (2000) The TSC1 tumour suppressor hamartin regulates cell adhesion through ERM proteins and the GTPase Rho. Nat Cell Biol 2:281–287
Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL (2005) Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8:1727–1734
Zhou L, Jones EV, Murai KK (2012) EphA signaling promotes actin-based dendritic spine remodeling through slingshot phosphatase. J Biol Chem 287:9346–9359
Wang Y, Shibasaki F, Mizuno K (2005) Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J Biol Chem 280:12683–12689
Zhou L, Martinez SJ, Haber M, Jones EV, Bouvier Doucet G, Corera ET, Fon EA, Zisch AH, Murai KK (2007) EphA4 signaling regulates phospholipase Cgamma1 activation, cofilin membrane association, and dendritic spine morphology. J Neurosci 27:5127–5138
Wang Y, Dong Q, Xu XF, Feng X, Xin J, Wang DD, Yu H, Tian T, Chen ZY (2013) Phosphorylation of cofilin regulates extinction of conditioned aversive memory via AMPAR trafficking. J Neurosci 33:6423–6433
Yuen EY, Liu W, Kafri T, van Praag H, Yan Z (2010) Regulation of AMPA receptor channels and synaptic plasticity by cofilin phosphatase Slingshot in cortical neurons. J Physiol 588:2361–2371
Waites CL, Leal-Ortiz SA, Andlauer TF, Sigrist SJ, Garner CC (2011) Piccolo regulates the dynamic assembly of presynaptic f-actin. J Neurosci 31:14250–14263
Witke W (2004) The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol 14:461–469
Bannerman DM, Sprengel R (2010) Multiple memory mechanisms? The long and the short of it. EMBO J 29:1790–1791
Ikeda S, Cunningham LA, Boggess D, Hawes N, Hobson CD, Sundberg LP, Naggert JK, Smith RS, Nishina PM (2003) Aberrant actin cytoskeleton leads to accelerated proliferation of corneal epithelial cells in mice deficient for destrin (actin depolymerizing factor). Hum Mol Genet 12:1029–1037
Shibasaki M, Mizuno K, Kurokawa K, Suzuki T, Ohkuma S (2011) Role of actin depolymerizing factor in the development of methamphetamine-induced place preference in mice. Eur J Pharmacol 671:70–78
Itohara S, Kobayashi Y, Nakashiba T (2015) Genetic factors underlying attention and impulsivity: mouse models of attention-deficit/hyperactivity disorder. Curr Opin Behav Sci 2:46–51
Frangiskakis JM, Ewart AK, Morris CA, Mervis CB, Bertrand J, Robinson BF, Klein BP, Ensing GJ, Everett LA, Green ED, Proschel C, Gutowski NJ, Noble M, Atkinson DL, Odelberg SJ, Keating MT (1996) LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell 86:59–69
Hoogenraad CC, Akhmanova A, Galjart N, De Zeeuw CI (2004) LIMK1 and CLIP-115: linking cytoskeletal defects to Williams syndrome. BioEssays 26:141–150
Gomez MR, Sampson JR, Whittemore VH (1999) Tuberous sclerosis complex. Oxford Univ Press, New York
Yeoh S, Pope B, Mannherz HG, Weeds A (2002) Determining the differences in actin binding by human ADF and cofilin. J Mol Biol 315:911–925
Yoo Y, Ho HJ, Wang C, Guan JL (2010) Tyrosine phosphorylation of cofilin at Y68 by v-Src leads to its degradation through ubiquitin-proteasome pathway. Oncogene 29:263–272
Minamide LS, Painter WB, Schevzov G, Gunning P, Bamburg JR (1997) Differential regulation of actin depolymerizing factor and cofilin in response to alterations in the actin monomer pool. J Biol Chem 272:8303–8309
Acknowledgments
I thank Dr. Walter Witke for discussion on the manuscript. This work was supported by a Research grant (24/2014 MR) of the University Medical Center Giessen and Marburg (UKGM).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rust, M.B. ADF/cofilin: a crucial regulator of synapse physiology and behavior. Cell. Mol. Life Sci. 72, 3521–3529 (2015). https://doi.org/10.1007/s00018-015-1941-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-1941-z