

Abstract
The mouse vomeronasal system controls several social behaviors. Pheromones and other social cues are detected by sensory neurons in the vomeronasal organ (VNO). Stimuli activate a transduction cascade that leads to membrane potential depolarization, increase in cytosolic Ca2+ level, and increased firing. The Ca2+-activated chloride channels TMEM16A and TMEM16B are co-expressed within microvilli of vomeronasal neurons, but their physiological role remains elusive. Here, we investigate the contribution of each of these channels to vomeronasal neuron firing activity by comparing wild-type (WT) and knock-out (KO) mice. Performing loose-patch recordings from neurons in acute VNO slices, we show that spontaneous activity is modified by Tmem16a KO, indicating that TMEM16A, but not TMEM16B, is active under basal conditions. Upon exposure to diluted urine, a rich source of mouse pheromones, we observe significant changes in activity. Vomeronasal sensory neurons (VSNs) from Tmem16a cKO and Tmem16b KO mice show shorter interspike intervals (ISIs) compared with WT mice, indicating that both TMEM16A and TMEM16B modulate the firing pattern of pheromone-evoked activity in VSNs.
Significance Statement
Vomeronasal sensory neurons (VSNs) express two Ca2+-activated chloride channels TMEM16A and TMEM16B, however their physiological role is still unclear. Using a loss of function approach, we found that TMEM16A modulates the pattern of VSN spontaneous spike activity, while TMEM16A and TMEM16B reduced the instant frequency of pheromone-evoked activity. These new findings call for a reconsideration of the patterns of the peripheral coding of sensory stimuli.
Introduction
Most mammals use at least two distinct olfactory systems to detect chemicals: the main and the accessory (vomeronasal) olfactory system. In general, pheromones affect the physiology and/or behavior of an individual by activating either one or both systems (Tirindelli et al., 2009; Touhara and Vosshall, 2009; Mohrhardt et al., 2018).
Chemical signals bind to chemosensory neurons in the vomeronasal organ (VNO), a bilateral cylindrical structure within a cartilaginous capsule located beneath the septum (Døving and Trotier, 1998). Vomeronasal sensory neurons (VSNs) are bipolar neurons with a single apical dendrite that has a terminal swelling (knob) with several microvilli, where vomeronasal receptors (V1Rs, V2Rs, FPRs) are located and sensory transduction occurs. Each VSN expresses one type or few types of vomeronasal receptors, and axons from VSNs expressing the same receptor project to various glomeruli in the accessory olfactory bulb (AOB; Rodriguez et al., 1999; Francia et al., 2014).
Vomeronasal receptor-ligand binding activates a G-protein mediated signaling pathway involving phospholipase C activation. Phosphoinositide turnover activates transient receptor potential canonical 2 (TRPC2) channels allowing the influx of Na+ and Ca2+ (Liman et al., 1999; Lucas et al., 2003; Leinders-Zufall et al., 2018). In parallel, Ca2+ release from intracellular stores has also been described (Kim et al., 2011). Ultimately, VSN signal transduction generates membrane depolarization and action potentials that are sent to the AOB.
The increase in intracellular Ca2+ concentration on activation of the transduction cascade has several physiological effects, including the activation of Ca2+-activated chloride channels (Yang and Delay, 2010; Kim et al., 2011; Dibattista et al., 2012; Amjad et al., 2015). At least two members of the TMEM16 family, TMEM16A and TMEM16B, form Ca2+-activated chloride channels and are expressed in several different tissues (Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008; Pifferi et al., 2009a; Stephan et al., 2009; Pedemonte and Galietta, 2014). Interestingly, although TMEM16A and TMEM16B are individually expressed in different cell types, they co-express in microvilli of VSNs (Dibattista et al., 2012; Münch et al., 2018). Not many other cell types are known to co-express TMEM16A and TMEM16B. Indeed, to our knowledge, the only other cells that have been shown to functionally express both proteins are pinealocytes (Yamamura et al., 2018). Therefore, studying the role of these channels in the VNO is helpful to understand how VSNs respond to pheromones and contributes to understanding their individual role in other cells co-expressing them.
In isolated VSNs, Amjad et al. (2015) have shown that the biophysical properties of Ca2+-activated chloride currents, measured both in whole-cell and in inside-out patches from the apical neuronal region, resemble those of TMEM16A, rather than TMEM16B channels. Moreover, Ca2+-activated chloride currents were abolished in VSNs from Tmem16a conditional knock-out mice (cKO), demonstrating that TMEM16A is a necessary component of Ca2+-activated chloride channels in mouse VSNs (Amjad et al., 2015; Münch et al., 2018). Different results were obtained in Tmem16b KO mice, with a study reporting the lack of Ca2+-activated chloride currents in VSNs (Billig et al., 2011) and another one showing that disruption of Tmem16b only insignificantly reduced those currents (Münch et al., 2018).
In this study, we investigated the individual roles of TMEM16A and TMEM16B in controlling physiological VSN activity. We compared spontaneous and stimulus-induced firing in wild-type (WT), cKO for Tmem16a or mCherry Tmem16b KO (Zhang et al., 2017) mice. We found that only TMEM16A is involved in spontaneous firing patterns, while both TMEM16A and TMEM16B modulate the firing pattern of pheromone-evoked activity in VSNs.
Materials and Methods
Animals
Mice were handled in accordance with the Italian Guidelines for the Use of Laboratory Animals and European Union on animal research, under a protocol approved by the ethics committee of Scuola Internazionale Superiore di Studi Avanzati (SISSA) and approved by Italian Ministry of Health.
The generation of Tmem16a cKO mice has been previously described in detail (Amjad et al., 2015). Tmem16a cKO mice were homozygous for the floxed Tmem16a alleles and heterozygous for Cre and Omp (Li et al., 2004; Faria et al., 2014; Schreiber et al., 2015). Tmem16afl/fl mice were used as controls.
mCherry Tmem16b KO mice were kindly provided by Lily Jan (University of California, San Francisco, Zhang et al., 2017) and were generated by inserting mCherry sequence with a farnesylation signal at the C terminus (mCherry-F) in frame with the alternative start ATG codon in the third exon of Tmem16b, therefore membrane associated mCherry marks neurons that normally express Tmem16b (Zhang et al., 2017; see their Fig. S2C).
V2r1b-tau-GFP mice were provided by Peter Mombaerts (Max Planck Research Unit Neurogenetics, Frankfurt, Germany; Rodriguez et al., 1999).
All experiments were performed on tissues from two to three months old mice of either sex. All experiments and data analysis were performed by researchers who were blind to the mouse genotype.
Immunohistochemistry
VNO sections and immunohistochemistry were obtained as described previously (Dibattista et al., 2012; Amjad et al., 2015). The following primary antibodies were used: polyclonal goat anti-TMEM16A (1:200; sc-69 343, Santa Cruz), polyclonal rabbit anti-TMEM16B (1:500; NBP1-90739, Novus), rabbit anti-RFP (1:500; 600-401-379, Rockland), goat anti-OMP (1:1000; 544-1001, Wako). The following secondary antibodies obtained from Life Technologies (dilution; catalog number) were used: donkey anti-rabbit Alexa Fluor 594 Plus (1:500; A32754), donkey anti-goat Alexa Fluor 488 Plus (1:500; A32814). Immunoreactivity was visualized with a confocal laser scanning microscope (Nikon A1R). Images were acquired using NIS-Elements Nikon software (at 1024 × 1024-pixel resolution) and were not modified other than to balance brightness and contrast. Nuclei were stained by DAPI. Control experiments without the primary antibodies gave no signal.
