

Abstract
Mutations in the voltage-gated sodium channel gene SCN1A are associated with human epilepsy disorders, but how most of these mutations alter channel properties and result in seizures is unknown. This study focuses on two different mutations occurring at one position within SCN1A. R1648C (R-C) is associated with the severe disorder Dravet syndrome, and R1648H (R-H), with the milder disorder GEFS+. To explore how these different mutations contribute to distinct seizure disorders, Drosophila lines with the R-C or R-H mutation, or R1648R (R-R) control substitution in the fly sodium channel gene para were generated by CRISPR-Cas9 gene editing. The R-C and R-H mutations are homozygous lethal. Animals heterozygous for R-C or R-H mutations displayed reduced life spans and spontaneous and temperature-induced seizures not observed in R-R controls. Electrophysiological recordings from adult GABAergic neurons in R-C and R-H mutants revealed the appearance of sustained neuronal depolarizations and altered firing frequency that were exacerbated at elevated temperature. The only significant change observed in underlying sodium currents in both R-C and R-H mutants was a hyperpolarized deactivation threshold at room and elevated temperature compared with R-R controls. Since this change is constitutive, it is likely to interact with heat-induced changes in other cellular properties to result in the heat-induced increase in sustained depolarizations and seizure activity. Further, the similarity of the behavioral and cellular phenotypes in the R-C and R-H fly lines, suggests that disease symptoms of different severity associated with these mutations in humans could be due in large part to differences in genetic background.
Significance Statement
Knock-in Drosophila lines were generated by CRISPR-Cas9 gene editing to explore how the R1648C (R-C) and R1648H (R-H) mutations in the SCN1A sodium channel gene contribute to distinct epilepsy disorders, Dravet syndrome, and GEFS+, respectively. Drosophila heterozygous for the R-C or R-H mutation displayed spontaneous seizures exacerbated at high temperature. GABAergic neurons in both mutants exhibited sustained depolarizations, interrupting neuronal firing, that increased in incidence at elevated temperature. A hyperpolarized deactivation threshold in R-C and R-H sodium currents was constitutive, implicating interaction with other heat-sensitive processes to alter firing properties. The similarity of the behavioral and cellular phenotypes in the R-C and R-H fly lines suggests that disorders of different severity in humans could be due in large part to differences in genetic background.
Introduction
Mutations in voltage-gated sodium channels are implicated in a range of human seizure disorders (Escayg and Goldin, 2010). In the sodium channel gene SCN1A, there are >1250 dominantly inherited mutations identified in patients with epilepsy. Though the loci of these mutations are known, how individual mutations lead to patient symptoms is largely unknown.
The NaV1.1 sodium channel, encoded for by SCN1A, is composed of four homologous domains (I–IV), each with six transmembrane-spanning segments (termed S1–S6; Catterall, 2017). The fourth segment of each domain (S4) has conserved positively charged amino acids at every third position, notably arginines, which are important in activating the channel in response to depolarizing membrane potential (Shen et al., 2017; Pan et al., 2018). The S4 segment in domain IV is unique in that its movement is slower, and the movement uncovers a binding site for the inactivation loop in the pore (Chen et al., 1996; Chanda and Bezanilla, 2002; Goldschen-Ohm et al., 2013). In this study, we focused on two mutations in the S4 segment of domain IV, R1648H (R-H) and R1648C (R-C). Although the mutations occur at the same amino acid location within the channel, they lead to different forms of epilepsy. R1648H causes generalized epilepsy with febrile seizures plus (GEFS+) in patients (Baulac et al., 1999), while R1648C leads to Dravet syndrome (DS; Ohmori et al., 2002; Striano et al., 2008). GEFS+ is usually a moderate form of epilepsy, characterized by febrile seizures that persist beyond the age of 6 years. DS is generally more severe and is characterized by delayed development and impaired motor functions in addition to seizures. Both types of epilepsy feature febrile as well as spontaneous seizures. In addition, patients with these types of epilepsy are frequently resistant to anticonvulsant drugs (Ogiwara et al., 2007; Escayg and Goldin, 2010). As the connection between individual mutations and patient symptoms remains largely unknown, we aim to model patient-specific epilepsies in the laboratory.
Studies of isolated sodium currents in oocytes, HEK cells, and computational models found that the R1648H mutation resulted in a decreased use dependence and faster recovery from inactivation. In addition, both R1648H and R1648C mutant sequences resulted in large persistent sodium currents during sustained depolarization not seen in the wild-type channels (Spampanato et al., 2001; Lossin et al., 2002; Rhodes et al., 2004; Vanoye et al., 2006; Kahlig et al., 2010). Later studies using a knock-in mouse model of R1648H demonstrated that this mutation resulted in animals that displayed reduced threshold to heat-induced seizures. Analysis of isolated neurons indicated decreased repetitive firing selectively in bipolar inhibitory neurons, which has been attributed to reduced sodium currents with slower recovery from inactivation and increased use dependence inactivation (Tang et al., 2009; Martin et al., 2010). Surprisingly, there were no changes in persistent current levels, which had been a key result from heterologous expression system studies. These findings demonstrate that a single mutation can result in distinct underlying mechanisms of cellular dysfunction in heterologous expression systems and excitable cells. However, since there is no R1648C mouse model to date, it has not been possible to explore similarities and differences in changes associated with R-H versus R-C in mouse neurons. In addition, it is unknown how temperature affects channel function for either mutation.
To evaluate the effect of the same mutation at a single locus in excitable cells, and to explore the effects of temperature on both cellular and behavioral phenotypes, we used the Drosophila model. One advantage of using Drosophila is that they have only a single sodium channel gene compared with mammals, which have multiple genes encoding voltage-gated sodium channels including three that are highly expressed in the adult brain (SCN1A, SCN2A, and SCN8A; Escayg and Goldin, 2010). The Drosophila voltage-gated sodium channel gene (para) encodes a protein (Para) similar in structure and function to NaV1.1 (Loughney et al., 1989; O’Dowd et al., 1989). Previous studies showed that knock-in animals homozygous for the SCN1A mutation K1270T causing GEFS+ or S1231R causing DS displayed heat-induced seizure phenotypes with distinct properties in the two mutant lines that were not seen in the controls (Sun et al., 2012; Schutte et al., 2014, 2016). It was also noted that the mutations caused distinct alterations to sodium channel activity in homozygous neurons. In the present study, CRISPR-Cas9 gene editing was used to insert R1648C, R1648H, or R1648R (R-R) into the Drosophila sodium channel gene. Behavioral and electrophysiological studies were performed on R-C and R-H heterozygous mutants and R-R controls.
Materials and Methods
Drosophila generation
A plasmid expressing the DsRed marker fused to an eye-specific promoter and flanked by worm sequences and two arms (2 and 1.4 kb) homologous to para, as well as plasmids expressing guide RNA (gRNA), were injected into fly embryos that expressed Cas9 under the germ cell promoter vas. Successfully transformed adults were selected based on DsRed expression in their eyes. The para gene is located on the X chromosome in Drosophila, so disruption of the para gene in DsRed flies resulted in lethality in hemizygous males and homozygous females. The line was maintained by balancing the mutant X chromosome with FM7.
