Article Text

Abstract
Background: Mental retardation (MR) affects 2–3% of the human population and some of these cases are genetically determined. Although several genes responsible for MR have been identified, many cases have still not been explained.
Methods: We have identified a pericentric inversion of the X chromosome inv(X)(p22.3;q13.2) segregating in a family where two male carriers have severe MR while female carriers are not affected.
Results: The molecular characterisation of this inversion led us to identify two new genes which are disrupted by the breakpoints: KIAA2022 in Xq13.2 and P2RY8 in Xp22.3. These genes were not previously fully characterised in humans. KIAA2022 encodes a protein which lacks significant homology to any other known protein and is highly expressed in the brain. P2RY8 is a member of the purine nucleotide G-protein coupled receptor gene family. It is located in the pseudo-autosomal region of the X chromosome and is not expressed in brain.
Conclusions: Because the haploinsufficiency of P2RY8 in carrier mothers does not have a phenotypic consequence, we propose that the severe MR of the affected males in this family is due to the absence of the KIAA2022 gene product. However, screening 20 probands from X linked MR families did not reveal mutations in KIAA2022. Nonetheless, the high expression of this gene in fetal brain and in the adult cerebral cortex could be consistent with a role in brain development and/or cognitive function.
- AR, androgen receptor
- BrdU, 5-bromodeoxyuridine
- FISH, fluorescent in situ hybridisation
- MR, mental retardation
- NS MR, non-syndromic mental retardation
- NS XLMR, non-syndromic X linked mental retardation
- PAR1, pseudoautosomal region 1
- chromosomal rearrangement
- KIAA2022
- P2RY8
- X chromosome
- X linked mental retardation
Statistics from Altmetric.com
- AR, androgen receptor
- BrdU, 5-bromodeoxyuridine
- FISH, fluorescent in situ hybridisation
- MR, mental retardation
- NS MR, non-syndromic mental retardation
- NS XLMR, non-syndromic X linked mental retardation
- PAR1, pseudoautosomal region 1
The genetic basis of non-syndromic mental retardation (NS MR) is complex and heterogeneous. In the past 5 years, several genes causing non-syndromic X linked mental retardation (NS XLMR) have been identified using positional cloning strategies.1 In addition to these findings, a number of genes involved in syndromic MR conditions have also been shown to cause NS MR. This is the case for genes such as RSK2,2 ATR-X,3 MECP2,4 FDG1,5 and ARX.6
However, despite important advances in the field of XLMR, two major problems still need to be addressed. First, the number of affected families by far exceeds the number of identified genes.7,8 This means that a significant number of disease causing genes remain to be identified. Second, recent data have shown that a mutation reported as disease causing in MRX families because it segregates with the disease can be a simple polymorphism, thereby increasing the number of families in which the gene defect remains to be identified.9,10
The strategy used to identify MR genes often takes advantage of linkage studies in large families followed by sequencing of positional candidate genes. In a number of other instances, the causative genes were cloned from or near the breakpoint of a chromosomal rearrangement. Indeed, X autosome translocation represents one of the most powerful means to achieve this goal, especially if a new class of genes is to be identified.11–13 In female patients, such rearrangements, when balanced, almost invariably lead to the inactivation of the normal X chromosome, the translocated X chromosome being active probably because of a strong selection against a possible inactivation of the autosomal material.14 Although less frequently reported, other chromosomal anomalies can be used to identify disease genes.
We report here on the molecular characterisation of an X chromosome pericentric inversion inv(X)(q13;p22). The inversion segregates in a family where two severely mentally retarded males are carriers of the inversion in two generations.15 Obligate carriers in this family are clinically normal. This characteristic led us to hypothesise that the causative gene (if any) would likely be located in the vicinity of the Xq13 breakpoint rather than in Xp22.3 which is the pseudoautosomal region 1 (PAR1) on the human X chromosome and in which genes are expressed from both the X and Y chromosomes. A gene defect in this region of the human X chromosome is known to have visible phenotypic effects in female patients.15 In addition, recent data show that at least one additional mental retardation gene lies in the Xq12–q21 region.16
The molecular characterisation of the inv(X)(q13;p22) chromosomal rearrangement led us to the identification of two new genes which are interrupted by the breakpoints: the KIAA2022 gene in Xq13.2 and the P2RY8 gene in Xp22.3. We have determined the genomic structure of these two genes and studied their expression. We show that the P2RY8 gene is located in the pseudoautosomal region, that carrier women are not affected in this family, and that the P2RY8 transcript is not expressed in human brain. These data led us to hypothesise that the phenotype of the mentally retarded males is caused by the disruption of the KIAA2022 gene in Xq13 since our data show that the corresponding transcript is highly expressed in fetal brain and the adult cerebral cortex. To further assess the role of the KIAA2022 gene in the aetiology of mental retardation, we screened 20 probands from Xq13 linked XLMR families for mutations. No mutations or nucleotide variants were identified.
METHODS
Cell culture, RNA isolation, and RT-PCR
All lymphoblastoid cell lines were grown in RPMI 1680 (Gibco BRL, Carlsbad, CA, USA) with 10% fetal bovine serum in the presence of 0.1 mg/ml of kanamycin at 37°C and 5% CO2. RNA was prepared using patient’s lymphocytes and the QuickPrep mRNA purification kit according to the instructions of the manufacturer (Pharmacia, Newark, NJ, USA). Human normal tissue RNA was purchased (BD Bioscience, San Jose, CA, USA). Reverse transcription of 5 μg of total RNA was performed in 50 μl of 1×Superscript reaction buffer (Gibco BRL) containing 3 ng/μl of dN6, 40 U of RNasin (Promega, San Luis Obispo, CA, USA), 10 mM dNTP, and 200 U of Superscript II reverse transcriptase (Gibco BRL). RT-PCR was performed using 1/10th of the first strand reaction.
