

Abstract
The V-ATPase is a highly conserved enzymatic complex that ensures appropriate levels of organelle acidification in virtually all eukaryotic cells. While the general mechanisms of this proton pump have been well studied, little is known about the specific regulations of neuronal V-ATPase. Here, we studied CG31030, a previously uncharacterized Drosophila protein predicted from its sequence homology to be part of the V-ATPase family. In contrast to its ortholog ATP6AP1/VhaAC45 which is ubiquitous, we observed that CG31030 expression is apparently restricted to all neurons, and using CRISPR/Cas9-mediated gene tagging, that it is mainly addressed to synaptic terminals. In addition, we observed that CG31030 is essential for fly survival and that this protein co-immunoprecipitates with identified V-ATPase subunits, and in particular ATP6AP2. Using a genetically-encoded pH probe (VMAT-pHluorin) and electrophysiological recordings at the larval neuromuscular junction, we show that CG31030 knock-down induces a major defect in synaptic vesicle acidification and a decrease in quantal size, which is the amplitude of the postsynaptic response to the release of a single synaptic vesicle. These defects were associated with severe locomotor impairments. Overall, our data indicate that CG31030, which we renamed VhaAC45-related protein (VhaAC45RP), is a specific regulator of neuronal V-ATPase in Drosophila that is required for proper synaptic vesicle acidification and neurotransmitter release.
- ATP6AP1L/ATP6AP1
- CG31030/VhaAC45RP
- Drosophila melanogaster
- neuronal V-ATPase
- quantal size
- synaptic vesicle acidification
Significance Statement
In this study, we provide evidence that a previously uncharacterized Drosophila protein, CG31030, is necessary for fly survival and expressed specifically in neurons, where it interacts with constitutive and accessory subunits of the V-ATPase. Physiologically, we show that this protein is required for synaptic vesicle acidification and to ensure proper synaptic transmission at the neuromuscular junction. This implies that CG31030, alias VhaAC45RP, is a novel synaptic protein essential to nervous system functioning and neurotransmitter release. This work therefore provides a new step toward a more exhaustive understanding of the regulations of neuronal V-ATPase and their potential repercussions on synaptic transmission and neurologic diseases.
Introduction
Many cellular processes require a specific electrochemical environment for correct functioning, such as posttranslational modifications of proteins in the Golgi apparatus, lysosomal degradation, endosomal ligand-receptor dissociation, or hormone concentration (for review, see Forgac, 2007). Eukaryotic cells use a highly conserved proton pump, called the vacuolar H+-ATPase (V-ATPase), to achieve the adequate level of acidity in different cellular compartments (Saroussi and Nelson, 2009). This large enzymatic complex must be tightly regulated, as it is essential for it to be localized on the right membrane, and to fit the different pH ranges specific to each organelle and cell type.
In neurons, the V-ATPase plays a crucial role at the synapse, being responsible for acidifying synaptic vesicles and thus providing the driving force for neurotransmitter loading (Moriyama et al., 1992). Recently, neuronal V-ATPase has also gained interest in the context of aging and neurodegenerative diseases, as its dysregulation, and resulting impairment of the autophagy-lysosomal pathway, have been linked to several pathologies such as Alzheimer’s and Parkinson’s diseases (Colacurcio and Nixon, 2016; Collins and Forgac, 2020). If the core mechanism of the proton pump is now well understood, the regulations conferring the cell-specific functions of neuronal V-ATPase remain largely unknown, considerably limiting its potential use as a therapeutic target.
The V-ATPase complex is composed of a cytoplasmic domain (V1) and a membrane-bound domain (V0). The V1 domain contains the catalytic unit responsible for ATP hydrolysis. The energy resulting from this reaction powers a rotational molecular motor spanning from V1 to V0, allowing protons to cross membranes through the port contained in V0 (Vasanthakumar and Rubinstein, 2020). The assembly of V1 to V0 is necessary for the pump to function, and reversible dissociation of the two domains has been shown to occur as a way to regulate V-ATPase activity (Collins and Forgac, 2020). Though the core mechanism stays the same, one V-ATPase can differ from another by its composition. In vertebrates, as well as in Drosophila, V0 is made of five subunits (a, c, c’’, d, and e), while V1 contains eight subunits (A–H; Allan et al., 2005). Each subunit can have several paralogs encoded by different genes, and each gene can produce several isoforms, allowing many different possible combinations to form the full V-ATPase complex. These differences of composition can also have regulatory effects on the complex, both on its localization and on its functional properties (Vasanthakumar and Rubinstein, 2020).
The V-ATPase can also be regulated by two accessory subunits, ATP6AP1/Ac45 and ATP6AP2/PRR (Jansen and Martens, 2012). Both proteins are found in the nervous tissue but are also required in other organs, and their mutations have been linked to cognitive impairments as well as systemic symptoms like immunodeficiency or hepatopathy (Jansen et al., 2016; Cannata Serio et al., 2018). These two subunits interact directly with the V0 domain and are believed to promote assembly of the membrane and soluble regions of the V-ATPase complex (Abbas et al., 2020). In addition, two homologs of ATP6AP1 have been described. The first one, called AC45-like protein (AC45LP), was identified in Xenopus where, unlike the ubiquitous AC45/ATP6AP1, it is expressed specifically in kidney and lung (Jansen et al., 2010). The second one, called ATP6AP1-like (ATP6AP1L) or Ac45-related protein (Ac45RP), was recently functionally characterized in mice where it also shows tissue-specificity, its expression being restricted to neurons (Jansen et al., 2021). These studies suggest that the AC45 family is larger than initially expected, and might play an important role in the tissue-specificity of the V-ATPase complex. Drosophila possesses identified orthologs of vertebrate ATP6AP1/Ac45 and ATP6AP2/PRR, named VhaAC45 and ATP6AP2, respectively. These proteins also seem to contribute to assembly of the V-ATPase in fly tissues (Schoonderwoert and Martens, 2002a; Guida et al., 2018).
In this study, we examined the localization and function of CG31030, a novel Drosophila ortholog of both ATP6AP1/Ac45 and ATP6AP1L/Ac45RP, whose characteristic is to be expressed selectively and ubiquitously in neurons. Whereas a complete deficiency of this protein is lethal, we found that partial CG31030 knock-down in larval motoneurons impaired synaptic vesicle acidification, reduced quantal size, which is the amplitude of the postsynaptic response to the release of a single synaptic vesicle, and induced severe locomotion defects. We also report that CG31030 from brain tissue co-immunoprecipitated with V-ATPase subunits of the V0 domain. Overall, our results indicate that CG31030 is a novel accessory subunit of the neuronal V-ATPase that appears to be involved in the regulation of synaptic activity.
Materials and Methods
Drosophila culture and strains
Flies were raised on standard agar-cornmeal-yeast medium, at 25°C in a 12/12 h light/dark cycle. CG31030 RNAi strains and mutants were obtained from the Bloomington Drosophila stock center (BDSC) and the Vienna Drosophila Resource Center (VDRC). Detailed genotypes and references of these lines are provided in Table 1. To construct the UAS-CG31030 strain, the CG31030 cDNA was PCR amplified from the clone RH09162 obtained from the Drosophila Genomics Resource Center, using the following primers with added restriction sites: P1-EcoRI (forward) 5′-CCATCCGAATTCAAAATGCAGCTGATTCTCGT and P2-XhoI (reverse) 5′-TGGCTGCTCGAGATCTATTGGGTTATGAGAGA. The 1160-bp PCR fragment was inserted into pUAST (Brand and Perrimon, 1993), verified by sequencing (GATC Biotech) and sent to BestGene, for P-element transformation by random insertion in w1118 background. A 2d-chromosome insertion of UAS-CG31030 that yielded strong expression of the transgene was used thereafter.
Drosophila strains used in this study
Reverse transcription-coupled PCR and qPCR
Total RNAs were extracted from 20 heads (or 15 thoraces or 15 abdomens) of 8-d-old flies using the QIAzol Lysis reagent (QIAGEN). The Maxima First Strand cDNA Synthesis kit (Thermo Fisher Scientific, K1671) was used with oligo(dT)20 primers to synthesize the cDNAs. Relative quantitative PCR assays were conducted using a LightCycler 480 and the SYBR Green I Master mix (Roche LifeScience), with Act5C as internal control for normalization of mRNA levels. All reactions were performed in triplicate. The specificity of amplification products was assessed by melting curve analyses. The following forward and reverse primers, were used: for CG31030, 5′-GGCTTCGTTGTAGGCCAACAGA and 5′-CACCAGGTATCCCAAGTTCCAGA; for Act5C, 5′-CGTCGACCATGAAGATCAAG and 5′-TTGGAGATCCACATCTGCTG; for ATP6AP2, 5′-ACGATCCCTTCAACCTGGC and 5′-CCTGTCAGCTCGTAGGTCT; for VhaAC45, 5′-TGTCCCAAGTGGAGTTTGCC and 5′-TCAGGCCATTTTCCTCGAAGG; for Vha100-1, 5′-GGCATCCCCATGTTGGACA and 5′-CGGCGTTCTGATTCACCTCC; for VhaAC39-1, 5′-GCAGGCTGACTATCTCAACCT and 5′-GCTCCACAGCATGGTTCCTC; for Vha16-1, 5′-AAGTCTGGTACCGGTATTGC and 5′-CCATGACCACAGGAATGATG. PCR assays were performed using the PrimeStar Max DNA polymerase (Takara) and the following primers: for CG31030-RA/RB, 5′- TGAGGTGTACGACTGCATAGGA and 5′- GAAAGAACTCGATGGCCAAGGT; for CG31030-RA, 5′-CAAGGGATACGGTCGATTGAGCATCAC and 5′- CTTACTCCTGTACGGTGAAGGTCAAC; for CG31030-RB, 5′- CAAGGGATACGGTCGATTGAGCATCAC and 5′- ATGAACATGGCTGCCCGAGATTGTTTGCT.
Protein extraction and Western blotting
A total of 30 Drosophila heads were homogenized in 300-μl RIPA buffer (Sigma-Aldrich) containing protease inhibitors (Roche Diagnostics, Complete Protease Inhibitor, Cocktail), using bead tubes and a Minilys apparatus (Bertin Technologies). Proteins samples and Western blotting were processed as previously described (Issa et al., 2018). Briefly, the extracted proteins were mixed with LDS sample buffer and reducing agent (Invitrogen, NuPAGE), heated at 70°C for 5 min and separated in 4–12% Novex NuPAGE Bis-Tris precast polyacrylamide gels (Life Technologies) following the manufacturer’s protocol in a MOPS-SDS running buffer. A semi-dry transfer was done onto polyvinylidene difluoride membranes (GE Healthcare Hybond P 0.45 μm) using a Hoefer TE77 apparatus. The mouse monoclonal anti-V5 (Thermo Fisher Scientific, R960-25) was used, diluted at 1:500. Immunolabeled bands were revealed by ECL RevelBlOt Intense (Ozyme, OZYB002-1000) as chemiluminescent HRP substrate and digitally acquired using the ImageQuant TL software (GE Healthcare Life Science).