In the second step, a plasmid that expresses gRNA targeted to the worm sequences flanking DsRed, along with a plasmid containing the para gene exon 30 containing a point mutation, were injected into vas-Cas9 embryos. The repair template carried at the 1648 aa codon position either the desired mutation R-C (CGA to TGT) and R-H (CGA to CAT) or a silent mutation R-R (CGA to CGT) that serves as our control. Injected fly embryos that matured may produce eggs with the restored but mutated para gene. Females were crossed with FM7/Y males.
For the R-C and R-R transformation, the presence of the mutated para gene rescued male lethality, so males with non-bar-shaped eyes (expressing mutated para on the manipulated X chromosome) were used as founders for line propagation and then were sequenced for genotype verification. For the R-H transformation, male lethality was not rescued. Female flies were screened for the loss of DsRed, then were used as founders and sequenced. Sequencing of ∼1.7 kb of DNA surrounding the targeted mutation site and gRNA cut sites confirmed the presence of the predicted base pair change in each line with no additional mutations, other than a few silent mutations where two different codons for the same amino acid were present in the population. Multiple, independent lines were obtained, backcrossed for five generations on a UAS-GFP; w+ background, and maintained. R-C and R-H mutations are homozygous lethal, so all lines are maintained as heterozygous stocks over the X chromosome balancer FM7.
Seizure behavior test
To simulate febrile conditions that induce seizures in individuals with GEFS+ and DS, flies 1 d posteclosion were placed in groups of five in empty vials that were then immersed in a water bath, first at room temperature and then at high temperature for 2 min. An external bath temperature of 40°C was selected as it was the lowest temperature that induced clear seizure-like behavior in all mutant flies within the 2 min trial period. The maximum temperature inside the vial after 2 min was ∼33°C (see Fig. 2B). Video recordings of the assay were evaluated, and the number of flies seizing in each vial at 20 s intervals was determined. A seizure was defined as a fly exhibiting a whole-body twitch or jump, or falling onto its back or side, often accompanied by leg twitching or wing flapping. Researchers were blinded to genotype during analysis. Data from multiple experiments were used to determine the average probability of seizure for a fly of a certain genotype at any second during the assay.
Life-span assay
Three groups of 10 newly eclosed female flies from both mutant lines, R1648C and R1648H, as well as the control line R1648R, were separated by genotype and placed in vials laid horizontally containing standard cornmeal-based food. The flies were housed under a 12 h light/dark cycle at 23°C. Fatalities were recorded every 1–3 d, and food vials were changed twice weekly. Survival curves were created using the Kaplan–Meier estimate and analyzed using the Mantel–Cox log-rank test.
Electrophysiology
Whole-brain dissections were conducted on adult female mutant or control flies 2 d posteclosion. Briefly, the fly was anesthetized on ice for 1 min, then decapitated. The eyes were cut and mouth parts removed, and the rest of the head was placed into an external solution (described below) also containing a papain suspension. After 5 min, the brain was removed from the cuticle and connective tissue was removed. Neurons targeted for recording were located in the dorsal antennal lobe. Local neurons (LNs) were distinguished from projection neurons based on the soma size (∼10 vs 6 μm) and action potential amplitude (∼30–40 vs 2–5 mV).
Whole-cell sodium currents and depolarization-evoked action potentials were recorded with whole-cell pipettes of 9–11 MΩ. Sodium currents were recorded using a pipette solution containing the following (in mm): 102 CsOH, 102 d-gluconic acid, 17 NaCl, 0.085 CaCl2, 1.7 MgCl2, 8.5 HEPES, 0.94 EGT, and 25 ATP. The pH was adjusted to 7.2 with CsOH, and the osmolarity was adjusted to 232–230 mOsm. The external solution contained the following (in mm): 122 NaCl, 3.0 KCl, 1.8 CoCl2, 0.8 MgCl2, 5.0 glucose, and 10 HEPES. It also contained 2.5 mm tetraethylammonium (TEA), 1.0 mm 1-aminopyridine (4-AP), 20 μm (+)-tubocurarine, and 10 μm picrotoxin. The pH was adjusted to 7.2 with NaOH, and the osmolarity was adjusted to 250–255 mOsm/L. Evoked action potentials were recorded using the same internal solution, replacing the CsOH with 102 mm potassium gluconate, and the d-gluconic acid was removed. The external solution was the same except that the CoCl2 was replaced by CaCl2 and the TEA and 4-AP were removed. Spontaneous action potentials were recorded using the same internal and external solutions as for evoked action potentials, without the presence of blockers. The chamber was continuously perfused at 0.75–1.0 ml/min, and the temperature in the chamber was controlled and monitored using a CL-100 Bipolar Temperature Controller (Harvard Apparatus). Data were acquired with an Axopatch 200B amplifier (Molecular Devices), a Digidata 1322A digital-to-analog converter (Molecular Devices), a Dell computer (Dimension 8200), and pClamp9 software (Molecular Devices).
For evoked electrophysiological recordings, baseline activity was recorded at 22°C, activity at elevated temperature was recorded at 30°C, and activity postcooling was recorded after the chamber had returned to 22°C for 5 min. Spontaneous burst characteristics were analyzed for each trace at sample intervals 90–110, 190–210, and 290–310 s for baseline 22°C data, and 690–710, 790–810, and 890–910 s for postcooling 22°C data. For elevated temperature, 60 s of data during heating was analyzed, starting at 26°C for each cell and reaching an average of 29.5°C at the end of the 60 s.
Statistics
Statistical significance was determined using ANOVA with Tukey’s post hoc test for multiple experimental groups. Paired t tests were used to determine significance between room temperature and high-temperature data within a single genotype. Data are shown as the mean ± SE.
Results
CRISPR-Cas9 generation of Drosophila carrying epilepsy-causing mutations
To create flies expressing patient-specific epilepsy-causing mutations, a two-step CRISPR-Cas9-mediated genome-editing strategy was used. In the first step, exon 30 of the para sodium channel gene, where the target missense mutations are located, was replaced by a DsRed marker flanked by arms including Caenorhabditis elegans sequences (Fig. 1A). In the second step, using gRNA targeted to the worm sequences, the DsRed marker and worm sequences were replaced by exon 30 of the para gene containing either the desired mutation R-C (CGA to TGT) and R-H (CGA to CAT), or a silent mutation R-R (CGA to CGT) that serves as the control (Fig. 1A). Sequencing of ∼1.7 kb of DNA surrounding the targeted mutation site and gRNA cut sites (Fig. 1B) confirmed the presence of the predicted base pair change in each line with no additional mutations. Multiple, independent lines were obtained, each resulting from a unique CRISPR insertion of R-C, R-H, or R-R. Both R-C and R-H mutations are homozygous lethal, so all lines were maintained as heterozygous stocks over the X-chromosome balancer FM7. Patients with these mutations are heterozygous, so all experiments were performed on heterozygous females.