Northern and southern blot hybridisations
We hybridised human fetal MTN blot II and human brain MTN blot II (BD Bioscience) with a KIAA2022 cDNA probe (nucleotides 2966–3735 of the cDNA sequence AY563507) and a probe for β-actin (BD Bioscience). These probes were labelled by random priming using [α-32P]dCTP. Hybridisation of northern blots were carried out in 50% formamide buffer at 42°C for 16 h. For Southern blot preparation, DNA samples were digested with EcoRV, electrophoresed on 1% agarose gel, and blotted onto Hybond N+ nylon membrane (Amersham Pharmacia Biotech, Buckinghamshire, UK). Hybridisation and washing were carried out, respectively, in 5×SSC/0.5%SDS/1×Denhardt and 0.1×SSC/0.1%SDS at 65°C according to standard procedures.
Fluorescent in situ hybridisation
The probes (DNA from BACs) were labelled by random priming with bio-16-dUTP and in situ hybridised at a final concentration of 20 ng/μl. The hybridisation signals were made visible with fluorescein labelled avidin following standard protocols.19 Chromosomes were counterstained with propidium iodine diluted in pH 11 antifade.
X chromosome inactivation assay
Primers were designed in the (CAG)n flanking sequences of the androgen receptor (HUMARA) gene intron 1.20 Forward primer AR-P1 was 5′ labelled (IRD800) and the reverse primer AR-P2 was unlabelled. Primers sequences were: AR-P1: IRD800 5′ TCCAGAATCTGTTCCAGAGCGTGC 3′; AR-P2: 5′ GCTGTGAAGGTTGCTGTTCCTCAT 3′. A 400 ng sample of DNA was digested by HpaII and ethanol precipitated. PCR reactions were performed with 100 ng of DNA both on HpaII digested and undigested DNA. Affected males in our study were also tested using the same conditions and were interpreted for allele size at the AR locus after PCR on undigested DNA. For these males, PCR reactions on HpaII digested DNA were also used as digestion quality control (no amplification product on HpaII digested DNA at the AR locus, Liacoln, NE, USA). PCR conditions were as follow: 1×PCR buffer, 0.2 mM dNTPs, 1.25 Mg2+, 0.5 U Taq (Gibco BRL) in 20 μl final volume. Annealing temperature was 60°C for 30 total cycles. PCR products were loaded on an automated sequencer (Li-Cor, Lincoln, NE, USA) and quantification of the relative intensity of each allele was performed using OneD Scan software (Scanalytics, Fairfax, VI, USA).
Mutation screening
A total of 23 familial cases of X linked mental retardation previously linked to the Xq13–q21 region were investigated (family references and linkage intervals are available from CES and JG upon request). In each case, DNA from an affected individual was used for direct sequencing of KIAA2022. We designed primer pairs for each of the four coding exons including exon–intron boundary sequences when applicable. We used the following primers: exon 2 340 bp product, Ex2-1F (5′-ACAGGTAAATCCCAGTGAGC-3′) and Ex2-1R (5′-ATCCTGGACTCAACCTGTCC-3′); exon 3 (part one) 869 bp product, Ex3-1F (5′-GTACCAGAAACTGATCAAGG-3′) and Ex3-1R (5′-AGAATAGGTTGACAGACAGC-3′); exon 3 (part two) 636 bp product, Ex3-2F (5′-TCAGGATTGGGGTTACTTCG-3′) and Ex3-2R (5′-AGTGTCCCGAGCCATATAGC-3′); exon 3 (part three) 715 bp product, Ex3-3F (5′-GGGAGTTTCAGTGATGATAG-3′) and Ex3-3R (5′-AGTGTACCTTTTAGGCCT CC-3′); exon 3 (part four) 893 bp product, Ex3-4F (5′-CATTTCTGCCACCTGCTCG-3′) and Ex3-4R (5′-CTCCAAATTCACTGGATTGG-3′); exon 3 (part five) 769 bp product, Ex3-5F (5′-CTCATCCTCTGACTCTGAGC-3′) and Ex3-5R (5′-GTGAAAGGGTACTGCAGTCC-3′); exon 3 (part six) 720 bp product, Ex3-6F (5′-GACACTAGGAACACTAAAGG-3′) and Ex3-6R (5′-CACATCTGCCATACCAGAGG-3′); exon 3 (part seven) 660 bp product, Ex3-7F (5′-CTTCTGGATGATGACCAACG-3′) and Ex3-7R (5′-TGGGACAGTTTCTTTCATGC-3′); exon 4 380 bp product, Ex4-1F (5′-CTACATCATACGGCATCTAG-3′) and Ex4-2R (5′-ATAGTGCATAAACTACTTGTGC-3′). We sequenced all exons in both forward and reverse directions. Sequencing was carried out by MWG Biotech, Ebersberg, Germany and Sequencher software (Gene Codes, Ann Arbor, MI, USA) was used to analyse sequences and chromatograms.