Clarity and imaging
Brains and VNOs from adult mice (V2R1b-GFP-TMEM16Afl/fl-OMP/OMP and V2R1b-GFP-TMEM16Afl/fl-OMP/Cre) of both sexes were dissected. Brains were further sliced in 3 mm coronal sections including the AOB. To maintain structural integrity, the tissue was fixed overnight at 4°C in hydrogel fixation solution containing 4% acrylamide, 0.05% bis-acrylamide, 0.25% VA-044 Initiator (Wako Chemicals), 4% PFA in PBS. After polymerization, lipids were removed by passive clearing. The samples were incubated at 37°C for two months in 4% SDS and 200 mm boric acid at pH 8.5. The buffer was changed every other week. During the last clearing cycle, the nuclear marker DRAQ5 (1:1000, Thermo Fisher Scientific) was added to each sample. After washing steps, samples were incubated for 24 h in RIMS80, containing 80 g Nycodenz (PROGEN Biotechnik), 20 mm PS (phosphate buffer, pH 7.5), 0.1% Tween 20, and 0.01% sodium azide. Imaging was performed using a Leica SP8 DLS microscope equipped with a 488 and 633 nm diode laser, an HC PL FLUOTAR 5×/0.15 IMM DLS observing objective, an L 1.6×/0.05 DLS illuminating objective and a DLS TwinFlect 7.8 mm Gly mirror cap that creates the lightsheet. Specimens were positioned in a custom-made 3D-printed chamber.
Data were analyzed using IMARIS 9.5.1 (Oxford Instruments). AOB glomerular volume was determined by measuring the volume of all fluorescently labeled glomerular structures within the AOB. VNO cell density was measured by counting the number of fluorescently labeled cells in a defined volume of vomeronasal sensory epithelium (VNE). MATLAB R2019b (MathWorks) was used for plotting and statistical analysis. CorelDRAW 2019 (Corel Corporation) was used for figure preparation.
Preparation of acute slices of mouse VNO
Acute slices of mouse VNO were prepared as previously described (Shimazaki et al., 2006; Dibattista et al., 2008; Wong et al., 2018). In brief, the VNO was removed and transferred to ice-cold artificial CSF (ACSF) containing the following: 120 mm NaCl, 20 mm NaHCO3, 3 mm KCl, 2 mm CaCl2, 1 mm MgSO4, 10 mm HEPES, and 10 mm glucose, pH 7.4. The capsule and all cartilaginous tissues were carefully removed and the two halves of the VNO were isolated from the vomer bone. Each half of the VNO was then separately treated. The VNO was embedded in 3% Type I-A agarose (Sigma) prepared in ACSF once the agar had cooled to 38°C. Upon solidification, the agar block was fixed in a glass Petri dish and sliced with a vibratome (Vibratome 1000 Plus Sectioning System) at 200 to 250 μm thickness in oxygenated ACSF solution. Slices were then left to recover for >30 min in chilled and oxygenated ACSF before electrophysiological experiments were initiated.
Electrophysiological recordings from VSNs and stimuli presentation
Slices were viewed with an upright microscope (Olympus BX51WI) by infrared differential contrast optics with water immersion 20× or 60× objectives. The slice preparation maintained the VNO cross-sectional structure and many individual VSNs could be clearly distinguished by their morphology. Whole-cell or loose-patch recordings were obtained using patch pipettes pulled from borosilicate capillaries (WPI) with a PC-10 puller (Narishige) with a resistance of 3–6 MΩ. Recordings were obtained with a Multiclamp 700B amplifier controlled by Clampex 10 via a Digidata 1440 (Molecular Devices). Data were low-pass filtered at 2 kHz and sampled at 10 kHz. Experiments were performed at room temperature (20–25°C). The recording chamber was continuously perfused with oxygenated (95% O2, and 5% CO2) ACSF by gravity flow. The slice was anchored to the base of the recording chamber using a homemade U-shaped silver wire, holding down the agar support without touching the slice itself.
Membrane properties and inward and outward voltage-gated currents were measured in the whole-cell configuration with a KCl based intracellular solution containing the following: 80 mm K-gluconate, 60 mm KCl, 10 mm NaCl, 1 mm MgCl2, 0.023 mm CaCl2, 10 mm HEPES, and 10 mm EGTA, adjusted to pH 7.2 with KOH.
For recordings of Ca2+-activated currents, we used a CsCl based intracellular solution containing the following: 140 mm CsCl, 10 mm HEPES, and 10 mm HEDTA, adjusted to pH 7.2 with CsOH. The nominally 0 Ca2+ solution contained no added Ca2+. The 1.5 μm free Ca2+ solution was obtained by adding 3.209 mm CaCl2. The added concentration was the same as that previously used to obtain 1.5 μm free Ca2+ experimentally determined by Fura-4F (Invitrogen) measurements by using a LS-50B luminescence spectrophotometer (PerkinElmer; Pifferi et al., 2006, 2009a,b) and based on calculation with the program WinMAXC (C. Patton, Stanford University, Stanford, CA; Patton et al., 2004).
Extracellular recordings from the soma of VSNs were obtained in the on-cell loose-patch configuration with seal resistances of 40–100 MΩ. Pipette solution was ACSF, as the bath solution, and the recordings were made in voltage-clamp mode with a holding potential of 0 mV.
Stimuli were focally delivered through a gravity-fed multivalve perfusion system (ALA-VM8, ALA Scientific Instruments). The tip of the perfusion head, with a diameter of 360 μm, was placed ∼500 μm away from the slice. The time delay of the stimulus was measured using a high-K+ ACSF solution. To avoid mechanical artifacts, the slice was constantly perfused with ACSF and the flow out of the pipette was switched between ACSF and stimulus solutions. This results in a constant flow across the epithelium and sharp concentration transients, undiluted by the bath ACSF.
Urine collection and stimulus solutions
Urine was collected from both sexes C57BL/6 mice, filtered with a 0.2-μm filter, and frozen at −80°C. Before use, male and female urines were mixed in a 1:1 ratio, and the mixture was diluted to 1:50 in ACSF (pH 7.4). As mouse urine contains urea and K+, which could potentially cause neurons to fire by direct membrane depolarization, we used artificial urine diluted 1:50 in ACSF as a negative control. Artificial urine contained the following: 100 mm NaCl, 40 mm KCl, 20 mm NH4OH, 4 mm CaCl2, 2.5 mm MgCl2,15 mm NaH2PO4, 20 mm NaHSO4, and 333 mm urea, pH 7.4 adjusted with NaOH (Holy et al., 2000). To test cell viability, we used a high K+ solution (25 mm KCl) by replacing equimolar amounts of NaCl with KCl in ACSF. Diluted urine or artificial urine stimuli were applied in 10-s pulses with an interstimulus interval of at least 4 min to avoid desensitization or adaptation processes (Spehr et al., 2009; Wong et al., 2018). High K+ solution was applied in 5-s pulses. All compounds and chemicals were obtained from Sigma-Aldrich, unless otherwise stated.