CRISPR/Cas9 gene tagging
The sequence of a V5 tag was inserted in frame after the coding sequence of the CG31030 gene, using a homology-directed repair CRISPR-Cas9 method (see scheme in Fig. 1D). The following guide RNA sequence: 5′-TTCACCGTACAGGAGTAAGG-3′ was cloned into the BbsI site of pCFD3-U6:3-gRNA plasmid (Port et al., 2014; kind gift of Hervé Tricoire, Université de Paris, Paris, France). This plasmid was then injected, at a concentration of 500 ng/μl, with the following single-stranded oligodeoxynucleotide (ssODN) donor repair template: 5′-GTTCGCGCAGCAAACAGTTGACCTTCACCGTACAGGAGTACGCAGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTAGATTCTACGTAAGGAGGTCATAAGTCTCTGATGAACCAATAGATCTCGGGC-3′ (synthetized by Integrated DNA Technology), also at a concentration of 500 ng/μl (the sequence underlined corresponds to the in-frame V5 tag), into nos-cas9 embryos [genotype y1, P(nos-cas9, w+), M(3xP3-RFP.attP)ZH-2A, w*; Port et al., 2014]. An alanine was added before the V5 tag to prevent the creation of a potential tyrosine phosphorylation site. Embryo injections were performed by BestGene. Single F0 flies were crossed over the TM6C(Sb) balancer to establish stable lines. DNA was then extracted from three flies of each of these independent lines, and V5 insertion events were detected by dot blot using a mouse anti-V5 tag monoclonal antibody (Thermo Fisher Scientific, R960-25). Positive strains were outcrossed in a w1118 background and their genomic DNA was sequenced to check for proper in-frame V5 integration in CG31030. One of these equivalent CG31030V5 mutant line was selected for further studies.

Structure of CG31030 compared with its paralog VhaAC45 and construction of CG31030V5. A, Localization of the primers used for RT-PCR analysis of the two predicted CG31030 transcripts (CG31030-RA and CG31030-RB) in Drosophila heads. B, Primers detecting both CG31030-RA (F1-R1, expected amplicon size 288 bp) and CG31030-RB (F1-R1, expected amplicon size 233 bp) only amplify a band corresponding to CG31030-RA (lane 1). CG31030-RA-specific primers (F2-R2) also amplify a fragment of the expected size (485 bp, lane 2), whereas CG31030-RB-specific primers (F3-R3) did not amplify any cDNA fragment (lane 3). C, VhaAC45 and CG31030 both have a signal peptide (SP; orange bars), a transmembrane domain (TM; blue bars) and predicted N-glycosylation sites according to NetNGlyc (http://www.cbs.dtu.dk/services/NetNGlyc/; green diamonds). They also present the pair of cysteine residues characteristic of the AC45 family (red triangles). In addition, VhaAC45 has a predicted furin cleavage site (yellow bar) which is absent in CG31030 according to ProP 1.0 (http://www.cbs.dtu.dk/services/ProP/). D, Construction of the CG31030V5 mutant strain. A 14-amino acid V5 tag (in red) was fused to the C-terminal end of the CG31030 protein by inserting the V5 coding sequence ended by a new stop codon (in blue) in place of the original stop codon (in green) in the CG31030 gene using the CRISPR-Cas9 technology. E, A Western blotting probed with anti-V5 antibody detected two protein bands at an apparent molecular weight of ∼32 kDa in head extracts of CG31030V5 flies (lane 2) and none in the w1118 control sample (lane 1).
Immunohistochemistry
Adult brains of 8-d-old females, or third-instar larva CNS, were dissected in Drosophila Ringer’s solution or hemolymph-like saline solution (HL3; 70 mm NaCl, 5 mm KCl, 1.5 mm CaCl2, 70 mm MgCl2, 10 mm NaHCO3, 115 mm sucrose, 5 mm trehalose, and 5 mm HEPES, with pH adjusted to 7.6), respectively, and fixed in 4% paraformaldehyde (Thermo Fischer Scientific) for 1 h. After three 20 min washes in PBS plus 0.5% Triton X-100 (PBT), brains were blocked in PBT + 2% bovine serum albumin for 2 h. They were then incubated in primary antibodies diluted in blocking solution for 24 h at 4°C. The primary antibodies used were: mouse monoclonal anti-V5 (Thermo Fisher Scientific, R960-25, 1:200) and rat anti-Cadherin-N (CadN; DSHB, DN-Ex #8, 1:20). Brains were then washed three times, for 20 min each, in PBT before being incubated in secondary antibodies for 2 h. Secondary antibodies used were: Alexa Fluor 488 anti-mouse and Alexa Fluor 555 anti-rat (Fisher Scientific, A11029 and A21434, respectively), all diluted at 1:1000. After two 20-min washes in PBT followed by two 20-min washes in PBS, brains were mounted in Prolong Gold Antifade Mountant (Thermo Fisher Scientific, P36930). Imaged were acquired on a Nikon A1R confocal microscope.
For immunostaining of the larval muscles and neuromuscular junctions, the Alexa Fluor 488 Tyramide SuperBoost kit (Thermo Fisher Scientific, B40912) was used to increase the V5 signal that was otherwise faint in this tissue. The working protocol was as recommended by the manufacturer, with anti-V5 diluted to 1:100, then followed by classical immunostaining, as described above, to co-stain for the nerve terminals with an anti-HRP antibody (Jackson ImmunoResearch, 323-005-021, 1:200) and an anti-Vha100-1 (courtesy of Dr. Peter Robin Hiesinger, 1:2000).
Longevity assay
Approximately 110 virgin females from each genotype were collected on their hatching day and placed in clean bottles, with no more than 25 flies per bottle. Flies were transferred in new clean bottles, and survivors were counted, every 2 d for 60 d.
Co-immunoprecipitation coupled to nano liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) analysis
Approximately 200 heads from 8-d-old CG31030V5 and w1118 flies were lysed using glass beads in 500 μl of ice-cold lysis buffer: 50 mm Tris-HCl pH 7.4, 2 mm EDTA, 150 mm NaCl, 0.5% (v/v) IGEPAL CA-630 (Sigma-Aldrich, I3021), 10% (v/v) glycerol, 1 mm PMSF protease inhibitor (Sigma-Aldrich, P-7626), and 1× cOmplete Mini Protease Inhibitor Cocktail (Roche, 11836153001). Samples were left on ice, with occasional gentle agitation, for 30 min before being centrifuged at 12,000 rpm (13,000 × g) at 4°C for 10 min to remove insoluble material; 400 μl of the supernatants were then added to 50 μl of Anti-V5-tag mAb-Magnetic Beads (MBL International, M167-11), that had been previously washed as recommended by the manufacturer. Samples were incubated with gentle agitation at 4°C for 4 h. The supernatants were removed using a magnetic rack, and beads were washed three times with 500 μm ice-cold lysis buffer before being resuspended in 10 μl of milliQ water.
Nano LC-MS/MS was performed at the Proteomics facility of the Institut Jacques Monod (Université de Paris). Proteins on beads were digested overnight at 37°C with trypsin (Promega) in 25 mm NH4HCO3 buffer (0.2 μg trypsin in 20 μl). The resulting peptides were desalted using ZipTip μ-C18 Pipette Tips (Pierce Biotechnology). Eluates were analyzed using either an Orbitrap Fusion or an Orbitrap Q-Exactive Plus, coupled, respectively, to a Nano-LC Proxeon 1200 or a Nano-LC Proxeon 1000, both equipped with an easy spray ion source (Thermo Fisher Scientific).
On the Orbitrap Fusion instrument, peptides were loaded with an online preconcentration method and separated by chromatography using a Pepmap-RSLC C18 column (0.75 × 750 mm, 2 μm, 100 Å) from Thermo Fisher Scientific, equilibrated at 50°C and operated at a flow rate of 300 nl/min. Peptides were eluted by a gradient of solvent A (H2O, 0.1% FA) and solvent B (ACN/H2O 80/20, 0.1% FA). The column was first equilibrated for 5 min with 95% of A, then B was raised to 28% in 105 min and to 40% in 15 min. Finally, the column was washed with 95% B during 20 min and re-equilibrated at 95% A for 10 min. Peptides masses were analyzed in the Orbitrap cell in full ion scan mode, at a resolution of 120,000, a mass range of m/z 350–1550 and an AGC target of 4.105. MS/MS were performed in the top speed 3s mode. Peptides were selected for fragmentation by higher-energy C-trap dissociation (HCD) with a normalized collisional energy of 27% and a dynamic exclusion of 60 s. Fragment masses were measured in an Ion trap in the rapid mode, with and an AGC target of 1.104. Monocharged peptides and unassigned charge states were excluded from the MS/MS acquisition. The maximum ion accumulation times were set to 100 ms for MS and 35 ms for MS/MS acquisitions, respectively.
On the Q-Exactive Plus instrument, peptides were loaded with an online preconcentration method and separated by chromatography using a Pepmap-RSLC C18 column (0.75 × 500 mm, 2 μm, 100 Å) from Thermo Scientific, equilibrated at 50°C and operated at a flow rate of 300 nl/min. Peptides were eluted by a gradient of solvent A (H2O, 0.1% FA) and solvent B (100% ACN, 0.1% FA), the column was first equilibrated 5 min with 95% of A, then B was raised to 35% in 93 min and finally, the column was washed with 80% B during 10 min and re-equilibrated at 95% A during 10 min. Peptides masses were analyzed in the Orbitrap cell in full ion scan mode at a resolution of 70,000 with a mass range of m/z 375–1500 and an AGC target of 3.106. MS/MS were performed in a Top 20 DDA mode. Peptides were selected for fragmentation by HCD with a normalized collisional energy of 27%, and a dynamic exclusion of 30 s. Fragment masses were measured in the Orbitrap cell at a resolution of 17,500, with an AGC target of 2.105. Monocharged peptides and unassigned charge states were excluded from the MS/MS acquisition. The maximum ion accumulation times were set to 50 ms for MS and 45 ms for MS/MS acquisitions, respectively.
Raw data were processed on Proteome Discoverer 2.4 with the mascot node (Mascot version 2.5.1) against the Swissprot/TrEMBL protein database release 2019_12 for Drosophila melanogaster. A maximum of two missed cleavages was authorized. Precursor and fragment mass tolerances were set to, respectively, 7 ppm and 0.5 Da (Orbitrap Fusion) and to 6 ppm and 0.02 Da (Orbitrap Q-exactive Plus). The following posttranslational modifications were included as variable: acetyl (protein N-term), oxidation (M), phosphorylation (STY). Peptide identifications were validated with a 1% FDR (false discovery rate) threshold calculated with the Percolator algorithm. Label-free quantification was done in TOP three abundance calculation mode with pairwise ratio-based calculation and t test (background based) hypothesis test. Only proteins identified in at least one group in two independent experiments were kept in the analysis. Missing values were set to the minimum abundance of the experiment.
VMAT-pHluorin experiments
Third-instar larvae expressing VMAT-pHluorin with or without CG31030 RNAi in motor neurons using the OK371-Gal4 driver, were dissected to expose the body wall muscles in Ca2+-free HL3 saline solution (70 mm NaCl, 5 mm KCl, 70 mm MgCl2, 10 mm NaHCO3, 115 mm sucrose, 5 mm trehalose, and 5 mm HEPES, pH 7.6). Two other solutions were used: an acidic Ca2+-free HL3 saline (pH 5.5) and a neutral HL3-NH4Cl saline (pH 7.6), in which 50 mm NaCl was replaced by 50 mm NH4Cl. After dissection, larvae fillets were allowed to settle in Ca2+-free HL3 saline for 10 min before being scanned a first time directly in a drop of the solution using a Nikon A1R confocal microscope. Ca2+-free HL3 saline was then replaced either by the acidic Ca2+-free HL3 saline for low pH-induced quenching experiments, or by the HL3-NH4Cl saline for pH gradient collapse experiments. In both cases, larval fillets were rinsed three times with the modified solution, and incubated for 3 min, before being scanned a second time, in a drop of modified solution. Quantification was done on Z-projections (set to maximal intensity) of confocal stacks. Using the Fiji software, a fixed threshold was applied to all images to get rid of the background and select only synaptic areas. Percentage of area over the threshold was used as a measure of the signal intensity and ratios of the values obtained in modified solutions over standard HL3, i.e., the second scan over the first scan, were calculated for quantifications.