RESULTS
Clinical histories
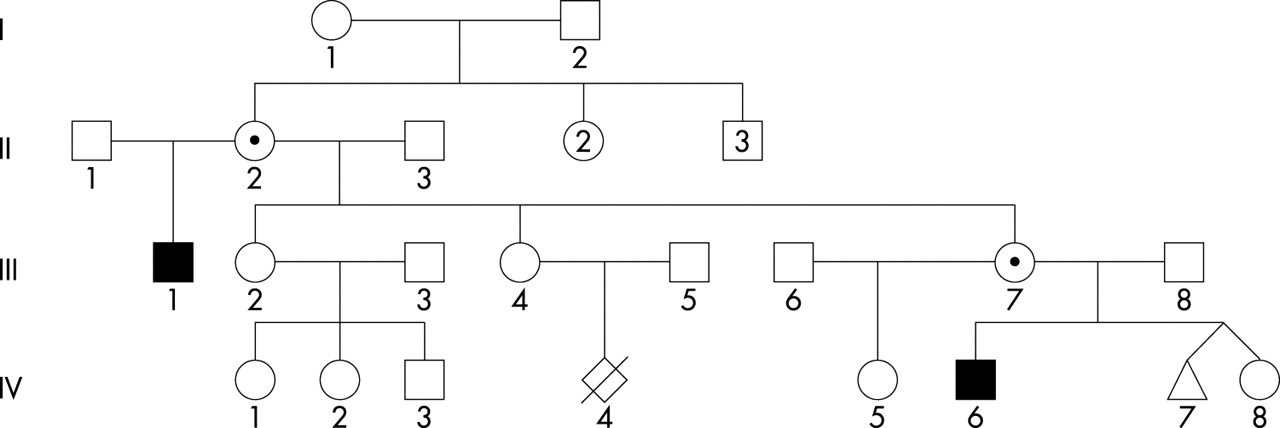
The first patient (fig 1, individual III-1) was born as the first child of unrelated Caucasian parents. His mother had three healthy girls from a first marriage (fig 1). Pregnancy was uneventful and delivery was induced at 38 weeks. The child was born in cephalic presentation with a birth weight of 2720 g and a height of 48.5 cm. In the first weeks, he was hypotonic and had poor visual pursuit. At 13 months, he had a first episode of tonic-clonic seizures and was placed on valproate therapy. EEG indicated a bilateral marked slow dysrhythmia. Seizures recurred in the context of hyperthermia. Gastroeosophageal reflux was noted. At 2 years of age, a gastric ulcer was diagnosed. He developed spastic quadriparesia and underwent surgery for pes calcaneovalgus at the age 3 years. He was severely hypotonic. A cerebral CT scan indicated enlarged ventricles. Complete blood count and serum electrolytes were normal, as were plasma amino acids, serum isoelectrofocussing of sialotransferrins, and urinary organic acids. Peripheral lymphocytes karyotype indicated 46,XY,inv(X)(p22.3;q13). His developmental milestones were delayed: he sat alone at 15 months and walked without assistance at 3 years. From the age of 2 years on, he started to exhibit hand stereotypic movements and his poor social interaction suggested a diagnosis of infantile autism. No language developed. When evaluated at the age of 12 years, he was profoundly retarded with permanent stereotypic movements, grunting, and drooling. On clinical examination, a mild facial dysmorphia was noted with short philtrum, short nose with thick tip, tentered upper vermilion border, and esotropia. Oculogyric crises and spastic quadriparesia with leg muscle wasting were also noted (figs 2 and 3). A shawl scrotum was present. Height was at – 2.6 SD, and weight and head circumference were normal (25th centile). Complete blood count, serum electrolytes, and liver enzymes were normal. Plasma amino acids were within the normal range. Brain MRI indicated moderate brain atrophy, as exemplified by enlarged ventricles, marked Virchow-Robin spaces, and a thin corpus callosum. Cerebellar vermis appeared somewhat small. A thick calvarium was also noted. Eye fundus and slit lamp examination were normal. He is attending a special school for the mentally handicapped.

Pedigree of the family. Individuals II-2 and III-7 are carriers of the inversion. IV-7 is a male fetus who was terminated because he was a carrier of the inversion. Affected individuals are represented with black symbols. Carrier mothers are shown with a black dot inside their symbol.
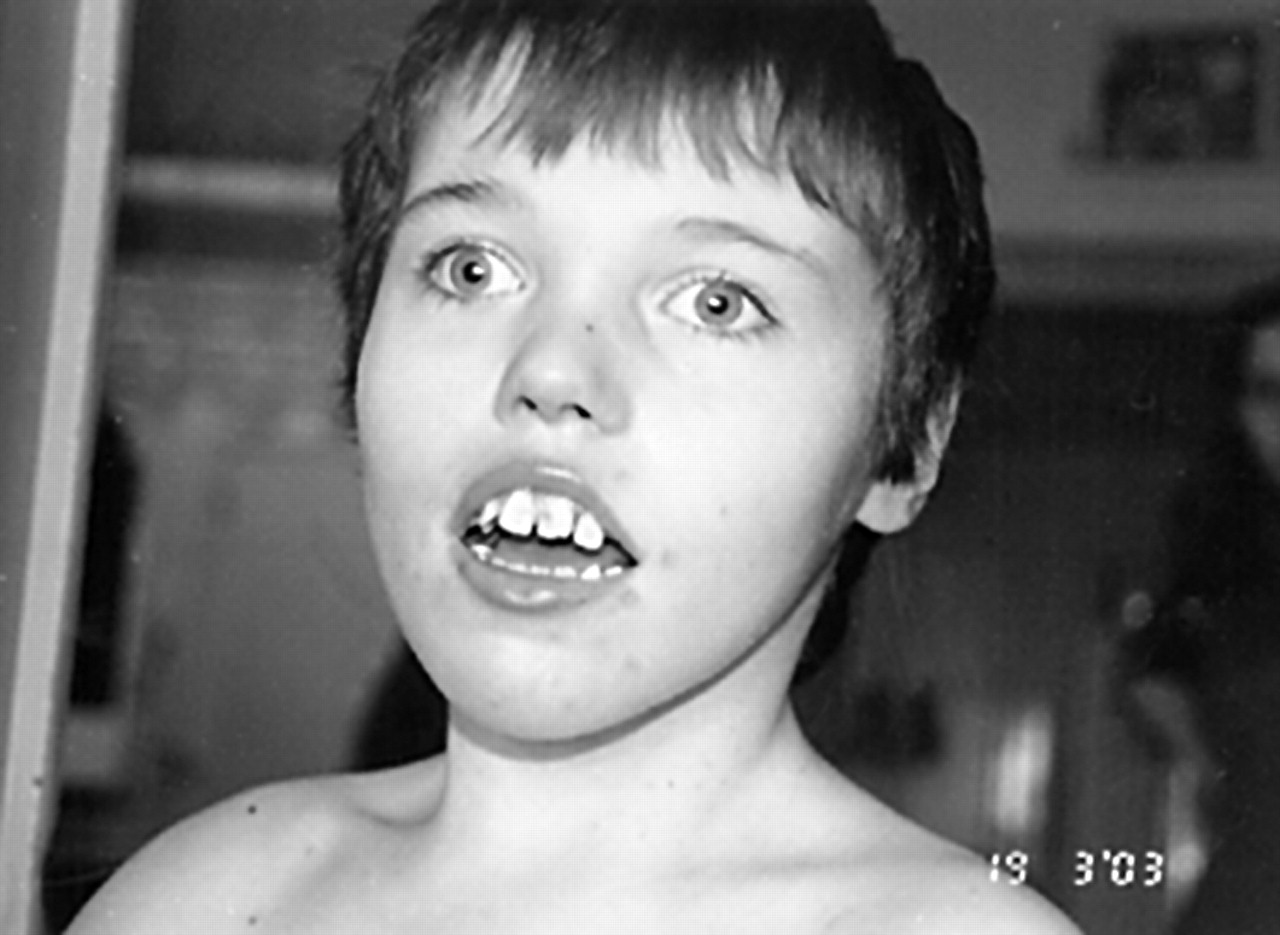
Facial appearance of patient III-1 at age 12. Note esotropia.

General view of patient III-1 at age 12. Note the dystonic attitude and muscle wasting of legs.
The second patient is the half-nephew of the first patient (fig 1, individual IV-6). He was born at term to a 27 year old G2 P2 Caucasian woman. She was unrelated to the father of the newborn. She was slightly obese (123 kg at delivery) and had been so since the end of her second decade. She had a normal girl aged 7 years from a first marriage. The patient was born at 38 weeks gestation after caesarean section for acute fetal distress. His birth weight was 2720 g for a height of 48.5 cm. Moderate hypotonia and major gastroeosophageal