

Visual Abstract

Abstract
Rett syndrome (RTT) is an X-linked neurodevelopmental disorder usually caused by mutations in methyl-CpG-binding protein 2 (MeCP2). RTT is typified by apparently normal development until 6–18 mo of age, when motor and communicative skills regress and hand stereotypies, autonomic symptoms, and seizures present. Restoration of MeCP2 function selectively to astrocytes reversed several deficits in a murine model of RTT, but the mechanism of this rescue is unknown. Astrocytes carry out many essential functions required for normal brain functioning, including extracellular K+ buffering. Kir4.1, an inwardly rectifying K+ channel, is largely responsible for the channel-mediated K+ regulation by astrocytes. Loss-of-function mutations in Kir4.1 in human patients result in a severe neurodevelopmental disorder termed EAST or SESAME syndrome. Here, we evaluated astrocytic Kir4.1 expression in a murine model of Rett syndrome. We demonstrate by chromatin immunoprecipitation analysis that Kir4.1 is a direct molecular target of MeCP2. Astrocytes from Mecp2-deficient mice express significantly less Kir4.1 mRNA and protein, which translates into a >50% deficiency in Ba2+-sensitive Kir4.1-mediated currents, and impaired extracellular potassium dynamics. By examining astrocytes in isolation, we demonstrate that loss of Kir4.1 is cell autonomous. Assessment through postnatal development revealed that Kir4.1 expression in Mecp2-deficient animals never reaches adult, wild-type levels, consistent with a neurodevelopmental disorder. These are the first data implicating a direct MeCP2 molecular target in astrocytes and provide novel mechanistic insight explaining a potential mechanism by which astrocytic dysfunction may contribute to RTT.
Significance Statement
Rett syndrome is a devastating neurodevelopmental disorder that affects 1 in 10,000–25,000 females. Mutations in methyl-CpG-binding protein 2 (MeCP2), a transcriptional regulator, are responsible for >95% of RTT cases. Recent work has shown that astrocytes contribute significantly to the disorder, although their contribution to this disease is not known. Here, we demonstrate that the critical astrocyte K+ channel Kir4.1 is a novel molecular target of MeCP2. MeCP2 deficiency leads to decreased Kcnj10/Kir4.1 mRNA levels, protein expression, and currents. These findings provide novel mechanistic insight and begin to elucidate the role of astrocytes in this disorder.
Introduction
Rett syndrome (RTT) is an X-linked neurodevelopmental disorder that affects 1 in 10,000–25,000 females (Percy, 2002). RTT is characterized by apparently normal development until 6–18 mo of age, when deficits in speech, ambulation, and hand use (e.g., hand wringing, clapping) become apparent (Neul et al., 2010). Breathing disturbances, scoliosis, diminished pain response, and seizures are also commonly associated with RTT (Jian et al., 2007; Glaze et al., 2010; Neul et al., 2010). More than 95% of girls with RTT have a mutation in the methyl-CpG-binding protein 2 (MECP2) gene located on the X-chromosome (Cuddapah et al., 2014). Knockout or mutation of Mecp2 in mice recapitulates symptoms associated with RTT (Chen et al., 2001; Guy et al., 2001). Functionally, MeCP2 is a methyl-CpG–binding protein, which binds methylated and unmethylated DNA to modulate gene activity, and can act as a repressor or an activator of gene transcription depending on the context (Chahrour et al., 2008).
MeCP2 is most highly expressed in neurons, but it is also expressed in glia cells (Ballas et al., 2009; Kifayathullah et al., 2010; Zachariah et al., 2012; Liu et al., 2015, 2017). In addition, there are indications that astrocytic dysfunction plays a role in the pathophysiology of RTT. RTT astrocytes are abnormal (Maezawa et al., 2009), with altered microtubule assembly (Nectoux et al., 2012) and glutamate clearance (Okabe et al., 2012). Also, when cultured with MeCP2-deficient glia, wild-type (WT) neurons display aberrant morphology (Ballas et al., 2009; Williams et al., 2014). Specific deletion of MeCP2 in astrocytes also resulted in disturbed breathing patterns (Garg et al., 2015). Importantly, postnatal re-expression of MeCP2 in astrocytes in globally Mecp2-deficient mice improved locomotion, decreased anxiety, increased lifespan, and normalized respiration, neuronal cell size, and dendritic morphology (Lioy et al., 2011). However, to date, few direct molecular links between astrocytes and astrocytic dysfunction in animal models of RTT have been demonstrated. Possibly shedding some light on this question, a recent transcriptomic study in MeCP2-deficient mice indicates that 46 genes that are unique to astrocytes are disrupted in RTT, one being Kcnj10, the gene that codes for Kir4.1 (Pacheco et al., 2017).
Kcnj10 expression is in the top 1% of all expressed genes in astrocytes (Zheng et al., 2014). Underscoring the relative importance of this channel, knockout or mutation of KCNJ10 causes seizures, ataxia, and developmental deficits in mice and humans alike (Djukic et al., 2007; Bockenhauer et al., 2009; Scholl et al., 2009). Direct evidence that loss of Kir4.1 alters neuronal function was recently demonstrated in a mouse model of Huntington’s disease, where AAV-mediated restoration of Kir4.1 expression specifically to astrocytes reduced the concentration of extracellular K+ ([K+]o), prolonged survival, and ameliorated motor abnormalities observed in these mice (Tong et al., 2014). Furthermore, a compound heterozygous change (two missense mutations) in KCNJ10 was reported in a patient with RTT-like symptoms who lacked a mutation in MECP2 (Sajan et al., 2017).
In the current study, we demonstrate that Kir4.1 is a direct molecular target of MeCP2. In situ, astrocytes from Mecp2-deficient mice show smaller astrocytic K+ currents, with concomitant significant reductions in Kir4.1 protein and transcription. These data suggest MeCP2 is a positive regulator of Kcnj10 gene expression through development and potentially provide insight explaining how astrocytic dysfunction may contribute to RTT.
Materials and Methods
Animals
All animal procedures were performed in accordance with the [Author University] animal care committee’s regulations. Every effort was made to minimize pain and discomfort. WT males were bred with heterozygous Mecp2tm1.1Jae (Jaenisch mutation) female mice, which lack exon 3 in the Mecp2 gene (Chen et al., 2001). Genotypes of offspring were confirmed by PCR of DNA isolated from tail clips. Mutant male mice (Mecp2–/y) were used for experimentation after postnatal day 50 (P50) when symptomatic, as demonstrated by hypoactivity and hindlimb clasping on suspension from tail compared with WT littermates of the same age. Symptomatic mutant female mice (Mecp2±) were used for experimentation at 7–8 mo of age and compared with WT female mice of the same age. All animals were on a C57BL/6 background.