Larval locomotion assays
Larval locomotion was assessed on an in-house made version of the FIMtable system (Risse et al., 2014). Third-instar larvae were collected and briefly rinsed in water to remove traces of food, before being gently placed on the recording table precoated with a thin layer of 1.2% agar gel. Only four larvae of the same genotype were recorded simultaneously, to avoid collisions between animals, for a period of 2 min with a Basler ace acA2040-25gm camera at 12.5 frames/s. Larvae that burrowed themselves into the agar plate or escaped the arena before the end of recording were excluded from the results. Tracking was done using the FIMtrack software, as described in (Risse et al., 2014). The number of peristaltic waves was computed from the variations in larval area. More precisely, the curves of area variation were first smoothed with a Savitzky–Golay filter to get rid of unwanted noise, then the number of waves was defined as the number of peaks on the curve (which was automatically computed by a custom-made Python script). Stride size was then calculated as the distance traveled by the larva divided by the number of peristaltic waves, while stride duration was defined as the recording time (i.e., 120 s) divided by the number of peristaltic waves.
Electrophysiological recordings and quantal analysis
Third-instar larvae expressing CG31030 RNAi in motor neurons using the OK371-Gal4 driver, and appropriate controls, were dissected to expose the body wall muscles, and the brain removed, in Ca2+-free HL3 saline solution (70 mm NaCl, 5 mm KCl, 70 mm MgCl2, 10 mm NaHCO3, 115 mm sucrose, 5 mm trehalose, and 5 mm HEPES, pH 7.6; Stewart et al., 1994; Cattaert and Birman, 2001). Spontaneous miniature EPSPs (mEPSPs) were recorded in the presence of tetrodotoxin (TTX) 10−6 m, so that no spike could occur, from the ventral longitudinal abdominal muscle 6 in segment A3.
Quantal analysis was performed following the theoretical background described in (Kuno, 1971) and (Castellucci and Kandel, 1974). Distribution histograms of mEPSP size were built from each muscle fiber recording with a 0.01-mV bin size. These histograms provided an estimate of the mean size of a unitary EPSP, since the peaks represent integer multiples of the unitary size. A theoretical distribution was then computed by convolving a binomial distribution, accounting for quantal content (number of quanta released), and a Gaussian distribution, allowing for variations in size of individual quanta. More precisely, the probability for an EPSP to contain
i quanta follows a binomial distribution:
The SD (σ) of each peak depends on the number of quanta it contains. It was calculated as follows:
Endocytosis and exocytosis measurements
We used fluorescent FM dye labeling to monitor synaptic vesicle cycling at the larval neuromuscular junction (Verstreken et al., 2008). Third-instar larvae expressing mCD8::GFP with or without CG31030 RNAi in motor neurons under control of the OK371-Gal4 driver, were dissected in a Ca2+-free HL3 saline solution (as described above), and incubated for 5 min in HL3 solution supplemented with 90 mm KCl, 1.5 mm CaCl2 and 10 μm fluorescent FM4-64 dye (Invitrogen, T13320) final concentrations. Preparations were then washed with Ca2+-free HL3 saline five times, for 2 min each, before being imaged on a Nikon A1R confocal microscope to measure the level of dye loading. Then the fillets were incubated for 2 min in HL3 solution supplemented with 90 mm KCl and 1.5 mm CaCl2 to allow the dye to be unloaded from the synapses. Preparations were then washed with Ca2+-free HL3 saline five times, for 2 min each, before being scanned once again to measure the level of dye unloading. Images were analyzed using the Fiji software. The fluorescence signal of the dye was measured in the synaptic area restricted to the signal of mCD8::GFP. To obtain the percentage of dye that was unloaded, the ratio of unloaded/loaded fluorescence intensities was calculated.
Synapse morphology analysis
Immunostaining was performed as described above in third-instar larvae using an antibody against Discs Large (Dlg; DSHB, catalog #4F3 anti-discs large-s, 1:500). Synaptic boutons were then counted manually on the ventral longitudinal abdominal muscle 6 of the segment A3, and their diameter measured in blind on randomized images using the Fiji software.
Statistical treatment
Statistical analysis was performed with the GraphPad Prism 6 software. The paired Student’s t test was used for comparison of two genotypes, while either paired or unpaired ANOVA, with Dunnett’s post hoc test for multiple comparisons, were used for three genotypes.
Results
Drosophila CG31030 encodes a single protein structurally similar to VhaAC45
Here, we studied the Drosophila gene CG31030, identified in FlyBase as a paralog of CG8029/VhaAC45, and as an ortholog of vertebrate ATP6AP1/AC45 and ATP6AP1L/AC45RP (Thurmond et al., 2019). In FlyBase, CG31030 is predicted to produce two different protein isoforms, CG31030-PA and CG31030-PB, which differ in the sequence of their C-terminal domain, the last 7 C-terminal amino-acids of CG31030-PA being theoretically replaced by a different segment of 46 amino-acids in CG31030-PB. We first investigated the existence of these two isoforms. Using primers framing the 55 bp difference between the predicted CG31030-RA and CG31030-RB transcripts (Fig. 1A), which encode CG31030-PA and CG31030-PB, respectively, we checked which isoform was present in Drosophila heads. Only a fragment with the size expected for CG31030-RA was amplified (Fig. 1B), suggesting that CG31030-PB is not expressed in this tissue. Consistent with this, PCR experiments using CG31030-RB-specific primers did not amplify any cDNA fragment, contrary to experiments using CG31030-RA-specific primers (Fig. 1B). Moreover, among the numerous cDNA clones matching CG31030 listed in FlyBase, either fully sequenced or only sequenced at the end (ESTs), most of which complement both isoforms, several were found to be specific to CG31030-RA. In contrast, none specifically corresponded to CG31030-RB, supporting the hypothesis that among the two predicted protein isoforms, only CG31030-PA is actually expressed in Drosophila. Therefore, we referred to CG31030-PA simply as CG31030 in the present study.
CG31030 and VhaAC45 share 69.9% similarity in amino-acid sequences. Like its paralog, CG31030 is also classified in the V-ATPase family group by the InterPro database (accession Q8IMJ0_DROME; Mitchell et al., 2019). They both have a transmembrane domain, as well as predicted N-glycosylation sites (Fig. 1C). They also present a pair of conserved cysteine residues typical of proteins from the AC45 family. Unlike Drosophila VhaAC45 and vertebrate ATP6AP1/AC45, CG31030 does not possess a potential furin cleavage site (Fig. 1C), similarly to vertebrate ATP6AP1L/AC45RP and Xenopus AC45LP (Jansen et al., 2010, 2021).
Using the CRISPR-Cas9 technology, we inserted a small V5 epitope tag in frame at the 3′ end of the CG31030 gene, thus disrupting the stop codon, to generate the CG31030V5 mutant line (Fig. 1D). A Western blotting on head extracts of CG31030V5 and control flies was then performed to determine the apparent size of the tagged protein. Two distinct bands, close to each other, were detected specifically in the CG31030V5 sample, consistent with the presence of glycosylated and unglycosylated forms of the same protein (Fig. 1E). While the predicted size of CG31030-V5 is 44 kDa, the observed bands corresponded to a size of ∼32 kDa. This could indicate that the protein is actually cleaved, like its paralog VhaAC45, despite the absence of a predicted cleavage site (Fig. 1C). However, it is known that membrane proteins migrate faster on SDS gels, leading to an underestimation of their size (Rath et al., 2009). This could also explain the smaller than expected apparent size of CG31030-V5 on Western blotting.
CG31030 is specifically expressed in neurons and addressed to synaptic areas
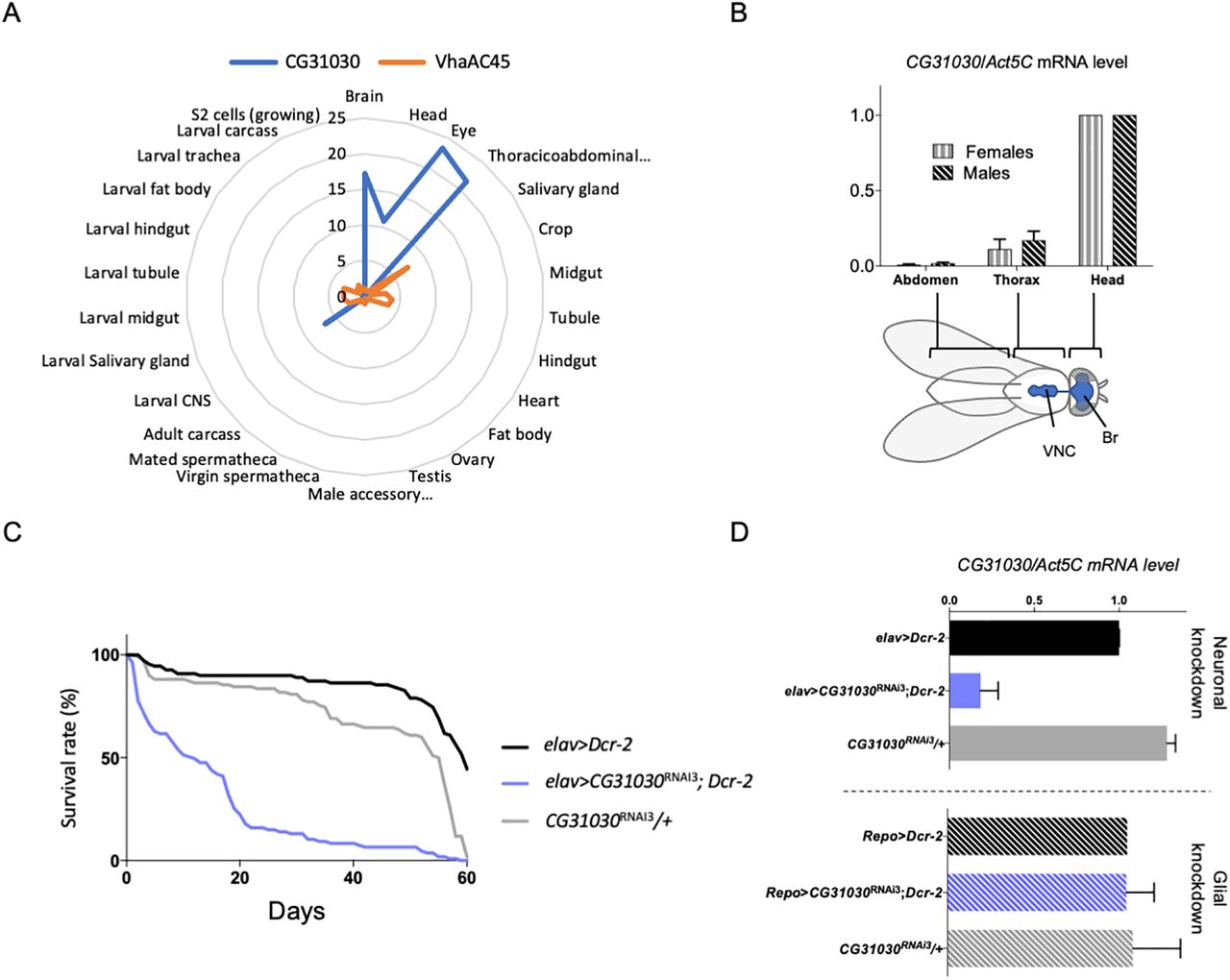
According to FlyAtlas (Chintapalli et al., 2007), VhaAC45 is expressed ubiquitously in Drosophila tissues. In contrast and interestingly, CG31030 seems to be specifically expressed in the nervous system in both larval and adult flies, making it a possible candidate for specific regulation of neuronal V-ATPase (Fig. 2A). To confirm this prediction, we checked by RT-qPCR the repartition of CG31030 transcripts in three parts of the fly body: the head and thorax, which contains the brain and ventral nerve cord (VNC), respectively, and the abdomen, which is relatively poor in nervous tissue. Both females and males showed highest expression in the head, minor expression in the thorax and no detectable expression in the abdomen (Fig. 2B). The expression of CG31030 therefore closely follows the repartition of the nervous system in adult flies.

