

Abstract
Several animal and clinical studies have highlighted the ineffectiveness of fear extinction sessions delivered shortly after trauma exposure. This phenomenon, termed the immediate extinction deficit, refers to situations in which extinction programs applied shortly after fear conditioning may result in the reduction of fear behaviors (in rodents, frequently measured as freezing responses to the conditioned cue) during extinction training, but failure to consolidate this reduction in the long term. The molecular mechanisms driving this immediate extinction resistance remain unclear. Here we present evidence for the involvement of the corticotropin releasing factor (CRF) system in the basolateral amygdala (BLA) in male Wistar rats. Intra-BLA microinfusion of the CRFR1 antagonist NBI30775 enhances extinction recall, whereas administration of the CRF agonist CRF6–33 before delayed extinction disrupts recall of extinction. We link the immediate fear extinction deficit with dephosphorylation of GluA1 glutamate receptors at Ser845 and enhanced activity of the protein phosphatase calcineurin in the BLA. Their reversal after treatment with the CRFR1 antagonist indicates their dependence on CRFR1 actions. These findings can have important implications for the improvement of therapeutic approaches to trauma, as well as furthering our understanding of the neurobiological mechanisms underlying fear-related disorders.
Significance Statement
Trauma-related disorders are costly, highlighting the need to understand the reduction of fear through extinction learning for the development of better therapies. When extinction programs are applied too soon after the traumatic event, numerous studies have found them to be ineffective, although the underlying mechanisms were unclear. Here we confirm that futility of immediate extinction and provide a mechanistic explanation. Using a pharmacological approach, we show evidence for the involvement of the corticotropin releasing factor (CRF) system in the basolateral amygdala in this extinction deficit. We link this involvement with downstream molecular targets of the CRF system that are critical in synaptic plasticity, thus explaining the futility of immediate extinction and providing further insight into fear-related disorders.
Introduction
Trauma-related disorders impose a high burden on both individuals and society (Kessler et al., 2012), inspiring numerous studies of the mechanisms underlying fear extinction learning (Pape and Pare, 2010; Milad and Quirk, 2012). Reduction of fear responses to previously fearful stimuli through fear extinction learning is a complex process involving a broad network of brain structures, including the basolateral nucleus of the amygdala (BLA; Sotres-Bayon et al., 2004; Quirk and Mueller, 2008, Herry et al., 2010). Several molecular targets have been identified for the development of therapeutic interventions for post-traumatic stress disorder (PTSD) and other fear-related disorders (Milad and Quirk, 2012; Singewald et al., 2015). Among them, emerging evidence points to a key role for the central corticotropin releasing factor (CRF) system, which is well known for its role in the regulation of stress, fear, stressful learning, and anxiety responses (Bale, 2005; Nemeroff et al., 2006; Regev and Baram, 2014). CRF, a 41–amino acid peptide involved in the activation of the hypothalamic-pituitary-adrenal axis, also exerts extrahypothalamic actions in different brain regions through activation of two G-protein–coupled receptors, CRFR1 and CRFR2. Both the BLA and the central nucleus of the amygdala contain CRF-expressing neurons, and the BLA presents particularly high CRFR1 densities (Korosi and Baram, 2008).
Recently, a key role for the CRF system in impaired extinction processes has been suggested. In humans, enhanced CRF levels found in the CSF of PTSD patients have indicated a link between increased CRF concentrations and disrupted fear extinction observed in PTSD and anxiety disorders (Bangasser and Kawasumi, 2015; Gafford and Ressler, 2015). In rats, administration of CRF into the lateral amygdala before fear recall testing in formerly fear-conditioned animals induced enhanced freezing responses to the conditioned stimulus (Isogawa et al., 2013). Additionally, pharmacological enhancement of CRF in the BLA impaired long-term retention of fear extinction, whereas CRFR1 antagonism had the opposite effect (Abiri et al., 2014), supporting a detrimental role of CRF in the BLA in the consolidation of cued fear extinction. Furthermore, cell-specific genetic disruption of GABAAα1 within CRF-expressing neurons in mice was found to specifically impair fear extinction (i.e., not affecting fear conditioning or retention) processes, whereas systemic administration of a CRF antagonist partially rescued the fear extinction deficit in these mice (Gafford et al., 2012). Accordingly, an overactive CRF system in the BLA seems to interfere with extinction processes.
Importantly, it is not known whether the CRF system is involved in a particularly challenging extinction case known as “immediate fear extinction deficit” (Maren, 2014). This refers to the ineffectiveness of fear extinction programs, frequently observed both in animals and humans, when extinction training is administered soon (e.g., from minutes to a few hours) after fear conditioning. Specifically, despite exhibiting decreased within-session freezing during extinction training, subjects fail to maintain this response over long-term retention intervals (Maren and Chang, 2006; Schiller et al., 2008; Woods and Bouton, 2008; Chang and Maren 2009; Archbold et al., 2010; but see Myers et al., 2006). Given that, in addition to its involvement in the acquisition and consolidation of fear conditioning (Sah et al., 2008), the BLA has been implicated in fear extinction (Sotres-Bayon et al., 2004; Quirk and Mueller, 2008; Herry et al., 2010), we hypothesized that mechanisms that contribute to fear conditioning in the BLA might underlie the effectiveness of immediate extinction trials. Converging lines of evidence support the involvement of the BLA CRF system in the immediate extinction deficit. First, acute stress has been shown to rapidly induce CRF release in the amygdala (Pich et al., 1995; Merali et al., 1998), leading to activation of CRFR1 in the BLA in the consolidation of fear learning (Roozendaal et al., 2002, 2008; Hubbard et al., 2007). Second, CRF increases excitability of CRFR1-containing BLA projection neurons (Rainnie et al., 1992) and induces long-lasting increases in the amplitude of field postsynaptic potentials in the BLA (Sandi et al., 2008; Ugolini et al., 2008). These effects are reversed by antagonizing CRFR1, but not CRFR2 (Rainnie et al., 1992; Ugolini et al., 2008). Finally, whereas CRF infusions in the lateral amygdala before long-term memory testing enhanced freezing responses to the conditioned stimulus (Isogawa et al., 2013), CRFR1 antagonism in the BLA facilitated long-term retention of fear extinction (Abiri et al., 2014). Therefore, we evaluated the involvement of the CRF system in the amygdala in the minimal fear suppression induced by extinction training given shortly after fear conditioning and explored the molecular machinery involved.
Materials and Methods
Subjects
Male Wistar rats (Charles River, L’Arbresle, France) weighing 250–300 g at the start of the experimentation served as subjects and were singly housed in polypropylene cages (34 × 29 × 17 cm) lined with abundant pine shavings. Animals had ad libitum access to food and water and were maintained in constant temperature (23°C) and lighting (0700–1900) conditions. All experiments were performed during the light phase. Animals were allowed to habituate to the vivarium for 1 week and were then handled for 2 min on 3 days before the beginning of all experiments.