Western blotting
Animals were anesthetized with CO2 and quickly decapitated. Brains were removed and placed in ice-cold PBS. Under a binocular microscope, the cerebellum and brainstem were removed, and the cortex was separated from hippocampus and midbrain. Cortex, hippocampus, midbrain, cerebellum, and brainstem were collected. Briefly, tissue was placed in ice-cold homogenization buffer (100 mm Tris, pH 7.5, 1% SDS at 50 mg/ml) supplemented with protease and phosphatase inhibitors (Sigma-Aldrich) and sonicated for 10 s. Tissue homogenates were centrifuged for 5 min at 12,000 × g at 4°C. Protein quantification was performed on the supernatant using a DC protein assay kit from Bio-Rad. Protein was heated to 60°C for 15 min in an equal volume of 2× sample buffer (100 mm Tris, pH 6.8, 10% SDS in Laemmli-SDS, and 600 mm β-mercaptoethanol). Equal amounts of protein were loaded into a 4%–20% gradient precast SDS gel (Bio-Rad). Gels were transferred at 100 V for 1 h at room temperature to PDVF membrane (Millipore). Membranes were blocked in blocking buffer (10% dried milk in TBST) for 1 h before probing with antibodies. The blots were probed with rabbit anti-Kir4.1 (Alomone, 1:1000 for 1 h), washed three times in TBST, and probed with an HRP-conjugated secondary antibody (Santa Cruz Biotechnology) for 1 h. After three 10-min washes, membranes were developed with Classico chemiluminescent reagent (Millipore) using a Kodak film developing system (Kodak). The blots were then stripped and reprobed for chicken anti-GAPDH (Abcam, 1:2000 for 1 h) or rabbit anti–β-tubulin (Millipore, 1:5000 for 30 min), which was used as a loading control. Protein expression was quantified using ImageJ. Target protein was normalized to GAPDH expression in the same lane. Both tetrameric (∼150-kDa) and monomeric (∼50-kDa) Kir4.1 isoforms were detected (e.g., Fig. 3A) that are specific to Kir4.1, as they are not present in Kcnj10 knockout animals or negative control lysates (Olsen et al., 2006). The analysis included the entire lane (both isoforms). Cortical and brainstem Western blots were run in triplicate or quadruplicate. Human embryonic kidney (HEK) cell lysates were used as a negative control for astrocytic proteins on Western blots. Relative amounts of protein are reported.
Quantitative PCR
mRNA was isolated from either cultured astrocytes or cortical and brainstem tissue collected as above using an RNA isolation kit (Qiagen). mRNA was converted to cDNA using the VILO Superscript kit (Life Technologies). Quantitative real-time PCR was run using TaqMan specific probes (all from Life Sciences) for Kcnj10 (Kir4.1, Mm00445028_m1), Gfap (GFAP, Mm01253033), Hexb (hexosaminidase B, Mm01282432_m1), Tmem119 (transmembrane protein 119, Mm00525305_m1), Mbp (myelin basic protein, Mm01266402_m1), Rbfox3 (NeuN, Mm01248771_m1), and Gapdh (GAPDH, Mm99999915_g1). All probes span exon boundaries and as such only amplify mRNA. Each sample was run in triplicate and normalized to Gapdh, and the comparative Ct method was used to calculate changes in gene expression.
Astrocyte isolation
Astrocyte isolation was performed according to Holt and Olsen (2016). Briefly, mice were anesthetized with CO2 and rapidly decapitated, and their cortices were microdissected in ice-cold cutting solution (120 mm NaCl, 3 mm KCl, 2 mm MgCl2, 0.2 mm CaCl2, 26.2 mm NaHCO3, 11.1 mm glucose, and 5 mm Hepes), supplemented with AP5 (3 mm) and CNQX (3 mm) and bubbled with 95% oxygen. The tissue was minced and enzymatically dissociated with a Papain Dissociation kit (Worthington) following manufacturer’s instructions. After dissociation, myelin debris and microglia were removed using a Myelin Removal Kit (Miltenyi Biotec) and Cd11b+ Microbeads (Miltenyi Biotec), respectively. Astrocytes were then acutely isolated using anti–ACSA-2+ (astrocyte cell surface antigen 2) MicroBead kit (Miltenyi Biotec). The ASCA-2+ epitope is from the astrocyte-specific β2 subunit of the sodium potassium exchanger (Batiuk et al., 2017). The anti–ACSA-2+ antibody is highly specific for astrocytes (Holt and Olsen, 2016), is robustly expressed in astrocytes (Kantzer et al., 2017), and is used as a first choice for isolating astrocytes (Batiuk et al., 2017). Manufacturer instructions were generally followed, except incubation times were extended to 25 min and total volume of microbeads was increased to 20–40 μl.
Primary astrocyte cultures
Astrocytes from RTT or WT littermates were isolated from P3–P6 pups as described above. Astrocytes were plated on 13-mm glass coverslips coated with poly-l-ornithine and laminin in 24-well plates at a density of 1.0 × 105 cells. Astrocytes were maintained in serum-free media (50% Neurobasal Medium (Thermo Fisher Scientific), 50% MEM, 1 mm sodium pyruvate, 2 mm glutamine, and B27). Medium was changed every day for 3 consecutive days, with subsequent media changes every 3–4 d. RNA was collected after 7 and 14 days in vitro (DIV) using Ambion’s PureLink RNA Isolation Kit.
Primary astrocyte culture immunohistochemistry
Astrocytes were fixed for immunofluorescence after 7 DIV. Cells were first washed with cold PBS, followed by fixation with 4% paraformaldehyde for 15 min at room temperature. Subsequently, cells were incubated for 1 h in blocking buffer (10% goat serum, 0.3% Triton X-100 in PBS). Astrocyte processes were stained with overnight incubation with Dako anti-rabbit GFAP (Z0334) at 1:1000 concentration. Fluorescent images were acquired with an Olympus VS-120 system.
Acute brain slice preparation
Mice were anesthetized with CO2 and rapidly decapitated. Brain was quickly isolated and placed in icy artificial cerebrospinal fluid (ACSF) cutting solution (see above). All solutions were continuously bubbled with 95% O2/5% CO2 to bring to pH 7.4. Coronal 300-μm slices were cut with a Leica VT1000A vibratome and placed in ACSF at room temperature containing the following in mm: 120 NaCl, 3 KCl, 1 MgCl2, 2 CaCl2, 26.2 NaHCO3, 11.1 glucose, and 5 Hepes. Slices were allowed to recover for 45 min at room temperature before use.
K+-sensitive microelectrodes
Borosilicate glass capillary tubes (Warner Instrument Corp.; GC200F-10; OD: 2.0 mm, ID: 1.16 mm) were washed in concentrated nitric acid, water, then 100% EtOH and dried overnight at 200°C. Pipettes were pulled on Sutter Instrument Model P-97 and then placed upright and incubated above a dish containing silane [bis(dimethylamino)dimethylsilane] for 5 min at 200°C. Using a fine-tipped syringe (Microfil 28 AWG, World Precision Instruments), a K+ ion exchanger (IE190, World Precision Instruments) was filled at the pipette tip, with care taken to ensure no bubbles. Pipettes were then backfilled with 100 mm KCl. K+-sensitive microelectrodes were calibrated with solutions containing in mm: 133 NaCl, 3 KCl, 1.5 CaCl2, 1.2 NaH2PO4, 1 MgCl2, 10 glucose, and 8.55 Hepes or 106 NaCl, 30 KCl, 1.5 CaCl2, 1.2 NaH2PO4, 1 MgCl2, 10 glucose, and 8.55 Hepes after every experiment. All experiments were performed using a World Precision Instruments electrometer (model FD 223), digitized with a Digidata 1440A (Molecular Devices), and stored and analyzed using Clampex 10.2. K+ responses were recorded, at depths 40–75 μm into the slice, in layer II/III of the cortex resulting from 3 consecutive 1-s stimulations of 100 μA at 20 Hz (0.1-μs pulse width) in layer IV/V of the cortex. Signals were low-pass filtered at 10 Hz.