Analysis of electrophysiological data
IgorPro software (WaveMetrics) and Clampfit (Molecular Devices) were used for data analysis and figure preparation. In loose-patch, slow changes in the baseline were corrected by filtering the recordings offline with a high-pass filter at 2 Hz. Spikes were initially detected using an event detection tool using an arbitrary threshold, and then confirmed by shape inspection. For spontaneous activity, the mean firing activity was estimated by dividing the number of spontaneous spikes by the duration of recordings in the absence of stimuli. Interspike interval (ISI) was calculated by measuring the time between each consecutive spike. Each ISI distribution was obtained by grouping the ISIs in bins, as indicated in the figures, and normalized by dividing the value for each bin by the total number of ISIs. The values in y-axes represent the percentage of spikes in each bin and the area under the curve is 100%.
For VSNs showing a high spontaneous activity, it was difficult to identify a urine response from a single recording. To avoid false positives, we applied at least three repetitive stimulations. Each recording was 90 s long divided into 40 s of prestimulus, 10 s of stimulus, and 40 s of poststimulus. To analyze the response to urine for a single cell we took the basal period (40 s prestimulation) and stimulus period (10 s of stimulation), and we calculated the basal and the stimulation frequency for each trace. We defined a threshold level as: BF + 2*σ2, where BF is the average of basal frequency for all the traces and σ2 is the SD of the basal frequency. A cell was considered responsive to urine if: (1) the average spike frequency was above threshold during stimulation; and (2) there was no response to diluted artificial urine.
Statistical analysis
Data are presented as mean ± SEM, with n indicating the number of neurons. VSNs were obtained from at least five (whole-cell recordings) or 10 (loose-patch recordings) different WT or mutant mice. Normal distribution of the data was tested with the Shapiro–Wilk test or with the Jarque–Bera test. For normally distributed data, statistical significance was determined using unpaired Student’s t tests. For multiple comparison of the data, we used Dunn–Hollander–Wolfe test after Kruskal–Wallis analysis. Kolmogorov–Smirnov test was used to compare cumulative distributions. p-values <0.05 were considered significant. Statistical analysis was performed with IgorPro software (Wavemetrics).
Results
TMEM16A and TMEM16B expression in the vomeronasal epithelium
TMEM16A and TMEM16B are co-expressed in microvilli of mouse VSNs (Dibattista et al., 2012; Münch et al., 2018) but their functional role is unclear. By immunohistochemistry, we confirmed that TMEM16A and TMEM16B are expressed at the apical surface of the vomeronasal epithelium of WT mice (Fig. 1A), whereas TMEM16A or TMEM16B immunoreactivity was absent, as expected, in Tmem16a cKO and Tmem16b KO mice, respectively.

TMEM16A and TMEM16B are expressed at the apical surface of the vomeronasal epithelium. A, Confocal micrographs of coronal sections of the VNO from WT, Tmem16a cKO, or Tmem16b KO mice, as indicated, immunostained with antibody against TMEM16A (green) and TMEM16B (red). No immunoreactivity to TMEM16A or TMEM16B was detectable in Tmem16a cKO or Tmem16b KO mice, respectively. B, Confocal micrographs of VNO sections from Tmem16b KO mice expressing mCherry in the membrane of cells that normally express TMEM16B, immunostained with antibody against mCherry (red) and OMP (green). At the bottom, a magnification showing the co-expression of mCherry and OMP. Note that mCherry is expressed, as expected, in the membrane of entire VSNs and not only at the apical side. Cell nuclei were stained by DAPI (blue).
mCherry Tmem16b KO mice (Zhang et al., 2017) allow visualization of those cells that normally express TMEM16B by membrane staining with farnesylated mCherry. Figure 1B clearly shows co-expression of OMP and mCherry in VSNs, confirming the expression of TMEM16B in VSNs and excluding TMEM16B expression in supporting cells of the VNO.
Ca2+-activated chloride currents in VSNs from WT, Tmem16a cKO, and Tmem16b KO mice
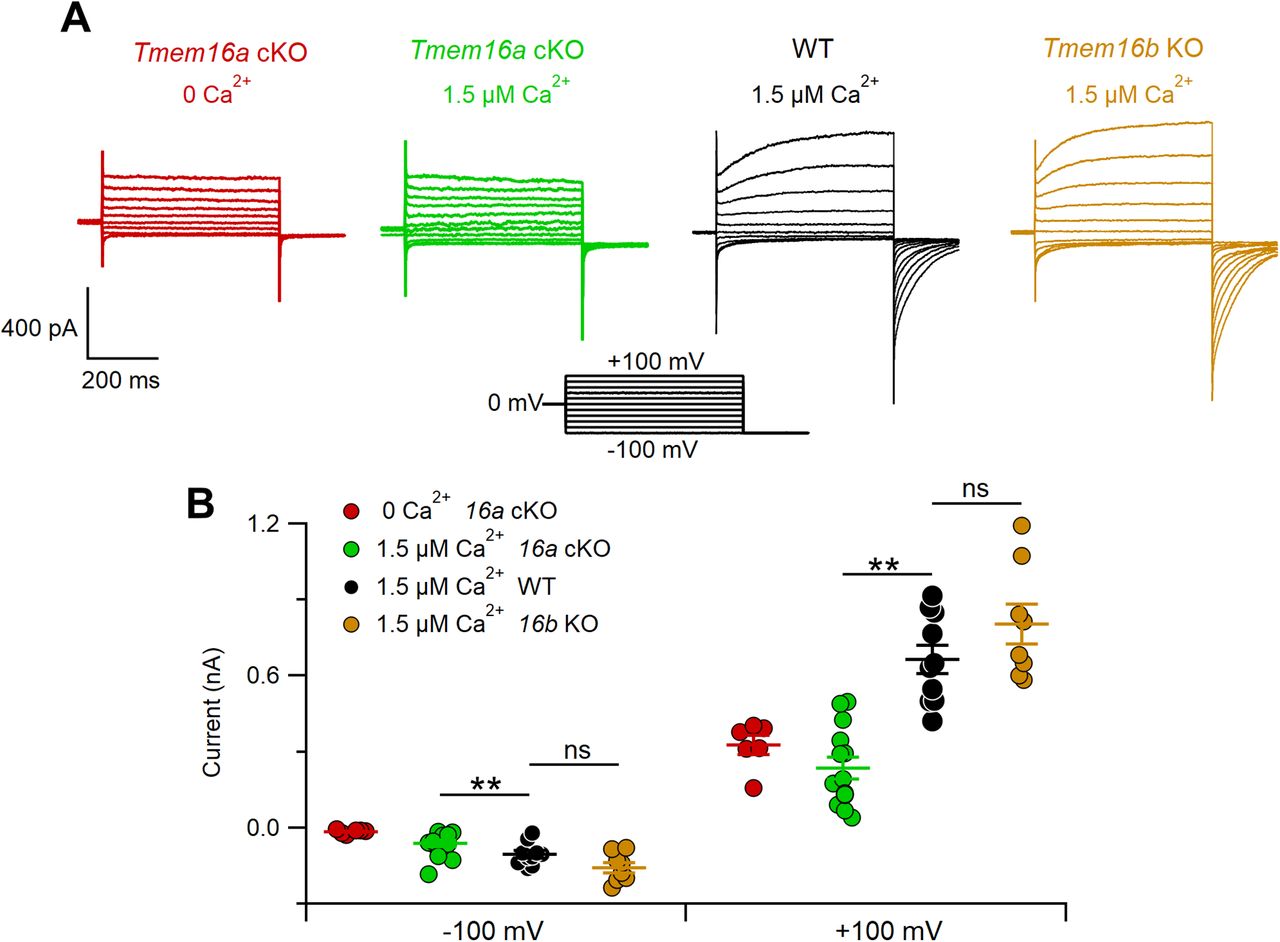