CG31030 is expressed specifically in neurons. A, Diagram representing expression levels of CG31030 and VhaAC45 in different tissues relative to the whole fly, according to FlyAtlas data (Chintapalli et al., 2007). CG31030 appears to be markedly enriched in the nervous system of larva and adult fly, whereas in contrast VhaAC45 seems to be uniformly expressed in all tissues in these two stages. B, In both males and females, CG31030 mRNA abundance follows the localization of the CNS (shown in blue on the fly sketch), with the highest expression in the head. Br, brain; VNC, ventral nerve cord. Results of three independent experiments. C, Knock-down of CG31030 expression in neurons decreases adult longevity. Pan-neuronal expression of CG31030RNAi3 and Dcr-2 with the elav-Gal4 driver led to a marked shortening of the lifespan of adult flies. Experiment conducted with 105–110 females per genotype. D, Expression of CG31030RNAi3 with Dcr-2 in all neurons using elav-Gal4 decreased CG31030 mRNA level in head by >80%, while expression of the RNAi construct and Dcr-2 in all adult glial cells with repo-Gal4 had not effect. Results of three independent experiments. Mean values with SD are reported on the graphs.
The single-cell RNA-Seq Scope database (Davie et al., 2018) furthermore indicated that, in Drosophila, CG31030 is expressed in all neurons, with few or no expression in glial cells. To verify this, we expressed three different CG31030 RNAi with either the pan-neuronal driver elav-Gal4 or the pan-glial driver repo-Gal4 (see Table 1 for genotypes of the different Drosophila lines used in this study). The expression of CG31030RNAi1 and CG31030RNAi2 was found to be lethal at embryonic and first larval stages, respectively, while CG31030RNAi3 produced viable adults with a shortened lifespan (Fig. 2C), and obvious locomotor impairments. This difference in phenotypes observed with different RNAi constructs could be attributed to a variation in residual levels of the CG31030 protein. RT-qPCR experiments showed that the pan-neuronal expression of CG31030RNAi3 together with the RNAi booster Dicer-2 (Dcr-2) was sufficient to decrease by >80% CG31030 transcripts abundance in extracts from the adult heads (Fig. 2D). On the other hand, glial expression of this RNAi construct with repo-gal4 had no significant effects on CG31030 transcript level (Fig. 2D), confirming that this gene is selectively expressed in neurons. It is interesting to note that both CG31030RNAi1 and CG31030RNAi2 also induced lethal phenotypes at various developmental stages when expressed either with a glutamatergic (VGlut-Gal4) or a cholinergic (Cha-Gal4) neuronal driver (data not shown), in accordance with the RNA-Seq data of the Scope website suggesting a pan-neuronal expression of this gene.
To validate these observations, we placed the available MIMIC allele CG31030MI107 that contains a stop codon inserted in the middle of the gene (Nagarkar-Jaiswal et al., 2015) over the deficiency Df(3R)Exel6214 encompassing CG31030 (Parks et al., 2004). The resulting mutant, likely to be a null, was found to be embryonic lethal, in agreement with the results obtained with two CG31030 RNAi lines. Remarkably, re-expressing the gene selectively in neurons, using a UAS-CG31030 construct driven by elav-Gal4, was sufficient to rescue this lethality, producing viable and fertile adults with no obvious behavioral defects (Table 2). These results convincingly indicate that CG31030 is an essential gene whose expression appears to be specifically required in neurons.
Pan-neuronal expression of CG31030 rescues the embryonic lethality of CG31030-deficient flies up to the adult stage
Next, we studied the cellular localization of this protein by performing immunostaining on CG31030V5 flies with an anti-V5 tag antibody. We observed that the general expression pattern of CG31030 in adult brain (Fig. 3A) and larval CNS (Fig. 3B) was widespread in the neuropile and quite similar to that of the synaptic marker CadN, consistent with a predominantly synaptic localization. No specific signal was detected in a control w1118 line that does not contain the V5-tagged protein (Fig. 3C). Neuronal cell bodies were also faintly marked with the V5 antibody in the CG31030V5 line, both in adult and larva, and a few of them displayed a signal intensity comparable to that of the synaptic areas (Fig. 3A,B, arrowheads). The synapse-containing neuropile areas (like the antennal lobes) were not as sharply defined as with the CadN antibody, suggesting that axons, like most cell bodies, also contains low levels of CG31030. Finally, co-immunostaining of larval body muscles wall with anti-horseradish peroxidase (HRP) antibodies, a marker of Drosophila neurons (Jan and Jan, 1982), showed precise co-localization with the V5 signal, indicating that CG31030 is addressed to synaptic boutons at the larval neuromuscular junction (Fig. 3D). The lack of V5 immunostaining at the neuromuscular junction of control w1118 flies is shown below in Figure 4D.

CG31030 is addressed to synaptic areas. A, B, Anti-V5 immunostaining in the CG31030V5 strain revealed that CG31030 is mainly addressed to synaptic areas in the adult brain (A) and larval CNS (B), as indicated by its colocalization with the presynaptic marker CadN. Green fluorescence outside synaptic areas reveal that cell bodies are also faintly marked by the V5 antibody, with a few of them showing a bright signal (arrowheads). OL, optic lobe; AL, antennal lobe; SO, subesophageal ganglion. C, CadN and V5 immunostaining of adult brain (left panels) and larval CNS (right panels) from w1118 control flies. No signal was detected with the anti-V5 antibody in the absence of V5-tagged protein. D, Anti-V5 antibody also labels the neuromuscular junction of CG31030V5 larvae, where it co-localizes with anti-HRP immunostaining that specifically marks neuronal membranes, confirming CG31030 synaptic localization. Scale bars: 100 μm (A–C) and 10 μm (D).

CG31030 co-immunoprecipitates with V-ATPase subunits. A, Scatter plot of proteins identified by nano LC-MS/MS in three independent co-immunoprecipitation experiments using anti-V5 antibodies. R1, R2 and R3 represent the abundance ratio of proteins identified in adult head extracts from CG31030V5 over w1118 control in experiments 1, 2 and 3, respectively. Log2(R) = 1 corresponds to a 2-fold abundance difference. A total of 12 proteins were found to be at least twice as abundant in CG31030V5 as in the control in all three experiments (red or blue dots on the graph). One of them is the immunoprecipitation target CG31030 (yellow dot) and three of these proteins belong to the V-ATPase complex (red dots). A list of these 12 proteins with their Log2(R) values is provided in Table 3. B, Standard model of the Drosophila V-ATPase complex showing structure of the V1 and V0 domains and the predicted localization of the three subunits that co-immunoprecipitated with CG31030. C, Co-localization of CG31030-V5 with Vha100-1 at the larval neuromuscular junction. Larval muscles were labeled with an anti-HRP antibody to mark neuronal membranes. CG31030 has a similar localization to that of Vha100-1, the synaptic isoform of subunit a of the V0 domain. D, In w1118 control flies, no specific signal could be detected at the neuromuscular junction with the anti-V5 antibody. Scale bars: 10 μm (C, D).
List of the genes encoding proteins that co-immunoprecipitated with CG31030 in Drosophila head extracts
CG31030 co-immunoprecipitates with V-ATPase subunits of the V0 domain
The CG31030 protein is predicted to be part of the InterPro V-ATPase family, but no experimental information is currently available regarding its potential interactors. To determine whether CG31030 could interact, directly or indirectly, with subunits of the V-ATPase complex, we conducted co-immunoprecipitation experiments using a V5 antibody on proteins extracted from heads of CG31030V5 mutants and w1118 control flies, followed by nano LC-MS/MS analysis of the precipitated proteins. Three independent experiments were performed to increase reliability, and in total, 410 proteins were identified in all three experiments. Among those, only 12 proteins had at least a 2-fold abundance difference with the control in all three experiments (Fig. 4A; Table 3), one of them being, as expected, the co-immunoprecipitation target CG31030-V5. Remarkably, three of the other proteins were identified as subunits of the V-ATPase complex: Vha100-1, VhaAC39-1, and ATP6AP2 (Fig. 4B).
Vha100-1 and VhaAC39-1 code for subunits a and d of the V0 domain, respectively (Vasanthakumar and Rubinstein, 2020). The subunit a is encoded by five different genes in Drosophila and constitutes the proton port of the pump (Collins and Forgac, 2020). Among these five isoforms, Vha100-1 has been shown to be specifically required in neurons and present at the synapse (Hiesinger et al., 2005). Co-immunostaining experiments confirmed that this protein colocalizes with CG31030 at the larval neuromuscular junction (Fig. 4C,D). Subunit d of V0 is encoded by two Drosophila genes: VhaAC39-1 and VhaAC39-2, and only the first co-immunoprecipitated with CG31030. According to FlyAtlas, VhaAC39-1 is expressed in many tissues and enriched in the brain, while VhaAC39-2 seems to be mostly found in testis and salivary glands. For both V0 subunits, CG31030 thus co-precipitated with the likely neuronal isoform. The co-immunoprecipitated V-ATPase subunit which appeared to be the most enriched in the CG31030V5 sample was, interestingly, the accessory subunit ATP6AP2, suggesting a possible direct interaction between this protein and CG31030 (Table 3). These experiments therefore reinforce the hypothesis that CG31030 directly interacts with the neuronal V-ATPase complex, and more specifically with V0 since all detected partners belong, or interact, with this domain.
Similarly, the mouse protein ATP6AP1L/AC45RP, a vertebrate homolog of CG31030, has recently been shown to interact selectively with V0 domain subunits of the neuronal V-ATPase (Jansen et al., 2021). In addition, these authors showed that strong in vitro upregulation (∼7-fold) of this gene resulted in an increased transcript level of some V0 subunit, while a moderate one (∼2.6-fold) had no effect except on ATP6AP2. We thus wondered whether CG31030 upregulation or downregulation could affect V0 subunit expression in vivo. Using a pan-neuronal driver, we were able to increase CG31030 transcript levels, but only up to ∼3.5-fold in an in vivo wild-type context, suggesting that transcript abundance is regulated posttranscriptionally. This moderate upregulation did not induce any significant changes in transcript levels of V0 subunits, but possibly it was not sufficient to see such effects (Fig. 5A). Conversely, we performed RT-qPCR experiments on extracts from Drosophila heads expressing CG31030RNAi3 in all neurons compared with controls, and here again, we could not find any significant change in V0 subunit expression level in knock-down context (Fig. 5B). Notably, VhaAC45 is expressed at a similar level in CG31030 knock-down flies and in controls. The homology between CG31030 and VhaAC45 could raise the question of a potential functional redundancy of the two proteins. However, the lethal phenotype and the highly reduced longevity occurring when CG31030RNAi.1/2 or CG31030RNAi.3 are expressed in all neurons, respectively, suggest that VhaAC45, though still present, is unable to substitute for CG31030.

Change in CG31030 expression does not alter V-ATPase subunit transcript levels. A, B, CG31030 upregulation (A) or knock-down (B) in vivo did not significantly affect adult head transcript levels of several V-ATPase V0 subunits. Results of three independent RT-qPCR experiments. Means and SD are displayed on the graph.