Whole-cell patch clamp electrophysiology
Patch pipettes were pulled from thin-walled borosilicate glass (World Precision Instruments, catalog no. TW150F-4) and filled with pipette solution containing the following in mm: 125 K-gluconate, 10 KCl, 10 Hepes (free acid), 10 disodium creatine phosphate, 2 MgATP, 0.2 NaGTP, and 0.5 EGTA. The pipette solution was brought to pH 7.3 with KOH and 285–290 mOsm with sucrose. After filling patch pipettes with pipette solution, final resistances were 6–8 MΩ. Slices were transferred to a Zeiss Examiner D1 equipped with a 40× water-immersion lens and Zeiss Axiocam MRm for imaging and constantly perfused with ∼30°C ACSF. Astrocytes were identified using morphologic features. Recordings were made with an Axopatch 200B amplifier (Molecular Devices), low-pass filtered at 1 kHz, and digitized with Digidata 1440A (Molecular Devices). Data were acquired and stored on a personal computer using Clampex 10.2. Cell capacitance and series resistance were compensated for using the amplifier. Cell capacitance was measured from the amplifier. Experiments with a series resistance up to 12 MΩ were used, and series resistance was compensated for by 80% to reduce voltage errors. Steady-state currents were measured.
Chromatin immunoprecipitation
Half-brains from 7–9-wk-old Mecp2+/y and Mecp2–/y mice were collected for chromatin immunoprecipitation (ChIP) analysis. Chromatin preparation was performed using the Magna ChIP G Tissue Kit (Millipore) according to manufacturer’s instructions. Briefly, samples were sheared in 1× Tissue Stabilizing Solution (Millipore) with protease inhibitors using a 1-ml pipette tip. Samples were then fixed in 1% formaldehyde for 10 min at 37°C with minor agitation. The fixation was quenched using 10× glycine (for final concentration at 1×) and incubated for 5 min at room temperature. Cross-linked samples were washed 3 times in ice-cold 1× PBS with protease inhibitors. Samples were then lysed on ice in Tissue Lysis Buffer with protease inhibitors for 15 min, with brief vortexing every 5 min. Samples were resuspended in 600 µl ChIP Dilution Buffer with protease inhibitors and sonicated on wet ice under the following conditions: 10 s pulse, 50 s rest, 20% amplitude, 5 total cycles. After sonication, samples were centrifuged at 15,000 rcf at 4°C for 10 min. The resulting supernatant was collected and distributed into 200-µl aliquots for immunoprecipitation (IP).
IP, washes, reverse cross-linking, and DNA collection and purification were performed as outlined in Li et al. (2012) with the following modifications. (1) Additional ChIP Dilution Buffer with protease inhibitors was added to each aliquot for a final volume of 500 µl. (2) Both IPs and inputs were digested using 10 µl of 0.5 m EDTA, 20 µl of 1 m Tris, pH 6.5, and 1 µl of 20 mg/ml proteinase K (Clontech) for 1 h at 55°C with minor agitation. DNA was collected and purified using the Qiagen PCR Purification kit according to manufacturer’s instructions with the following modifications. (1) At the final elution step, EB buffer was incubated on the column for 10 min before centrifugation. 2) Final DNA was eluted in 50 µl. Quantitative PCR (qPCR) was performed on the Bio-Rad CFX96 machine using the Applied Biosystems SYBR Select Master Mix for CFX. qPCR cycling parameters were as follows: 95°C for 3 min, followed by 40 cycles of 95°C for 10 s, 60°C for 30 s, and 72°C for 45 s, and a final incubation at 70°C for 10 min. Primer efficiency was verified through melting-curve analysis (60°C for 1 min, and 0.5°C increments starting at 60°C up to 95°C for 15 s each) and running qPCR products on a 1.5% agarose gel. Primers used to amplify specific regions within Kcnj10 promoter region are listed in Table 1. The ddCT method was used to determine fold change expression in Mecp2+/y relative to Mecp2–/y mice. Briefly, the ddCT was calculated as the following: where 6.644 represents 1% starting input;
where 6.644 represents 1% starting input; Fold change expression was calculated using the formula 2–ddCT for both Mecp2+/y and Mecp2–/y mice.
Fold change expression was calculated using the formula 2–ddCT for both Mecp2+/y and Mecp2–/y mice.
Targeted Kir4.1 CpG island I (promoter) PCR amplification sites
Data analysis
Data were organized in Microsoft Excel or Origin 8.5. Unless stated otherwise, two-tailed t tests were performed as appropriate, except for non–normally distributed data, in which case Mann–Whitney nonparametric test was performed. In Fig. 5, two-way ANOVA was performed with Tukey post hoc comparison. The statistical tests were performed using InStat 3 (GraphPad Software) or MYSTAT (Systat Software). All data are reported as average ± SEM.
Results
Kir4.1 currents are significantly decreased in Mecp2–/y mice
Astrocytes contribute to symptomology of RTT (Lioy et al., 2011). However, few studies have examined the functional differences between WT and RTT astrocytes. Kir4.1, a glia-specific inward rectifying channel, is critical for normal CNS function. Mutations in KCNJ10 have been linked to developmental disorders characterized by early onset seizures, ataxia, epilepsy, and profound developmental delay (Kofuji et al., 2000). Of note, these symptoms are also observed in RTT. We therefore sought to investigate the properties of Kir4.1 channel function and expression in RTT.
MeCP2 expression is high in cortical tissue and apparent cortical dysfunction is observed in Mecp2-deficient mice (Kishi and Macklis, 2004; D’Cruz et al., 2010). Kir4.1 currents were measured in layer I and layer II/III cortical astrocytes of symptomatic Mecp2 mutant male (Mecp2–/y) mice and littermate age- and sex-matched controls. To isolate Kir4.1 currents, we stepped cortical astrocytes from a holding potential of –80 to 0 mV, and then from –180 to 100 mV in 20-mV increments. We washed on 100 μM BaCl2, a concentration that specifically blocks Kir channels (Ransom and Sontheimer, 1995), and subtracted out currents that were sensitive to BaCl2. As there was no significant difference between astrocytes in layer I and layer II/III (data not shown), we pooled both layers together. Astrocyte recordings obtained from WT mice (n = 24) displayed large-amplitude, linear currents typical of passive astrocytes (Zhou et al., 2006). As depicted in Fig. 1E, the “passive” Ba2+-sensitive Kir4.1 currents were smaller in Mecp2–/y astrocytes (n = 19). Although whole-cell currents were significantly smaller in Mecp2–/y astrocytes (Fig. 1C), which could be partially explained by smaller Ba2+-sensitive Kir4.1 currents, there was also a decrease in other unidentified Ba2+-insensitive currents in Mecp2–/y astrocytes (Fig. 1D). In an attempt to identify other K+ channels disrupted in the cortex of MeCP2-deficient mice, we mined RNA sequencing data from a recent transcriptomic study performed in the cortex of WT and symptomatic MeCP2-deficient mice that were age matched with the current study. Of the 15 Kcnj genes identified in that RNA sequencing study, only Kcnj10 (Kir4.1) and Kcnj16 (Kir5.1, a channel thought to form heteromers with Kir4.1, but not homomeric channels) were significantly downregulated in the cortex of MeCP2-deficient mice (Table 2). Of the 14 Kcnk genes identified, only Kcnk2 (Trek-1) was differentially expressed, and it was modestly upregulated. Together, these data suggest that a loss of Kir4.1 homomers and possibly Kir4.1/Kir5.1 heteromeric channels contribute to the reduced K+ conductance observed in MeCP2-deficient mice.