To determine the respective contributions of TMEM16A and TMEM16B to Ca2+-activated chloride currents in VSNs, we used intracellular solutions with nominally 0 Ca2+ or with 1.5 μm free Ca2+ and obtained whole-cell voltage-clamp recordings from the soma of neurons in acute VNO slices from WT, Tmem16a cKO, and Tmem16b KO mice (Fig. 2). To avoid contributions from K+ currents (Ca2+-activated or voltage-activated), the pipette solution contained Cs+ instead of K+. Figure 2A shows representative VSN recordings (holding potential Vhold = 0 mV) from each mouse line at voltage steps between −100 and +100 mV, followed by a step to −100 mV. In agreement with previous results (Amjad et al., 2015), we confirmed that VSNs from Tmem16a cKO mice lacked a significant Ca2+-activated chloride current in the presence of 1.5 μm Ca2+. Indeed, the average currents measured at steady state in 0 Ca2+ (−15 ± 3 pA at −100 mV and 326 ± 38 pA at +100 mV, n = 6) or in 1.5 μm Ca2+ (−62 ± 13 pA at −100 mV and 236 ± 41 pA at +100 mV, n = 14) were not significantly different (Fig. 2B). Furthermore, VSN currents activated by voltage steps in Tmem16a cKO mice did not show any time dependence (red and green traces), whereas WT currents in 1.5 μm Ca2+ (black traces) showed the typical kinetics characterized by an instantaneous current followed by an outward or inward relaxation at the most depolarizing or hyperpolarizing voltage steps, respectively. Recordings from VSNs of Tmem16b KO mice (brown traces; −157 ± 20 pA at −100 mV and 805 ± 79 pA at +100 mV, n = 8) revealed Ca2+-activated chloride currents similar to WT mice (−105 ± 14 pA at −100 mV and 666 ± 55 pA at +100 mV, n = 10), further corroborating previous findings that TMEM16A largely contributes to Ca2+-activated chloride currents in VSNs (Fig. 2B).

Ca2+-activated chloride currents in VSNs. A, Representative whole-cell recordings obtained with an intracellular solution containing 0 or 1.5 μm Ca2+ from VSNs from WT, Tmem16a cKO, or Tmem16b KO mice, as indicated. The voltage protocol is in the center of the figure. B, Scatter dot plot with average ± SEM of steady-state current amplitudes measured at −100 or +100 mV with intracellular pipette solution containing nominally 0 Ca2+ from Tmem16a cKO (red, n = 6), or 1.5 μm free Ca2+ from Tmem16a cKO (green, n = 14), WT (black, n = 10), and Tmem16b KO (brown, n = 8) mice (**p < 0.01; Dunn–Hollander–Wolfe test after Kruskal–Wallis analysis at –100 and +100 mV), ns: not significant.
Membrane properties and voltage-gated currents of VSNs from KO mice are not altered
To test whether Tmem16a or Tmem16b deletion changes VSN passive membrane properties and/or voltage-gated currents, we performed whole-cell recordings in the presence of K+ in the patch pipette.
Representative recordings of voltage-activated inward and outward currents from each mouse line are shown in Figure 3A. For I-V relations, we plotted the minimum values of inward currents during the first 20 ms after each voltage pulse (Fig. 3B, circles) and the outward current value at the end of each voltage pulse (Fig. 3B, triangles). Figure 3B shows that I-V relations of voltage-gated currents were similar in VSNs from WT, Tmem16a cKO, and Tmem16b KO mice.
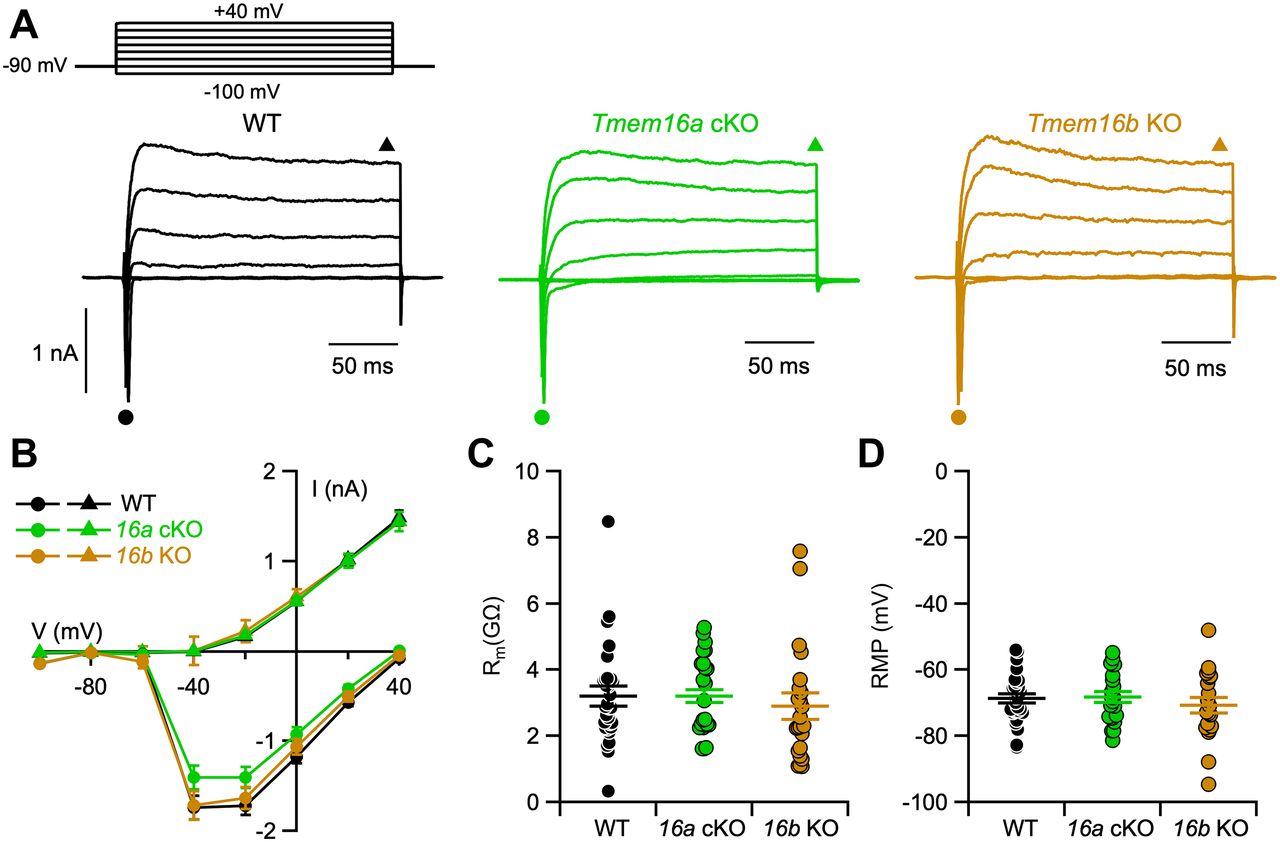
Tmem16a or Tmem16b deletion does not affect voltage-gated currents and membrane properties of VSNs. A, Representative whole-cell recordings of VSNs obtained from the indicated mouse lines. The holding potential was −90 mV and voltage steps from −110 to +30 mV with 20-mV increment were applied. B, Average ± SEM of the IV relationships of inward currents (circles) and steady-state outward currents (triangles) from WT (black, n = 31), Tmem16a cKO (green, n = 23), or Tmem16b KO (brown, n = 23) VSNs. Values were taken at the negative peak and at the end of voltage step as indicated by the symbols in A. C, Scatter dot plot with average ± SEM of the membrane resistance (Rm) recorded in voltage-clamp mode (WT, n = 31; Tmem16a cKO, n = 23; Tmem16b KO, n = 23. Kruskal–Wallis analysis p = 0.21). D, Scatter dot plot with average ± SEM of resting membrane potential (RMP) recorded in current-clamp configuration (WT, n = 28; Tmem16a cKO, n = 22; Tmem16b KO, n = 20. Kruskal–Wallis analysis p = 0.68).