reflux were noted on follow up. He was admitted on five occasions to the hospital for bronchitis. A cerebral CT scan indicated frontal cortical atrophy. Karyotype was 46,XY,inv(X)(p22.3;q13). Serum electrolytes, plasma amino acids, lactic acid, complete blood count, serum isoelectrofocussing of sialotransferrins, very long chain fatty acids, and urinary organic acids were within normal range. Developmental milestones were severely delayed. At 5 years of age, height was at −2.5 SD, weight at the 25th centile, and head circumference at −2 SD. No language developed and stereotypic movements of the hands appeared. He sat at 18 months and walked without aid at 3 years. Only mild dysmorphic features, similar to those observed in his uncle, were noted.
In summary, both patients had marked neonatal hypotonia, severely delayed developmental milestones with walking acquired at 3 years of age, progressive quadriparesia, gastroeosophageal reflux, and a diagnosis of infantile autism. Stereotypic movements of hands were very similar in both children.
Karyotype and X chromosome inactivation
Chromosome analysis of cultured blood lymphocytes from the two affected patients III-1 and IV-6 (fig 1) revealed the presence of a pericentric inversion of the X chromosome 46,XY,inv(X)(p22;q13). Mothers of the patients are both carriers of this chromosomal rearrangement (data not shown). X chromosome inactivation was assessed using both 5-bromodeoxyuridine (BrdU) incorporation analysis in cultured lymphocytes and determination of the methylation status at the androgen receptor (AR) locus. The results of these analyses showed that one of the carrier females (III-7) has a random X chromosome inactivation pattern (data not shown). DNA was not available for II-2.
FISH mapping of the breakpoint
In order to localise the breakpoint in Xp22 and Xq13, we initially performed systematic fluorescent in situ hybridisation (FISH) using genomic clones originating from these two chromosomal regions. For this purpose, we used the available physical maps and sequence information (see Methods) to establish anchored BAC contigs in the regions of interest (data not shown). After several rounds of hybridisation which reduced the critical interval by half at each step (data not shown), we identified two genomic clones spanning the inversion breakpoints. These clones are RP11-79C13 localised in Xq13 and RP11-261P4 localised in the pseudoautosomal region 1 (PAR1) of the human X chromosome in Xp22.3 (fig 4).

Fluorescent in situ hybridisations of the two BACs spanning the X chromosome inversion breakpoints. (A) Hybridisation of clone RP11-79C13 (Xq13.2) on a metaphase of carrier mother III-7. The normal X chromosome and the chromosome carrying the inversion are marked with arrows. On the inv(X), two FISH signals are detectable suggesting that the genomic interval covered by this BAC is interrupted by the presence of the breakpoint. (B) Hybridisation of clone RP11-261P4 (Xp22.3) on a metaphase of patient IV-6; the Y chromosome and the X chromosome carrying the inversion are marked with arrows. Two signals are visible on the inv(X) and two additional signals are seen on chromosome 6 (cross hybridisation).
Cloning and sequencing of the breakpoints
Because the Xp22 breakpoint is localised in the pseudoautosomal region which is present in two copies in males, we decided to focus on the Xq13 breakpoint first.