All procedures were conducted in conformity with the École Polytechnique Fédérale de Lausanne’s guidelines for animal experimentation. All efforts were made to minimize suffering and reduce the number of animals used.
Elevated plus maze
Before experiments, anxiety-related behavior was measured using the Elevated Plus Maze (EPM) according to the procedure described in Herrero et al. (2006). As previous reports indicate that CRF antagonist NBI30775 affects subjects differently depending on their natural anxiety level (Sandi et al., 2008), all groups were matched according to similar scores in this test. EPM sessions for all experiments were conducted 4–7 d before the first fear conditioning session.
Fear conditioning
Conditioning session
The training cage (Context A) consisted of a Plexiglas transparent chamber (30 × 37 × 25 cm; Panlab, Barcelona, Spain) that was positioned inside a sound-attenuating chamber. This chamber was constructed of black stainless steel walls of smooth texture, with a ceiling and door made of Plexiglas. The floor consisted of 20 steel rods wired to a shock source and solid-state scrambler for the delivery of foot shocks. Conditioning took place in a single session. After 3 min of free exploration, rats received five pairings of a 2-s conditioned-stimulus (CS) tone (80 dB, 2000 Hz) and a 0.5-s unconditioned-stimulus foot shock (0.6 mA). The intershock interval was 60 s. Subjects were removed from the chambers 58 s after the final shock presentation (thus, the training session lasted 8 min) and left undisturbed in their home cage until the extinction session.
Extinction session
Extinction of cued fear learning took place in a different context (Context B). The context shape was modified, the grid was replaced by a plastic smooth floor, and visual and odor cues were changed. Animals were free to explore the environment during the first 3 min, and then 70 CS were presented every 40 s. Depending on the protocol, the extinction session took place 30 min, 3 h, or 24 h after training. For each behavioral experiment, separate groups of animals were placed in the extinction context without any CS presentation as controls.
Testing session
Forty-eight hours after training, extinction memory was assessed in Context B. After 3 min of free exploration, the rats received five CS presentations with an intertrial interval of 60 s. Rats were removed from the chambers 58 s after the final CS presentation (8 min total duration)
In all sessions, behavior was monitored with a camera connected to a videorecorder for offline analysis, which was performed by an experimenter blind to the animal’s experimental condition. Fear was assessed by measuring the percentage of time spent freezing, characterized by a crouching posture and an absence of any visible movement except breathing.
Surgery and amygdala microinfusions
Animals were anaesthetized with i.p. ketamine (70 mg/kg) and xylazine (6 mg/kg). They were then implanted with two 18-mm stainless steel guide cannulae (23-gauge; Plastic One, Roanoke, VA) using a standard stereotaxic frame (Kopf Instruments, Bioseb, France). Cannulae were bilaterally implanted at the BLA coordinates (anteroposterior, –2.8 mm relative to bregma; lateral, ±5.1 mm from midline; ventral, –5.5 mm from dura). The tips of the cannulae were aimed 2 mm above the intended area. The cannulae were fixed to the skull with dental acrylic cement. Stylets were inserted into the guide cannulae to prevent clogging. Rats were given 1 week to recover from surgery, after which they received the EPM and fear sessions as described above.
All animals were handled individually for approximately 1–2 min each day during the 2–3 days preceding infusion to habituate them to the infusion procedure. Immediately after training, rats were gently restrained, stylets were removed, and injection needles (30 gauges) were inserted, extending 2 mm from the tip of the guide cannula. The injection needles were connected via polyethylene tubing to two 10-μl Hamilton microsyringes driven by an automated microinfusion pump (Harvard Apparatus). The needles were then left in position for an additional minute to enable diffusion of the solution into the tissue and minimize dragging of the liquid along the injection track.
Drugs
NBI30775 (3-[6-(dimethylamino)-4-methyl-pyrid-3-yl]-2,5-dimethyl-N,N-dipropyl-pyrazolo[2,3-a]pyrimidin-7-amine), a nonpeptide CRFR1 antagonist (also known as R121919) was a gift from Dimitri Grigoriadis (Neurocrine Inc., San Diego, CA). It was dissolved in dimethylsulfoxide (DMSO) and bilaterally infused in the BLA at different concentrations (0.1, 0.3, 1, or 10 µg in 0.5 µl) at a rate of 0.3 µl/min immediately after fear conditioning. Vehicle infusions of DMSO were administered in a similar manner. The infusion volume for NBI30775 experiments was 0.5 µl/hemisphere. Depending on the experiment, animals were subject to extinction 30 min thereafter or left undisturbed until the testing session 48 h and 7 d after training.
The CRF agonist CRF6–33 (Sigma-Aldrich, St. Louis, MO) was dissolved in saline and infused bilaterally in the BLA at a concentration of 0.1 µg in 0.2 µl, at a rate of 0.3 µl/min just before a delayed (24 h after training) extinction session. The infusion volume for agonist experiments was 0.2 µl/hemisphere. Animals were tested 48 h after training. Vehicle infusions of saline were administered in a similar manner.
After completion of behavioral experiments, animals were overdosed with sodium pentobarbital (100 mg/kg i.p.). The brains were removed and immediately frozen at –50°C in isopentane and stored at –20°C. Coronal sections (40 µm thick) were stained with thionine for histological checking. Of the 84 implanted animals, 12 were discarded due to misplacement of one or both cannulae.
Western blot
Tissue preparation
Immediately after decapitation, the amygdala was rapidly dissected out and frozen at –80°C until processing. Tissue was homogenized in 10 volumes of ice-cold sucrose (0.32 m) and HEPES (5 mm) buffer that contained a cocktail of protease inhibitors (Complete TM, Roche, West Sussex, UK) with 16 strokes and centrifuged at 1000 × g for 5 min. The resulting total fraction pellet was resuspended in Krebs buffer with 1% NP40, incubated at 4°C for 40 min, and centrifuged at 10,000 × g for 20 min at 4°C. Protein concentration for each sample was estimated by bicinchoninic acid protein analysis (Bio-Rad, Hercules, CA).