Membrane input resistance was evaluated in voltage-clamp mode with a negative pulse of 20 mV from a holding potential of −90 mV, as in these conditions voltage-gated currents are not activated (Dibattista et al., 2008). The average membrane input resistances of VSNs from WT, Tmem16a cKO, and Tmem16b KO mice were 3.2 ± 0.3 GΩ (n = 31), 3.2 ± 0.2 GΩ (n = 23), and 2.9 ± 0.4 GΩ (n = 23), respectively (Fig. 3C). These values were not statistically different, indicating that membrane input resistance is not altered by either the lack of TMEM16A or TMEM16B.
The resting membrane potential was measured in current-clamp mode (Iinject = 0 pA) and was not statistically different between genotypes (Fig. 3D), with values of −69 ± 1 mV (n = 28) for WT, −68 ± 2 mV (n = 22) for Tmem16a cKO, and −71 ± 2 mV (n = 20) for Tmem16b KO VSNs.
These results indicate that the Ca2+-activated chloride channels TMEM16A and TMEM16B do not contribute to passive membrane properties of mouse VSNs and do not alter major voltage-gated inward and outward currents in the range from −100 up to +40 mV. As TMEM16A can also be activated by high positive voltages in absence of intracellular Ca2+ (Xiao et al., 2011; Segura-Covarrubias et al., 2020), these results show that TMEM16A does not alter passive properties at membrane potentials up to +40 mV.
Spontaneous firing pattern is altered in VSNs from Tmem16a cKO
It is well known that VSNs display spontaneous firing activity in the absence of stimuli (Holy et al., 2000; Arnson and Holy, 2011) and that changes in sensory transduction components may modify this activity, as for example VSNs from Trpc2 KO mice showed reduced spontaneous firing (Arnson and Holy, 2011). Moreover, experiments in the main olfactory epithelium have shown that deletion of TMEM16B, which is highly expressed in the cilia of olfactory sensory neurons (OSNs), caused a reduction of spontaneous firing activity in those neurons (Pietra et al., 2016). Based on these findings, we hypothesized that TMEM16A and TMEM16B might play a role in the spontaneous VSN firing.
By performing extracellular loose patch recordings from individual neurons in VNO slices from WT and mutant mice, we found that VSNs have a highly variable spontaneous activity among neurons of the same genotype with frequencies varying from 0 Hz (no spontaneous activity) to ∼4 Hz (Fig. 4A). The viability of each neuron was tested by depolarization with a high K+ solution to verify the neurons firing capability, and only VSNs responding to high K+ were included in the analysis. VSNs that did not show spontaneous activity were: six out of 50 for WT, five out of 52 for Tmem16a cKO, and one out of 34 for Tmem16b KO. Thus, at least 88% VSNs from each genotype showed spontaneous activity.

Tmem16a deletion modifies VSNs spontaneous firing activity. A, Raster plots of recordings of the spontaneous activity of VSNs from WT (black, n = 50), Tmem16a cKO (green, n = 52), or Tmem16b KO (brown, n = 34) mice. Each row shows spike activity from a different VSN. B, Mean frequency of spontaneous activity from the cells shown in A. C, ISI distributions of spontaneous firing from a subset of cells with mean frequency >0.1 Hz (bin = 10 ms). Values were normalized to the area under each curve to show the spike percentages for WT (black, n = 25), Tmem16a cKO (green, n = 18), or Tmem16b KO (brown, n = 20) mice. D, Cumulative fraction of ISI distributions (***p < 0.001, Kolmogorov–Smirnov test).
Although the average spontaneous frequency was not significantly different among the three genotypes (0.32 ± 0.08 Hz, n = 50 for WT; 0.16 ± 0.04 Hz, n = 52 for Tmem16a cKO, and 0.65 ± 0.18 Hz, n = 34 for Tmem16b KO; Fig. 4B), we measured differences in firing patterns. To analyze activity patterns, we calculated ISI distributions from neurons with a mean frequency >0.1 Hz and plotted the distributions for each mouse line (Fig. 4C). To avoid overrepresentation of neurons with higher versus those with lower spontaneous activity, we normalized the ISI distribution by dividing the number of spikes for each bin by the total number of spikes for each single VSN before adding it to create the ISI distribution for each genotype. Moreover, to compare data from the three genotypes, data were normalized to the area under the curve (Fig. 4C). Corresponding cumulative fraction plots are shown in Figure 4D. We found that ISI distributions of spontaneous firing were statistically different (p < 0.01) between WT and Tmem16a cKO VSNs, while they did not differ between WT and Tmem16b KO VSNs. Indeed, in WT and Tmem16b KO VSNs, ∼80% of ISIs range between 50 and 200 ms (black and brown traces, respectively). By contrast, the ISI distribution for Tmem16a cKO neurons is broader showing only ∼50% of activity in the 50- to 200-ms ISI range (green traces). These results show that short ISIs were reduced in Tmem16a cKO compared with WT neurons (Fig. 4C,D), indicating that TMEM16A, but not TMEM16B, exerts firing pattern control during spontaneous activity of VSNs.
Deletion of TMEM16A and resulting changes in spontaneous firing do not modify axonal targeting to AOB glomeruli
In the main olfactory epithelium, spontaneous activity of OSNs is essential for both formation and maintenance of the glomerular map in the OB (Yu et al., 2004; Lodovichi and Belluscio, 2012; Lorenzon et al., 2015). Furthermore, deletion of TMEM16B, the Ca2+-activated chloride channel in OSNs, altered spiking and affected the glomerular formation of I7-expressing OSNs (Pietra et al., 2016). While the relation between spiking pattern and glomerular formation has been investigated in the OB, similar studies in the AOB were missing. To investigate whether spontaneous firing pattern alteration, resulting from TMEM16A deletion, affects VSN density or glomeruli formation in the AOB, we crossed Tmem16a cKO with V2R1b-GFP (Del Punta et al., 2002) mice to generate a mouse line in which V2R1b expressing VSNs were fluorescently labeled, but lack TMEM16A (V2R1b-GFP-TMEM16A-cKO). V2R1b-GFP-TMEM16A cKO mice were homozygous for V2R1b-GFP and Tmem16a-Flox genes, and heterozygous for OMP/Cre, while control animals were homozygous for V2R1b-GFP, Tmem16a-Flox and OMP genes, lacking the expression of Cre recombinase. We performed VNO and forebrain tissue clearing to quantify and compare (1) the number of fluorescently labeled neurons per VNO, and (2) the V2R1b-specific axonal glomerular architecture in the AOB, between TMEM16A-deficient V2R1b-GFP mice and corresponding controls (Fig. 5). Figure 5A–C shows an example of a cleared VNE observed with a light sheet microscope and the corresponding 3D reconstruction (Movie 1). Single VSNs were readily observed, and cellular density was calculated (Fig. 5D). We found no significant statistical difference in VSN density between V2R1b-GFP-WT and V2R1b-GFP-TMEM16A-cKO mice (Fig. 5D, n = 5–7). These results indicate that deletion of TMEM16A does not affect the number of V2R1b-expressing VSNs.