For this purpose we carried out long range PCR reactions using primers designed to amplify overlapping 10 kb fragments in the critical interval (defined as the interval covered by the BAC clone RP11-79C13) using the DNA of patient IV-6 as a template. All reactions but one yielded a PCR product of the expected size (data not shown). The 10 kb fragment which could not be amplified from the patient’s DNA was presumed to contain the breakpoint and it was again divided into approximately 1 kb sub-fragments to perform PCR amplifications. Again, all but one primer pairs amplified the expected size fragment (data not shown). The absence of amplification of this particular PCR product on the patient’s DNA localised the Xq13 breakpoint within a putative 1 kb fragment.
Next, we used a probe localised in this 1 kb fragment to hybridise a southern blot of the patient’s DNA. An abnormal EcoRV restriction fragment of 7 kb (that is, the junction fragment) was detected using this probe instead of the wild type 8.5 kb restriction fragment detected on a control DNA (fig 5A). This abnormal restriction fragment was excised from the gel, cloned, and sequenced. Analysis of the sequence showed that it originated from Xq13 on one side and Xp22 on the other side. Direct sequence comparisons allowed us to map precisely the Xq13 breakpoint at position 8703 in the sequence of clone RP13-9D14 (GenBank accession number AL390035) and at position 99184 in the sequence of clone RP11-261P4 (GenBank accession number AL683870). Using this information, we designed PCR primers on both sides of each breakpoint to amplify these regions using the genomic DNA of the patients as a template. Products of the expected size were obtained in both cases (fig 5B). Sequencing of these two PCR fragments revealed that the pericentric inversion occurred without loss of genetic material except for the presence of an insertion of 6 bp on the short arm of the inv(X) (data not shown).
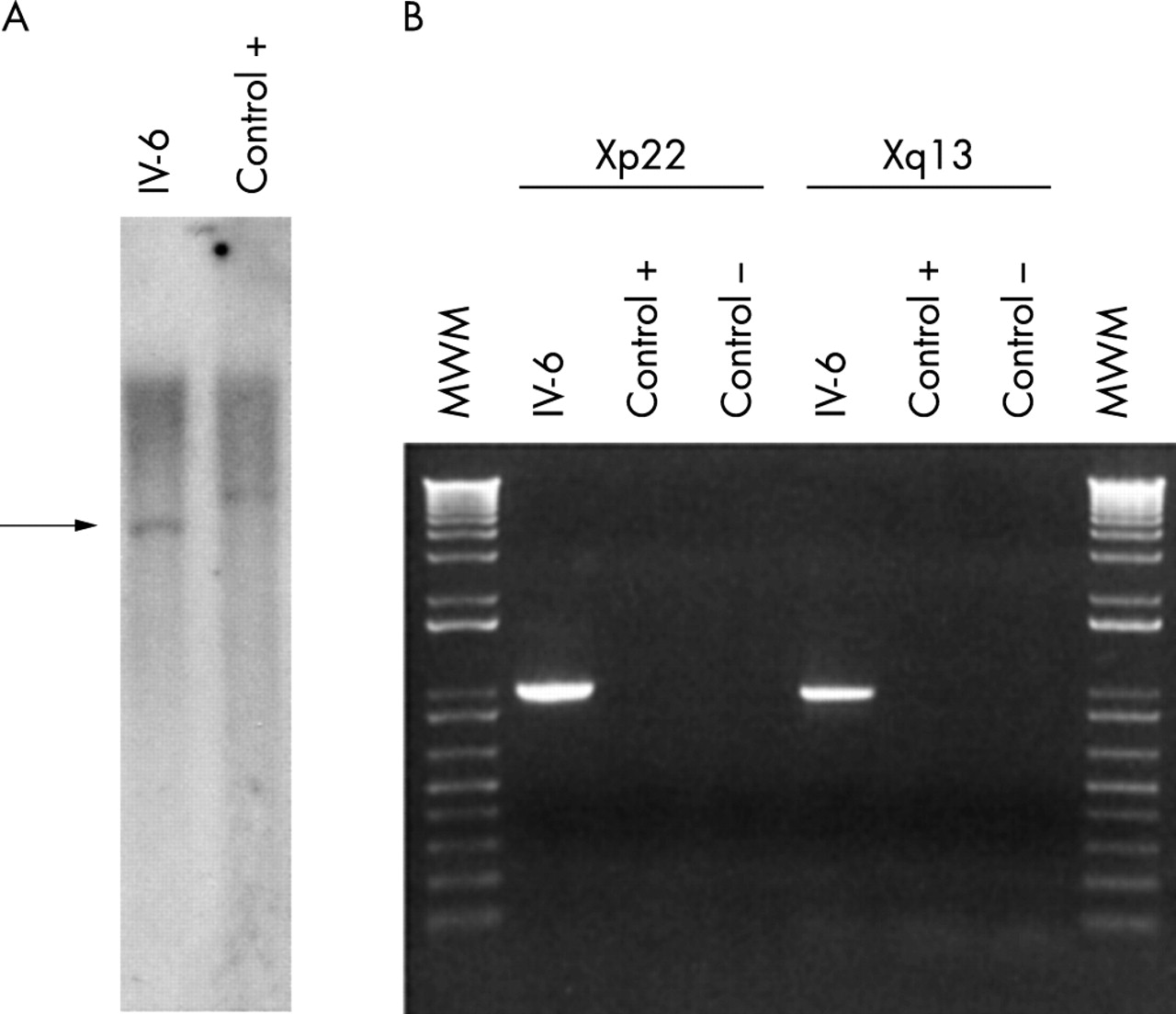
(A) Southern blot of a control DNA (Control +) and the DNA of patient IV-6 hybridised with a probe corresponding to nucleotides 6559–8265 of the RP11-9D14 clone. An abnormal restriction fragment is detected on the patient’s DNA (arrow). (B) PCR amplification of the two breakpoints using the genomic DNA of the patient with the inversion (IV-6), a control DNA (C +), or no DNA (C −). Left: Amplification of the Xp22 breakpoint with flanking primers (986 bp product). Right: Amplification of the Xq13 breakpoint with flanking primers (963 bp product). MWM, molecular weight marker. A specific PCR product is obtained using these primer combinations on the patient’s DNA only. The two products were sequenced to definitively confirm their origin.
