

Abstract
Long-term depression (LTD) and long-term potentiation (LTP) at cerebellar parallel fiber-Purkinje cell (PF-PC) synapses play critical roles in motor learning. The 1 Hz stimulation at PF-PC synapses induces a postsynaptically expressed LTP that requires a postsynaptic Ca2+ transient, phosphatases, and nitric oxide (NO). However, the mechanism underlying 1 Hz PF-LTP remains unclear because none of the known events is related to each other. Here, we demonstrated that 1 Hz PF-LTP requires postsynaptic cytosolic phospholipase A2 α (cPLA2α)/arachidonic acid (AA) signaling and presynaptic endocannabinoid receptors. Using patch-clamp recording in cerebellar slices, we found that 1 Hz PF-LTP was abolished in cPLA2α-knock-out mice. This deficit was effectively rescued by the conjunction of 1 Hz PF stimulation and the local application of AA. 2-Arachidonoylglycerol and the retrograde activation of cannabinoid receptor 1 (CB1R) were also involved in 1 Hz LTP because it was blocked by the hydrolysis of 2-AG or by inhibiting CB1Rs. The amount of NO released was detected using an NO electrode in cultured granule cells and PF terminals. Our results showed that the activation of CB1Rs at PF terminals activated NO synthetase and promoted NO production. The 1 Hz PF-stimuli evoked limited NO, but 100 Hz PF stimulation generated a large amount. Therefore, 1 Hz PF-LTP, distinct from classical postsynaptically expressed plasticity, requires concurrent presynaptic and postsynaptic activity. In addition, NO of sufficient amplitude decides between the weakening and strengthening of PF-PC synapses.
Introduction
Long-term use-dependent alterations in synaptic strength underlie information storage in the brain. In the cerebellum, long-term depression (LTD) at parallel fiber-Purkinje cell (PF-PC) synapses promotes adaptation of the vestibulo-ocular reflex and eye-blink conditioning (De Zeeuw et al., 1998; Ito, 2001; Feil et al., 2003; Boyden et al., 2006; Hansel et al., 2006). Meanwhile, long-term potentiation (LTP) at PF-PC synapses causes the extinction of learned associations in trained animals (Han et al., 2000; Medina et al., 2000; Schonewille et al., 2010). The reversible change in synaptic efficacy resulting from PF-LTD and PF-LTP is suggested to be the resetting mechanism for motor learning (Sejnowski, 1977; Houk and Wise, 1995; Medina et al., 2000). Evidence from fish and rat cerebellum demonstrated that PF-LTD and PF-LTP can reverse each other (Han et al., 2000; Lev-Ram et al., 2003).
PF-PC synapses undergo two forms of LTP, which are expressed either presynaptically or postsynaptically depending on stimulus frequency (Jörntell and Hansel, 2006). Upon PF stimulation at 4–8 Hz, presynaptically expressed LTP is evoked via presynaptic cyclic adenosine monophosphate signaling (Salin et al., 1996; Chen and Regehr, 1997; Linden and Ahn, 1999). Distinctly, 1 Hz PF stimulation induces postsynaptically expressed LTP because presynaptic glutamate release does not change and the serine/threonine phosphatases at postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) are required (Lev-Ram et al., 2002; Belmeguenai and Hansel, 2005; Schonewille et al., 2010). The postsynaptically expressed LTP is suggested to be the extinguishing mechanism for LTD because it reverses LTD and vice versa, whereas the presynaptically expressed LTP leaves LTD still saturated (LevRam et al., 2003). Thus, investigating how 1 Hz PF-LTP is induced is important for understanding the mechanisms of motor learning.
The 1 Hz PF stimulation evokes a small Ca2+ transient (Coesmans et al., 2004), which is required for the induction of LTP because the Ca2+chelator, BAPTA, blocks such LTP (Coesmans et al., 2004). The 1 Hz LTP also depends on nitric oxide (NO) because an inhibitor of nitric oxide synthase (NOS) or trapping NO blocks it (Lev-Ram et al., 2002, 2003). Despite these findings, most of the signaling underlying 1 Hz PF-LTP remains unclear. For example, the link between postsynaptic Ca2+ transient and NO production is unknown. In the present work, we aimed to investigate the signaling molecules involved in 1 Hz LTP. Our results indicate that the induction of 1 Hz PF-LTP requires both presynaptic and postsynaptic events, distinct from previously recognized plasticity. Interestingly, 1 Hz PF-LTP and PF-LTD share several signaling molecules, including Ca2+, cytosolic phospholipase A2α (cPLA2α), and NO, that may work as bidirectional messengers to balance the induction of LTP and LTD.