Ba2+-sensitive Kir4.1 currents are significantly reduced in layer II/III astrocytes of Mecp2–/y mice. A, Representative traces of pre-Ba2+, post-Ba2+, and Ba2+-sensitive currents in WT and Mecp2–/y astrocytes. B, Mecp2–/y astrocytes show higher input resistance than their WT littermates. C–E, Current–voltage graphs (n = 19–24) demonstrate that pre-Ba2+, post-Ba2+, and Ba2+-sensitive currents are significantly reduced in Mecp2–/y astrocytes [except at the –80-mV step in the pre-Ba2+ currents (C) and the Ba2+-sensitive currents (E)]. In all, results are consistent with a reduction of Kir4.1 currents in the plasma membrane. Mann–Whitney test was conducted for each voltage step between the two genotypes. **, p < 0.01.
RNA sequencing indicates 14 kcnj and 15 kcnk potassium channels identified in the cortex of symptomatic MeCP2–/y mice and WT age-matched littermates
Kir4.1 channels contribute to the intrinsic membrane properties of astrocytes (Kofuji et al., 2000; D’Ambrosio et al., 2002; Neusch et al., 2006; Olsen et al., 2006; Djukic et al., 2007; Chever et al., 2010; Ma et al., 2014). Therefore, we examined input resistance and resting membrane potential (RMP) in WT and Mecp2–/y cortical astrocytes. Kir4.1 has a high open probability at rest (Nwaobi et al., 2016), and the relative expression levels of Kir4.1 correlate with the astrocyte input resistance. Lower Kir4.1 channel activity in Mecp2–/y mice was associated with significantly higher membrane resistance (Fig. 1B; 15.9 ± 2.8 MΩ in WT vs. 33.2 ± 5.6 MΩ in Mecp2–/y; p = 0.006; n = 19–23). However, both genotypes showed similar hyperpolarized resting membrane potential (–77.9 ± 0.7 mV in WT vs. –76.6 ± 0.9 mV in Mecp2–/y; p = 0.23; n = 19–24) and whole-cell capacitance, a measure of cell size (18.5 ± 1.5 pF in WT vs. 20.3 ± 2.9 pF in Mecp2–/y; p = 0.56; n = 19–24). The lack of a change in RMP is in contrast to previous reports regarding the importance of Kir4.1 to the RMP of astrocytes (Olsen et al., 2006; Djukic et al., 2007; Seifert et al., 2009). The lack of a change in resting membrane potential in RTT astrocytes may be explained by the partial reduction in current in Mecp2–/y mice as opposed to complete knockout (Kir4.1 KO mice) or knockdown (siRNA knockdown in cultured astrocytes) as published in previous reports. Astrocytes are thought to be selectively permeable to K+ ions; as such, the remaining Kir4.1 and leak channels would maintain the resting membrane potential near the K+ equilibrium potential. Alternatively, the increased Kcnk2 gene expression in the cortex of MeCP2–/y mice may compensate for lower levels of Kir4.1 expression.
[K+]o homeostasis is dysregulated in MeCP2–/y mice
Given the previously demonstrated role of astrocytes in the buffering and maintenance of [K+]o (Kofuji and Newman, 2004), we assessed whether K+ homeostasis is disrupted in Mecp2–/y mice. To test this, we stimulated neuronal activity in layer IV/V of the cortex with a 1-s stimulus (20 Hz, 100 μA) followed by a 30-s recovery, performed 3 times in series. Changes in [K+]o were measured in layer II/III with a calibrated K+-sensitive microelectrode. Upon insertion of the K+-sensitive microelectrode into the slice, we detected a spike in [K+]o followed by a fall to a steady-state level. Consistent with the possibly that MeCP2 regulates Kir4.1 expression and consequentially K+ homeostasis, we found the peak of this K+ spike to be larger in Mecp2–/y mice compared with WT littermates (5.2 ± 0.5 mm in WT vs. 7.9 ± 1.2 in Mecp2–/y; Fig. 2B, left; p < 0.05; n = 7–8). Additionally, steady-state [K+]o was higher in Mecp2–/y mice compared with WT littermates (4.2 ± 0.2 mm in WT vs. 5.1 ± 0.4 in Mecp2–/y; Fig. 2A, dashed line, Fig. 2B, middle; p < 0.05; n = 7–8). Intriguingly, a similar elevation in baseline K+ was observed in a murine model of Huntington disease, which showed similar reductions in astrocyte Kir4.1-mediated currents (Tong et al., 2014). We also observed an increase in the amplitude of the K+ undershoot after the second stimulation (Fig. 2A, arrows). The amplitude of the undershoot under baseline increased from 0.03 ± 0.02 mm in WT to 0.08 ± 0.02 mm in MeCP2–/y mice (Fig. 2B, right; p < 0.05; n = 6–9). Using a similar approach, Chever et al. (2010) demonstrated the appearance of a significant undershoot after stimulation in the hippocampus of GFAP-targeted Kir4.1 knockout mice. Together, these data indicate that K+ homeostasis is disrupted in MeCP2–/y mice and support the possibility that loss of MeCP2 disrupts Kir4.1 function.

[K+]o is elevated and [K+]o undershoots after stimulation in MeCP2–/y cortex. A, Representative changes in [K+]o after three successive stimulations in WT and Mecp2–/y slices. Baseline [K+]o (dashed horizontal line) is elevated in Mecp2–/y slices. B, Left, peak [K+]o increase after insertion of K+-sensitive microelectrode into slice is greater in Mecp2–/y slices. Middle, steady-state [K+]o is elevated in Mecp2–/y slices. Right, K+ undershoot after the second stimulation is greater in Mecp2–/y slices. *, p < 0.05.
Kir4.1 protein expression is significantly downregulated in Mecp2–/y mice
To investigate whether decrease in currents stems from decreased Kir4.1 protein expression, we examined Kir4.1 expression in the cortex. We isolated the cortex through microdissection, and Western blot analysis revealed 63% downregulation of Kir4.1 protein expression in Mecp2–/y mice compared with littermate controls, with GAPDH and tubulin used as loading controls (Fig. 3A, B; p < 0.05; n = 10–12). The bands on the Western blot represent both tetrameric (∼150-kDa) and monomeric (∼50-kDa) isoforms of Kir4.1 (Fig. 3A), which are commonly observed when blotting for Kir4.1 and are not observed in negative control samples (HEK cells or Kir4.1 KO mice; Olsen et al., 2006). Intriguingly, higher multimers of Kir4.1 are associated with developmental upregulation of the channel that occurs during postnatal development and astrocytic maturation (Dibaj et al., 2007), and these higher molecular weight bands are reduced in MeCP2-deficient mice.