CRISPR strategy and validation. A, Scheme of the two-step CRIPSR-Cas9-mediated genome editing for targeting the mutations to the Drosophila para gene. B, Mutant Drosophila lines were confirmed by Sanger sequencing. The wild-type codon is CGA. Step 2 repair plasmids carried the silent mutation CGT for R-R controls, TGT for R-C mutants, and CAT for R-H mutants.
Mutants display seizure-like behavior and reduced life span
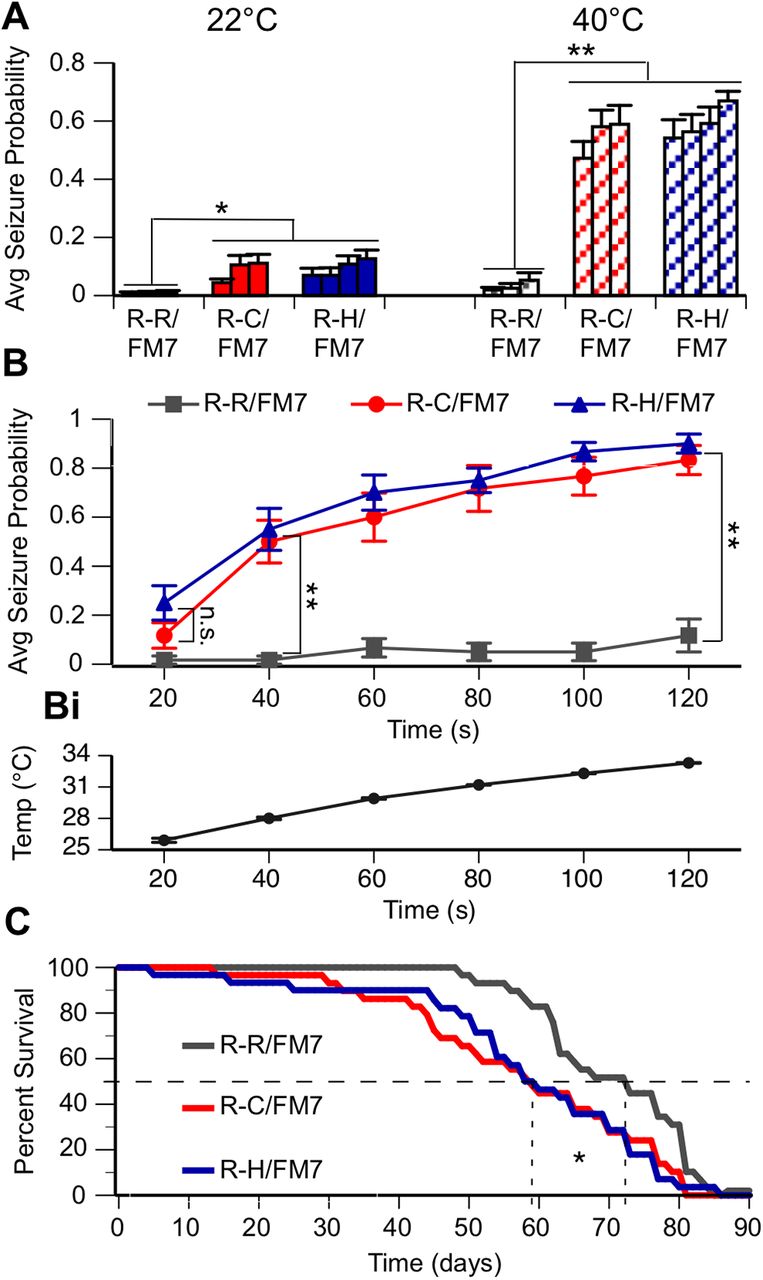
As individuals with DS and GEFS+ frequently have febrile-induced seizures in addition to spontaneous seizures, three fly lines heterozygous for the R-C mutation and four lines heterozygous for the R-H mutation were monitored for the presence of spontaneous and/or heat-induced seizure behavior. Groups of five flies were collected 1 d posteclosion and placed in a vial. A 2 min video was recorded immediately following immersion of the vial into a water bath, first at room temperature (22°C) and then at high temperature (40°C). Videos were later evaluated for seizure activity. A seizure was defined as the fly exhibiting a whole-body twitch or jump, or falling onto its back or side, often accompanied by leg twitching or wing flapping. Analysis was conducted blinded with respect to genotype.
Results are reported as the average probability of seizure for a fly of a certain genotype at any point in time during the assay. The four R-R control fly lines did not exhibit seizure activity in water baths at room temperature or high temperature (Fig. 2A). In contrast, both R-C and R-H heterozygous flies had a significantly increased seizure probability at room temperature compared with the R-R/FM7 control. At high temperature, seizure probability increased significantly in both R-C and R-H when compared with their behavior at room temperature, and with control R-R flies at high temperature (Fig. 2A; p < 0.05, ANOVA, Tukey’s post hoc test). There was no difference in seizure probability between lines with the same mutation. These data indicate that the seizure activity is associated with the introduced mutations, either R-C or R-H, and is not influenced significantly by the specific insertion events during the CRISPR process that resulted in the generation of each line.

R-C and R-H mutants display seizure-like behavior and reduced life span. A, Average seizure probability at any point in a 2 min trial for each independent line (3 R-R/FM7, 3 R-C/FM7, 4 R-H/FM7). B, Average seizure probability for each genotype shown at 40°C over time. At both 22°C and 40°C, R-C and R-H mutants have a significantly greater average seizure probability compared with R-R controls. Comparisons between R-C and R-H flies or between lines of one genotype are not significant: *p < 0.05; **p < 0.01; two-way ANOVA, Tukey's post hoc test. n > 12 trials/group. Bi, Average temperature inside vial over time of vial in 40°C bath. C, Kaplan–Meier survival curve estimates of R-R, R-C, and R-H flies over time. Median survival (50%) line is shown. Median percentage of survival analyzed using the Mantel–Cox log-rank test. Life span is significantly longer in R-R flies compared with R-C and R-H flies. *p < 0.05. n = 30 flies/genotype.
To evaluate the time course of heat-induced seizure activity associated with the different mutations, multiple flies in single R-H, R-C, and R-R lines were examined at 20 s intervals over a 2 min period following immersion of the vial in water bath of 40°C. The actual temperature inside the vial was continuously monitored during heating, reaching a temperature of 33°C at 2 min (Fig. 2Bi). The onset of heat-induced seizures was similar in the R-C and R-H lines, with maximal seizure probability occurring between 100 and 120 s (Fig. 2B). The similarity in the spontaneous and heat-induced seizure phenotypes in the R-C and R-H lines was unexpected since the R-C mutation is associated with the more severe seizure disorder Dravet syndrome, while R-H is associated with GEFS+.