Identification and characterisation of KIAA2022 in Xq13
The sequences of the Xq13 BACs in the critical region were analysed using the NIX interface at HGMP (see Methods). This analysis revealed that the Xq13 breakpoint fell inside a predicted 180 kb intron inside a gene called KIAA2022. In order to determine the genomic structure of this gene, we initially compared the genomic sequence of the BACs in the vicinity of the breakpoint (clones RP11-130N24 and RP11-79C13) with the KIAA2022 cDNA (GenBank accession number XM_291326). To confirm these predictions, we used RNA isolated from lymphoblastoid cell lines and from different human tissues to perform RT-PCR experiments with various primer combinations (data not shown). These experiments allowed us to determine the exact genomic structure of the KIAA2022 gene (fig 6). The gene is composed of four exons spanning 192 kb of genomic DNA. It has an open reading frame of 4551 bp encoding a putative protein with 1516 amino acids. It has a very long 3′ untranslated region spanning 5828 bp. The sequence of KIAA2022 previously deposited in GenBank with the accession number XM_291326 does not contain the correct sequence in the 5′ untranslated region. Using 5′-RACE experiments we determined that the first exon of KIAA2022 has a size of 327 bp (data not shown). The sequence resulting from our analysis is deposited with GenBank accession number AY563507.
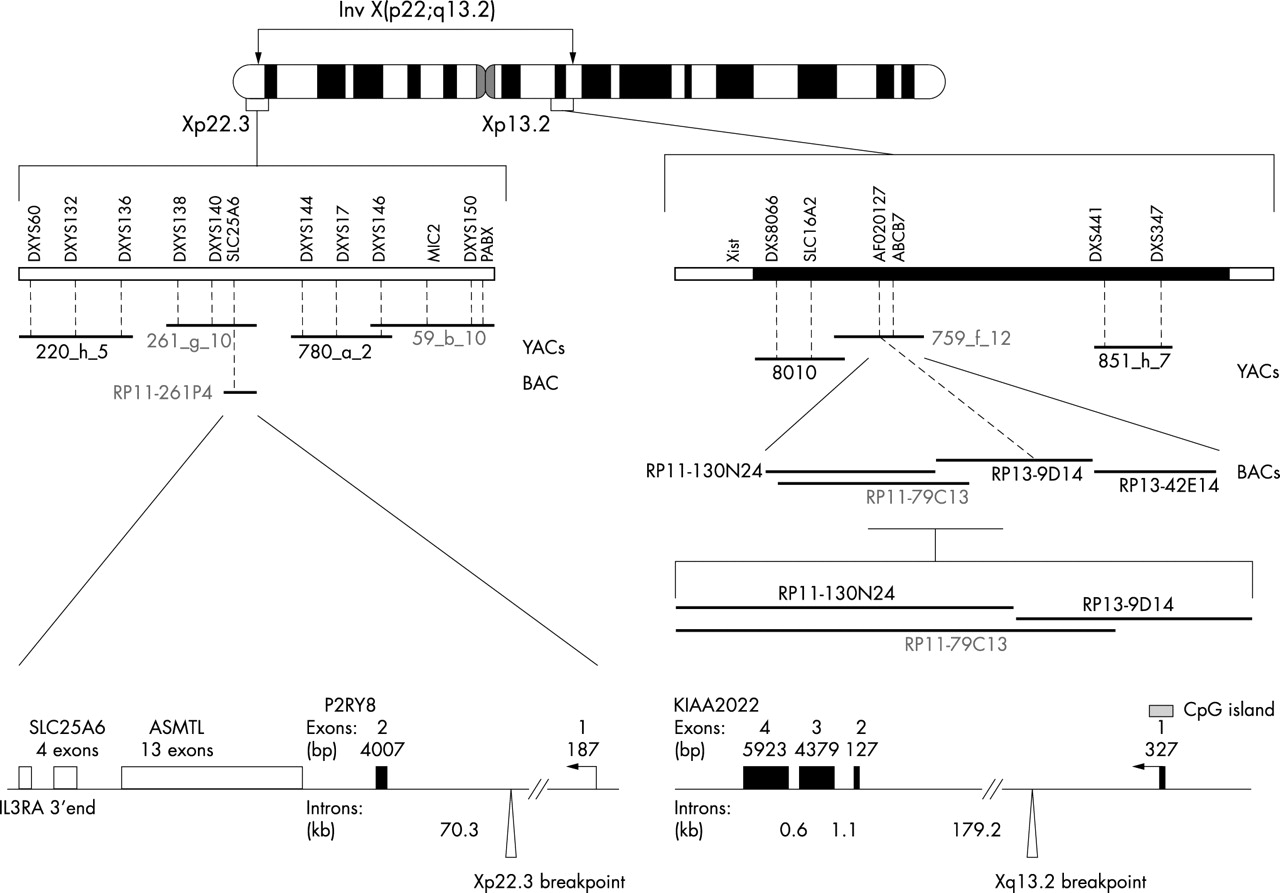
Drawing of the human X chromosome showing the structure of the two breakpoint regions.