Materials and Methods
Slice preparation.
All experiments were approved by the Animal Experimentation Ethics Committee of Zhejiang University and were specifically designed to minimize the number of animals used. Original breeding pairs of the cPLA2α knock-out (KO) strain were obtained from Dr. Adam Sapirstein (Johns Hopkins University, Baltimore, MD) and maintained at the Experimental Animal Center of Zhejiang University. Mice were kept under temperature-controlled conditions on a 12:12 h light/dark cycle with food and water ad libitum. Sagittal slices of cerebellar vermis (250 μm) were prepared from P17-P23 anesthetized mice of either sex using a vibrating tissue slicer (Leica VT1000S) and ice-cold standard artificial CSF (aCSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 26 NaHCO3 and 25 d-glucose, bubbled with 95% O2/5% CO2. After recovery for 30 min at 37°C, slices were placed in a submerged chamber that was perfused at 2 ml/min with aCSF supplemented with 10 μm GABAzine to block GABAA receptors. Recording electrodes (3–5 mΩ) were filled with a solution containing the following (in mm): 120 K gluconate, 9 KCl, 3.5 MgCl2, 4 NaCl, 4 Na2ATP, 0.4 Na3GTP, 10 HEPES, and 17.5 sucrose (pH 7.2 was adjusted with KOH and 300 Osm). All drugs were from Sigma and Tocris Bioscience, with the exception of fatty-acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) from Cayman Chemical. When chemicals were supplemented in the internal saline, the concentration of K gluconate was reduced accordingly to maintain the desired osmolarity (Vapro Osmometer 5520, Wescor).
Electrophysiology.
Electrophysiological experiments were as in our previous work (Su and Shen, 2009; Su et al., 2010; Sun et al., 2011; Wang et al., 2012). PCs were visualized under an upright microscope (BX51; Olympus Optical) with a 40× water-immersion objective and equipped with infrared differential interference contrast enhancement. Whole-cell recordings were obtained with an Axon MultiClamp 700B amplifier (Molecular Devices). Currents were digitized at 10 kHz and filtered at 3 kHz. For PF stimulation, standard patch pipettes were filled with aCSF and placed in the middle third of the molecular layer. Synaptic responses were evoked every 20 s (0.05 Hz) using ∼10 μA pulses (100 μs duration). The series resistance for whole-cell recordings was typically 7–10 mΩ after 70% compensation and monitored throughout. Recordings were excluded from analysis if series resistance, input resistance, or holding current varied by >15% over the course of an experiment. To minimize the spontaneous spike activity that might escape voltage-clamp resulting from the poor space-clamp characteristics of PCs, baseline EPSC amplitude (holding potential −70 mV) was kept relatively small (typically <400 pA). All electrophysiological experiments were done at room temperature.
Granule cell culture.
Dissociated cultures of cerebellar granule cells were made as previously described with some modifications (Gallo et al., 1982; He et al., 2010). Briefly, the cerebellar cortex was dissected on ice from anesthetized P0-P1 Sprague Dawley rats, digested with 0.1% trypsin for 7–9 min at 37°C, and dissociated into single cells by gentle trituration. Cells were plated on coverslips coated with 100 g/ml poly-d-lysine in Neurobasal medium (Invitrogen) supplemented with 10% FBS and 2% B27 (Invitrogen). Cells were grown in basal modified Eagle's medium containing 10% heat-inactivated FCS, gentamycin (100 μg/ml), and 5 mm KC1. The culture dishes were incubated at 37°C in humidified 95% O2/5% CO2. After 18–20 h, 10 μm cytosine arabinoside was added to the medium to prevent the replication of non-neuronal cells. The culture medium was renewed at DIV2 and DIV5.