Our initial observations suggested that the mutant lines were more difficult to maintain over time than the control lines. Since seizure activity in humans can be associated with premature death and homozygous knock-in mice with the R-H mutation have reduced life span (Tang et al., 2009; Martin et al., 2010), this property was evaluated in the R-C, R-H, and R-R lines maintained at 22°C. Since individual lines from the same genotype displayed similar seizure phenotypes, one line from each genotype was chosen and was used exclusively for the life-span assay and the remainder of the studies. Both the heterozygous R-H and R-C flies display a markedly decreased median survival time of ∼60 d when compared with R-R flies with a median survival of 73 d (Fig. 2C; p < 0.05, Mantel–Cox log-rank test). This similarity in severity between R-C and R-H was again unexpected because the R-H mutation is associated with the less severe GEFS+ seizure disorder.
Constitutive and heat-induced alterations in firing properties in R1648C and R1648H inhibitory neurons
Although behavioral studies facilitate an exploration of organismal dysfunction in the presence of R1648C and R1648H sodium channel mutations, studying neuronal firing and sodium current activity directly in the neurons of mutated flies is necessary to establish the underlying cellular mechanisms of dysfunction. In both mouse and fly knock-in models of SCN1A epilepsy, alterations in sodium currents and excitability have been primarily observed in inhibitory neurons (Ogiwara et al., 2007; Schutte et al., 2016). Therefore, we focused on evaluating the effects of R-C and R-H mutations in inhibitory neurons. Though the site of seizure generation in flies is unknown, LNs in the fly antennal lobes are a readily accessible population of GABAergic inhibitory neurons in the important and well characterized olfactory circuit within the Drosophila brain (Seki et al., 2010). To observe neuronal firing and sodium channel activity in these neurons, the whole brain was dissected out of adult flies (Roemmich et al., 2018), dorsal lateral local neurons were identified by their location and morphology, and whole-cell electrophysiological recordings were performed.
Evoked firing was evaluated by subjecting each cell to a series of depolarizing current steps, first at 22°C, following an increase in bathing solution temperature to 30°C, and finally after return of the bath solution to 22°C. A temperature of 30°C was chosen for high-temperature electrophysiology recordings as a temperature at which mutant flies exhibited seizure-like behavior during the water bath assay (Fig. 2Bi), and as the highest recording temperature at which quality cell recordings could be maintained. We measured the threshold, amplitude, half-width, and maximum rise slope for the first evoked action potential in each cell, the spike frequency adaption ratio (at half-maximal firing, first interspike interval over final interspike interval), and peak instantaneous firing frequency at a baseline of 22°C, during heating to 30°C, and after cooling back to 22°C for each genotype (Table 1). There were no significant differences between genotypes for any of these characteristics.
Properties of evoked firing in R-R, R-C, and R-H LNs
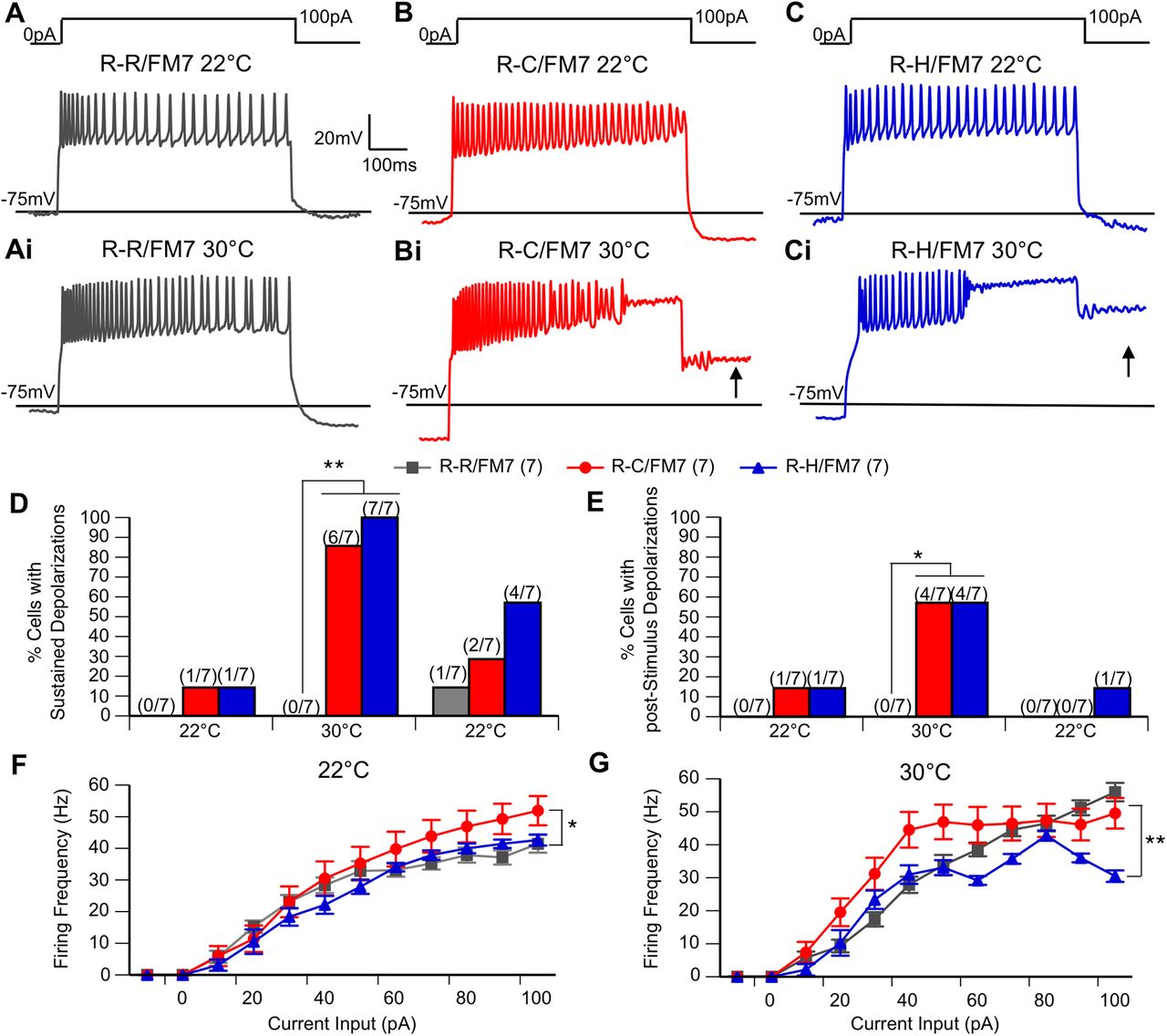
Before heating, the majority of neurons in all three genotypes fired action potentials throughout the 600 ms current step (Fig. 3A,3B,3C). While a similar firing pattern persisted in the control LNs (Fig. 3Ai) at 30°C, heating resulted in sustained depolarizations and cessation of action potential firing in R-C and R-H neurons. In some cases, depolarizations persisted after the current step (poststimulus depolarization; Fig. 3Bi,Ci). Quantitative analysis of these events demonstrates that at 22°C preheating, sustained depolarizations and poststimulus depolarizations occurred in a minority of mutant neurons but were never seen in control neurons (Fig. 3D,E). At 30°C, the incidence of sustained depolarizations increased dramatically in the mutant neurons such that nearly all of the R-C and R-H neurons exhibited these events (Fig. 3D). In contrast, there was no evidence of sustained depolarization or poststimulus depolarization in the control R-R neurons even at elevated temperature. Poststimulus sustained depolarization incidence increased as well, seen in the majority of mutant neurons at elevated temperature (Fig. 3E). The incidence of sustained depolarizations and poststimulus sustained depolarizations decreased postcooling, and there are no significant differences in these measures between genotypes at the return to 22°C. Importantly, the reversal of the temperature-induced changes in firing demonstrate that these alterations are associated with the mutation and are not simply a result of irreversible damage to the cell induced by heating.There were also differences in the firing frequency and response to temperature increase observed between control and mutant neurons. In control LNs, the firing frequency increased with higher current inputs at both 22°C and 30°C (Fig. 3F,G). At 22°C, R-C LNs displayed increased firing frequency compared with controls. However, for R-C and R-H LNs the firing frequency increased with higher current inputs only at 22°C. At 30°C, particularly for R-H LNs, the firing frequency began to decrease at the larger current steps corresponding with the appearance of sustained depolarizations in which the firing of action potentials stopped but the cell remained depolarized, either for the remainder of the stimulus, or even after the current injection was over (poststimulus sustained depolarizations; Fig. 3F,G; p < 0.05, two-way ANOVA).