Quantification of phosphorylation of the AMPA GluA1 subunits
Ten micrograms of protein were loaded in each well and separated on 10% (w/v) SDS-PAGE and transferred (70V, 90 min) to a nitrocellulose membrane (Whatman, Maidstone, UK). Membranes were incubated overnight at 4°C with a rabbit anti–phospho-Ser845 GluA1 polyclonal antibody (detects phosphorylation on GluA1 serine-845 only, 1:5000, cat. P1160-845, RRID: AB_2492128; PhosphoSolutions, Aurora, CO), a rabbit anti–phospho-ser831 GluA1 monoclonal antibody (detects phosphorylation on GluA1 serine-831 only, 1:5000, cat. 04-823, RRID: AB_1977218; Merck-Millipore, Billerica, MA), and a rabbit anti-GluA1 polyclonal antibody (detects GluA1 irrespective of modifications, 1:10,000, cat. ADI-905-416-1, RRID: AB_2039139; Assay Designs). Monoclonal mouse anti-actin (1:5000, cat. A3853, RRID: AB_262137; Sigma-Aldrich), and a mouse monoclonal anti-GAPDH (1:40,000, cat. Ab8245, RRID: AB_2107448; Abcam, Cambridge, UK) were incubated as loading controls. The blots were washed with PBS plus Tween, incubated for 1 h with a secondary antibody, a goat anti-rabbit IgG horseradish peroxidase conjugate (1:5000, cat. G-21234, RRID: AB_1500696; Thermo Fisher Scientific, Waltham, MA), and a goat anti-mouse IgG peroxidase conjugate for loading controls (1:5000, cat. 401215, RRID: AB_10682749; Calbiochem) and developed using an enhanced chemiluminescence system (Pierce, Rockford, IL). Bands were revealed with a ChemiDoc imaging system (Bio-Rad) for optimum exposure time. Images were analyzed using QuantityOne software v4.6.3 (Bio-Rad), where the adjusted volume was calculated for each band. For each group, the value of pGluA1 Serine subunit was normalized to total GluA1 after normalization to loading controls. To assess changes relative to the basal state, each experimental group is reported as a percentage of home cage group values.
Calcineurin activity ELISA assay
Immediately after rapid decapitation, brains were dissected out and flash-frozen in ice-cold isopentane. The basolateral amygdala was tissue punched on a freezing cryostat and stored at –80°C for further processing. Samples were homogenized and processed according to the manufacturer’s protocol for the Calcineurin Cellular Activity kit (BML-AK816; Enzo Life Sciences, Lausen, Switzerland), with the following adjustments. Punches were homogenized in 200 µl lysis buffer with protease inhibitor using a motorized pestle and passed through a desalting column to remove excess phosphate. Calcineurin activity was then measured according to manufacturer’s specifications.
Data analyses
Intergroup comparisons were evaluated using Student’s unpaired t-test and one- or two-way ANOVA followed by the Fisher test for post hoc analysis where appropriate. Differences were considered significant if p < 0.05. Superscript letters listed with p-values correspond to the statistical tests shown in Table 1. For Western blot data and calcineurin activity, data are shown as percentage of home cage controls.
Summary of statistics for all experiments.
Results
In all experiments, we verified that freezing levels during fear conditioning did not differ for the different experimental groups included. For the sake of clarity, these analyses are reported in Table 2.
Summary of statistics for fear conditioning sessions.
Immediate extinction sessions result in inefficient extinction
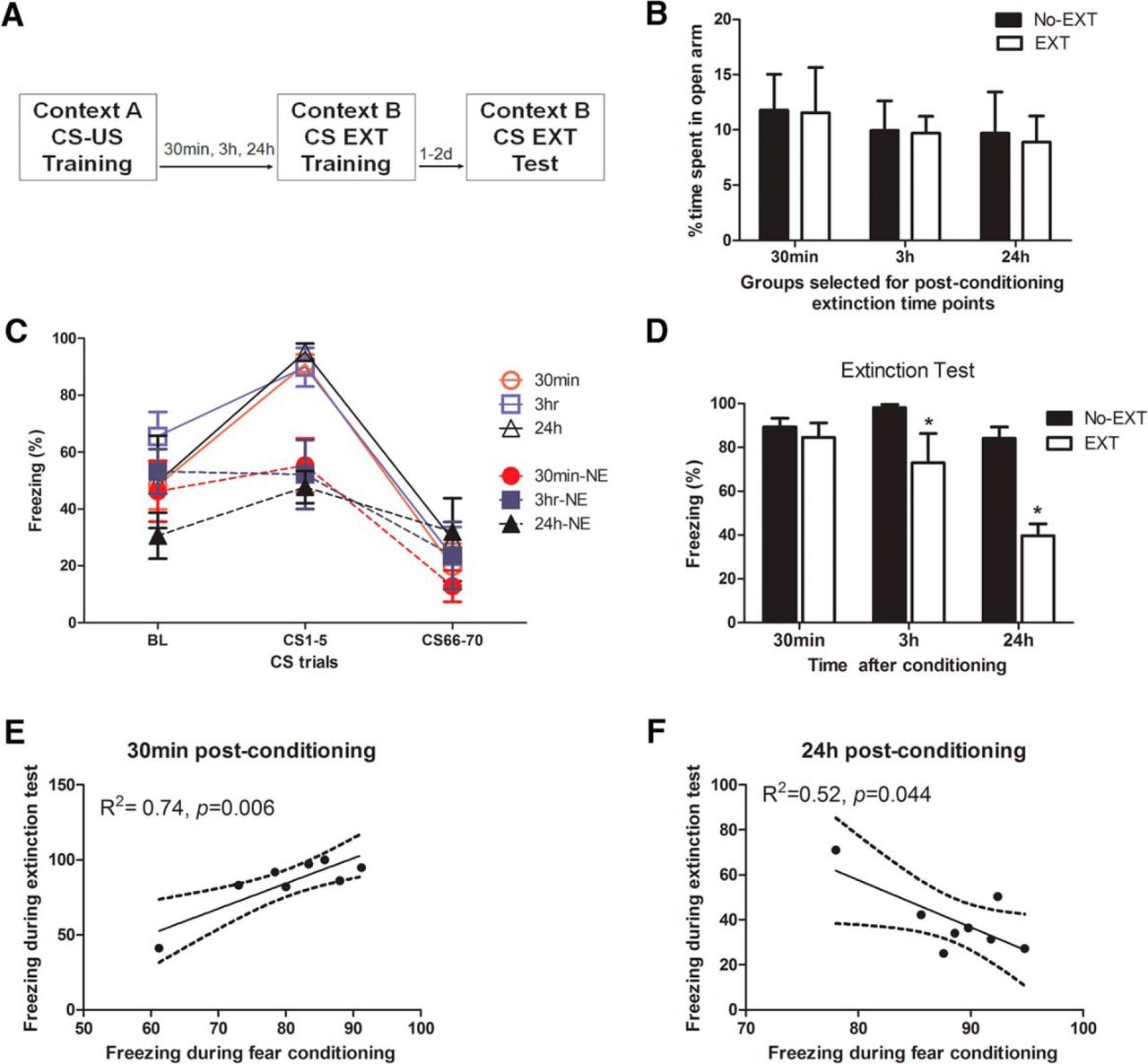
To identify appropriate experimental conditions to evaluate mechanisms underlying the immediate fear extinction deficit phenomenon, we first examined the efficiency of performing extinction training at several time points post-conditioning (Fig. 1A). Here, efficiency means that we examined whether animals exposed to extinction training at particular post-conditioning intervals were able to demonstrate significantly reduced freezing levels during a test, compared to corresponding non-extinguished (No-EXT) groups. All groups were balanced for trait anxiety using the EPM such that there were no a priori significant differences in the time spent on the open arms (F(1,38) = 0.03; p = 0.86a; Fig. 1B). Although pre-tone freezing levels for the immediate extinction groups were high, we found this behavior to be common in the literature when extinction training sessions were given shortly after fear conditioning (Maren and Chang, 2006; Chang and Maren, 2009; Chan et al., 2010; Goode et al., 2015). A three-way repeated-measures general linear model (extinction × interval × trial) for the percentage of time spent freezing during exposure to either the extinction training (EXT) or context B (No-Ext) groups found significant, but equivalent changes in freezing behavior, as indicated by a significant main effect of trial (F(2,36) = 47.8; p < 0.0001b) and extinction × trial interaction (F(2,36) = 8.47; p = 0.001c), but no significant interval × trial (F(4,74) = 1.53; p = 0.21d) or extinction × interval × trial (F(4,74) = 0.322; p = 0.86e; Fig. 1C) interaction. During the extinction test performed 48 h after conditioning, a two-way ANOVA (extinction × interval) revealed a significant effect of extinction (F(1,42) = 18.85, p < 0.001f), interval (F(2,42) = 8.05, p = 0.001g), and extinction × interval interaction (F(2,55) = 3.22, p = 0.026h; Fig. 1D). Fisher’s post hoc tests revealed that extinction applied 30 min after conditioning was ineffective in suppressing CS-elicited fear responses since the level of freezing of animals with extinction was not significantly different than non-extinguished control animals (p = 0.63i). However, animals were able to extinguish fear responses when extinction training was given after a post-conditioning delay of 3 h (p = 0.01j) and 24 h (p < 0.001k). The extinction was more efficient for a delay of 24 h than for 3 h, since animals extinguished 24 h after conditioning exhibited significantly less freezing than animals of the 3-h group (p < 0.01l) during the test.