Kir4.1 protein is downregulated in Mecp2–/y brains. A, Representative Western blot of cortical protein lysates from 5 wild-type and 6 symptomatic Mecp2–/y males demonstrates a significant loss of Kir4.1 protein expression in cortex (∼150 kDa tetramer, 37 kDa monomer) when normalized to a loading control (GAPDH or tubulin). B, Quantification of ImageJ analysis normalizing Kir4.1 protein expression to GAPDH in cortex. Similar loss of Kir4.1 immunoreactivity in Mecp2-deficient male mice is seen in brainstem and hippocampus. In isolated astrocytes, Kir4.1 expression is lower in Mecp2–/y mice compared to WT, as in whole-cortex lysate (C, D).
We have recently shown that Kir4.1 is expressed at significantly higher levels in the brainstem relative to other CNS structures (Nwaobi et al., 2014). Therefore, we also examined whole-brainstem homogenates from WT and Mecp2–/y to determine whether Kir4.1 was affected in this structure. Western blots indicate a 20% downregulation of Kir4.1 in the brainstem (Fig. 3B; p < 0.05; n = 9–12). Given that a deficiency of Kir4.1 may also contribute to hyperexcitability in the hippocampus (Calfa et al., 2011b), we probed for Kir4.1 expression and found a 36% downregulation (Fig. 3B; p < 0.05; n = 6–7). Loss of Kir4.1 in cortex, brainstem, and hippocampus may contribute to altered function in these brain regions in Mecp2–/y mice.
Kir4.1 is most highly expressed in astrocytes but is also expressed in oligodendrocytes and NG2+ glia (Nwaobi et al., 2016). To evaluate Kir4.1 expression in WT and Mecp2–/y in astrocytes specifically, versus a whole-cortical homogenate, magnetic cell separation (MACS) was used (Holt and Olsen, 2016). Astrocytes were isolated from symptomatic MeCP2-deficient males and their age-matched littermates. Western blot analysis from isolated astrocytes in WT and Mecp2-deficient mice indicated that Kir4.1 is significantly decreased (Fig. 3C, D; p < 0.01). The reduction is similar in both whole cortex and isolated astrocytes (Fig. 3D).
Kir4.1 (Kcnj10) RNA expression is significantly downregulated in Mecp2–/y and Mecp2–/+ mice
As we demonstrated a global decrease in expression of Kir4.1 protein in Mecp2-deficient mice (Fig. 3), we next examined whether Kcnj10 transcription is altered in the RTT brain. As a first step, we performed quantitative PCR (qPCR) on microdissected samples isolated from symptomatic Mecp2–/y mice and WT littermates to examine Kcnj10 gene expression. In all brain regions, there was a significant loss of Kcnj10 transcription (Fig. 4A): midbrain (41 ± 15%, p < 0.05) cerebellum (32 ± 3%, p < 0.001), brainstem (31 ± 5%, p < 0.001), cortex (31 ± 5%, p < 0.001), and hippocampus (18 ± 8%, p < 0.05). Additionally, MACS-isolated astrocytes from P60 symptomatic Mecp2–/y mice cortex also showed reduction in Kcnj10 transcription, same as in the whole brain (Fig. 4B, 33 ± 25%, p < 0.05).

Decreased Kir4.1 transcription in Mecp2-deficient mice. A, Kcnj10 mRNA is significantly downregulated in microdissected cerebellum, midbrain, cortex, hippocampus, and brainstem of Mecp2–/y mice. B, Similar reduction is observed in isolated cortical astrocytes from Mecp2–/y mice. C, D, Kcnj10 is significantly downregulated in the cortex and brainstem of Mecp2± symptomatic female mice. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Most research involving murine models of RTT involve mutant male mice, although RTT typically affects females. Female mice present a slightly less severe and more heterogeneous symptomology that has a delayed onset, due to variable X-chromosome inactivation (Calfa et al., 2011a). The mutation in our mice, the Jaenisch mutation, expresses in half of the neurons, except at 9-mo-old females in the dentate gyrus, which tend to express less (∼40%) of the chromosome with the mutation (Smrt et al., 2011). Nonetheless, there is no data concerning the effect of X-linked inactivation in glial cells. However, because RTT typically affects females, we asked whether loss of MeCP2 function is also associated with decreased Kcnj10 mRNA expression in Mecp2± mice. Symptomatic Mecp2± mice (7–8 mo of age), which displayed motor abnormalities including hindlimb clasping, were used for these studies. We observed a significant decrease in Kcnj10 gene expression in Mecp2± symptomatic females in the cortex (Fig. 4C, 1.01 ± 0.08 WT vs. 0.61 ± 0.07 Mecp2±) and brainstem (Fig. 4D, 1.01 ± .07 WT vs. 0.67 ± 0.11 MeCP2±). Interestingly, the decrease in mRNA in female Mecp2± mice was significantly larger than that seen in males, although females presumably maintain ∼50% MeCP2 expression. The decrease in Kcnj10 mRNA expression suggests a transcriptional mechanism of regulation.
MeCP2 regulates Kcnj10 transcription in a cell-autonomous mechanism
The question remains whether MeCP2 controls Kcnj10 expression via a cell-autonomous mechanism (i.e., direct effect of MeCP2 in astrocytes) or via an indirect effect. To attempt to discern between the two possibilities, we used neuronal-free primary cultured astrocytes. Cortical astrocytes from either WT or Mecp2–/y P3 mice pups (n = 8–12 samples, with 4–6 total cultures, 2 biological replicates per culture) were isolated using the MACS astrocyte isolation as described previously. These astrocytes were cultured for either 7 or 14 DIV, when cells were collected and RNA transcription was measured using qPCR. The cultured astrocytes retained their shaped as assessed by GFAP staining (Fig. 5A), and the RNA expression profile fitted that of isolated astrocytes: Gfap and Kcnj10 expression matching whole-cortex expression, whereas markers for other cell types were depleted (Fig. 5B). We observed no difference in Kcnj10 transcription between the two genotypes at 7 DIV (Fig. 5, p = 0.98), when Kcnj10 transcription levels in situ are also relatively low. In contrast, Kcnj10 transcription increases significantly in WT astrocytes after 14 DIV, indicative of normal developmental upregulation (Fig. 5, p < 0.001). Transcription in Mecp2–/y astrocytes did not increase (p = 0.99) and was significantly reduced compared with WT astrocytes (p < 0.01). These data suggest that loss of Kcnj10 transcription is autonomous to the mutant astrocyte and results from loss of astrocytic MeCP2.