NOS activity assay.
The activity of NOS was quantified as the nitrite concentration in granule cell cultures according to the well-described Griess reaction. In brief, supernatant (100 ml) from the control or WIN55,212–2 (WIN55)-treated group was used to measure the nitrite concentration according to the manufacturer's instructions (Jian Cheng Bioengineering). The Griess reagent contained 1% sulfanilamide in 5% phosphoric acid and 0.1% naphthylethylenediamine dihydrochloride in water. The absorbance of the mixture at 550 nm was determined with an ELISA plate reader (Dynatech Labs). All measurements of NOS activity were normalized for total protein content using a BCA protein assay kit (Pierce).
NO detection.
NO efflux was monitored using an NO-selective amperometric microprobe (WPI), similar to previous work (Stefano et al., 1995). An L-shaped microprobe with a tip diameter of 30 μm was used in cultured granule cells, and another straight sharp microprobe with a tip diameter of 7 μm was used in cerebellar slices. The redox current was continuously recorded by a current–voltage converter circuit with DUO 18 software (WPI). Calibration of the electrochemical sensor was performed using different concentrations of the nitrosothiol donor S-nitroso-N-acetyl-dl-penicillamine to generate a standard curve. The probes were routinely cleaned in 0.1 m H2SO4 to remove the cellular debris that tended to accumulate. For NO recording in cultured cells, a micromanipulator attached to the stage of an inverted microscope (Olympus) was used to position the L-shaped microprobe 10 μm above the cell surface (see Fig. 5C). To record the intercellular NO in slices, a sharp microprobe was positioned 5 μm away from the tip of the stimulating electrode using a fine micromanipulator (see Fig. 5E). Both probes were allowed to equilibrate for 40 min in the incubation medium before being transferred to cultures or tissues. Manipulation of tissues was performed only with glass instruments.
Statistics.
Data analysis was performed using Excel 2003 (Microsoft), Clampfit 10 (Molecular Devices), and Igor Pro 6.0 (Wavemetrics). Error bars indicate the mean ± SEM. p values were determined by Student's t test. The accepted level of significance was p < 0.05. n represents the number of cells or cultures tested.
Results
The 1 Hz PF-LTP is cPLA2α-dependent
Whole-cell patch-clamp recordings were made from mouse PCs to investigate the induction of 1 Hz PF-LTP according to the protocol described previously (Lev-Ram et al., 2002; Coesmans et al., 2004; Belmeguenai and Hansel, 2005). In brief, PF-EPSCs were monitored in voltage-clamp mode, and LTP was obtained when the PF was stimulated at 1 Hz for 5 min in current-clamp mode (Fig. 1A). The amplitude of LTP varies among laboratories (Lev-Ram et al., 2002; Belmeguenai and Hansel, 2005). In our hands, the potentiation of EPSCs recorded from wild-type (WT) mice reached 142 ± 3% of baseline (t = 38 min; n = 20, p < 0.01 compared with baseline; Fig. 1B). The paired-pulse facilitation (PPF) ratio was measured by applying two pulses at an interval of 80 ms, and it was unaltered by 1 Hz stimulation (t = 38 min: 1.67 ± 0.06; p > 0.05 compared with baseline, 1.66 ± 0.02; n = 20; Fig. 1C), indicating that the presynaptic release was not affected (Lev-Ram et al., 2002; Coesmans et al., 2004; Belmeguenai and Hansel, 2005). cPLA2α is highly sensitive to Ca2+ and moves to the membrane when activated (Evans and Leslie, 2004). The same protocol for LTP induction was then performed in cPLA2α KO mice. Interestingly, 1 Hz LTP was blocked in KO cells (Fig. 1D,E) with PPF ratios unaffected (Fig. 1F). For comparison, WT cells were pretreated with a specific cPLA2α inhibitor, arachidonyl trifluoromethyl ketone (AACOCF3, 10 μm), which significantly reduced the LTP (Fig. 1G). Likewise, the PPF ratios did not change after induction (Fig. 1H). These results suggested that 1 Hz PF-LTP is cPLA2α-dependent.