R-C and R-H mutants display sustained depolarizations during evoked firing, even after stimulus has ended. A–Ci, Representative trains of action potentials evoked by illustrated stimulus protocol at 22°C (A,B,C) and 30°C (Ai,Bi,Ci). Arrow indicates the appearance of poststimulus depolarization. D, E, Percentage of cells in each genotype with sustained depolarizations during stimulus (D) and poststimulus depolarizations (E) at 22°C, at 30°C, and after cooling back to 22°C. R-C and R-H mutants have increased the incidence of SDs and poststimulus SDs at 30°C. *p < 0.05, **p < 0.01, χ2 test. Any differences before or after heating are not significant. F, G, Average spike frequency shown as a function of current input at ambient temperature (F) and elevated temperature (G). At 22°C, R-Cs have generally increased firing frequency. At 30°C, R-Hs have generally decreased firing frequency compared with R-Cs. n = 7 groups/genotype. *p < 0.05; **p < 0.01, two-way ANOVA, with Tukey’s post hoc test.
Although evoked firing frequency in mutant LNs is generally comparable to that in controls, the presence of sustained depolarizations in the mutants could cause overall decreased excitability in interneurons. Therefore, we evaluated the spontaneous activity in LNs in the absence of any current injection. To explore the effect of temperature, spontaneous neuronal activity in each cell was recorded continuously for 14 min, as follows: 5 min at 22°C, 4 min during heating to 30°C, and 5 min during cooling back down to 22°C. The long traces in Figure 4A,4B,4C represent typical responses to the heating curve from each of the three genotypes. Demonstrated by the expanded timescale in Figure 4Ai, the traces consist of underlying oscillations in membrane potential with action potentials that fire at the peak of each oscillation. Both control and mutant neurons exhibited a regular burst firing pattern at 22°C (Fig. 4Ai,4Bi,4Ci) that ceased during heating at ∼30°C (Fig. 4Aii,Bii,Cii). Elevated temperature data were thus taken during the minute of heating between 26°C and 29°C (Fig. 4D–F). Burst firing similar to that seen before heating resumed following the return to 22°C, as illustrated in Figure 4A,B,C and Aiii,Biii,Ciii.
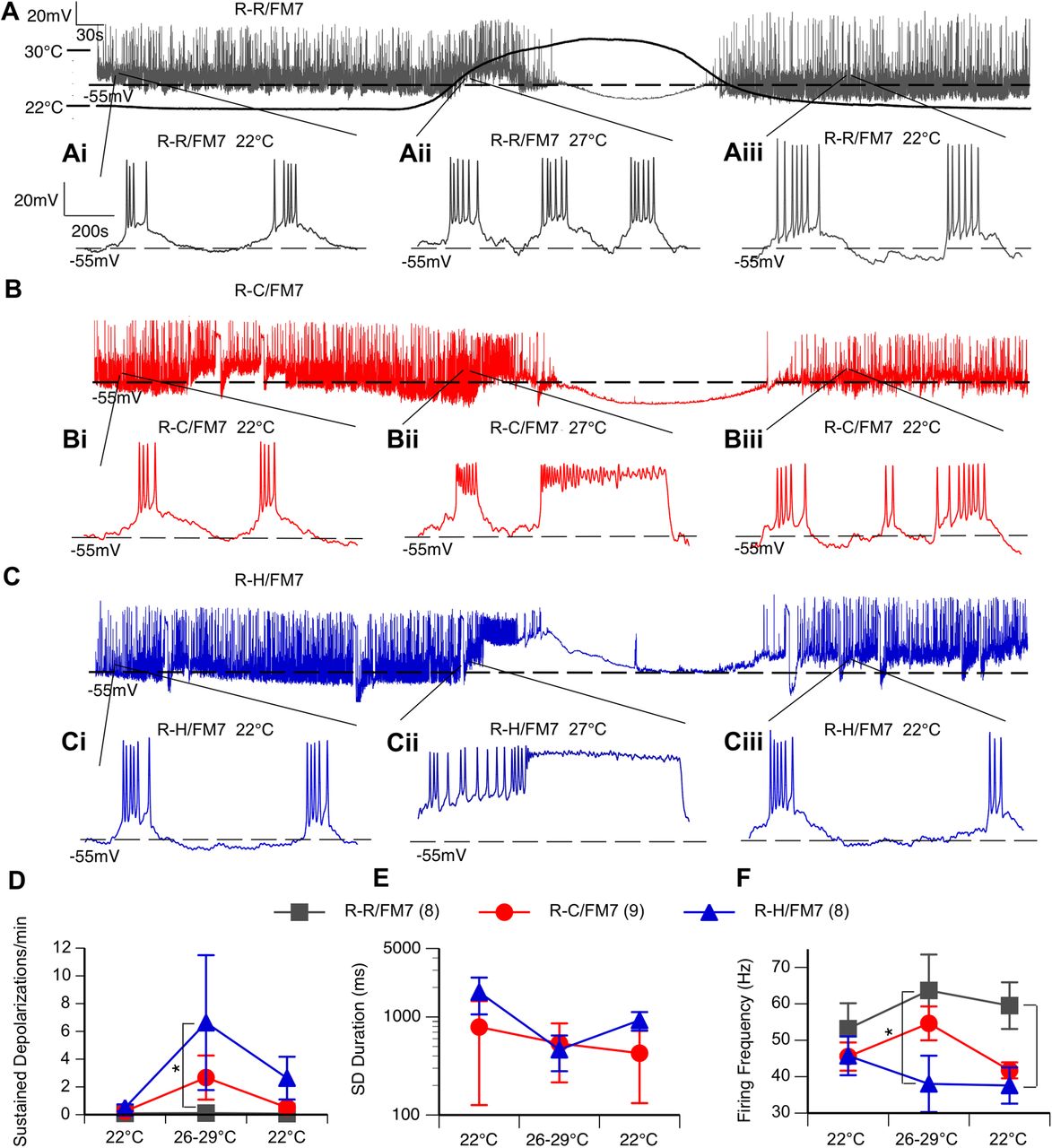
R-C and R-H mutants display spontaneous sustained depolarizations and reduced inhibitory firing frequency at elevated temperatures. A, B, C, Representative continuous recording of neuron subjected to heating protocol. Chamber temperature over heating protocol shown on A. Ai–Aiii, Bi–Biii, Ci–Ciii, The 1.1 s interval within the labeled part of the temperature curve at 22°C preheating (Ai, Bi, Ci), 27°C mid-heating (Aii, Bii, Cii), and 22°C postcooling (Aiii, Biii, Ciii). D–F, Average frequency and duration of sustained depolarizations (D, E) and firing frequency of action potentials (F) during baseline (22°C), heating (26–29°C), and cooling (22°C). n = 8–9 cells/genotype. *p < 0.05, one-way ANOVA, with Tukey’s post hoc test.