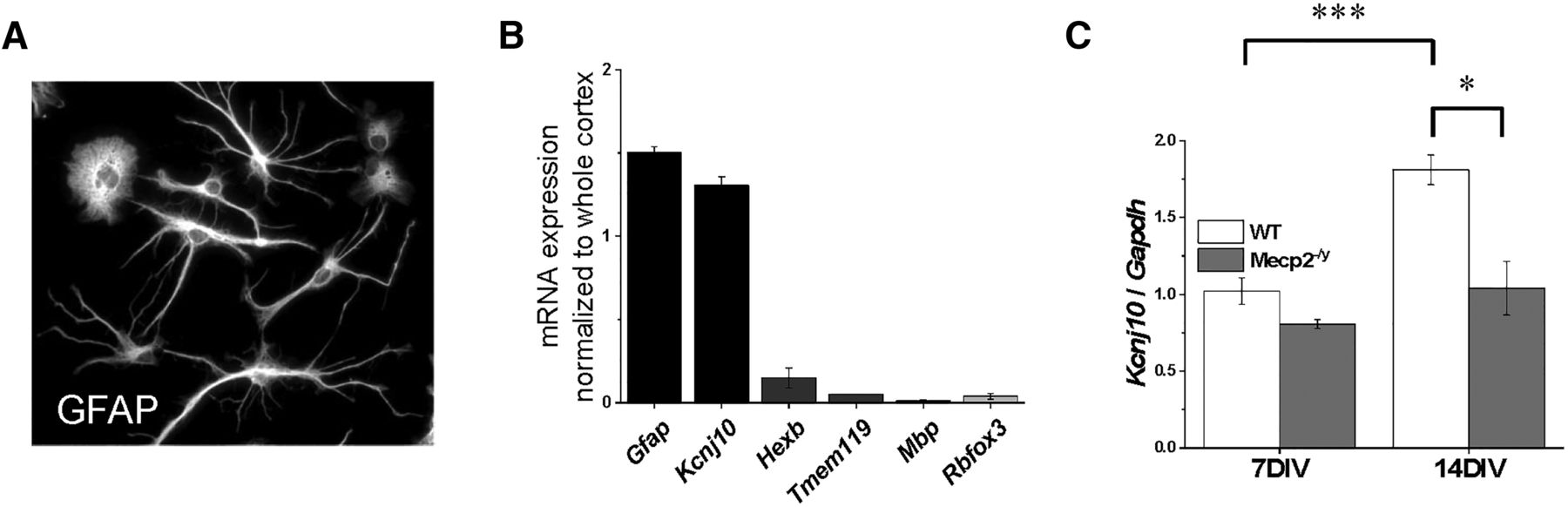
Loss of Kcnj10 is autonomous to astrocytes. With a magnetic-bead sorting technique, isolated astrocytes were plated and kept in serum-free medium for 7–14 d. Four to six cultures were used, with two biological replicates per culture, a total of 8–12 samples. A, By 7 d, WT astrocytes attain a stellate morphology as indicated by GFAP staining. B, By 14 d in culture astrocytes, Gfap and Kcnj10 expression is enriched compared to age-matched cortex. Cultures express low levels of mRNA for other cell types including microglia (HexB, Tmem119), oligodendrocytes (Mbp), and neurons (Rbfox3), indicating relative purity of the astrocyte cultures. C, When examining the transcription of Kcnj10, there is a significant effect for both genotype (p < 0.001) and DIV (p < 0.001). There was no difference at 7 DIV between WT and Mecp2–/y astrocytes in transcription of Kcnj10. Transcription of Kcnj10 shows a typical developmental increase in WT astrocytes at 14 DIV, while such an increase was not seen with the Mecp2–/y astrocytes. Two-way ANOVA was performed with Tukey post hoc comparison. *, p < 0.05; ***, p < 0.001.
Kir4.1 is a direct molecular target of MeCP2
Previous work has demonstrated that Kcnj10 gene expression is robustly, developmentally up-regulated and dependent on the degree of methylation at its promoter (Nwaobi et al., 2014). We postulated that decreased Kcnj10 transcription might be due to a loss of interaction between MeCP2 and the Kcnj10 gene in Mecp2-deficient mice. To test this, we performed a chromatin immunoprecipitation (ChIP) assay. MeCP2 binds DNA in cytosine-phosphodiester-guanine (CpG) islands that are either methylated or unmethylated to modulate gene expression (Chahrour et al., 2008). To determine whether the entire Kcnj10 gene contained CpG islands, we performed an in silico analysis of mouse Kcnj10 gene (Fig. 6A). Two CpG islands were identified: the first spanned the promoter region and the 5′ UTR, and the second spanned the transcriptional start site and the initial segment of the exonic region. Because MeCP2 is often bound to actively transcribed promoters (Yasui et al., 2007), we designed four (100–150-bp) overlapping primers to amplify the promoter of Kcnj10 (Table 1). The ChIP analysis was performed by probing for a MeCP2 protein interaction with regions 1–4 in the Kcnj10 promoter. For these experiments, IgG served as a negative control, and RNA polymerase II (RNA Pol II) served as a positive control. Results from these experiments show significant interactions between MeCP2 and sites 1, 2, and 4 of Kcnj10 in WT mice (Fig. 6B). To confirm the specificity of the immunoprecipitation, we performed the ChIP assay with Mecp2–/y and did not detect any amplification (Fig. 6C). These data are the first demonstrating a direct molecular interaction between MeCP2 and an astrocyte gene target, providing possible mechanistic support for the loss of Kcnj10 mRNA and its subsequent protein product in Mecp2-deficient mice.
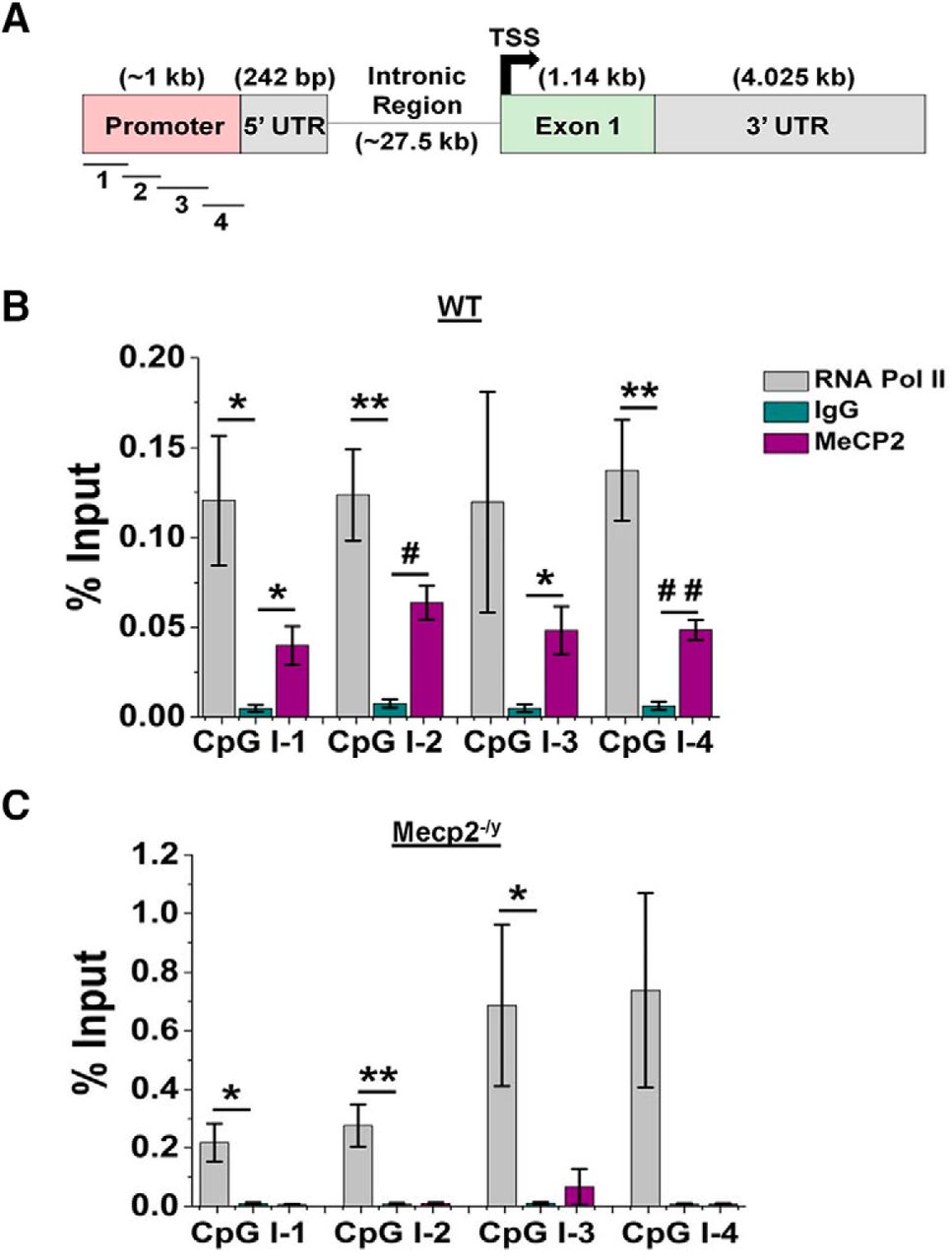
Kcnj10 is a direct molecular target of MeCP2. A, A schematic of the mouse Kcnj10 gene indicating the promoter, 5′ UTR, intronic, and exonic region of the gene. The relative locations of primers used to query MeCP2 binding are indicated. B, C, ChIP results indicate significant physical interaction between MeCP2 and Kcnj10 in three sites in the promoter of the Kcnj10 gene (B, sites 1, 2, and 4, p < 0.05, n = 5), while no interactions are found in the Mecp2-null mice (C). *, p < 0.05; **, p < 0.01; #, p < 0.0005; ##, p < 0.0001.
Kir4.1 gene and protein expression are downregulated early in disease progression in MeCP2–/y mice
Kir4.1 protein levels are markedly increased during early postnatal development, correlating with increased gene transcription and reduced methylation levels of the Kcnj10 gene (Nwaobi et al., 2014). The most significant increases in gene and protein expression occur from in postnatal wk 2–4 (Nwaobi et al., 2014), when astrocytes undergo significant morphological refinement and maturation (Bushong et al., 2004). To determine whether MeCP2 deficiency altered normal developmental patterns of Kcnj10 gene expression, we performed qPCR at two developmental time points (P10 and P21), which are early in the disease progression in RTT mice. No difference in Kcnj10 transcription was observed at P10 (Fig. 7A), but we observed a ∼30% decrease in transcription in the Mecp2–/y mice by P21 (Fig. 7B; p < 0.05; n = 6). Western blot analysis indicates a significant deficit in Kir4.1 protein levels in Mecp2–/y (Fig. 7C, D; p < 0.05; n = 6) at both time points. These data demonstrate that normal protein levels of Kir4.1 are not achieved at any time point in Mecp2-deficient animals, which may be the result of a direct positive interaction between MeCP2 protein and Kcnj10 gene promoter that is absent in MeCP2–/y mice.