The 1 Hz PF-LTP is deficient in cPLA2α KO mice. A, PF-LTP was induced by PF stimulation at 1 Hz for 5 min. Example EPSCs before (baseline) and after (t = 38 min) stimulation are shown. Both EPSC1 and EPSC2 were potentiated after stimulation. B, Time course of percentage change in EPSC1 amplitude (n = 20). Each data point indicates the average of three successive EPSCs evoked at 0.05 Hz. C, Time course of the PPF ratio (EPSC2/EPSC1) from a subset of the cells shown in B. D, Example EPSCs from one KO cell before (baseline) and after (t = 38 min) stimulation. EPSC amplitude was unaltered by the stimulation. E, Time course of percentage change of EPSC1 amplitude (n = 14). Each data point indicates the average of three EPSCs. F, Time course of the PPF ratio from cells shown in E. G, Time course of percentage change of EPSC1 amplitude when WT cells were locally perfused with 100 μm AACOCF3 before and during stimulation (n = 11), indicating that AACOCF3 blocked the LTP induction (n = 11). H, Time course of the PPF ratio of EPSCs from cells shown in G.
cPLA2α contributes to PF-LTP by providing AA
Because cPLA2α selectively liberates AA (Evans and Leslie, 2004), the effect of exogenous AA was tested in PF-LTP. We first asked whether AA is able to rescue PF-LTP in the weaker stimulation. Consistent with previous work (Lev-Ram et al., 2002), 1 Hz stimulation for 2.5 min (150 spikes) failed to induce PF-LTP (Fig. 2A,B; 99 ± 8% at t = 38 min; n = 12, p > 0.05 compared with baseline). In contrast, local application of 2 μm AA overlapping this weaker stimulation in WT cells was sufficient to potentiate PF-EPSCs (Fig. 2A,B; 126 ± 5% of baseline at t = 38 min; n = 13, p < 0.01 compared with baseline). Neither weak stimulation nor conjunction changed the PPF ratio (Fig. 2C). The next experiment was to apply exogenous AA (2 μm) to KO cells simultaneously with the strong stimulation. As expected, local AA perfusion (2 μm) overlapping 5 min stimulation robustly rescued the LTP in KO cells (Fig. 2D,E; 134 ± 5% of baseline at t = 38 min; n = 14, p < 0.01 compared with baseline). These results supported the view that AA is involved in 1 Hz PF-LTP induction. Alternatively, cPLA2α activated by the stimulation subsequently releases AA and contributes to LTP induction.

AA is involved in PF-LTP. A, Example PF-EPSCs from WT cells stimulated for 2.5 min at 1 Hz (stim) or locally perfused with 2 μm AA overlapping the 1 Hz stimulation (stim+AA). Black and gray traces represent EPSCs before (baseline) and after (t = 38 min) the stimulation, respectively. EPSCs were potentiated in the stim+AA group but not in the stim group. B, Time course of percentage changes of EPSC amplitudes in “stim” and “stim+AA” groups. Each data point indicates the average of three EPSCs. C, Time course of the PPF ratio from cells shown in B. D, KO cells were locally perfused with 2 μm AA overlapping the 1 Hz stimulation for 5 min. Example PF-EPSCs before (baseline) and after (t = 38 min) stimulation are shown. E, Time course of percentage change of EPSC amplitude. Each data point indicates the average of three EPSCs. F, Time course of PPF ratio from cells shown in E. G, Time course of percentage change of PF-EPSC amplitude when KO cells were locally perfused with 2 μm AA overlapping 1 Hz stimulation for 5 min. These cells were internally perfused with 10 mm BAPTA. The mean EPSC amplitude at 38 min was 100 ± 4% of baseline (n = 9). p > 0.05 compared with baseline. H, Time course of PPF ratio of EPSCs from cells shown in G. I, Example EPSCs at 0 and 45 min when WT cells were perfused with 2 μm AA. J, Time courses of percentage change of EPSC amplitude (filled circles) and PPF ratio (open circles). Black bar represents the presence of AA.