There were no differences in the average frequency of bursts, action potential threshold, action potential amplitude, or average number of action potentials within these bursts between genotypes (Table 2). There was also no significant effect of temperature on any of these properties.
Properties of spontaneous burst firing in R-R, R-C, and R-H LNs
However, the occurrence of spontaneous sustained depolarizations (>100 ms in duration) was a characteristic of both mutant lines (Fig. 4Aii–Cii,D,E). While both R-C and R-H mutants occasionally exhibited sustained depolarizations at room temperature (Fig. 4E), the average frequency of sustained depolarizations was increased at elevated temperatures in both mutants, but the increase was only significantly different in the R-H mutants compared with controls (Fig. 4D; p < 0.05, two-way ANOVA). The duration of the sustained depolarizations was similar in the two mutants, and the duration did not further increase with temperature (Fig. 4E). These prolonged sustained depolarizations contributed to decreased firing frequency within bursts for R-H neurons, particularly at elevated temperature (Fig. 4E; p < 0.05, two-way ANOVA).
At elevated temperatures, resting membrane potential (RMP) first depolarized, then hyperpolarized above 30°C as cells ceased firing. While RMP did not fully recover over the length of the recording, R-H neurons displayed a significant depolarizing shift in resting membrane potential at elevated temperatures (Table 2; two-way ANOVA).
Alterations in sodium currents in R1648C and R1648H inhibitory neurons
To evaluate changes in the underlying sodium currents, voltage-clamp recordings were conducted on inhibitory neurons in R-R, R-C, and R-H flies at both ambient and elevated temperatures. Sodium currents were activated by holding the cell below firing threshold at −75 mV and applying a depolarizing voltage step in increasing 5 mV steps. The sodium current waveform is similar in all three cells lines with an initial transient current (INaT) and a smaller persistent current at both 22°C and 30°C (Fig. 5A). The significant delay between the stimulus onset and sodium current activation at all recording temperatures is because of the recording electrode being located on the LN cell body, which is electronically distant from the sodium channels located on the distal axonal segments in fly neurons (Ravenscroft et al., 2020). While there were no genotype- or temperature-induced changes in latency between stimulus and response onset, this recording configuration prohibits adequate space clamp, preventing accurate assessment of the time course of activation and inactivation. Additionally, the transient sodium current amplitude in many neurons showed a decrease during the heating regime (Fig. 5A–C) that did not recover when cooled (Fig. 5D). Therefore, we did not further evaluate the potential effects of the mutation on peak current amplitude. In contrast, the average activation threshold, defined as the least depolarized voltage step that activates the first current, was stable throughout the recording. This property was significantly more hyperpolarized in R-H compared with R-C and R-R, but there was no effect of temperature (Fig. 5E; p < 0.05, two-way ANOVA).
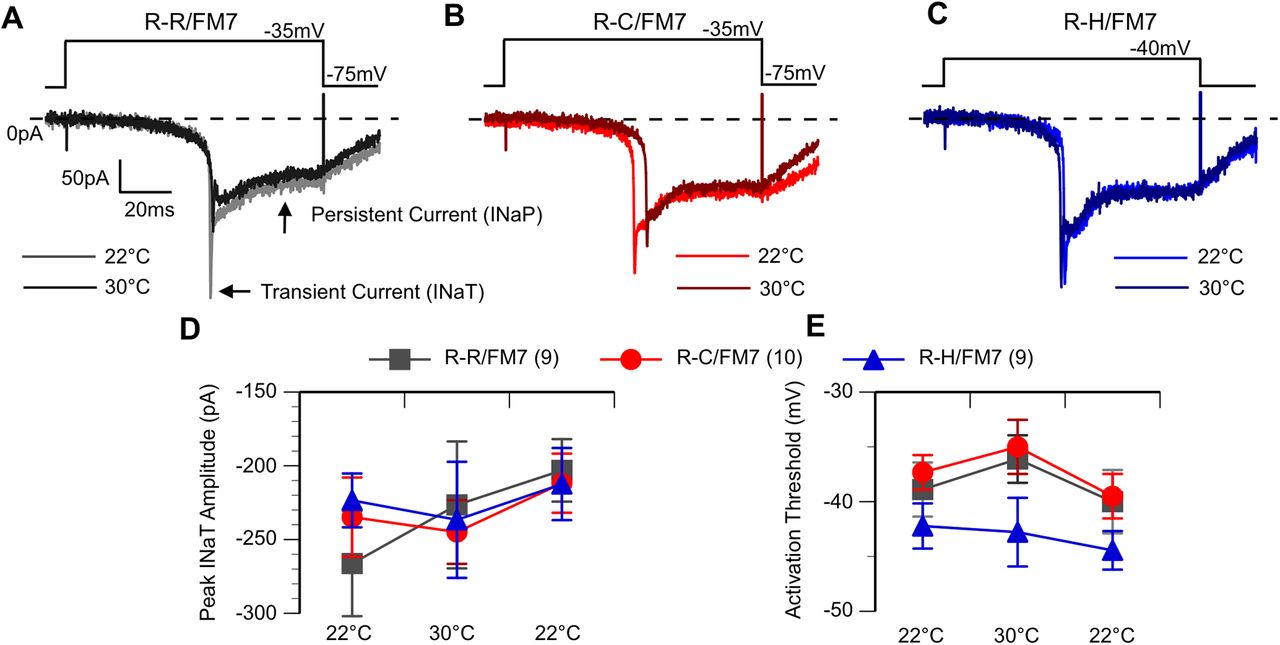
R-H neurons exhibit hyperpolarized sodium current activation threshold. A–C, Voltage step-elicited currents at 22°C and 30°C. The first voltage step to elicit current shown. Cells exhibit both an initial transient and then a persistent current. D, Average peak INaT amplitude. E, Average activation threshold at 22°C, 30°C, and following cooling (second threshold at 22°C). R-H mutants have an average activation threshold hyperpolarized to R-C and R-R, but no effect by temperature. *p < 0.05, two-way ANOVA, with Tukey’s post hoc test.