Efficient fear extinction is dependent on the interval between conditioning and extinction sessions. Animals were separated into EXT and No-EXT (NE) groups (A) balanced for anxiety-like behavior on the elevated plus maze (B) and conditioned to similarly associate a foot shock with a tone assessed by levels of freezing (see also Table 2). Animals in the EXT group were then exposed to an extinction session (30 min, 3 h, or 24 h after conditioning), with all interval groups showing similar patterns of extinction (C, open symbols). Animals in the No-EXT group were exposed to the Context B arena without tones, and each subgroup showed similar levels of freezing during this period (C, filled symbols). Animals exposed to extinction sessions immediately after conditioning (30 min) exhibited levels of freezing similar to those of the No-EXT group during the extinction test (D). Animals exposed to delayed extinction sessions (3 or 24 h) exhibited significantly reduced freezing levels. BL, baseline freezing during the first 3 min pretone. The freezing behavior during the extinction test in animals exposed to an extinction session 30 min after conditioning was positively and significantly correlated with the amount of fear shown during the initial fear training sessions (E), whereas those receiving an extinction session 24 h after conditioning had a significant negative correlation between freezing behavior in the extinction test versus fear training (F). Data are depicted as mean percentage of time spent freezing ± SEM. *Significant difference (p < 0.05) with the corresponding No-EXT group; n = 8/group.
Then, to examine the relationship between fear training and extinction, we performed Pearson’s correlational analyses between the percentage of time spent freezing during fear conditioning and during the extinction test. We found a significant positive correlation in animals who received extinction 30 min after conditioning, such that those that exhibited the greatest fear during conditioning also exhibited the greatest deficit (R 2 = 0.74, p = 0.006m; Fig. 1E). Conversely, in animals who received extinction 24 h after conditioning, we found the opposite correlation, with those who exhibited the least freezing during conditioning exhibiting the highest levels of freezing during the extinction test (R 2 = 0.52, p = 0.044n; Fig. 1F). Animals that received extinction 3 h after conditioning had no significant correlation between freezing behaviors during the fear conditioning and the extinction test (data not shown; R 2 = 0.28, p = 0.17o).
Intra-BLA infusion of a CRFR1 antagonist after conditioning facilitates immediate extinction without interfering with fear learning consolidation
Given the above behavioral results, we chose to perform pharmacological experiments (intra-BLA infusion of NBI30775 at doses of 0.1, 0.3, 1, and 10 µg; Fig. 2A,C) at a postconditioning interval of 30 min with groups balanced for their a priori anxiety-like behavior on the EPM (F(4,33) = 0.33; p = 0.86p; Fig. 2B). During extinction sessions, all groups significantly decreased their freezing across extinction training trials (F(2,34) = 294.8; p < 0.0001q), with no significant differences in freezing between vehicle- and drug-treated animals (F values < 1; Fig. 2D). For the extinction test performed 48 h after fear conditioning, a one-way ANOVA revealed a significant effect of the drug infusion (F(4,34) = 3.51, p = 0.01r; Fig. 2E). Fisher’s post hoc tests showed that infusion of the highest dose of NBI30775, 10 µg, promoted extinction efficiency, since these animals exhibited significantly less freezing than vehicle-treated control animals (p = 0.009s; Fig. 2E). For the second test performed 1 week after fear conditioning, a one-way ANOVA revealed a significant effect of the prior drug infusion (F(4,34) = 3.51, p = 0.017t). Fisher’s post hoc tests showed that infusion of both 1 µg/0.5 µl and 10 µg/0.5 µl promoted long-term extinction efficiency, since these animals exhibited significantly less freezing than vehicle-treated control animals (p = 0.04u and p = 0.004v, respectively; Fig. 2F).

Impaired extinction efficiency due to immediate post-conditioning interval is restored by a post-conditioning bilateral infusion of CRFR1 antagonist NBI30775 in the BLA. Animals were separated into groups (A) balanced for anxiety-like behavior on the elevated plus maze (B). After successful fear conditioning they received a bilateral infusion of NBI30775 or vehicle immediately at the end of the session (C) and were given an extinction training session 30 min later (D). Animals infused with the 10-µg dose of NBI30775 showed significantly reduced freezing levels during the first testing session 48 h post-conditioning (E). Both 1- and 10-µg doses showed significantly reduced freezing levels when tested 1 week later (F). A separate group of animals were fear conditioned and received NBI or vehicle afterward and were left undisturbed until testing 48 h later. Treated animals showed similar levels of freezing in the test, suggesting that infusion of NBI does not affect memory consolidation (G). BL, baseline freezing during the first 3 min pretone. Data depicted as mean percentage of time spent freezing ± SEM. *Significant difference (p < 0.05) with the DMSO vehicle group; n = 6–10/group.
Subsequently, we performed a follow-up experiment to investigate whether the observed effects of the CRFR1 antagonist could have been due to interference with the consolidation of fear conditioning. In this experiment, animals were infused with 10 µg of NBI 30775 into the BLA immediately after conditioning and did not receive any extinction session. When they were tested 48 h afterward, NBI-treated animals did not differ in their freezing levels from the vehicle-infused group (Student’s t-test t = 1.32, p = 0.21w; Fig. 2G).
Taken together, these results reveal that blocking CRF activity in the BLA immediately after fear conditioning facilitates an immediate extinction carried out 30 min after training but does not interfere with normal consolidation processes.