It has been shown that internal Ca2+ is increased by 1 Hz stimulation and is involved in the induction of PF-LTP (Coesmans et al., 2004). We next investigated whether the activation of cPLA2α/AA signaling is sufficient to induce PF-LTP in a manner independent of internal Ca2+. To do so, BAPTA (10 mm), at a concentration lower than previously used (Coesmans et al., 2004), was added to the internal saline. We found that BAPTA completely blocked LTP in KO cells, even if these cells were given a conjunction of 2 μm AA and PF stimulation (Fig. 2G,H). AA was also applied alone to test whether it is sufficient to potentiate PF-EPSCs. Against this assumption, we found that bath application of 2 μm AA in WT cells did not induce an appreciable LTP-like potentiation of PF-EPSCs (Fig. 2I,J; 106 ± 3% at t = 45 min; n = 10, p > 0.05 compared with t = 1 min). The PPF ratio also did not change during the AA application (Fig. 2J). Together, these results suggested that cPLA2α/AA signaling is necessary but not sufficient for the induction of 1 Hz LTP. Ca2+ is the critical regulator of this LTP through regulating not only cPLA2α but other events, such as the phosphorylation of AMPARs. Notably, the effects of AA on PF-EPSC amplitude may be dose-dependent because higher concentrations of AA decrease PF-EPSC amplitude (Le et al., 2010; Wang et al., 2012), whereas lower concentrations have no effect (Le et al., 2010) (Fig. 2I).
2-Arachidonoylglycerol/cannabinoid receptor 1 signaling is required for 1 Hz PF-LTP
Endocannabinoids, N- arachidonoylethanolamine (AEA) and 2-arachidonoylglycerol (2-AG), are derivatives of AA (Malcher-Lopes et al., 2008). AEA is selectively hydrolyzed by FAAH (Cravatt et al., 2001; Okamoto et al., 2004) and 2-AG is selectively hydrolyzed by MAGL (Dinh et al., 2002; Bisogno et al., 2003). To determine the downstream effector of cPLA2α/AA signaling in 1 Hz LTP, 0.7 μg/ml MAGL or 1 μg/ml FAAH was added to the pipette solution and 1 Hz LTP was induced in WT cells. We found that 1 Hz LTP was blocked by MAGL (Fig. 3A). On the contrary, FAAH did not affect the LTP induction (Fig. 3A). The mean amplitudes of EPSCs at 38 min were 97 ± 5% (MAGL; n = 13, p > 0.05 compared with baseline) and 138 ± 6% (FAAH; n = 14, p < 0.01 compared with baseline) (Fig. 3B). The PPF ratio was not affected in either condition (Fig. 3C). These results indicated that 2-AG, but not AEA, is required for LTP induction. CB1R, the receptor for endogenous 2-AG, mainly occurs at PF terminals (Diana et al., 2002). Thus, we investigated the role of CB1Rs in PF-LTP. AM251 (5 μm), a CB1R antagonist, was locally applied to cells before and during stimulation. We found that PF-LTP was blocked by AM251 (Fig. 3D,E). Therefore, 2-AG and CB1R are needed for 1 Hz PF-LTP.

PF-LTP requires 2-AG production and CB1R activation. A, Example PF-EPSCs from WT cells that were stimulated for 5 min at 1 Hz. Cells were filled with either MAGL or FAAH. Black and gray traces represent EPSCs before (baseline) and after (t = 38 min) stimulation, respectively. EPSCs were potentiated in the FAAH group but not in the MAGL group. B, Time course of percentage change of EPSC amplitude in the MAGL and FAAH groups. Each data point indicates the average of three consecutive EPSCs. Black bar above represents the presence of MAGL or FAAH in the pipette. C, Time course of PPF ratio from cells shown in B. D, Example EPSCs derived from a WT cell before (baseline) and after (t = 38 min) stimulation with bath application of AM251. Cells were stimulated for 5 min at 1 Hz. E, Time course of percentage change of EPSC amplitude. AM251 was locally applied before and during stimulation. Each data point indicates the average of three consecutive EPSCs. The mean EPSC amplitude at 38 min was 98 ± 6% of baseline (n = 12). p > 0.05 compared with baseline. F, Time course of PPF ratio from EPSCs shown in E.