CG31030 knock-down impairs synaptic vesicle acidification
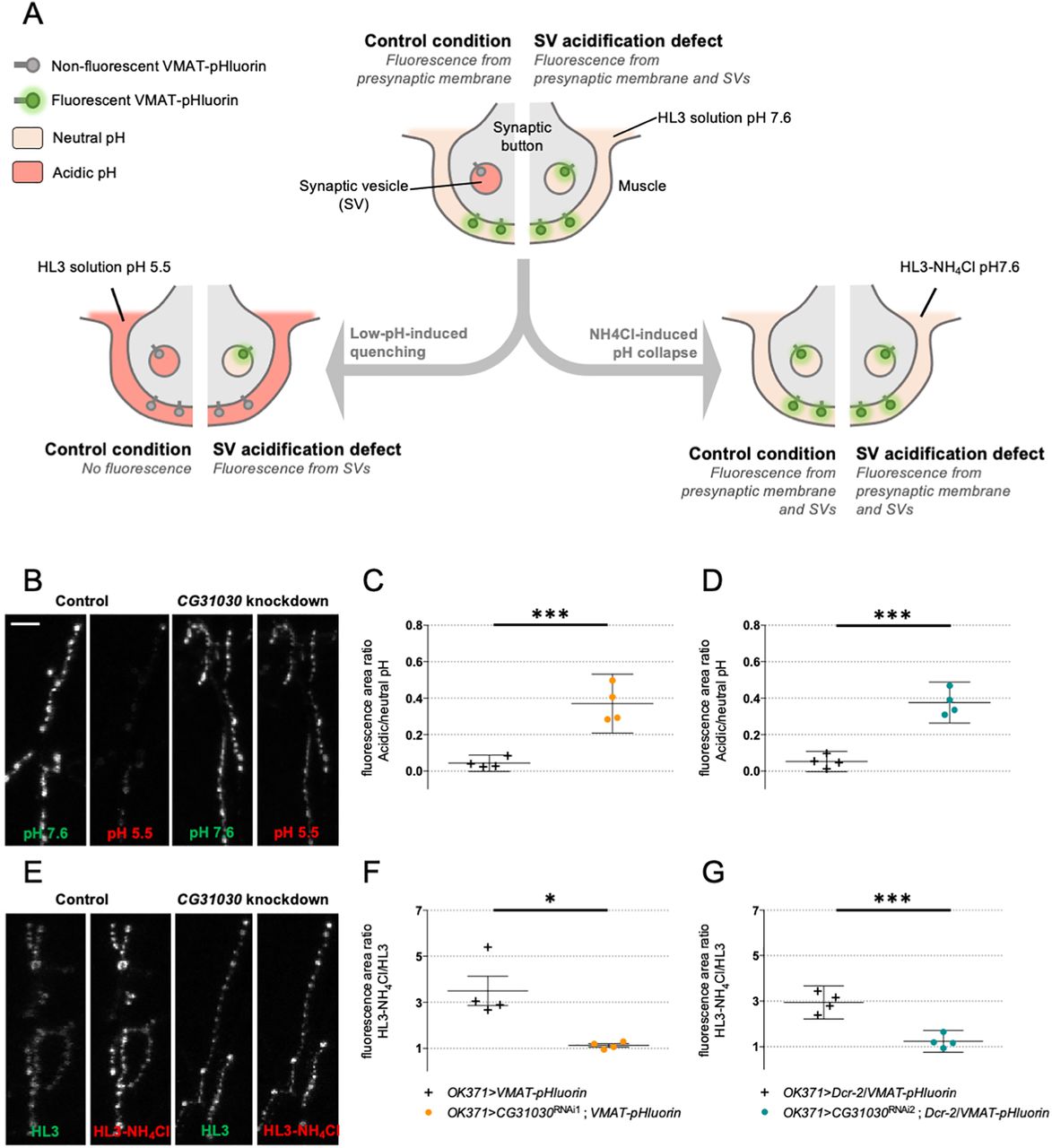
Because CG31030 appeared to be mainly localized in synaptic areas, we chose to look at the physiological effect of its disruption at the Drosophila larval neuromuscular junction, a model that has contributed to elucidate many essential synaptic processes. At synaptic nerve endings, a prominent role of the V-ATPase is to acidify the lumen of synaptic vesicles, the electrochemical gradient generated providing the driving force to load and concentrate the neurotransmitters. Thus, a malfunction of synaptic V-ATPase should induce a decrease of neurotransmitter concentration inside the vesicles, potentially resulting in an altered synaptic transmission. To test this hypothesis, we co-expressed each of the two strongest CG31030 RNAi constructs together with VMAT-pHluorin, a pH-sensitive probe targeted to synaptic vesicles (Wu et al., 2013), in larval motoneurons using the glutamatergic driver OK371-Gal4. Both RNAi1 and RNAi2 induced a lethal phenotype at pupal stage in these conditions. VMAT-pHluorin is an ecliptic pHluorin that is fluorescent at neutral pH, and gets quenched, by protonation, at acidic pH (Miesenböck et al., 1998). Thus, in control condition, VMAT-pHluorin should not be fluorescent in synaptic vesicles, whose pH is at ∼5.5, but only when externalized on the presynaptic membrane during exocytosis, and so, in contact with the more neutral synaptic cleft milieu. In the case of an acidification defect of synaptic vesicles, the probe could be fluorescent both in synaptic vesicles, where pH would be abnormally high, and on the presynaptic membrane (Fig. 6A, central panel).

CG31030 knock-down larvae have a synaptic vesicle acidification defect. A, Schematic representation of the protocols used to assess relative acidity levels of synaptic vesicles at the larval neuromuscular junction. Top center diagram, In control conditions, fluorescence can be emitted by VMAT-pHluorin in the presynaptic membrane but not in synaptic vesicles since their lumen is acidified. In case of defective synaptic vesicle acidification, both the presynaptic membrane and synaptic vesicles should emit fluorescence. Left diagram, The fluorescence emitted by VMAT-pHluorin in the presynaptic membrane can be quenched by replacing the extracellular medium with an acidic HL3 solution. This quenching should result in an almost complete extinction of the signal in control flies, in which synaptic vesicles are normally acidified, while a residual signal is expected to be visible in flies having a synaptic vesicle acidification defect. Right diagram, Replacement of 50 mm NaCl by 50 mm NH4Cl in the neutral HL3 solution should lead to a collapse of the pH gradients because of the free diffusion of NH3 in membranes, so that fluorescence will be emitted both by the presynaptic membrane and synaptic vesicles both in control and mutant conditions. B, Representative pictures showing the effect of perfusing an acidic HL3 solution on VMAT-pHluorin fluorescence in control and CG31030 knock-down larvae. C, D, Quantification of the ratio of the fluorescence level at pH 5.5 over the original signal at pH 7.6. Whereas the low pH extinguished fluorescence in control flies, ∼37% of the signal persisted after quenching in CG31030 knock-down larvae using two different RNAi constructs. E, Representative pictures showing the effect of collapsing the synaptic vesicle pH by perfusing HL3-NH4Cl in control and CG31030 knock-down larvae. F, G, Quantification of the ratio of the signal in HL3-NH4Cl over the original signal in HL3 showed that fluorescence increased ∼3-fold in controls while it only rose by 10–20% depending on the RNAi in CG31030 knock-down larvae. Results of four independent experiments, with three to five larvae analyzed per genotype in each experiment. Unpaired Student’s t test; *p < 0.05, ***p < 0.001. Mean values with 95% confidence intervals are reported on the graphs. Scale bar: 15 μm (B).
In order to evaluate the ratio of the internal fluorescence (from synaptic vesicles) over the external fluorescence (from the presynaptic membrane) at the neuromuscular junction of CG31030 knock-down larvae compared with controls, we first quenched the external signal by replacing the physiological milieu by an identical one with pH adjusted to 5.5 (Fig. 6A, left panel). This operation resulted in only the internal signal being conserved. In controls, this meant that all signal was abolished, as expected because the synaptic vesicles were normally acidified (Fig. 6B, left panel). In contrast and strikingly, a residual signal was still visible in this acidic milieu in both RNAi1 and RNAi2 knock-down larvae (Fig. 6B, right panel). Quantification of the ratio of fluorescence area in acidic milieu over neutral milieu showed that ∼37% of the total signal remained visible in the RNAi larvae after external quenching (Fig. 6C,D).
To verify that the residual signal seen in knock-down larvae was indeed coming from inside vesicles, the opposite strategy was used: instead of quenching the outside signal, we revealed all the internal one by collapsing the pH gradient of synaptic vesicles (Fig. 6A, right panel). To do so, we replaced the physiological milieu by an ammonium solution, as previously described (Poskanzer and Davis, 2004). This solution had the same composition except that 50 mm NaCl were replaced by 50 mm NH4Cl. Ammonium and ammonia being in equilibrium (NH4+ ⇄ NH3 + H+), uncharged ammonia crosses membranes and binds to protons leading to an alkalization of vesicle lumen pH. This increase of vesicular pH is maintained during NH4Cl exposure. In control condition, this gradient collapse should reveal the VMAT-pHluorin probe present in synaptic vesicles and thus highly increase the fluorescent signal. On the contrary, in the case of an acidification defect, the signal should remain fairly stable since synaptic vesicles are already fluorescent. Results were consistent with this hypothesis: while the fluorescence of controls increased ∼3-fold in the pH collapsing ammonium solution, the signal in CG31030 knock-down larvae hardly rose by 10–20% depending on the RNAi construct (Fig. 6E–G). Taken together, these results indicate that CG31030 knock-down significantly decreases protons concentration in synaptic vesicles of motoneuron terminals.