Intra-BLA infusion of a CRF agonist immediately before a delayed extinction impairs extinction
Given the above findings, we then wanted to investigate whether CRH activation was sufficient to produce an extinction deficit. We focused on a delayed extinction protocol (24 h after fear conditioning) in which we found no evidence of an extinction deficit (Fig. 1). We reasoned that if, in immediate extinction protocols, endogenously shock-induced activation of CRH is sufficient for the extinction deficit, then enhancing CRH activation in the BLA before a delayed extinction protocol should also induce a deficit. Thus, we infused a CRF agonist into the BLA 30 min before a delayed extinction session given 24 h after fear conditioning (Fig. 3A) in groups balanced for their a priori anxiety-like behavior on the EPM (t = 1.7; p = 0.12x; Fig. 3B). During extinction training, both groups exhibited significant decreases in freezing behavior, indicated by a main effect of trial (F(2,8) = 9.94; p = 0.007y) at a similar level (interaction and treatment effect F values < 1; Fig. 3C). As hypothesized, pre-extinction training infusion of 1 µg/0.2 µl of CRF6–33 into the BLA altered extinction efficiency, as these animals exhibited significantly more freezing than vehicle-infused animals in the extinction test given 24 h afterward (Student’s t-test t = 2.90, p = 0.028z; Fig. 3D). These results indicate that infusion of a CRF agonist just before extinction training reduces subsequent fear extinction efficiency.

Infusion of CRF agonist CRF6–33 in the BLA immediately before delayed extinction session alters extinction efficacy. Animals were separated into groups (A) balanced for anxiety-like behavior on the elevated plus maze (B), successfully fear conditioned to a similar level (C), and infused with CRF6–33 or saline 30 min before a delayed (24 h) extinction session. Treated animals showed significantly increased freezing levels compared with vehicle (D). BL, baseline freezing during the first 3 min pretone. Data depicted as percentage of time spent freezing ± SEM. *Significant difference (p < 0.05) with the CRF agonist CRF6–33 group (0.1 µg/0.2 µl); n = 5–6/group.
Alteration of extinction is correlated with alteration of phosphorylation of the AMPA GluA1 subunits
To uncover possible mechanisms underlying the involvement of the CRF system in the BLA in impaired extinction learning, we examined phosphorylation of AMPA receptors at specific serine residues, as they have been previously linked with extinction learning (Monfils et al., 2009). A new cohort of animals was conditioned and received an extinction session (CtxB CS) either immediately (30 min: CtxB CS 30 min) or after a delay (24 h: CtxB CS 24 h) after the fear conditioning session. Animals were sacrificed at the end of the session (Fig. 4A), and Western blots were performed against phosphorylated AMPA receptor subunits. To compare our results with others who have examined phosphorylated AMPA receptor subunit changes after 3 min of CS extinction exposure, we included an additional group (CtxB 3 min CS) in which animals were exposed to 3 min of extinction either 30 min or 24 h after training and took brain samples immediately afterward. To control for possible effects of the new context, an additional group of animals was exposed to Context B without any CS presentations (CtxB no CS). All groups were balanced for their a priori anxiety-like behavior on the EPM (F(2,31) = .0002; p = 0.99aa; Fig. 4B). During extinction training, all groups exhibited significant decreases in freezing behaviors, indicated by a main effect of trial (F(1,31) = 103.1, p < 0.0001bb), but no significant effects of training interval (F(1,31) = 2.18; p = 0.15cc), group (F(2,31) = 2.37; p = 0.11dd), or interaction (F(2,31) = 1.45; p = 0.25ee; Fig. 4C). For the 30-min post-conditioning interval, i.e., when animals exhibited a deficit in fear extinction efficiency, a one-way ANOVA revealed a significant effect of condition (F(3,19) = 4.35, p = 0.017ff). Fisher’s post hoc tests showed that the percentage of phosphorylation of the AMPA GluA1 Ser845 subunit in home cage was not significantly different from the CtxtB no CS control group (p = 0.75gg) but was significantly higher than CtxB CS 30 min and CtxB 3 min CS groups (p = 0.042hh and p = 0.019ii, respectively; Fig. 4D), indicating that AMPA GluA1 Ser845 phosphorylation is decreased 30 min after fear conditioning. For the 24 h postconditioning interval, i.e., when animals did not show any deficit in fear extinction learning, a one-way ANOVA did not reveal any significant effect (F values < 1). Analysis of phosphorylation at another serine residue, GluA1 Ser831, found no significant alterations in phosphorylation compared to home cage controls in any condition (F values < 1; Fig. 4E). Importantly, analysis of the total GluA1 receptors also revealed no significant differences between groups (F values < 1; Fig. 4F).
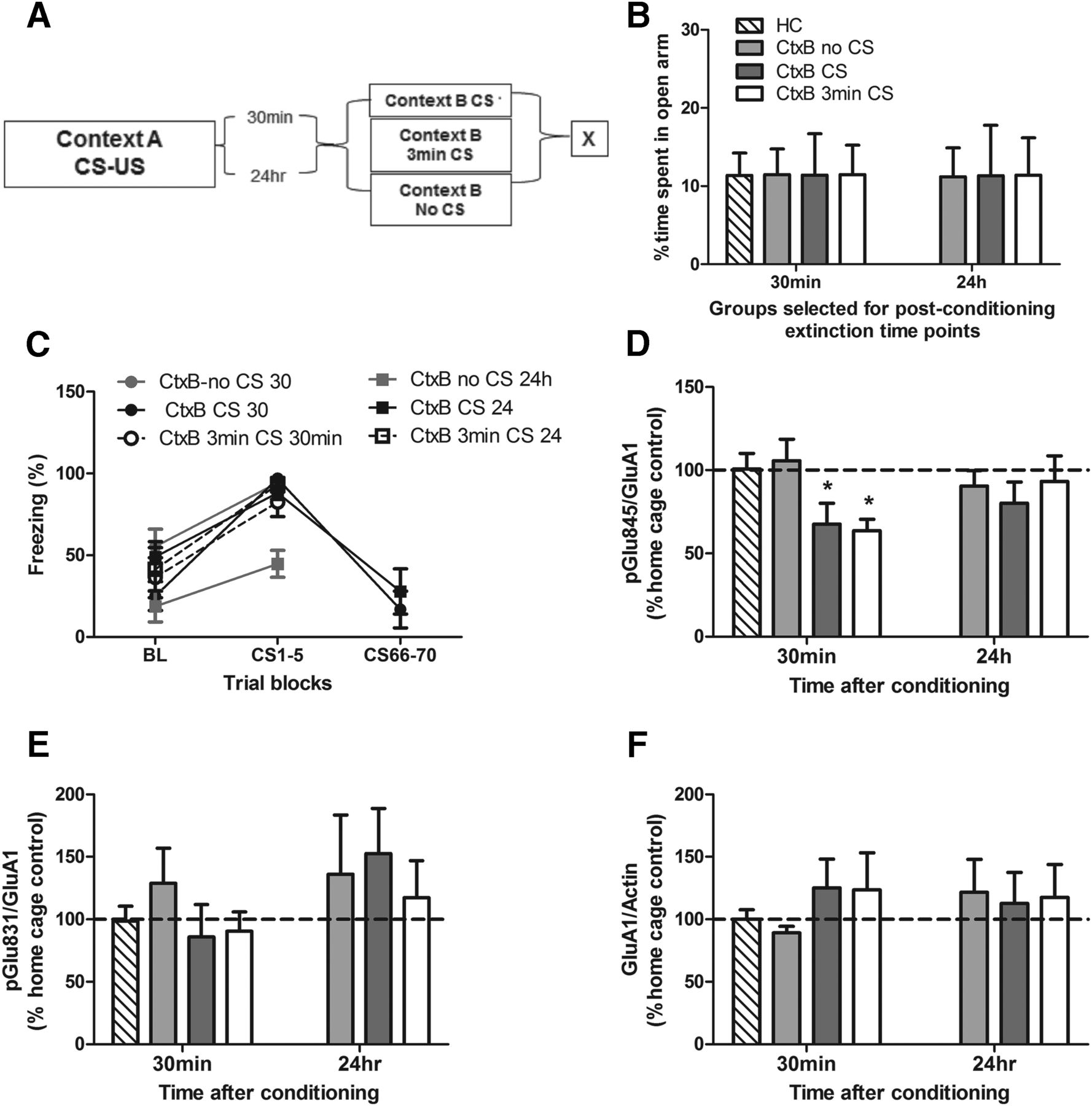
Immediate extinction impairment is associated with reduced phosphorylation of the AMPA GluA1 Ser845 subunit. Animals were separated into groups (A) balanced for anxiety-like behavior on the elevated plus maze (B), fear conditioned and sacrificed (X) immediately after various context B (CtxB) exposures either 30 min or 24 h after training, with each group showing similar levels of freezing during CtxB exposure (C). Phosphorylation of AMPA GluA1 Ser845 was significantly decreased in animals exposed to CS presentations 30 min post-conditioning but not in those with a delayed (24 h) exposure (D) compared with the home cage controls (HC). There were no significant differences in the phosphorylation levels of AMPA GluA1 Ser831 (E) or total GluA1 receptor protein (F). Data depicted as percentage of control group ± SEM. *Significant difference (p < 0.05) with HC group; n = 6/group.
Modulation of AMPA GluA1 Ser845 phosphorylation by CRFR1 antagonist in the BLA
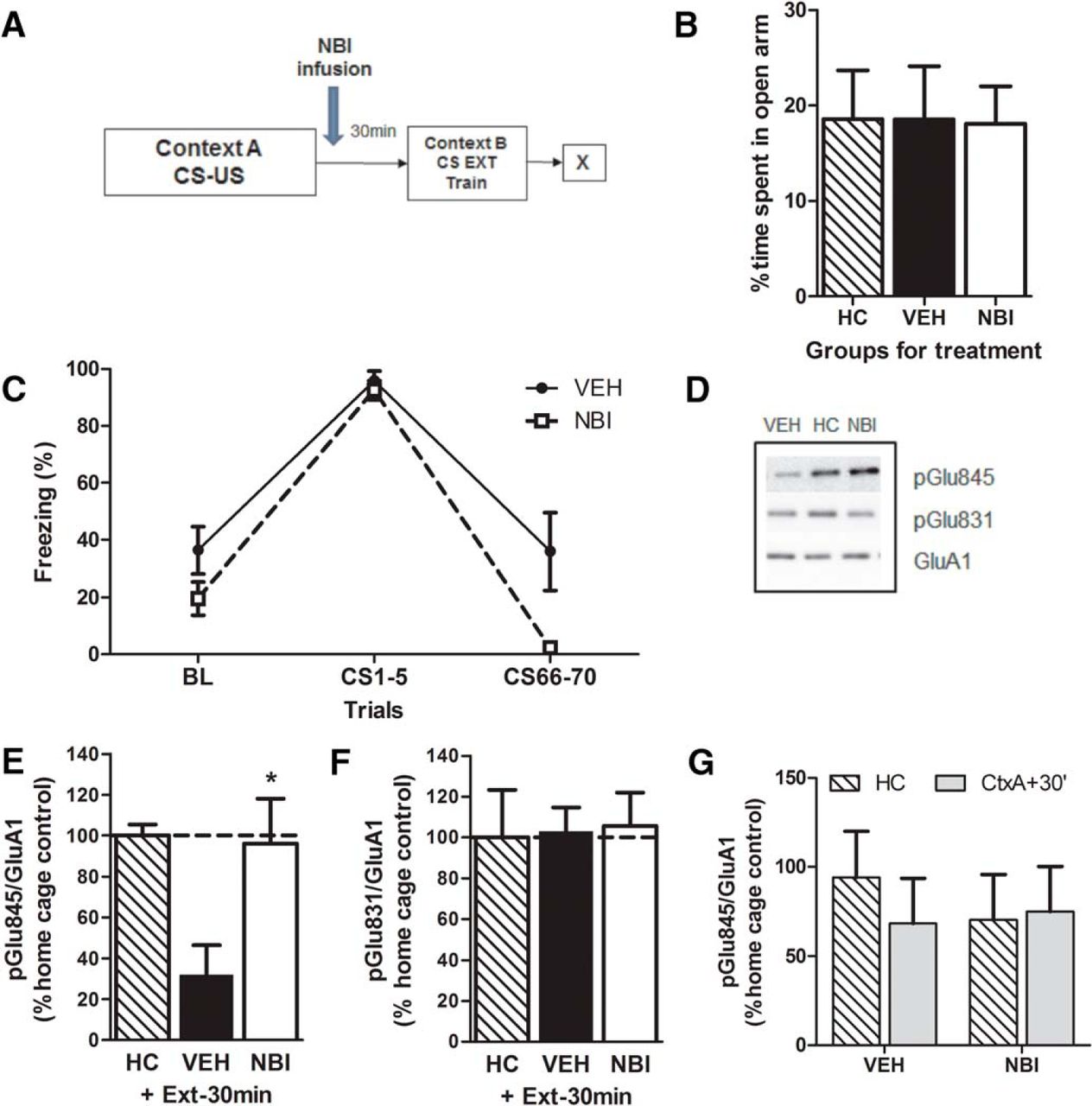
To determine whether AMPA GluA1 Ser845 phosphorylation levels were modulated by the actions of CRF after fear conditioning, we examined phosphorylation in the amygdala in animals infused with either vehicle or NBI30775 and sacrificed after an extinction session delivered 30 min postconditioning (Fig. 5A) in groups balanced for their a priori anxiety-like behavior on the EPM (F(2,27) = 0.004; p = 0.99jj; Fig. 5B). During extinction training, there was a significant main effect of trial (F(2,26) = 89.4; p < 0.0001kk), indicating that both groups decreased their freezing over time. There was also a significant effect of treatment (F(1,13) = 7.26; p = 0.02ll) and a trend for an interaction (F(1.6,20.7) = 3.06; p = 0.08mm; Fig. 5C), in which NBI treatment reduced freezing during extinction training. Western blots were performed against phosphorylated AMPA receptor subunits in samples from the amygdala (Fig. 5D). Vehicle-treated animals exhibited significantly reduced phosphorylation on the Ser845 subunit compared to home cage controls (Student’s t-test, t = 4.68; p < 0.01nn; Fig. 5D, E). Infusion of the CRFR1 antagonist NBI30775 restored the phosphorylation levels of this subunit to those of home cage levels (Student’s t-test, t = 0.18; p = 0.86oo). Notably, there was no effect of either vehicle or NBI treatment on GluA1 Ser831 phosphorylation (Fig. 5D, F), nor was there an effect of NBI treatment alone on GluA1 Ser845 phosphorylation after fear conditioning (F(1, 56) = 0.388; p = 0.54pp; Fig. 5G).