To examine the current deactivation properties, a multistep protocol was used. The cell was first depolarized to a fixed voltage of −5 mV to activate sodium currents and then returned to different resting potentials that varied in 10 mV steps between −95 and −25 mV (Fig. 6A–Ci). Deactivation threshold was defined as the least negative voltage step resulting in current returning to baseline at the end of the stimulus. Both the R-C and R-H neurons had a significant hyperpolarizing shift in deactivation threshold (−55 mV) compared with that of R-R control neurons (−40 mV; Fig. 6D; p < 0.05, two-way ANOVA). There was no effect of temperature on the deactivation. The deactivation data indicate that mutant sodium channels in inhibitory neurons are open over a wider voltage range, resulting in increased influx of sodium ions for the same stimulus in mutants compared with controls. However, unlike the abnormal firing properties, the sodium current alterations in deactivation were present at similar levels at both room temperature and high temperature.

R-C and R-H neurons exhibit hyperpolarized sodium current deactivation threshold. A–Ci, Voltage-step sodium channel currents for deactivation at room temperature (22°C; A, B, C) and high temperature (30°C; Ai, Bi, Ci), with the current steps in bold. Cells subjected to a multistep protocol, as shown. Selected sweeps shown to illustrate differences in deactivation properties. D, Average deactivation threshold at 22°C, 30°C, and after cooling back to 22°C. R-C and R-H mutants had hyperpolarized deactivation threshold compared with R-Rs. *p < 0.05, two-way ANOVA, with Tukey’s post hoc test.
Discussion
For patients with genetic epilepsy, the connection between a specific mutation and behavioral symptoms is largely unknown. Our study focused on SCN1A-mediated genetic epilepsies that arise from mutations at the same amino acid position (1648). The R-C mutation is associated with the more severe disorder Dravet syndrome, while the R-H mutation is associated with the milder disorder GEFS+ (Baulac et al., 1999; Ohmori et al., 2002; Striano et al., 2008; Escayg and Goldin, 2010). This leads to the question of whether the differences in severity arise from distinct changes in channel function in the two mutants or whether similar channel changes result in different behavioral outcomes depending on genetic background. Using independently generated Drosophila lines carrying either the R-H or R-C mutation, we were able to compare the effects of the mutations on cellular physiology and organismal behavior in an identical genetic background. We observed that both R1648C and R1648H mutations resulted in largely similar spontaneous and temperature-sensitive seizure activity, decreased life span, and several patterns of cellular dysfunction. This suggests that it is likely that genetic background contributes to difference in severity in patients with these mutations.
While animal behavior was essentially indistinguishable between R-C and R-H lines, there were instances of significant electrophysiological alterations in one, but not both, mutant lines. R-C mutants displayed increased evoked firing frequency at ambient temperatures. R-H mutants displayed decreased evoked firing frequency at elevated temperatures, decreased spontaneous firing frequency at elevated temperatures, depolarized resting membrane potential at elevated temperatures, and a constitutively hyperpolarized sodium channel activation threshold. While R-H local neurons displayed more significant differences from controls than R-C local neurons, these cellular changes surprisingly did not result in a more severe behavioral profile. It seems that the changes shared by both mutant lines—a hyperpolarized sodium current deactivation threshold and increased incidence of sustained depolarizations seen most clearly with evoked firing—have the greatest effect on the behavioral phenotypes in R-C and R-H SCN1A Drosophila.
Additionally, many of the changes attributed to the mutation were observed in only a minority of mutant neurons, such as the appearance of sustained depolarizations in only one of seven LNs in each of the mutant lines at 22°C. This is most likely because of this being a low-frequency event in any one neuron at this temperature. Thus, there is a low probability that an event will be detected during the one or two trials during which the data are collected. If this were true, then increasing the number of trials in each cell at 22°C would increase the percentage of LNs in which at least one event was detected. At high temperature, the majority of neurons in both mutant lines exhibit these events, demonstrating that the alteration that leads to these existed in all the neurons but was only manifest at high temperature. It is possible that larger sample sizes could even out some of these smaller changes between R-C and R-H mutants. It is also possible that there are different subclasses of LNs with distinct distributions of sodium channels in the cell, such that some are more susceptible to the mutation resulting in sustained depolarizations at 22°C.
Given that the R-C DS mutation did not result in increased behavioral severity in Drosophila, and, given that several smaller changes in R-H sodium channel physiology did not result in increased seizures in Drosophila, there could be other background alterations that contribute to disease phenotype in humans. A role for genetic background in contributing to disease phenotype is not inconsistent with clinical data indicating that the severity of seizure disorders can vary even between individuals with the same SCN1A mutation. For example, in a single multigenerational French family, R1648H was identified in 13 individuals whose symptoms ranged from experiencing a few febrile seizures in early childhood to intractable epilepsy throughout adulthood associated with moderate or severe intellectual disability (Baulac et al., 1999). This suggests that interplay between the mutation and other factors are involved in determining disease severity. This could include genetic background or environmental/developmental differences. Recent studies in human induced pluripotent stem cell-derived neurons demonstrate that the GEFS+ causing the K1270T mutation has differential effects on sodium currents and neuronal firing properties in different genetic backgrounds (Xie et al., 2020). Transgenic Drosophila will be valuable in assessing the interaction between specific epilepsy causing missense mutations and genetic background in manifestation not only of cellular dysfunction but also of behavioral phenotypes.
Previous studies using mice have been important in understanding cellular mechanisms associated with several epilepsy-causing sodium channel mutations, including a knock-in model of the missense mutation R1648H (Tang et al., 2009; Martin et al., 2010). While the phylogenetic distance between humans and flies is greater than that between humans and mice, there are some distinct advantages to conducting parallel studies in Drosophila. The DNA sequences that encode voltage-gated sodium channels are highly conserved between flies and mammals, but Drosophila has only one voltage-gated sodium channel gene (para; Loughney et al., 1989; O’Dowd et al., 1989). In contrast, there are nine different voltage-gated sodium channel α-subunit isoforms in mice and other mammals, three of which are highly expressed in the nervous system (SCN1A, SCN2A, and SCN8A; Catterall, 2017). These isoforms can and do interact in models of patient epilepsies. For example, mice that were double heterozygous mutants for SCN1A and SCN8A had restored seizure susceptibility and life span compared with mice heterozygous for only SCN1A (Martin et al., 2007). In addition, creation and maintenance of multiple different mouse lines carrying distinct missense mutations in different genetic backgrounds is a time- and resource-intensive proposition. In the present study, the transgenic flies used were created using a two-step CRISPR/Cas9 gene-editing method that involved introducing a visible marker in the first step. This resulted in efficient detection of multiple founder flies with the desired mutations in an identical genetic background. In addition, the low cost of maintaining multiple lines, including control substitutions in addition to the mutants, was an attractive reason to use Drosophila. While there is only one sodium channel gene in flies, worm sequences not present elsewhere in the genome flanked the replacement sequence in the first step targeted for excision in the second step. Sequencing through the excision sites revealed identical sequences where additional mutations were most likely to be introduced, decreasing any concerns about off-target effects. Further confirmation of the specificity of mutants could be done by determining whether a genetic rescue, such as using CRISPR to replace the mutation in the R-C or R-H lines, restores wild-type function.