CG31030 downregulation decreases larval locomotor performance
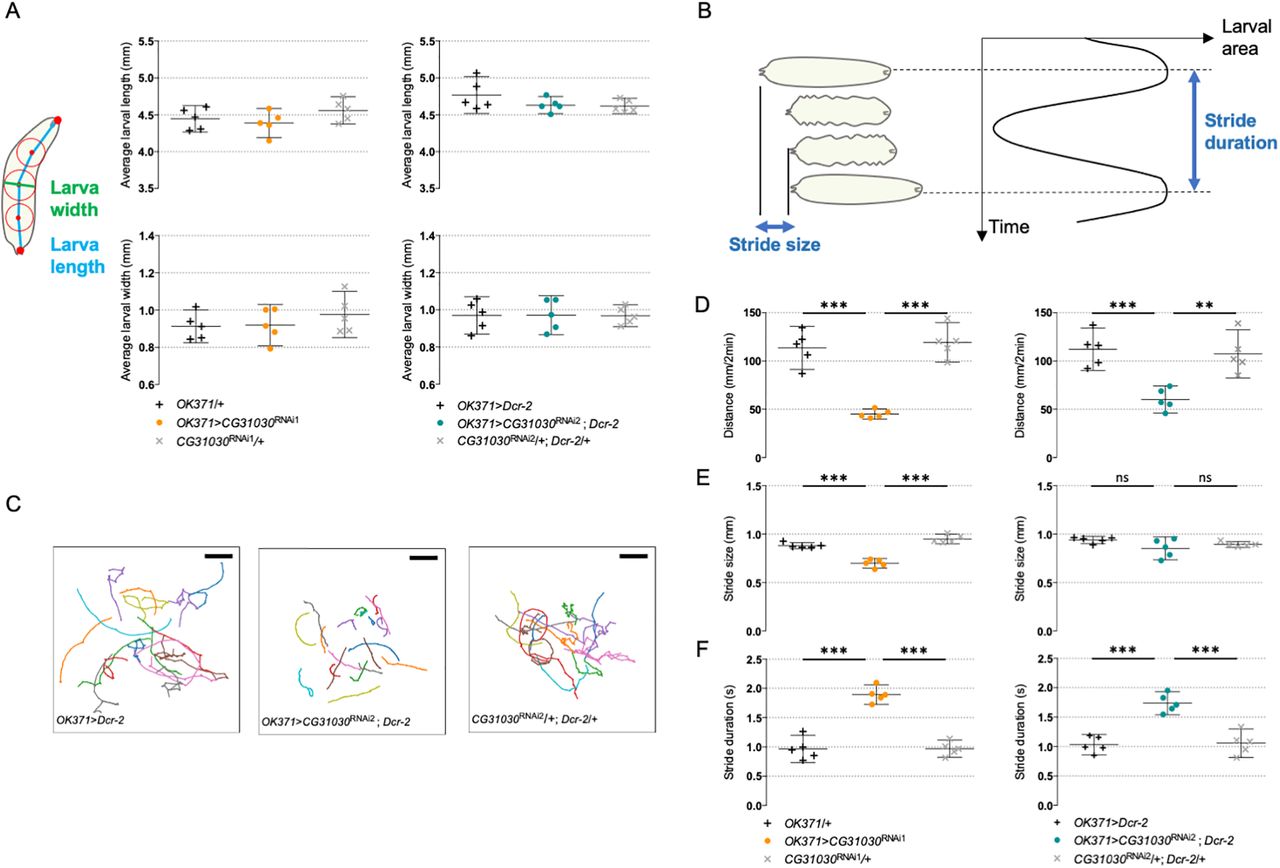
A reduced pH gradient of synaptic vesicles in larval motoneurons could alter synaptic transmission and, consequently, the larval locomotor behavior. To assess locomotion, we recorded the spontaneous crawling of third-instar larvae expressing CG31030 RNAi1 or RNAi2 in motoneurons with OK371-Gal4 on an agar plate with no food source for 2 min periods. We first checked that the average length and width of knock-down larvae were not significantly different from those of control larvae (Fig. 7A). Tracking was then performed using the FIMtrack software (Risse et al., 2014), allowing measurement of the total distance traveled and of the stride size, defined as the distance crawled during one peristaltic wave of muscle contraction, and duration (Fig. 7B). The knock-down larvae obviously moved less than controls on the plate, as confirmed by their trajectory maps (Fig. 7C), traveling a distance only half as long as controls with both RNAi (Fig. 7D).

CG31030 downregulation in motoneurons decreases larval locomotor performance. A, CG31030 knock-down in motoneurons did not significantly alter larval size. Larval length is defined as the spine length from head to tail, while larval width is the diameter of the mid-spine circle. Average larval length and width were not significantly different between animals expressing CG31030 RNAi1 (left panels) and RNAi2 (right panels) in motoneurons and controls. B, Schematic representation of the successive phases of larval locomotion. Stride size is defined as the distance crawled during one peristaltic wave of muscle contraction, while stride duration is the time necessary for the completion of one peristaltic wave. C, Locomotor trails of individual larvae recorded over a period of 2 min. Larvae expressing CG31030 RNAi in motoneurons show reduced spontaneous movements (middle panel) compared with the driver (left panel) and effector (right panel) controls, respectively. Scale bar: 25 mm. D, Quantification of the traveled distances confirmed that knocking down CG31030 in motoneurons with two different RNAi induced significant locomotor defects. E, F, Stride size (E) appears to be less affected than stride duration (F) in the knocked-down larvae. Result of five independent experiments, with three to four larvae analyzed per genotype in each experiment. One-way ANOVA with Dunnett’s post hoc test for multiple comparisons; **p < 0.01, ***p < 0,001. Mean values with 95% confidence intervals are reported on the graphs.
In contrast, the stride size actually showed little or no difference, depending on the RNAi, between knock-down larvae and controls (Fig. 7E). Instead, we found that the stride duration, which is the time necessary to accomplish one peristaltic wave, was significantly longer in knock-down animals compared with controls for both RNAi (Fig. 7F). These results suggest that the knock-down larvae may need about twice as much time as controls to reach the required level of muscular contraction to accomplish one stride. This could be a compensatory mechanism to adapt to a lower amount of neurotransmitter released in response to motor nerve stimulation, as would be expected if the synaptic vesicles were less filled in CG31030 knock-down context.
Quantal size is reduced in CG31030 knock-down larvae
The locomotor deficit of CG31030 knock-down larvae could originate from an impairment of synaptic transmission at the neuromuscular junction or a defect in the central control of motor behavior, or both. Fluorescent FM-dye loading/unloading experiments can be used to monitor potential defects in endocytosis or exocytosis of synaptic vesicles at the larval neuromuscular junction (Verstreken et al., 2008). We therefore assessed whether the synaptic vesicle cycle induced by KCl stimulation was affected by CG31030 downregulation, as this could well have an impact on neurotransmission and locomotion. Representative images of neuromuscular junctions from control and knock-down larvae after FM-dye loading and unloading are shown in Figure 8A,B, respectively. The larvae expressing CG31030 RNAi1 or RNAi2 in motoneurons, were apparently able to load and unload the dye in synaptic junctions similarly as controls (Fig. 8C,D). The percentage of previously loaded FM4-64 dye released during the unloading phase was also comparable for control and knock-down larvae (Fig. 8E,F). This result indicates that the synaptic vesicle cycle, and in particular the exocytosis machinery, were apparently not impacted by CG31030 inactivation.
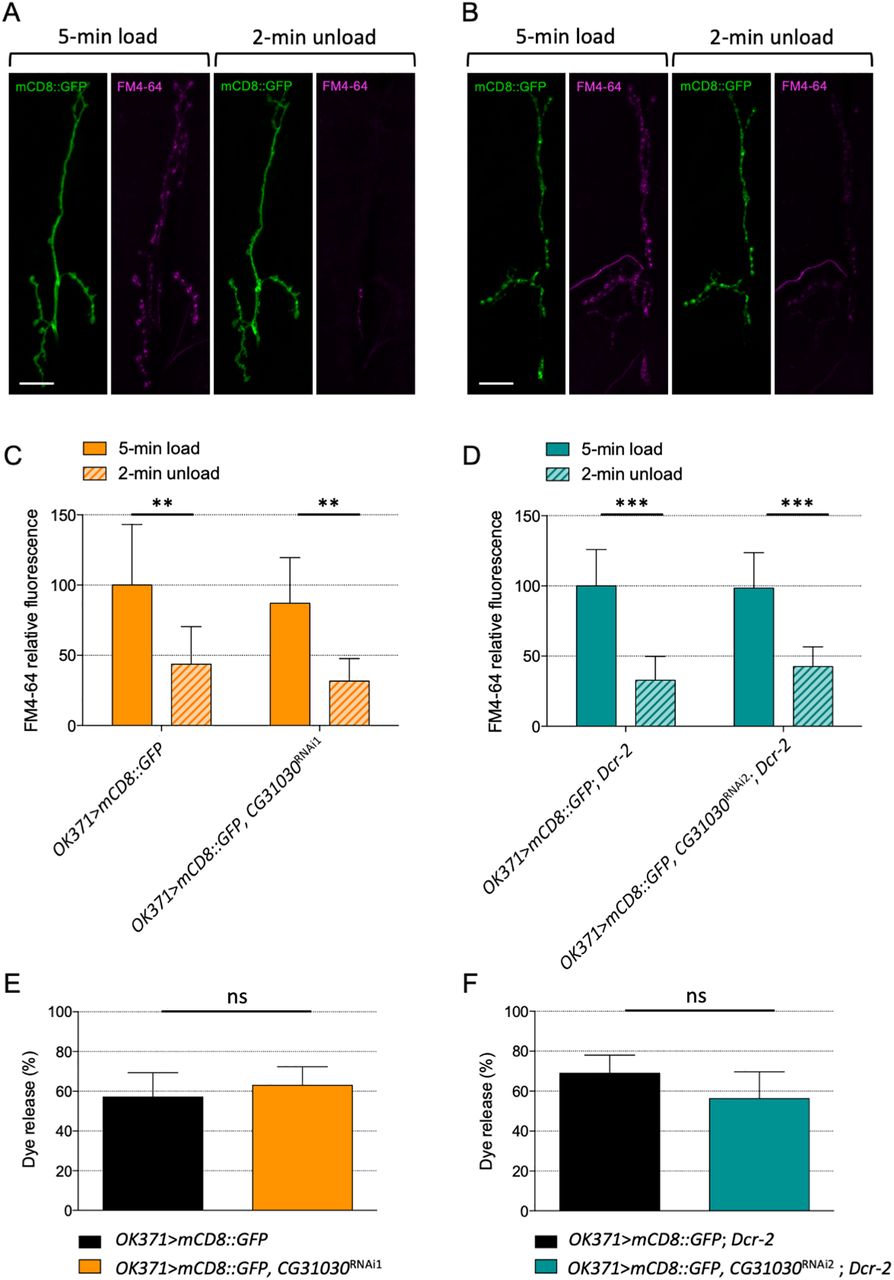
Lack of effect of CG31030 knock-down on the synaptic vesicle cycle at the larval neuromuscular junction. A, B, Representative images of mCD8::GFP-labeled neuromuscular junctions of a control (A) and a knock-down (B) larva after loading and unloading of the fluorescent FM4-64 dye. C, D, Nerve terminals from larvae expressing CG31030 RNAi1 (C) or RNAi2 (D) were able to both load and unload the fluorescent FM4-64 dye as efficiently as controls. No significant difference was observed between controls and knock-down animals during the loading and the unloading phases. Fluorescence was normalized relative to the level in control larva synapses after 5-min loading. E, F, The percentage of previously loaded FM4-64 dye released during the unloading phase was similar for control and knock-down larvae when using either RNAi1 (E) or RNAi2 (F); **p < 0.01, ***p 0.001, ns: not significant.