Tmem16a cKO effect on VSN density and glomerular volume. A–C, top, Representative 3D projection image of a VNE from a V2R1b-WT mouse. Zoom-in indicated by white rectangles. Arrows indicate example fluorescently labeled V2R1b neurons. D, Scatter dot plot (average ± SEM) of VSN density in V2R1b-WT and V2R1b-Tmem16a cKO mice (Student’s t test, p = 0.1876). E–G, top, Representative 3D projection image of AOB from V2R1b-WT mouse. Zoom-in indicated by white rectangles. Arrows indicate exemplary fluorescently labeled V2R1b glomerular structures. H, Scatter dot plot with average ± SEM of glomerular volume in V2R1b-WT and V2R1b-Tmem16a cKO mice (Student’s t test, p = 0.2573).
Next, we evaluated the glomerular architecture in the AOB. We identified several glomeruli innervated by V2R1b axons, confirming previous reports (Del Punta et al., 2002). Given the variability in glomerular number between animals from the same mouse line, ranging between 2 and 10 glomeruli per AOB (Del Punta et al., 2002), we decided to evaluate whether the total volume of AOB glomeruli was affected by TMEM16A deletion. Figure 5E–G shows a representative 3D reconstruction of the AOB with glomeruli formed by V2R1b-GFP axons (Movie 2). Single glomeruli were discernable (Fig. 5G, white arrows). A comparison of GFP-positive glomerular volume between V2R1b-WT and V2R1b-cKO mice show no significant statistical difference (Fig. 5H, n = 5–6). These results indicate that deletion of TMEM16A protein in VSNs and resulting alterations in firing do not affect general glomerular development in the AOB.
Light-sheet fluorescence microscopy of cleared VNOs. Examples show intact transparent organs from wild type control (V2R1b-GFP - TMEM16Afl/fl - OMP/OMP; left) and TMEM16A knockout (V2R1bGFP - TMEM16Afl/fl - OMP/Cre; right) animals. Three-dimensional rendering displays the VNOs rotating around their longitudinal axes.
Light-sheet fluorescence imaging of cleared AOBs. Examples show intact transparent olfactory bulb samples from wild type control (V2R1b-GFP - TMEM16Afl/fl - OMP/OMP; left) and TMEM16A knockout (V2R1bGFP - TMEM16Afl/fl - OMP/Cre; right) mice. Nuclei are stained with DRAQ5 (grey scale). Green fluorescence of V2R1b-positive vomeronasal sensory neuron axons becomes apparent upon fiber coalescence and convergence in AOB glomeruli.
Response to sensory stimulation is altered in VSNs from KO mice
Next, we asked whether TMEM16A or TMEM16B deletion affects VSN responses to sensory stimulation. Thus, we stimulated VSNs with diluted mouse urine, a natural source of pheromones that has been frequently used to activate VSNs (Mohrhardt et al., 2018). While recording in loose-patch configuration, we stimulated VSNs with diluted urine and recorded the evoked firing activity. Stimulus application was repeated at least three times to avoid the detection of false-positive responses in spontaneously active neurons (Fig. 5). The viability of each VSN was tested by using a short depolarizing high K+ solution stimulus (Fig. 6A), where unresponsive neurons were discarded. Furthermore, as urine also contains various ions including K+ that could depolarize neurons (see Materials and Methods), we stimulated VSNs with diluted artificial urine (Fig. 6B) Neurons responding to the artificial urine control stimulus were excluded from the analysis. Finally, as VSNs display spike frequency adaptation to repetitive stimulation (Wong et al., 2018), we selected an interstimulus interval between urine application of 4 min to avoid adaptation.

Evoked firing activity. A–C, Representative loose-patch recordings of a VSN from each indicated mouse line stimulated with high K+ (A), diluted artificial urine as control (B), or diluted urine (C). Black bars indicate the time of stimulus presentation. D, Average firing activity (1-s bin width) for the responses to diluted artificial urine (empty circles) or urine (filled circles) from B, C. Error bars represent SEM.
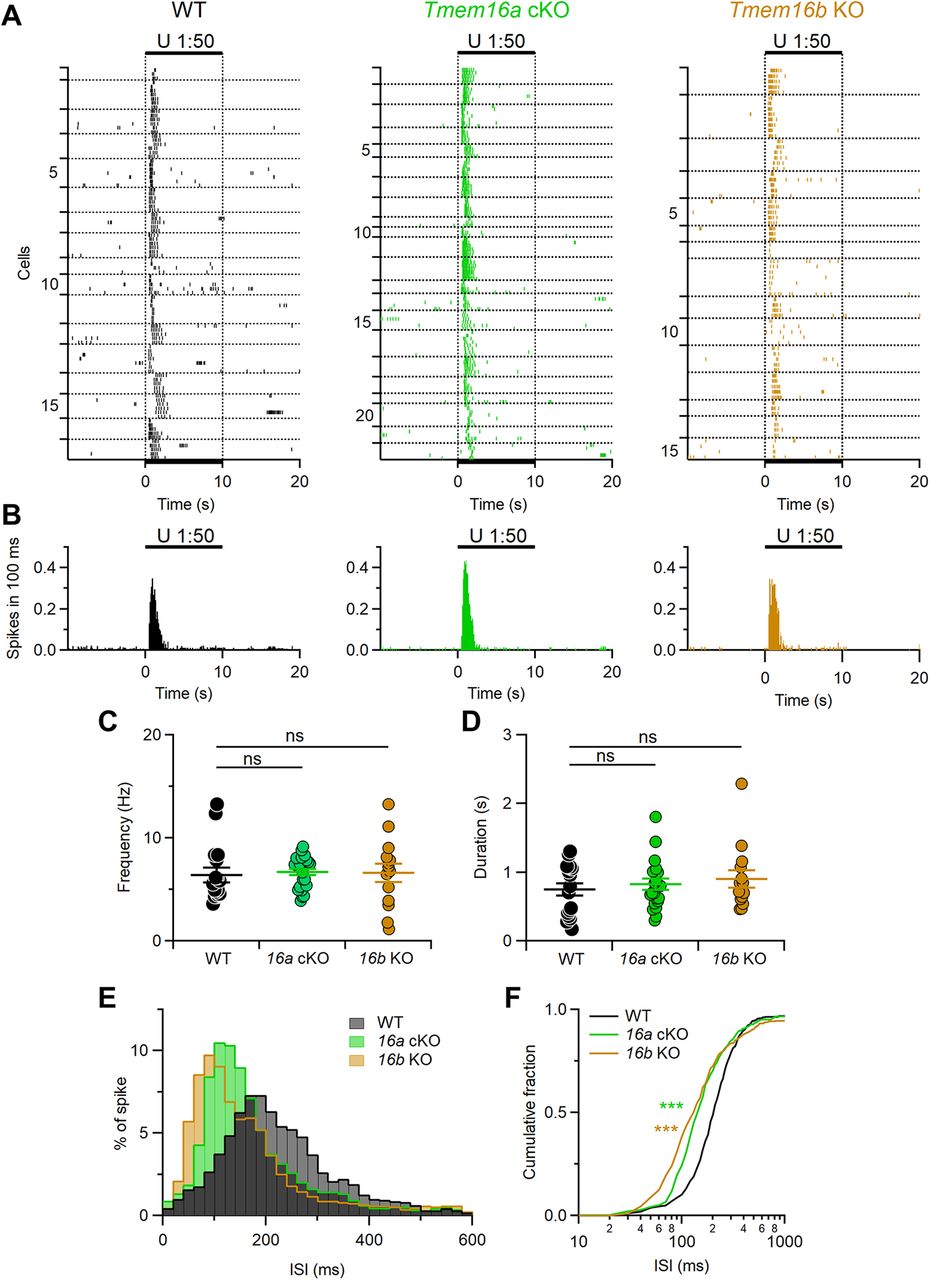
Patterns of evoked firing activity are modulated by TMEM16A and TMEM16B. A, Raster plots showing the responses of VSNs from each indicated mouse line to diluted urine. Black bars indicate the time of stimulus presentation (10 s). Every neuron was tested at least three times. Horizontal dotted lines delimit multiple recordings from each VSNs. Numbers on the left indicate the neuron number. B, Histograms of the number of spikes from neurons in A (100-ms bin width). Each neuron was averaged to equally contribute to spikes count. C, D, Scatter dot plots with average ± SEM of the mean frequency (C, Kruskal–Wallis analysis p = 0.37) and duration (D, Kruskal–Wallis analysis p = 0.79) of the responses to urine of VSNs from WT (black, n = 17), Tmem16a cKO (green, n = 22), or Tmem16b KO (brown, n = 15) mice. E, Normalized ISI distributions of firing activity (20-ms bin width). F, Cumulative fraction of the ISI distributions shown in D (***p < 0.001, Kolmogorov–Smirnov test).