GluA1 Ser845 phosphorylation levels are reversed with administration of NBI. Animals were separated into groups (A) balanced for anxiety-like behavior on the elevated plus maze (B) and received a post-conditioning infusion of either the CRFR1 antagonist NBI30775 or vehicle and an immediate extinction session 30 min later (C). Treatment with NBI30775 restored GluA1 Ser845 phosphorylation to vehicle home cage (HC) levels (D,E) but had no effect on GluA1 Ser831 (D,F) or on its own (G). Data depicted as percentage of vehicle HC control group (hatched bar and also dotted line) ± SEM. *Significant difference (p < 0.05) with the vehicle-treated HC group; n = 8–12/group.
Calcineurin modulates AMPA GluA1 Ser845 phosphorylation in the BLA
We next investigated whether activity of the phosphatase calcineurin might be mediating the actions of CRF on AMPA GluA1 Ser845 phosphorylation during extinction. In the 30-min postconditioning interval, as both CtxB 3 min CS and CtxB CS 30 min groups exhibited significantly decreased phosphorylation (see Fig. 4D), we examined whether treatment with NBI30775 reduced calcineurin activity, which would allow for the restoration of phosphorylation levels. Animals were divided into groups (Fig. 6A) balanced for their a priori anxiety-like behavior on the EPM (F(3,69) = 0.07; p = 0.97qq; Fig. 6B). Then, they were treated with either NBI30775 or vehicle and sacrificed 30 min postconditioning after either 3 min (CtxB 3 min CS) or a full (CtxB CS 30 min) extinction exposure. A second group of animals received the same behavioral and pharmacological treatments but were sacrificed from the home cage as controls. Treatment with NBI30775 significantly reduced calcineurin activity in the BLA compared with vehicle-treated extinction (CtxB CS 30 min and CtxB 3 min min CS) groups (two-way ANOVA, F(1,67) = 6.17; p = 0.015rr; Fig. 6C), indicative of a link between CRF levels and calcineurin activity. Post hoc tests revealed a significant increase in calcineurin activity in the vehicle-treated CtxB CS 30 min group (p = 0.02ss) that was blocked by NBI treatment (p = 0.75tt) compared with vehicle-treated home cage controls.

Enhanced calcineurin activity after extinction is reversed by NBI administration. Animals were fear conditioned and infused post-conditioning with either NBI30775 or vehicle, separated into groups (A) balanced for anxiety-like behavior on the elevated plus maze (B), and sacrificed (X) for calcineurin activity assessment after either 3 min or a full extinction session 30 min after conditioning. Calcineurin activity tended to increase after the extinction session in vehicle-treated animals (CtxB CS group) but was blocked by post-conditioning treatment with NBI30775 (C). Data depicted as percentage of vehicle home cage control group (hatched bar and dotted line) ± SEM. *Significant difference (p < 0.05) with the vehicle-treated home cage group; n = 8–10/group.
Discussion
Previous work has highlighted a deficit for extinction programs that are delivered shortly after fear conditioning, as opposed to a better efficiency of delayed extinction sessions (Maren, 2014). Similarly, psychotherapeutic interventions provided soon after a traumatic event have been reported to be rather ineffective in reducing long-term fear responses (Rothbaum and Davis, 2003; Gray and Litz, 2005), although the underlying mechanisms were unclear. Here, we provide strong evidence implicating the CRF system in the BLA as a key mechanism mediating this immediate extinction deficit and identify the phosphorylation of GluA1 and enhanced calcineurin activity as potential downstream mechanisms for CRF actions.
First, in agreement with substantial work in the literature (Maren and Chang, 2006; Woods and Bouton, 2008; Chang and Maren, 2009; Maren 2014), we show here in rats that long-term extinction efficiency is impaired when extinction training occurs shortly (30 min) after fear conditioning, but correctly retained when extinction training is given after a longer delay period (24 h; note that some effectiveness of the extinction training can already be observed at the 3-h time point). Furthermore, individuals exhibiting the greatest amount of freezing during conditioning also show the greatest extinction impairment when the extinction training occurs shortly after fear conditioning, as opposed to the opposite relationship between freezing during conditioning and extinction efficiency when extinction occurs.
In our effort to investigate underlying mechanisms, we reasoned that mechanisms facilitating fear conditioning in the BLA might be at the core of the immediate extinction deficit and postulated that a fear conditioning-activated CRF system in the BLA interferes with the immediate extinction process. This hypothesis was based on previous work implicating CRF in delayed extinction deficits (Gafford et al., 2012) and evidence for a rapid activation of CRF in the amygdala elicited by acute stress, including foot shock (Merali et al., 1998; Yamano et al., 2004), which facilitates fear consolidation processes (Roozendaal et al., 2008; Pitts and Takahashi, 2011; but see Isogawa et al., 2013) through the activation of CRFR1 in the BLA (Roozendaal et al., 2002; Hubbard et al., 2007). In agreement with our hypothesis, we found that intra-BLA post-training infusion of the CRFR1 antagonist NBI30775 (0.1–10 µg) given shortly after fear conditioning promoted, at the higher doses tested, long-term extinction learning in animals submitted to extinction training 30 min postconditioning and tested for extinction 48 h and 7 d postconditioning. Importantly, the reduced long-term freezing exhibited by animals treated with the CRFR1 antagonist is not simply the result of disrupted fear consolidation, as animals just treated with NBI30775 post-conditioning (i.e., not submitted to extinction learning) show freezing levels comparable to vehicle-infused animals when tested 48 h afterward. In supplementary experiments using delayed extinction procedures, we obtained further evidence in support of a causal link between shock-immediacy and/or increased CRF in the BLA and extinction deficits. As with the immediate extinction deficit, we found that the effectiveness of extinction training was impaired when an intra-BLA infusion of the CRF agonist CRF6–33 (0.1 µg) was applied just before a delayed (i.e., 24 h postconditioning) extinction training session. Although this evidence supports our line of reasoning, our data do not allow us to exclude the alternative possibility that CRF treatment could have acted as a CS on the extinction session, inducing some sort of aversive conditioning to context B, which would then be manifested as increased freezing during the extinction testing session. Further experiments including extinction testing in a different “C” context are warranted for unambiguously concluding the impact of increased CRF on extinction efficiency.