Kir4.1 is not sufficiently upregulated through development in Mecp2-deficient mice. A, Kcnj10 mRNA is not significantly different in the cortex of p10 Mecp2–/y mice. B, Kcnj10 mRNA is significantly decreased in the cortex of p21 Mecp2–/y mice. C, D, Representative Western blots and ImageJ quantification demonstrate loss of Kir4.1 protein expression in cortex in both p10 (C) and p21 (D) mice when normalized to a loading control (GAPDH). *, p < 0.05.
Discussion
Several recent reports have implicated a role of glia in the progression of RTT (Ballas et al., 2009; Maezawa and Jin, 2010; Lioy et al., 2011; Derecki et al., 2012). Mechanistically, it was hypothesized that RTT astrocytes inhibited neuronal maturation through release of a molecule that stunted dendritic maturity or through deficient release of pro-growth soluble molecule. Previous in vitro work also suggests that excess glutamate contributes to abnormal neuronal morphology (Maezawa and Jin, 2010). In the current study, we demonstrate reduced Kir4.1 channel activity in cortical astrocytes. This decrease in channel activity is associated with significant reductions in Kir4.1 protein and mRNA levels and may result from the loss of a direct positive interaction between the MeCP2 protein and the Kcnj10 gene promoter in MeCP2 knockout mice. Supporting this notion, astrocyte cultures derived from MeCP2–/y mice fail to upregulate Kir4.1 compared with cultures derived from WT mice. These results indicate that the loss of Kir4.1 is autonomous to the astrocyte and provide a mechanism for failure of Kir4.1 developmental upregulation in RTT mice. These data suggest that MeCP2 is a positive regulator of Kcnj10 gene expression through development and potentially provide insight into how astrocytic dysfunction may contribute to RTT.
The role of Kir4.1 in RTT and normal brain
Accumulating evidence indicates that Kir4.1 plays an integral role in the CNS. Decreased Kir4.1 currents are associated with various pathologies such as epilepsy, cerebral trauma, cerebral ischemia, cerebral inflammation, Alzheimer’s disease, amyotrophic lateral sclerosis, and Huntington’s disease (Nwaobi et al., 2016). Here, we demonstrate that astrocyte Kir4.1 currents are significantly reduced in symptomatic MeCP2-deficient mice. Of note, the extent to which astrocytes are clamped is uncertain because of the leaky membrane and the complex morphology. Reduction in Kir4.1 function may contribute to neuronal dysfunction. After firing of an action potential, K+ is extruded from neurons. Because of the small size of the extracellular space, small fluxes in K+ can lead to large concentration changes that are sufficient to modulate the efficacy of neuronal transmission (Ransom et al., 2000). Regulation of extracellular potassium ([K+]o) is the most well-recognized function of astrocytes (Kuffler and Potter, 1964; Orkand et al., 1966; Kuffler, 1967; Kofuji and Newman, 2004). Basal [K+]o in rat hippocampal slices was increased when Kir channels were blocked with BaCl2 (D’Ambrosio et al., 2002). Loss of Kir4.1, with a similar increase in [K+]o, was reported in a mouse model of Huntington’s disease (Tong et al., 2014) and is associated with medium spiny neuron dysfunction. AAV-mediated rescue of Kir4.1 specifically to astrocytes rescued elevated K+, prolonged survival, and ameliorated motor deficits in these mice.
Our data demonstrate that loss of Kir4.1 in MeCP2-deficient mice is associated with an increase in [K+]o and K+ undershoot (Fig. 2). Inhibition of Kir4.1 with 200 μM barium in rat hippocampus led to elevated [K+]o and K+ undershoot (D’Ambrosio et al., 2002), as in our work. The undershoot under normal conditions is explained by a model in which Kir4.1 maintains [K+]o by efflux of K+ from astrocytes to balance excessive K+ uptake by the ATPase (D’Ambrosio et al., 2002). Thus, loss of Kir4.1 leads to a larger K+ undershoot and perturbation of [K+]o.