Figure 6 shows representative responses of single VSNs from WT or mutant mice. A 10-s stimulus of diluted urine was applied four times and, each time, clearly elicited an increase in firing frequency (Fig. 6C), whereas application of diluted artificial urine did not alter spiking activity (Fig. 6B). The plot of average firing frequency (1-s-bin width) shows a clear frequency increase during urine application in VSNs from either genotype (Fig. 6D).
Figure 7 shows reliable responses to repeated urine stimulation in 17 out of 30 WT, 22 out of 30 Tmem16a cKO, and 15 out of 34 Tmem16b KO VSNs, indicating that >45% VSNs from each genotype can respond to urine (Fig. 7A,B).
Although the mean firing frequency and response duration to urine were not significantly different among genotypes (Fig. 7C,D) a more detailed analysis showed that patterns of firing responses were altered by deletion of TMEM16A or TMEM16B (Fig. 7E,F). Indeed, ISI distributions and cumulative fractions of mutant mice were different from those of WT. ISI distribution for WT showed a peak ∼200 ms, while in the ISI distribution from Tmem16a cKO and Tmem16b KO the peak was shifted to a shorter ISI of ∼100 ms. These results indicate that both TMEM16A and TMEM16B modulate firing activity during signal transduction in mouse VSNs.
Discussion
In this study, we investigated the individual roles of TMEM16A and TMEM16B in regulating the physiological activity of VNO neurons. We found that TMEM16A, but not TMEM16B, modulates spontaneous spike patterns by increasing activity, while both channels shape pheromone-mediated spike activity by increasing ISIs.
Expression of TMEM16A and TMEM16B in olfactory systems
While the presence of Ca2+-activated chloride currents in neurons of the main olfactory system is known since the early 1990s (Kleene and Gesteland, 1981; Kleene, 1993; Kurahashi and Yau, 1993; Lowe and Gold, 1993; Boccaccio and Menini, 2007; Pifferi et al., 2012; Dibattista et al., 2017), the first recordings of Ca2+-activated currents from VSNs have been obtained more recently (Yang and Delay, 2010; Kim et al., 2011; Dibattista et al., 2012; Amjad et al., 2015).
Ca2+-activated chloride channels encoded by TMEM16A and TMEM16B are characterized by functional differences. These include slower activation kinetics and higher Ca2+ sensitivity for TMEM16A compared with TMEM16B. The complexity is further increased by the existence of several spice variants with different properties (Ferrera et al., 2009; Huang et al., 2012; Ponissery Saidu et al., 2013; Pedemonte and Galietta, 2014).
In the main olfactory epithelium, TMEM16A and TMEM16B are expressed in different cell types: TMEM16B is expressed in the dendritic ends and cilia of OSNs (Stephan et al., 2009; Sagheddu et al., 2010; Billig et al., 2011), whereas TMEM16A is present in apical membranes and microvilli of a subpopulation of supporting cells (Henriques et al., 2019; Maurya and Menini, 2014). In the vomeronasal epithelium, however, both TMEM16A and TMEM16B are found in VSNs (Billig et al., 2011; Dibattista et al., 2012; Amjad et al., 2015; Münch et al., 2018), where they co-express in microvilli (Dibattista et al., 2012; Münch et al., 2018). Here, we confirmed previous results that TMEM16A is present only in mature VSNs, but not in VNO supporting cells (Amjad et al., 2015; Münch et al., 2018). By taking advantage of mCherry Tmem16b KO mice (Zhang et al., 2017), we showed that TMEM16B is also selectively expressed in mature VSNs. Thus, vomeronasal TMEM16A and TMEM16B are exclusively expressed in mature VSNs. The relative expression level of the two channels has been evaluated in the transcriptomic study of Ibarra-Soria et al. (2014). They found high expression for both transcripts, with Tmem16a levels about twice that of Tmem16b, without significant difference between males and females (Ibarra-Soria et al., 2014; see their Dataset S1 and Dataset S3).
In OSNs, deletion of TMEM16B completely abolished Ca2+-activated chloride currents measured in whole-cell recordings (Billig et al., 2011; Pietra et al., 2016). In VSNs, the deletion of TMEM16A also completely abolished the current activated by 1.5 μm Ca2+ and measured with voltage steps up to +100 mV. As VSNs also express TMEM16B we expected to measure substantial residual current in the absence of TMEM16A. Indeed, although TMEM16B is less sensitive to Ca2+ than TMEM16A, it should be activated by 1.5 μm Ca2+ at +100 mV, as an EC50 of ∼2 μm has been reported at high voltages (Cenedese et al., 2012; Adomaviciene et al., 2013). We also attempted to increase intracellular Ca2+ to 13 and 100 μm, respectively, but VSNs in slices became leaky with these solutions. However, previous experiments in isolated VSNs successfully increased intracellular Ca2+ up to 2 mm without finding any measurable Ca2+-activated chloride current in VSNs from Tmem16a cKO mice (Amjad et al., 2015). Yet, as the average whole-cell leak current at +100 mV was ∼300 pA, we cannot exclude that we missed a small current activated by TMEM16B. Lack of current in Tmem16a cKO mice was also measured by Münch et al. (2018) who suggested the possibility that Ca2+ concentration in the microvilli does not reach the concentration necessary to activate TMEM16B. We found that deletion of TMEM16B did not significantly reduce Ca2+-activated chloride currents in VSNs confirming that most current results from TMEM16A activation. Our result thus agrees with recently published findings (Münch et al., 2018). By contrast, our data contradict a previous report showing a lack of Ca2+-activated chloride currents in VSNs from Tmem16b KO mice (Billig et al., 2011).