Analysis of two previous Drosophila knock-in models of SCN1A missense mutations, one causing Dravet syndrome (S1231R) and one causing GEFS+ (K1270T) exhibited alterations in sodium current and firing properties only in animals homozygous for the mutations (Schutte et al., 2016). Heterozygotes exhibited moderate seizure behavior at high temperatures but no obvious alterations in neuronal activity (Sun et al., 2012; Schutte et al., 2014). Therefore, the finding that fly lines homozygous for the R-C or R-H mutations were lethal, and that heterozygotes displayed significant seizure activity and reduced life span, was somewhat unexpected. The location of the 1648 mutation on the voltage-sensing region of SCN1A, which is particularly crucial for inactivation, may explain this increased level of severity compared with the previously examined DS (S1231R) and GEFS+ (K1270T) mutations. The R-C mutation substitutes a neutral amino acid for a positive gating charge. R-H conserves charge, but adds steric hindrance. Interestingly, in a study investigating the effects of an R-H mutation in the S4 segment of domain IV of a human skeletal sodium channel expressed in HEK cells, changing the pH to protonate the histidine did not change the functional alterations of the mutation (mixed inactivation defects; Alekov et al., 2000). This suggests that the steric effects of the histidine side chain may be the primary molecular culprit behind the phenotype associated with the R1648H mutation. Changing the size and/or charge of one of the gating charges on the S4 segment in domain IV likely affects its ability to exchange ion-binding partners with the environment. Given our observations that R1648H or R1648C mutations in inhibitory neurons result in sustained depolarizations and require repolarization to more negative voltages to deactivate the sodium current, both mutations likely impact the ability of the S4 segment in domain IV to move back into place, closing the channel (Ahern et al., 2016). The S4 segment in domain IV of SCN1A may therefore play a previously undescribed role in deactivation, as well as inactivation.
The R-C and R-H inhibitory neurons exhibited heat-induced alterations in neuronal firing properties including the appearance of sustained depolarizations, poststimulus sustained depolarizations, and decreased firing frequency during both evoked and spontaneous activity. The temperature-dependent aspect of these changes suggests that they are likely to contribute to the heat-induced increases in seizure activity. Analysis of the underlying sodium currents in mutant neurons revealed a hyperpolarized deactivation threshold in R-C and R-H neurons even at room temperature. This is consistent with the R-H and R-C sodium channels being more difficult to close once activated, which would result in increased ion flow because of longer channel openings and/or channel opening to a greater extent. Dravet syndrome has most often been attributed to loss-of-function mutations, as was the case in the previous S1231R Dravet syndrome knock-in model in Drosophila. The current result indicates that mutations resulting in increased sodium influx can also contribute to Dravet syndrome. However, unlike the abnormal firing properties, mutant sodium current alterations were present at both room temperature and high temperature. This suggests that the increased inward flow of sodium ions is not sufficient to trigger the sustained depolarizations and reduced firing frequency seen at high temperature by themselves but through an interaction with a different and as yet unidentified heat-induced change.
One heat-induced aspect of cellular physiology that could be involved are calcium currents. Previous studies have found that calcium currents exhibit increased conductance at elevated temperature in rat brain slices (Radzicki et al., 2013). Previous studies have shown that calcium currents also contribute to membrane depolarization in Drosophila neurons, including in the neurons located in the adult fly brain (Peng and Wu, 2007; Gu et al., 2009). In projection neurons in the adult antennal lobes, the calcium currents have both a transient and sustained component. The alpha1T gene encodes a Cav3-type calcium channel mediating the transient current (Iniguez et al., 2013), and the cac gene encodes a Cav2-type calcium channel mediating the sustained current (Gu et al., 2009). If similar calcium currents are present in antennal lobe LNs, it is possible that potassium currents are sufficient in most cases for repolarizing the action potential, even with the increased sodium conductance in the mutants because of hyperpolarized deactivation of the channels at room temperature, resulting only rarely in sustained depolarizations. However, at high temperature an increased magnitude of calcium currents, in conjunction with an increased magnitude of the sodium currents in the mutants, could more severely impede the ability of potassium currents to repolarize effectively, resulting in more sustained depolarizations. Future studies, taking advantage of both forward and reverse genetic approaches that are relatively easy to perform in Drosophila, will be important in identifying the interacting variables associated with the heat-induced change in firing properties.
In the mouse model, the primary effect of the R1648H mutation is on inhibitory neurons, where a decrease in inhibitory neuron activity can lead to unbalanced overexcitability in the brain and seizures (Martin et al., 2010). The present study focused exclusively on LNs, inhibitory neurons in the antennal lobe that process olfactory information in Drosophila. While the para sodium channel in Drosophila undergoes extensive alternative splicing (Loughney et al., 1989; Lin et al., 2009), the R1648 amino acid is located in an obligate exon, so mutations at this site will be present in all neurons (Lin et al., 2009; Ravenscroft et al., 2020). Therefore, it will be interesting in future studies to also evaluate the effect of this mutation on excitatory neurons in the same antennal lobe circuit.
Together, the results of the present study demonstrate that R1648C and R1648H SCN1A mutations result in improper deactivation in inhibitory neuron sodium channels in Drosophila. The incomplete closing and/or extended opening of the sodium channels could cause an inappropriate influx of sodium ions, which in turn causes impaired inhibitory neuron firing. Neuronal network activity is based on a balance between inhibitory and excitatory activity, and with the impaired firing of inhibitory neurons, the balance is tilted in favor of overexcitation, which can lead to seizures. CRISPR-generated Drosophila lines represent low-cost platforms that can be used to elucidate neuronal mechanisms of seizure generation and guide the development of new patient-specific therapies for epilepsy. Patient-specific studies using model organisms, including Drosophila, will be an important tool in our quest to improve our mechanistic understanding of genetic epilepsy and develop better patient-specific treatments.
Acknowledgments
Acknowledgements: We thank Dr. Debra Mauzy-Melitz for the GAANN (Graduate Assistance in Areas of National Need) program that supported A.J.R.
Footnotes
The authors declare no competing financial interests.
This work was supported by the National Institutes of Health Grant R01-NS-083009 (D.K.O.), the Graduate Assistant in Areas of National Need Grant P200A120207, and a Graduate Dean’s Dissertation Fellowship, Graduate Division, University of California, Irvine (to A.J.R).
This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International license, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