Therefore, we identify here the activation of CRFR1 in the BLA as a key mechanism interfering with the effectiveness of immediate fear extinction training. This novel finding fits with previous reports that have implicated the BLA CRF system in impaired extinction in delayed extinction training paradigms (Gafford et al., 2012; Abiri et al., 2014) and fits with the view that the degree of fear individuals experience just before the onset of an extinction session might determine the efficacy of extinction learning (Maren and Chang, 2006; Maren, 2014). Although whether BLA CRFR1 activation precisely reflects the CS-related degree of amygdalar excitation remains to be established, intra-BLA CRF infusions were shown to lead to robust increases in anxiety behaviors (Sajdyk et al., 1999) and to induce long-lasting sensitization of noradrenergic substrates and PTSD-like symptoms (Rajbhandari et al., 2015) in rodents. Moreover, in vivo release of CRF in the BLA has been found to correlate with the level of freezing behavior in response to fear conditioning experiments (Mountney et al., 2011).
In addition, we show evidence implicating a decrease in phosphorylation of the GluA1 glutamate receptors at Ser845, but not Ser831, as a downstream mechanism of BLA CRFR1 implication in the immediate fear extinction deficit. GluA1 membrane insertion was shown to be required for fear conditioning–induced synaptic plasticity and consolidation (Rumpel et al., 2005) and regulated by GluA1 phosphorylation at Ser845 (Blackstone et al., 1994). Importantly, a transient up-regulation of GluA1 phosphorylation at Ser845 has been critically implicated in the susceptibility of long-term expressed memories to fear erasure by manipulations involving reconsolidation (Clem and Huganir, 2010) or extinction (Monfils et al., 2009) protocols after a brief retrieval of the fear memory. Conversely, administration of two retrieval sessions of a long-term established auditory fear memory close in time (i.e., the second within 1 h after the first retrieval session) led to a rapid dephosphorylation of GluA1 at Ser845 that was associated with the inability to induce memory-impairing effects (i.e., fear memory reconsolidation) by a protein synthesis inhibition (Jarome et al., 2012). These data suggest that the second retrieval rapidly altered the phosphorylation state of GluA1. This fits with our findings that, whereas fear-conditioned animals placed in a novel context 30 min post-training showed similar levels as home cage controls, those exposed to either a short (3-min) or long (30-min) extinction protocol displayed dephosphorylation of GluA1 at Ser845 in the BLA. Importantly, the same extinction treatments did not affect the phosphorylation rate when they were given 24 h after fear conditioning, further supporting a potential role of dephosphorylation of GluA1 at Ser845 in the BLA in the immediate extinction deficit. As observed in our study, the proximity of the CS application in the short and long extinction sessions to fear conditioning in the immediate fear extinction deficit phenomenon mimics mechanisms underlying the repetition of stimuli and conditions described by Jarome et al. (2012) that make the fear memory resistant to erasure. The rapid dephosphorylation of GluA1, as observed in our study, has been linked to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor endocytosis, leading to alterations in synaptic strength (Ehlers 2000) and shown to depend on increased activity of the protein phosphatase 2B or calcineurin (Ehlers 2000; Snyder et al. 2003). In full agreement with these findings, we observed increased calcineurin activity in the BLA in the groups submitted to short (3-min) or long (30-min) extinction protocols starting 30 min after fear conditioning, the same time points that also display a dephosphorylation of GluR1 at Ser845. Calcineurin has been previously linked to the regulation of anxiety and fear conditioning in the amygdala (Lin et al., 2003; Baumgärtel et al., 2008). Importantly, intra-BLA infusion of NBI30775 immediately after fear training (at the dose of 10 µg that enables efficient extinction in the immediate extinction protocol) in animals that were exposed to extinction training 30 min after fear conditioning prevented (1) the dephosphorylation GluA1 at Ser845 and (2) the increase in calcineurin activity in the BLA observed in vehicle-treated animals after extinction. Therefore, our findings suggest a possible mechanism whereby fear conditioning–induced enhancement of CRF and activation of CRFR1 in the BLA may act to prevent immediate extinction learning by blocking GluA1 insertion into the synapse via targeted dephosphorylated GluA1 AMPA subunits by enhanced calcineurin activity.
Although CRFR1 is primarily associated with increased production of cyclic AMP (cAMP) through adenyl cyclases, studies have shown that it can also interact and influence other G-protein systems, such as those of protein phosphokinases, modifying the balance between several signaling cascades rather than just one pathway, in a tissue-specific manner (Grammatopoulos and Chrousos, 2002; Gallagher et al., 2008). Evidence from the literature points to spiny pyramidal glutamatergic projection neurons in the BLA as particularly involved in the CRFR1-mediated effects reported in this study. For example, administration of CRF into the BLA produced a specific dose-dependent increase in the expression of cFos-ir in pyramidal neurons (Rostkowski et al., 2013). In addition, calcineurin is predominantly found in pyramidal neurons in the BLA (Leitermann et al., 2012). The involvement of BLA projection neurons is particularly relevant in the context of extinction learning, as substantial work shows that the BLA regulates the consolidation of fear extinction not only through local mechanisms, but also through reciprocal projections to other brain regions, particularly the medial prefrontal cortex (Akirav and Maroun, 2007; Quirk and Mueller, 2008; Herry et al., 2010; Pape and Pare, 2010). In fact, the medial prefrontal cortex has been critically implicated in the encoding and retrieval of extinction (Maren, 2014) and with stress-induced morphological changes associated with impaired extinction (Izquierdo et al., 2006; Miracle et al., 2006; Wilber et al., 2011). Furthermore, studies have identified links between BLA activity and medial prefrontal cortex function (Dilgen et al., 2013). Thus, in the case of immediate extinction deficit, it has been proposed that amygdalar hyperexcitability may inhibit medial prefrontal cortex circuitry and interfere with extinction retrieval. Given the ability of CRF to render the BLA excitable for long periods of time (Rainnie et al., 1992; Sandi et al., 2008), our data support this hypothesis via a CRF-mediated mechanism.
Immediate extinction therapies have been offered as a potential solution to combat the development of PTSD in individuals exposed to traumatic events. Studies from animal research have demonstrated that these kinds of therapies may not be effective. In this study, we go beyond the behavioral level and identify the activation of CRFR1 in the BLA as a critical mechanism underlying this phenomenon. Our findings highlight the treatment with CRFR1 antagonists as a potential adjuvant capable to improve the effectiveness of behavioral therapies given shortly after exposure to trauma.
Acknowledgments
Acknowledgments: We thank D. Grigoriadis (Neuroendocrine, Inc., San Diego, CA) for the generous gift of the NBI30775 CRFR1 antagonist.
Footnotes
Authors report no conflict of interest.
This work was supported by grants from the Swiss National Science Foundation (31003A-152614; NCCR Synapsy) and intramural funding from the École Polytechnique Fédérale de Lausanne.
This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