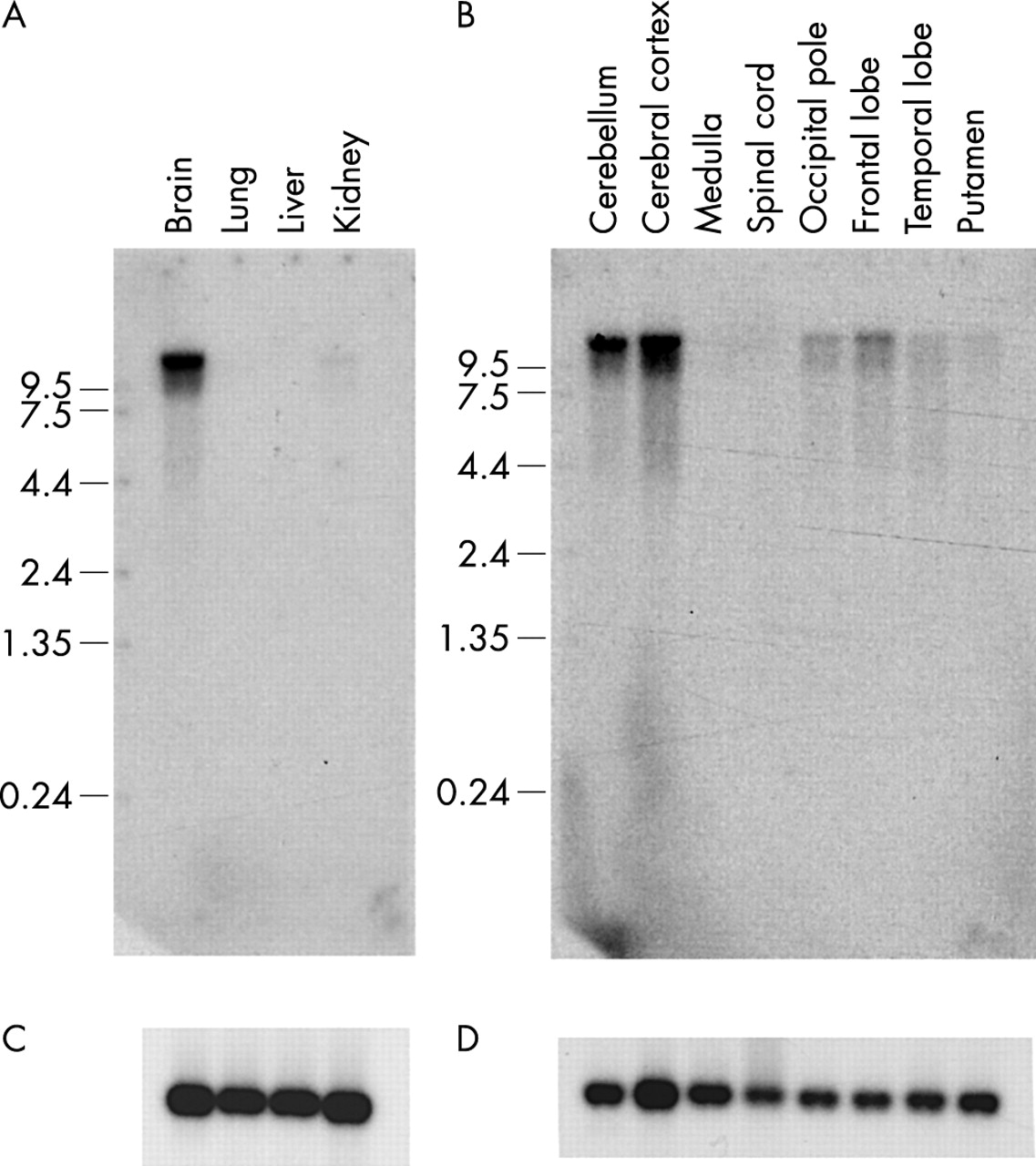
In order to examine the expression of the KIAA2022 gene, we performed RT-PCR experiments using RNA extracted from various human tissues (fig 7) and we hybridised adult and fetal northern blots with a specific probe (fig 8). These experiments revealed that KIAA2022 is highly expressed in fetal and adult brain and that it is expression in adult brain is predominantly in the cerebral cortex and the cerebellum. It is also expressed in other tissues but to a lesser extent.

Expression of KIAA2022, P2RY8, and G3DPH in several human adult tissues and in fetal brain. G3PDH was used as a control to check that the same amount of cDNA was present in each sample. Expression of the same genes was tested in cDNA prepared from the lymphocyte RNA of patients III-1, IV-6, and the carrier mother III-7. A control male and female cDNA were also used. In each case, the primers were selected in two different exons of the tested genes and we checked that they did not yield amplification products if genomic DNA was used as a template instead of cDNA. KIAA2022 is highly expressed in brain, but is no longer expressed in the patients’ lymphocytes. P2RY8 is not expressed in brain but is highly expressed in control lymphocytes and in the lymphocytes of the patients and one carrier mother.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hybridisation of a KIAA2022 probe (nucleotides 2966–3735 of the cDNA sequence AY563507) on northern blots prepared with RNA isolated from (A) fetal tissues and (B) different regions of the human adult brain. (C, D) Control hybridisations with an actin probe. The KIAA2022 gene is highly expressed in fetal brain and adult cerebellum and cerebral cortex.
Identification and characterisation of P2RY8 in Xp22
Using the same strategy as for Xq13.2, we cloned and sequenced the Xp22.3 breakpoint. We found that the breakpoint lies within the single intron of a gene called P2RY8 (fig 6). This gene has not previously been fully characterised in humans. It is composed of two exons and has a coding region of 1077 bp. Its expression was studied using the same RT-PCR experiments as those performed for KIAA2022. Using P2RY8 specific primers, we show that the corresponding transcript is highly expressed in lymphocytes (fig 7). A weaker expression is seen in heart, kidney, and lung.