It is of interest to note that TMEM16A and TMEM16B are co-expressed in pinealocytes where they can form both homomeric and heteromeric channels (Yamamura, 2018), raising the possibility that VSNs also express heteromeric channels.
Spontaneous and stimulus-evoked spike activity
In both OSNs and VSNs, Ca2+-activated chloride channels are expected to play a role in the response to odorants or pheromones, as they are located close to transduction sites, i.e., in OSN cilia and VSN microvilli. This subcellular distribution is well suited to sense high Ca2+ concentrations produced in microdomains by transduction cascades downstream odorant or vomeronasal receptor activation (Tirindelli et al., 2009; Pifferi et al., 2010). Indeed, although the transduction cascade components in the two olfactory systems are very different, transduction currents produce an increase in intracellular Ca2+ in both cases. The resulting membrane depolarization triggers generation of action potentials that are conducted along OSN or VSN axons to the main or AOB, respectively. To understand the physiological role of TMEM16 channels in peripheral sensory neurons of olfactory systems, it is critical to note that both OSNs and VSNs maintain high internal chloride concentrations (Reuter et al., 1998; Kaneko et al., 2004; Kim et al., 2015; Untiet et al., 2016). Therefore, Ca2+-activated chloride channels are well suited to play an excitatory transduction role by amplifying the depolarizing cation current through chloride efflux. In addition, these channels might also play a stabilizing role by setting the membrane potential close to the chloride equilibrium potential, preventing excessive depolarization.
In OSNs, extracellular loose patch recordings of spontaneous firing activity showed that TMEM16B increased spontaneous firing, as TMEM16B-deficient OSNs exhibited a reduction in instantaneous frequency (inverse of ISI) of spontaneous activity (Pietra et al., 2016). This likely results from random activation of signal transduction proteins that produces a Ca2+ increase sufficient to open ciliary TMEM16B channels that contribute small depolarizations and firing activity. As VSNs also display a spontaneous activity that is dependent on signal transduction cascade components (Arnson and Holy, 2011), we hypothesized that TMEM16 channels could also affect spontaneous VSN firing. Indeed, deletion of TMEM16A, but not of TMEM16B, caused differences in ISI distribution: the percentage of ISIs between 50 and 200 ms was reduced to 50%, compared with 80% in WT VSNs. The decrease in spontaneous activity after TMEM16A deletion indicates that TMEM16A favors spike generation, probably by mediating a chloride efflux that further depolarizes the membrane after spontaneous activation of signal transduction components, similarly to TMEM16B in OSNs. As TMEM16A has a higher Ca2+ sensitivity than TMEM16B, a possible explanation for the lack of contribution of TMEM16B in VSNs is that the spontaneous activation of the signal transduction cascade leads to intracellular Ca2+ increase, which is insufficient to activate TMEM16B. Moreover, Tmem16b transcript expression level has been reported to be half of that of Tmem16a in VSNs (Ibarra-Soria et al., 2014) indicating that TMEM16B, in addition to being less sensitive to Ca2+, may also have a lower expression than TMEM16A.
In the main olfactory system, TMEM16B is also involved in correct OSN glomerular targeting, as its deletion caused the formation of supernumerary glomeruli for neurons expressing the I7 odorant receptor (Pietra et al., 2016). In the vomeronasal system, deletion of TMEM16A did not produce modifications of glomerular targeting for VSNs expressing the V2R1b receptor, although we cannot exclude the possibility that this deletion may affect glomerular targeting of VSNs expressing other receptors.
In OSNs, TMEM16B has been shown to have a dual role in the control of firing, by increasing basal spiking but reducing stimulus-induced firing activity. In VSNs, we found that TMEM16A also plays a similar dual role. Indeed, individual loss of TMEM16A (or TMEM16B) strongly affected urine-evoked firing patterns by shifting the peak in the ISI distribution to ∼100 ms from the value of 200 ms in WT. Thus, both TMEM16A and TMEM16B channels play a role in regulating VSN firing activity evoked by natural stimuli by doubling ISI. Stimulation of VSNs with diluted urine can forcefully activate the signal transduction cascade generating a stronger depolarization and higher cytosolic Ca2+ increase compared with spontaneous activation; under these conditions, TMEM16A and/or TMEM16B could first mediate a chloride efflux that amplifies the depolarization followed by a chloride influx, when the membrane potential becomes more positive than the chloride equilibrium potential, leading to membrane repolarization and increase in the ISI. Indeed, the internal chloride concentration in dendritic knobs of VSNs at rest has been estimated to be 42 mm (Untiet et al., 2016). In our experimental conditions, this would produce an equilibrium potential for chloride near −30 mV. Consequently, opening of Ca2+-activated chloride channels would lead to depolarization at lower membrane potential and to repolarization at increased membrane potential.
Another study used Tmem16a/Tmem16b double KO mice (Münch et al., 2018) and reported a loss of spontaneous spiking and drastically diminished stimulus evoked spiking in VSNs indicating a relevant physiological role of these channels. Recordings and/or data analysis were somehow different from ours as they reported that only ∼30% of VSNs had spontaneous activity while we measured spontaneous activity in almost 90% of VSNs from WT mice, in agreement with previous reports (Holy et al., 2000; Nodari et al., 2008; Arnson and Holy, 2011; Wong et al., 2018). This difference could stem from either sample viability, as depolarization controls were not reported, or different recording conditions. A more detailed study of firing activity should be performed in VSNs from the double KO model to better elucidate firing patterns. The same study performed a behavioral test showing that male-male territorial aggression, an innate VNO-dependent behavior in rodents, remained unaltered in double KO Tmem16a/Tmem16b mice.
It is important to note that our criteria for selecting neurons responding to urine are more stringent with respect to those of other studies (Kim et al., 2011; Münch et al., 2018) and could have excluded some VSNs with very high spontaneous firing frequencies. We argue that our method, similar to that used by Arnson and Holy (2011), is suitable to limit the analysis to a subset of clearly viable neurons.
In summary, our data provide evidence that both TMEM16A and TMEM16B modulate the pheromone-mediated spike activity of VSNs and provide the foundation for future work investigating the precise physiological role of the Ca2+-activated chloride currents in VSNs.
Acknowledgments
Acknowledgements: We thank Anna Boccaccio (CNR, Genova), Michele Dibattista (University of Bari A. Moro), and all members of the laboratory for discussions; Elettra Grdina, Angel Pascual Camerota, Lorenzo Maschietto, Giovanni Tamburin, and Cristina Degrassi (SISSA) for mice handling and Helena Krmac and Christina Vlachouli for mice genotyping; Corinna H. Engelhardt, Stefanie Kurth, and Jessica von Bongartz (RWTH-Aachen University) for excellent technical assistance; and Paul A. Heppenstall (SISSA, Trieste) for providing mCherry antibody.
Footnotes
The authors declare no competing financial interests.
This work was supported by Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) Grants 368482240/GRK2416 (to R.D. and M.S.) and the German Academic Scholarship Foundation (to R.D.).
This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International license, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