Given the role that Kir channels play in the regulation of K+ concentration, loss of Kir4.1 in RTT may predict a change in neuronal excitability. There are conflicting reports regarding the excitability levels in the Mecp2-mutant brains. Some studies show hyperexcitability in various regions of mutant Mecp2 mice from the level of the neuron (Zhang et al., 2010; Calfa et al., 2011b) up to the level of the neuronal network (Asaka et al., 2006; Moretti et al., 2006; Zhang et al., 2008; D’Cruz et al., 2010; Calfa et al., 2011b; Colic et al., 2011; McLeod et al., 2013). Other studies have not demonstrated the same hyperexcitability in RTT (Dani et al., 2005; Dani and Nelson, 2009; Wood et al., 2009). These discordant findings may be a result of examination at different stages of disease development. Studies reporting measurements from presymptomatic to early symptomatic animals (P14–P35) show hypoactivity, while studies demonstrating network hyperexcitability use tissue from older mice, which clearly exhibit symptoms associated with RTT (D’Cruz et al., 2010; Colic et al., 2011). Thus, excitability of cortical neurons may dramatically change during disease progression. Supporting this, measurements of cellular density in cortical layers II/III, IV, V, and VI in Mecp2–/y mice demonstrate that no abnormalities are observed at P14, but by P56 changes in all cortical layers are apparent (Kishi and Macklis, 2004). This fits the findings in this study, in which Kir4.1 expression is normal in early development, and thus [K+] regulation by astrocytes may not be disrupted. Failure to upregulate Kir4.1 expression in astrocytes may contribute to the hyperexcitability in aged, symptomatic animals.
Supporting the importance of Kir4.1 in normal brain function, patient populations who carry loss-of-function mutations in the Kcnj10 gene have been identified. Intriguingly, CNS symptoms in these patients, including early-onset seizures, ataxia, profound lower motor extremity weakness, and severe cognitive deficits (Scholl et al., 2009; Sicca et al., 2011), are also commonplace in RTT patients. Of note, Mecp2–/y cortex demonstrated a significant reduction in Kir4.1 expression, not a complete loss, as would be the case in a Kcnj10-knockout animal. Although Kir4.1 currents are downregulated in Mecp2–/y mice, the resting membrane potential of the astrocytes appear unaltered. This may be due to other compensatory K+ channel regulation in astrocytes.
Impaired developmental regulation of Kir4.1 in RTT mice
Kir4.1 protein and mRNA expression are strongly developmentally up-regulated in all brain regions, with highest expression observed in caudal brain structures (Nwaobi et al., 2014). Throughout the CNS, the increased expression of Kir4.1 parallels the increase in MeCP2 expression and coincides with the onset of symptoms in RTT. Upregulation of Kir4.1 is directly related to the methylation status of several CpG islands found in the promoter and intronic regions of the gene (Nwaobi et al., 2014). Furthermore, the Kcnj10 promoter is methylated at a low level and highly expressed. Given that MeCP2 is a methyl-binding protein, which is often found bound to transcriptionally active genes with low-methylated promoters (Yasui et al., 2007; Chahrour et al., 2008), we speculated that MeCP2 was a positive transcriptional regulator of the Kir4.1 gene. Indeed, loss of MeCP2 leads to decreased Kir4.1 expression. Also, we found direct physical interaction between MeCP2 and a CpG-rich island in the promoter of Kcnj10 using ChIP (Fig. 6). The data from the cultured isolated astrocytes (Fig. 5) suggests that loss of Kcnj10 mRNA is autonomous to the astrocyte and not dependent on development in the diseased CNS. This is the first example of a glia-specific gene targeted by MeCP2 and suggests that MeCP2 positively regulates Kcnj10 gene transcription.
We demonstrate that Kir4.1 expression does not undergo a typical robust developmental upregulation when MeCP2 is absent. Kir4.1 is not downregulated, but instead never reaches normal levels, suggesting that astrocytic maturation may be altered in the absence of MeCP2. In vivo, the loss of Kir4.1 that exists before the onset of severe symptomatology in Mecp2-deficient mice indicates that it may contribute to disease progression, and not simply an epiphenomenon of a “diseased” brain.
The vast majority of individuals with RTT are females, yet most animals studies focus on male Mecp2 mutants. The reasons for this are that (1) male mice present with earlier symptoms and more severe symptoms, and (2) the loss of MeCP2 expression is less variable in male mice. Because the Mecp2 gene is on the X chromosome, mutant female mice express one WT copy of Mecp2, leading to less severe and later symptoms. Additionally, random X chromosome inactivation may lead to skewed expression of the mutant or WT allele, which can cause variable disease progression. Nevertheless, given that RTT largely affects females, we investigated Kir4.1 mRNA expression in aged symptomatic female animals and show a loss of Kcnj10 transcript similar to that observed in males.
Kir4.1/Kcnj10 dysregulation in Mecp2–/y mice: from direct interaction to function
Transcriptomic and, more recently, proteomic studies have begun to shed light on gene and protein disruption in human patients and animal models of RTT (Colantuoni et al., 2001; Jordan et al., 2007; Chahrour et al., 2008; Urdinguio et al., 2008; Ben-Shachar et al., 2009; Gibson et al., 2010; Gabel et al., 2015; Gabel et al., 2015; Lin et al., 2016). Although no study has systematically evaluated astrocytes, a recent publication indicates that 46 of 391 differentially expressed genes are uniquely astrocytic, including Kir4.1 (Pacheco et al., 2017). The changes in astrocyte gene expression do not indicate a typical reactive gliosis as seen in most neurologic diseases, because in RTT tissue many markers associated with reactive gliosis are downregulated. Intriguingly, several disrupted genes are developmentally regulated, possibly indicating alterations in astrocyte maturation in the MeCP2-deficient cortex. Although numerous studies have demonstrated that MeCP2 has many neuronal targets, including Bdnf, Gabr3, Reln, Sst, and Creb1 (Singh et al., 2008), here we show for the first time that one such critical astrocytic gene, Kcnj10, is a direct molecular target of MeCP2.
Importantly, we also show the first electrophysiological studies performed in astrocytes indicating that normal membrane currents are affected in a mouse model of RTT. These data may shed light on the role of astrocytes in RTT and indicate that astrocytes and Kir4.1 may represent novel therapeutic targets for the treatment of RTT.
Acknowledgments
Acknowledgments: We thank Lucas Pozzo-Miller, PhD, and Wei Li, PhD, for animals.
Footnotes
The authors declare no competing financial interests.
This work was supported by International Rett Syndrome Foundation for Basic Research Grant (#2916) to MLO, National Institutes of Health grants NS075062 (MLO) and HL104101 (MLO, DKM), the UAB CCTS (grant number 5UL1RR025777) for a pilot grant to MLO, and the Civitan International Research Center for funding to VAC. NLP was funded in part by a grant to the University of Alabama at Birmingham from the Howard Hughes Medical Institute through the Med into Grad Initiative. Additional funds were also provided by Connecticut Department of Public Health Grant 150263 (DKM).
This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International license, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