Expression of KIAA2022 and P2RY8 in the carriers of the inversion
We used RNA extracted from lymphocytes of the two patients and one of the carrier females (III-7) to perform RT-PCR experiments and test the expression of the two genes (fig 7). This analysis showed that the KIAA2022 transcript is no longer expressed in the patients’ cells whereas the P2RY8 transcript is detected, although apparently in smaller amounts than in control samples (fig 7). The amount of P2RY8 is similar in the cells of the affected patients and the tested carrier female. This observation is consistent with the fact that a normal copy of the gene is present on the Y chromosome in males and that this gene is expressed from both X chromosomes in females. The carrier mother of patient IV-6 also expresses KIAA2022, a finding which is in good agreement with its X chromosome inactivation pattern (see above).
The direction of transcription of the two genes (telomere to centromere for KIAA2022 on the long arm and centromere to telomere for P2RY8 on the short arm) prevents the putative constitution of a fusion transcript after the occurrence of the inversion.
Mutation screening in unrelated XLMR families
The fact that the KIAA2022 gene was no longer expressed in the patients’ cells and the fact that it is highly expressed in fetal and adult brain made it a good candidate for X linked mental retardation. Conversely, the fact that P2RY8 is not expressed in brain and is located in the pseudoautosomal region made it a poor candidate for this phenotype. We thus decided to focus our attention on KIAA2022 and to screen a cohort of 20 unrelated XLMR families linked to the same interval of the human X chromosome for mutations in the gene (see Methods). We sequenced the KIAA2022 gene using genomic DNA of one affected individual from each family. This analysis failed to reveal any disease causing mutations or polymorphisms.
DISCUSSION
We cloned the breakpoints of a pericentric inversion which segregates in a family where two males are affected by severe mental retardation. Two genes are disrupted by these breakpoints. One is the KIAA2022 gene in Xq13.2 and the other is the P2RY8 gene in Xp22.3. The P2RY8 gene is not expressed in the brain and is located in a region of the human X chromosome where gene defects often cause a phenotype in carrier females.15 These data led us to hypothesise that the phenotype of the affected boys in the present family could be due to the lack of expression of KIAA2022. Our results indicate that KIAA2022 is no longer expressed in the cells of the affected male patients carrying the inversion whereas its expression is indistinguishable from the wildtype in the cells of the carrier mothers.
We sequenced the KIAA2022 gene in 20 probands from Xq13 linked non-syndromic XLMR families without identifying any mutation. This negative result might be explained in several ways. First, the gene itself may be a rare cause of X linked mental retardation. Since several new non-syndromic MR genes are found to be responsible for less than 2% of the cases which are screened,11,13 testing a larger sample of families will be necessary before drawing any conclusion. The second hypothesis is that the phenotype of the patients is not caused by the absence of expression of KIAA2022 but rather by the haploinsufficiency of P2RY8. In this case, it would be very difficult to explain why the carrier mothers in this family do not have any clinical signs. The fact that they are clinically normal rather indicates that the haploinsufficiency of P2RY8 is not deleterious. The third hypothesis would be that the phenotype of the patients is caused by a combination of the two gene deficits. In this case, it will probably be impossible to find another case where these two genes are simultaneously mutated.
Very little information is available on the function of the two genes disrupted by the chromosomal breakpoints. KIAA2022 encodes a large protein of 1516 amino acids. No known functional motif or significant homology to other proteins was found after a careful search in available databases. The genomic structure of this gene is quite unusual with a first intron of 180 kb and a very large third exon of more than 4 kb where the vast majority of the coding region is located. It also has an unusually large 3′ untranslated region of 5923 bp. Although KIAA2022 is easily amplified using human adult or fetal brain RNA in RT-PCR experiments, no human expressed sequence tag (EST) is present in the databases for its coding region. In the corresponding Unigene cluster (Hs.124128), the 25 ESTs all originate from the 3′ UTR. This is also a very unusual finding for which we currently have no explanation.
More information is available for P2RY8. It was classified as a member of the purine nucleotide G-protein coupled receptor family of proteins on the basis of amino acid sequence homologies.17 It encodes a small protein of 359 amino acids. The biology of the P2Y proteins is complex and these molecules are known to be involved in a large number of physiological processes ranging from blood platelet aggregation to the control of chloride ion fluxes in airway epithelia.18 We show here that this gene is composed of two exons separated by a large (70 kb) intron which contains the breakpoint of the inversion in our family. We also show that the P2RY8 transcript is not expressed in the brain, and that it is easily detected in blood lymphocytes in contrast to previous reports for this tissue.17 The absence of detectable expression in brain makes it a poor candidate to be responsible for the neurological phenotype of the patients carrying the pericentric inversion.
Since the data obtained for the KIAA2022 gene make it the more likely candidate to be involved in XLMR, it will now be necessary to study its expression during the development and in different regions of the cerebral cortex. In parallel, more patients with XLMR will need to be screened for mutations in this gene.
Acknowledgments
We thank Carlos Cardoso, Mike Mitchell, and Anne Moncla for helpful discussions. We thank Frank Kooy for sharing some of the data from his laboratory during the course of this work, WB Dobyns (Chicago, IL) for interpreting the brain MRI, and G Bourrouillou (CHU Toulouse, France) for providing clinical information on remote members of the family.
REFERENCES
Footnotes
-
↵* These two authors contributed equally to this work.
-
Financial support to VC from the French Ministry of Research is gratefully acknowledged. This study was supported in part by a grant from NICHD (HD26202) to CES.
-
Conflict of interest: none declared.