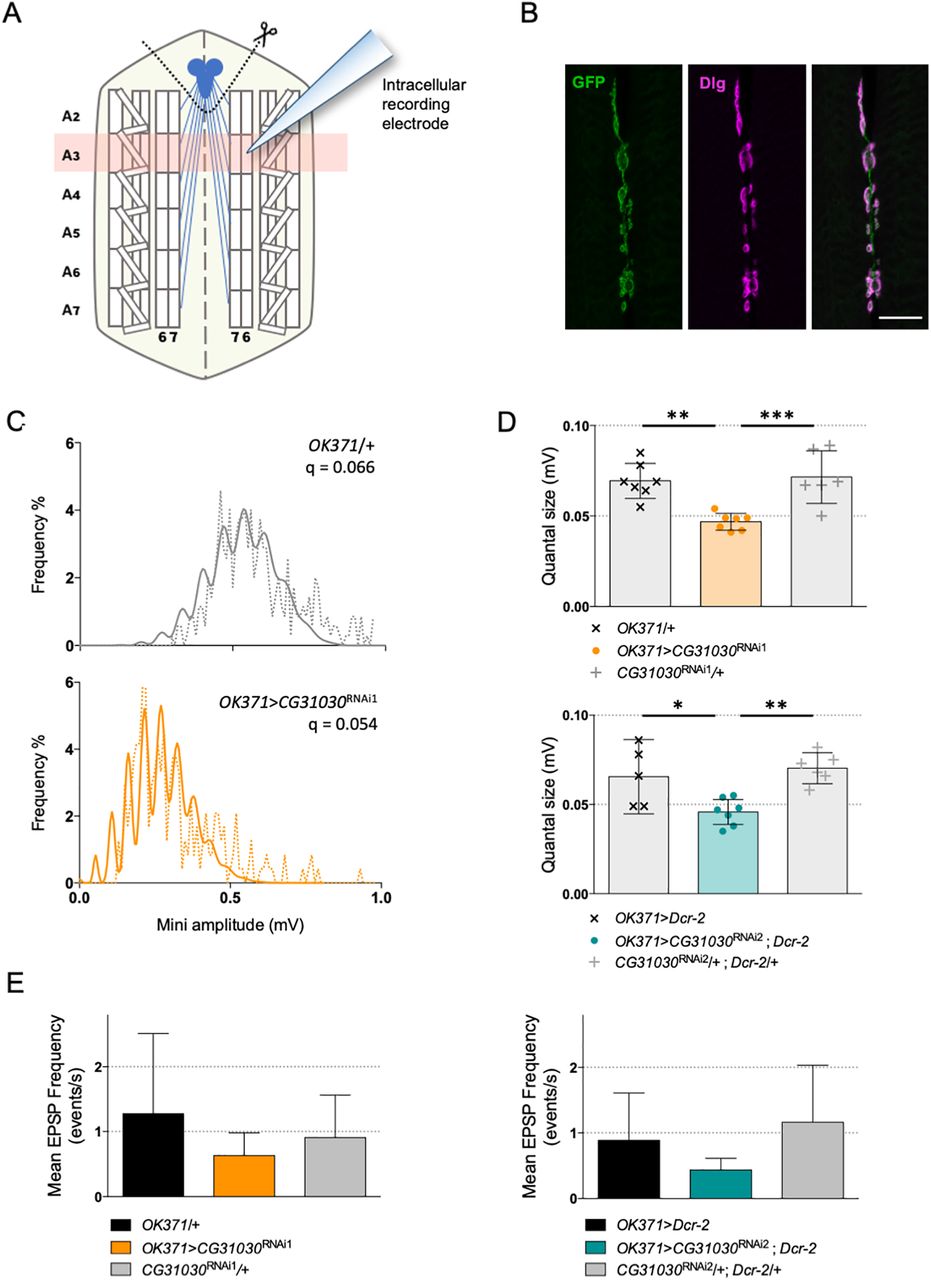
To determine whether neurotransmission was otherwise affected by CG31030 knock-down, we conducted electrophysiological recordings to measure the quantal size, which is the postsynaptic response to the release of one synaptic vesicle. We expressed CG31030 RNAi1 or RNAi2 in motoneurons of third-instar larvae with the OK371-Gal4 driver, and recorded spontaneous mEPSPs intracellularly from ventral longitudinal abdominal muscle 6 of segment A3 (Fig. 9A). These muscles are innervated by synaptic boutons that are clearly marked by OK371-Gal4, as shown by the co-localization of membrane-associated GFP and the postsynaptic marker Dlg in OK371>mCD8::GFP flies (Fig. 9B). Representative amplitude distributions of mEPSPs in a control and RNAi1 larvae are shown in Figure 9C. Quantal analysis of recorded events confirmed that both RNAi1 and RNAi2 knock-down larvae have a significantly reduced quantal size compared with controls (Fig. 9D), suggesting a decrease in glutamate vesicular uptake, and potentially linking vesicle acidification defect and locomotor impairment. The V-ATPase dysfunction in synapses of CG31030 knock-down larvae could apparently lead to incomplete loading of vesicles, thus decreasing the amount of neurotransmitter released per unit of time during a peristaltic wave, and potentially slowing down larval locomotion. Although the mean frequency of mEPSPs of both RNAi1 and RNAi2 larvae appeared lower than controls, this effect was not statistically significant (Fig. 9E). This may suggest that CG31030 knock-down did not significantly increase the number of unacidified vesicles empty of neurotransmitter.

Synaptic quanta size is reduced in CG31030 knock-down larvae. A, Schematic representation of a dissected larval fillet. Spontaneous miniature EPSPs (mEPSPs) were recorded intracellularly from the ventral longitudinal abdominal muscle 6 in segment A3. B, Expression of membrane-associated mCD8::GFP with the glutamatergic driver OK371-Gal4 strongly labels the presynaptic nerve endings at the neuromuscular junction of muscles 6–7 in segment A3. The scaffolding protein Dlg was used as a postsynaptic marker. Scale bar: 20 μm. C, Representative distributions of spontaneous mEPSPs recorded in a control larva (top panel, in gray) and a CG31030 RNAi knock-down larva (bottom panel, in orange). The dotted lines represent actually recorded amplitudes and the plain lines the computed theoretical distributions. Genotypes and quantal size (q) are indicated on each graph. D, Quantal analysis of recorded events showed that knock-down larvae have a significantly reduced synaptic quanta size compared with controls, both with CG31030 RNAi1 (top panel, in orange) or RNAi2 (bottom panel, in blue). E, Although both RNAi1 (left panel) and RNAi2 (right panel) larvae had a lower mean EPSP frequency than controls, this difference was not statistically significant. Six to seven cells recorded per genotype. One-way ANOVA with Dunnett’s post hoc test for multiple comparisons; *p < 0.05, **p < 0.01, ***p < 0.001. Mean values with 95% confidence intervals are reported on the graphs.
Finally, we also looked at the morphology of the neuromuscular junctions in CG31030 knock-down and control animals by measuring the number and diameter of synaptic boutons innervating the ventral longitudinal abdominal muscle 6 in the segment A3. The overall number and mean size of boutons were not significantly affected between the different genotypes (Fig. 10A–D). However, we observed that knock-down larvae exhibited a reduced number of boutons with a diameter superior to 4 μm for both RNAi1 and RNAi2 (Fig. 10E,F). The fact that these large boutons only represented a small fraction of the total explains why the average number or mean diameter of boutons were not altered in knock-down larvae. It is therefore unlikely that this size variation could result, by itself, in the significant decrease in quantal size and strong locomotor impairment induced by CG31030 downregulation. Neuronal activity is known to influence synaptic growth at the neuromuscular junction (Menon et al., 2013), so the lower number of larger boutons in knock-down animals could be a consequence of the impaired synaptic transmission resulting from the quantal size decrease. Alternatively, the lower number of high diameter boutons could also be a modest contributor to the observed quantal size decrease, as larger boutons have been shown to harbor bigger synaptic vesicles associated with higher quantal content (Karunanithi et al., 2002).
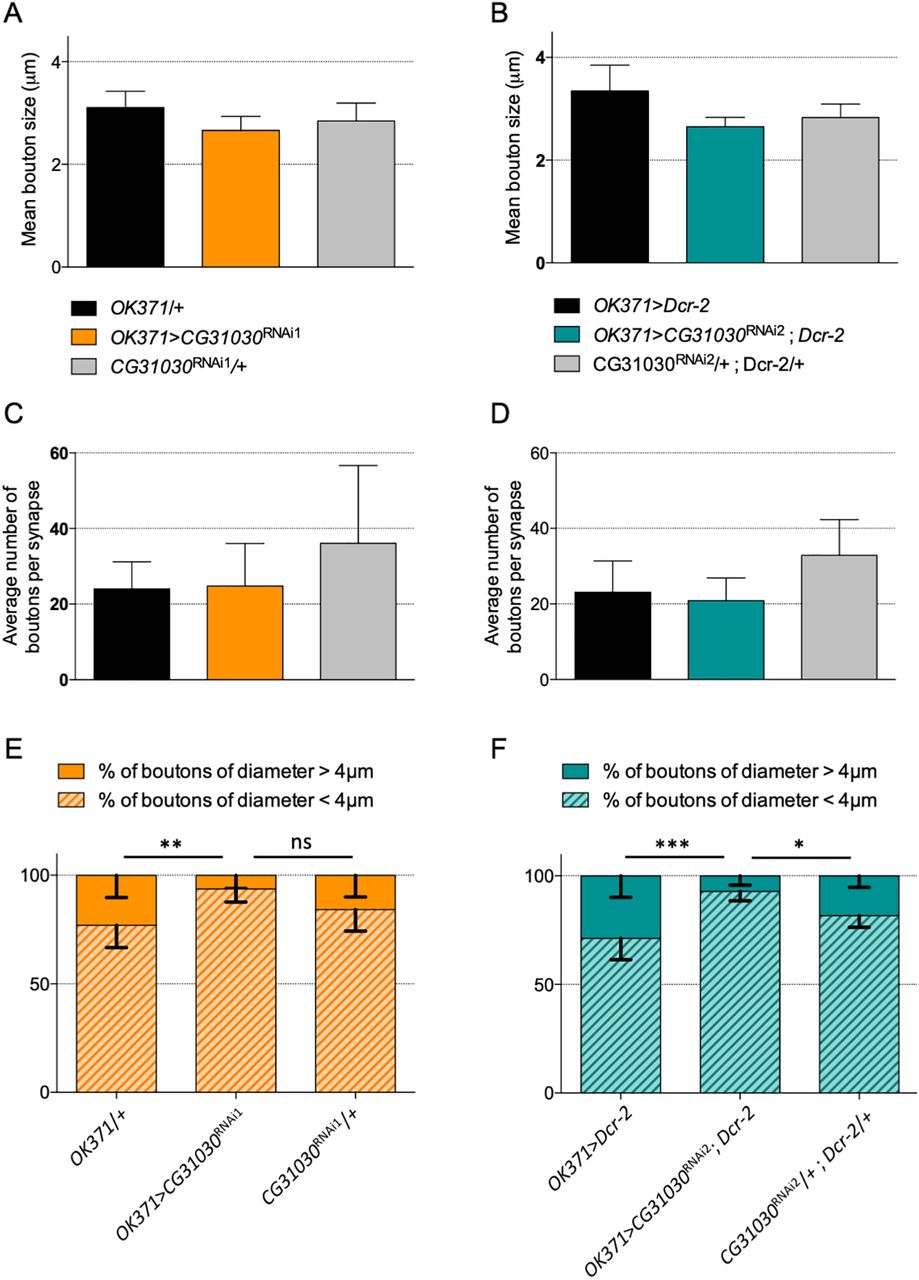
Effect of CG31030 knock-down on synapse morphology at the larval neuromuscular junction. A–D, No significant difference was observed in the mean synaptic bouton diameter (A, B) or the average number of boutons per synapse (C, D) between larvae expressing CG31030 RNAi1 (A, C) or RNAi2 (B, D) in motoneurons and the controls. E, F, Knock-down larvae showed a smaller percentage of boutons with a diameter larger than 4 μm for both RNAi1 [E; but not significant (ns) compared with the effector control] and RNAi2 (F), n = 6–8 synapses per genotype, two-way ANOVA with Dunnett’s correction for multiple comparisons; *p < 0.05, **p < 0.01, ***p < 0.001, ns: not significant.
Discussion
In this study, we investigated the hypothesis that the previously uncharacterized Drosophila protein CG31030 is a specific regulator of the neuronal V-ATPase. At variance with its broadly expressed paralog VhaAC45, we have shown that CG31030 is found mainly, if not only, in neurons. We also provide evidence that CG31030 interacts with two constitutive subunits and one accessory subunit of the V-ATPase, the constitutive ones being also enriched in neurons, and that it is required to have properly acidified synaptic vesicles. This implies that CG31030 is an essential protein for nervous system functioning in Drosophila.
CG31030 is an essential synaptic protein
In yeast, all V-ATPase subunits are coded by a single gene, with the exception of the V0 subunit a. The knock-out of any of the single-gene subunits all present a similar phenotype: the inability to survive in a neutral pH environment (Nelson, 2003). For subunit a, the same phenotype was only achieved in a double-mutant of both isoforms (Manolson et al., 1994). In multicellular organisms, mutations of V-ATPase subunits, or accessory proteins, also often lead to a lethal phenotype at various developmental stages, whether in mice (Inoue et al., 1999; Sun-Wada et al., 2000; Schoonderwoert and Martens, 2002b), Caenorhabditis elegans (Lee et al., 2010), or flies (Davies et al., 1996; Allan et al., 2005). Similarly, we found a CG31030 null mutant to be embryonic lethal, and, interestingly, this lethality could be rescued to the adult stage by re-expressing CG31030 in all neurons, showing that the protein is specifically required in this cell type for fly survival. The percentage of adult survivors was about three times less than what would be expected in case of a full rescue (Table 2) and the missing two-thirds likely died at an early developmental stage as no larval lethality was observed. This could be explained by the fact that CG31030 protein re-expression in rescued knock-out flies first required the expression of Gal4 under regulation of the elav promoter, which starts to express rather late in embryos, followed by activation of the UAS sequence upstream the CG31030 insert. So, it is possible that part of the mutant embryos did not survive the delay inherent to this ectopic expression process.
We observed that CG31030 transcripts follows the repartition of the nervous system, with the highest relative abundance in the head. Moreover, pan-neuronal expression of CG31030RNAi3, the RNAi construct with the weakest effect on fly survival, was sufficient to decrease by >80% its level in the brain of adult escapers. These results indicate that CG31030 expression is mainly neuronal, if not even entirely restricted to neurons. In addition, CG31030 cellular localization shows similarity with a synaptic pattern. Cell bodies were also marked, although generally less strongly. Synaptic protein complexes can be assembled in the cell bodies before being transported in axons, and it is the case for the soluble and membrane-bound domains of synaptic V-ATPase, as was shown in Torpedo (Morel et al., 1998). It is difficult to decide whether the signal coming from cell bodies corresponds to a functional site for CG31030 or rather to newly produced proteins that have not yet been targeted to synapses. However, the prominent signal in synaptic areas led us to assume that CG31030 plays an important role in the synaptic process.
CG31030 is required for proper synaptic vesicle acidification
In presynaptic terminals, the most abundant organelles are synaptic vesicles, which require acidification to provide the driving force for neurotransmitter loading. This acidification is ensured by the V-ATPase pump, which generates an electrochemical gradient by importing protons into the vesicle lumen, thus creating both a membrane potential (ΔΨ, inside positive voltage) and a pH gradient (ΔpH, acidic lumen). Our experiments showed that the knock-down of CG31030 at the glutamatergic larval neuromuscular junction increased the internal pH of synaptic vesicles, and so decreased the ΔpH component of the electrochemical gradient generated by the V-ATPase. This result is however by itself insufficient to conclude to a dysfunction of the proton pump, since other players have been shown to influence synaptic vesicle pH gradient downstream V-ATPase activity. For example, cation/H+ exchangers, found on synaptic vesicles, can decrease ΔpH while increasing ΔΨ by exchanging cations, like Na+ or K+, against luminal H+ (Takamori, 2016). This activity can be upregulated by increased intracellular concentration of these cations (Huang and Trussell, 2014). Glutamate being negatively charged, its transport across vesicular membrane is predominantly driven by ΔΨ (Maycox et al., 1988; Tabb et al., 1992). As a consequence, the increased membrane potential resulting from upregulation of cation/H+ exchangers actually facilitates glutamate uptake (Goh et al., 2011; Huang and Trussell, 2014). We measured synaptic quanta size by quantal analysis of mEPSPs of CG31030 knock-down larvae and found that it was decreased, contrary to what would be expected in the case of an upregulation of cation/H+ exchanger activity. This apparent decrease of vesicular glutamate concentration was corroborated by its phenotypical consequence, namely the locomotion defect exhibited by knock-down larvae. This suggests that the observed diminution of ΔpH is correlated with a diminution of ΔΨ, thus pointing toward a V-ATPase malfunction. This hypothesis is further supported, physiologically, by the lack of defect in the endocytosis/exocytosis cycling of synaptic vesicles in knock-down larvae, making it unlikely that such a defect could be at the origin of the locomotor impairment, and, at the molecular level, by the co-immunoprecipitation of CG31030 with three V-ATPase subunits of the V0 domain (Vha100-1, VhaAC39-1, and ATP6AP2), all of them being associated with the neuronal V-ATPase. Altogether, these results strongly suggest that CG31030 is a neuronal protein necessary for V-ATPase function in synaptic nerve endings.
Some V-ATPase subunits isoforms in other species have been shown to target specific subcellular compartments, like the Torpedo V0 a1 isoform which is found in synaptic V-ATPase and not in neuronal cell bodies (Morel et al., 2003). Similarly, CG31030 could be specifically involved in the regulation of synaptic V-ATPase, or, alternatively, globally act on all neuronal V-ATPase. Specificity level could even be pushed further, since synaptic vesicles are not the only organelles requiring acidification that are localized in synapses. Thus, a synaptic V-ATPase regulator could be devoted to synaptic vesicles as it could be affecting more generally all synaptic V-ATPase complexes, indifferently of their membrane localization. The answer to such questions could help better understand regulations of synaptic transmission, since synaptic V-ATPase activity is one of the presynaptic modulators of quantal response (Takamori, 2016; Gowrisankaran and Milosevic, 2020).
CG31030 interacts with ATP6AP2 and may regulate V-ATPase domain dynamics
While accessory subunits directly interact with V-ATPase domains, other stimulus can indirectly affect activity of the complex, like glucose concentration or serotonin (Sautin et al., 2005; Voss et al., 2007). Co-precipitation of CG31030 with V-ATPase subunits does not necessarily imply a direct interaction, but suggests a minima an indirect association with the complex, possibly in a non-transient manner. The impairment in V-ATPase activity induced by CG31030 downregulation rules out the hypothesis of an inhibitory action of this protein on the proton pump. Nevertheless, the precise requirement of CG31030 for activity of the V-ATPase complex still remains unknown to date. Lethality of the mutant could indicate an essential role of CG31030 in V-ATPase function. However, some V-ATPase regulators and accessory subunits have been found in other signaling pathways, like ATP6AP2 in the renin-angiotensin system (Nguyen, 2010), so we cannot exclude that lethality could be because of CG31030 playing a part in other vital neuronal functions. Signal in axons could also point to a role in targeting the complex to the synapses. The two domains of the V-ATPase, V0 and V1, are believed to be assembled in cell bodies before being separately transported to synaptic area, V0 by fast axonal anterograde transport, most likely directly on new synaptic vesicles, and V1 by slow axonal transport like other cytoplasmic synaptic proteins (Morel et al., 1998). CG31030 could be involved in the transport of one of the two domains, and effects of CG31030 knock-down could then result from a decreased synaptic abundance of the affected V-ATPase domain. However, it is believed that only one copy of the V-ATPase complex is sufficient to properly acidify one synaptic vesicle (Takamori, 2016), a consequence of this being that neurotransmitter loading would be an all-or-none process. Consistent with this, mutation of the Drosophila synaptic V0 subunit Vha100-1 does not seem to impact quantal size but rather mEPSP frequency, potentially reflecting the presence of an increased number of empty synaptic vesicles (Hiesinger et al., 2005). Similarly, a reduction in the frequency of spontaneous quantal events with no change in quantal size was observed when the Drosophila vesicular glutamatergic transporter (VGlut) was downregulated, also suggesting that a single copy of VGlut is sufficient for proper loading of a vesicle (Daniels et al., 2006). The fact that CG31030 knock-down changes the quantal size but not significantly mEPSP frequency seems to point toward a role of this accessory protein in V-ATPase efficiency, rather than in a process affecting the abundance of the complex such as synaptic targeting.
In this respect, it is interesting to note that the V-ATPase protein that more consistently co-immunoprecipitated with CG31030 in our experiments is the accessory subunit ATP6AP2, the fly homolog of human ATP6AP2/PRR. Interestingly, the vertebrate homolog of CG31030, ATP6AP1/Ac45, has also been shown to interact with ATP6AP2, and the complex they form has been proposed to enable the assembly and disassembly of the catalytic and membrane domains of the V-ATPase in the mammalian brain (Rujano et al., 2017; Abbas et al., 2020). This suggests that CG31030 could similarly play a role in the regulation of these dynamic processes. Indeed, it was shown that the dissociation of the two V-ATPase domains on synaptic vesicles is a necessary step before exocytosis (Bodzęta et al., 2017). When new vesicles are formed through endocytosis, the two domains then reassemble to start re-acidifying the vesicular lumen, allowing neurotransmitter uptake. Thus, during the synaptic vesicles recycling cycle, V0 and V1 dynamically alternate between assembled and disassembled states. CG31030 could then be important to facilitate the assembly of the two domains or maintain the assembled state, which may also influence neurotransmitter loading and release in a more continuous way. Our results suggest indeed that modulating V-ATPase activity in presynaptic terminals can finely affect quantal size. Whether such regulation actually occurs in physiological, or pathologic, conditions remains to be established.
Human ATP6AP2/PRR is known to be a Parkinsonism candidate gene and its mutations in humans, mice or flies can lead to cognitive impairment, neurodegeneration and epilepsy (Korvatska et al., 2013; Dubos et al., 2015; Ichihara and Yatabe, 2019). It is therefore of major interest to identify a potential new interactor of ATP6AP2 that is specific to the nervous system, as it could help better understand the consequences of V-ATPase dysregulation on synaptic transmission in pathologic contexts. Accordingly, it would be very interesting to determine whether human ATP6AP1 and/or ATP6AP1L share a conserved function with Drosophila CG31030 in the nervous system. Like CG31030, the mouse ATP6AP1L/AC45RP was recently reported to be restricted to neurons (Jansen et al., 2021), and they are, to date, the only known V-ATPase genes with such tissue specificity. Both proteins also have the particularity of not having a predicted furin cleavage site. ATP6AP1L/AC45RP was shown to be involved in neuronal growth (Jansen et al., 2021), while we observed that CG31030 downregulation also decreased the number of large synaptic boutons at the neuromuscular junction, hinting at a potential functional homology. Owing to its structural and functional similarity with its closest Drosophila homolog, VhaAC45/ATP6AP1, and vertebrates ATP6AP1L/AC45RP, we propose therefore that CG31030 be named VhaAC45-related protein (VhaAC45RP).
In conclusion, our present work identified a specific regulator of neuronal V-ATPase in Drosophila that is required for synaptic vesicle acidification, nervous system functioning and survival. Further study on this protein should provide a more detailed picture of the complex regulations surrounding neuronal V-ATPase specificity, revealing potential therapeutic targets and a better understanding of fundamental processes such as synaptic transmission.
Acknowledgments
Acknowledgements: We thank Dr. Philippe Marin for helpful advice about the quantitative analysis of mass spectrometry experiments, Dr. Hervé Tricoire and Dr. Elodie Martin for helpful suggestion on the CRISPR/Cas9 mutagenesis, Dr. Peter Robin Hiesinger for the gift of the anti-Vha100-1 antibody, Dr. David Krantz for providing the UAS-VMAT-pHluorin strain, Dr. Hermann Aberle for the OK371-Gal4 strain, Dr. Hervé Tricoire for the pCFD3-U6:3-gRNA plasmid, and Dr. Céline Petitgas and Manon Dobrigna for participating in preliminary experiments during their student internships.
Footnotes
The authors declare no competing financial interests.
This work was supported by the Fondation de France Grant Subvention 0086407 (to S.B.) and recurrent funding to S.B.’s laboratory from the École Supérieure de Physique et de Chimie Industrielles de la Ville de Paris and Centre National de la Recherche Scientifique. A.D., A.R.I., and J.S. were recipient of PhD fellowships from PSL University and Labex MemoLife, the Doctoral School 3C, and the China Scholarship Council, respectively.
This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International license, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