Production and anterograde diffusion of NO are required for PF-LTP
It is suggested that endogenous NO is necessary for the induction of both forms of synaptic plasticity at PF-PC synapses (Lev-Ram et al., 1995, 1997, 2002; Safo and Regehr, 2005). In the current work, several experiments were performed to assess the necessity of NO for the induction of 1 Hz LTP. First, slices were incubated with a competitive inhibitor of NOS, l-NAME (100 μm), for 1 h before recordings. PCs were then stimulated in the continued presence of l-NAME. Our results demonstrated that 1 Hz LTP was blocked by l-NAME, showing that the average EPSC amplitude at 38 min was 100 ± 3% of baseline (Fig. 4A,B, black traces; n = 12, p > 0.05 compared with baseline), similar to previous work (Lev-Ram et al., 2002). Second, whether NO has to cross the intercellular space at PF-PC synapses in LTP was tested using oxymyoglobin, a high-affinity NO trap. Oxymyoglobin (10 μm) was locally added to the cells throughout experiments with the duration longer than previous work (Lev-Ram et al., 2002). After the 1 Hz stimulation for 5 min, EPSCs transiently increased but immediately declined back to baseline (Fig. 4A,B, gray traces). The mean amplitude of EPSCs at 38 min was 101 ± 5% of baseline (n = 13, p > 0.05 compared with baseline).

NO is required for PF-LTP. A, LTP induction in WT cells perfused with l-NAME or oxymyoglobin (myoglobin) in the bath. Both groups were stimulated for 5 min at 1 Hz. Black and gray traces represent EPSCs before (baseline) and after (t = 38 min) stimulation, respectively. B, Time course of percentage change of EPSCs in l-NAME and oxymyoglobin groups. Each data point indicates the average of three consecutive EPSCs. Black bars represent the duration of application of l-NAME and myoglobin. C, Time course of PPF ratio from cells shown in B. D, The 1 Hz LTP was blocked by the inclusion of oxymyoglobin inside PCs. Representative traces show EPSCs before (baseline) and after (t = 38 min) stimulation. E, Time course of percentage change of EPSCs with internal oxymyoglobin (n = 10). The mean EPSC amplitude at 38 min was 100 ± 5% of baseline (n = 13). p > 0.05 compared with baseline. Each data point indicates the average of three consecutive EPSCs. The bar above represents the inclusion of oxymyoglobin in PCs (internal myoglobin). F, Time course of PPF ratio from cells shown in E. G, Example traces show EPSCs before (baseline) and after (t = 38 min) stimulation. The 1 Hz LTP was normal when 5 μm arginine was included in the recording pipettes. H, Time course of percentage change of EPSCs with internal arginine (n = 9). The mean EPSC amplitude at 38 min was 134 ± 3% of baseline (n = 13). p < 0.01 compared with baseline. Each data point indicates the average of three consecutive EPSCs. Bar indicates the duration of arginine included in PCs (internal arginine). I, Time course of PPF ratio from cells shown in H.
Because oxymyoglobin is membrane-impermeable, these results implied that the diffusion of NO in the intercellular space is important for LTP. We next considered whether the target of NO action is inside the PC. If so, one would predict that intracellular oxymyoglobin should prevent the induction of LTP because of its membrane impermeability. To trap NO and inhibit its mediated activation of guanylate cyclase in PCs, 100 μm oxymyoglobin was included in the patch pipettes. As predicted, 1 Hz LTP was blocked by the inclusion of oxymyoglobin in PCs (Fig. 4D,E), confirming that the target of NO is within the PC. Meanwhile, the PPF ratio was not affected after LTP induction (Fig. 4F). To further identify the source of NO in LTP induction, an NOS inhibitor, NG-nitro-arginine (arginine), was applied intracellularly in PCs. Arginine is a zwitterion and does not passively leak out of clamped cells to influence neighboring cells (Lev-Ram et al., 1995). Because arginine blocks NOS with an IC50 of ≤1 μm (East and Garthwaite, 1990; Dwyer et al., 1991; Furfine et al., 1993), 5 μm arginine was included in the patch pipettes to block NOS. Under these conditions, 1 Hz PF stimulation induced LTP normally (Fig. 4G,H), suggesting that such LTP does not need NOS activity within the PC.
Activation of CB1Rs at PF terminals produces NO
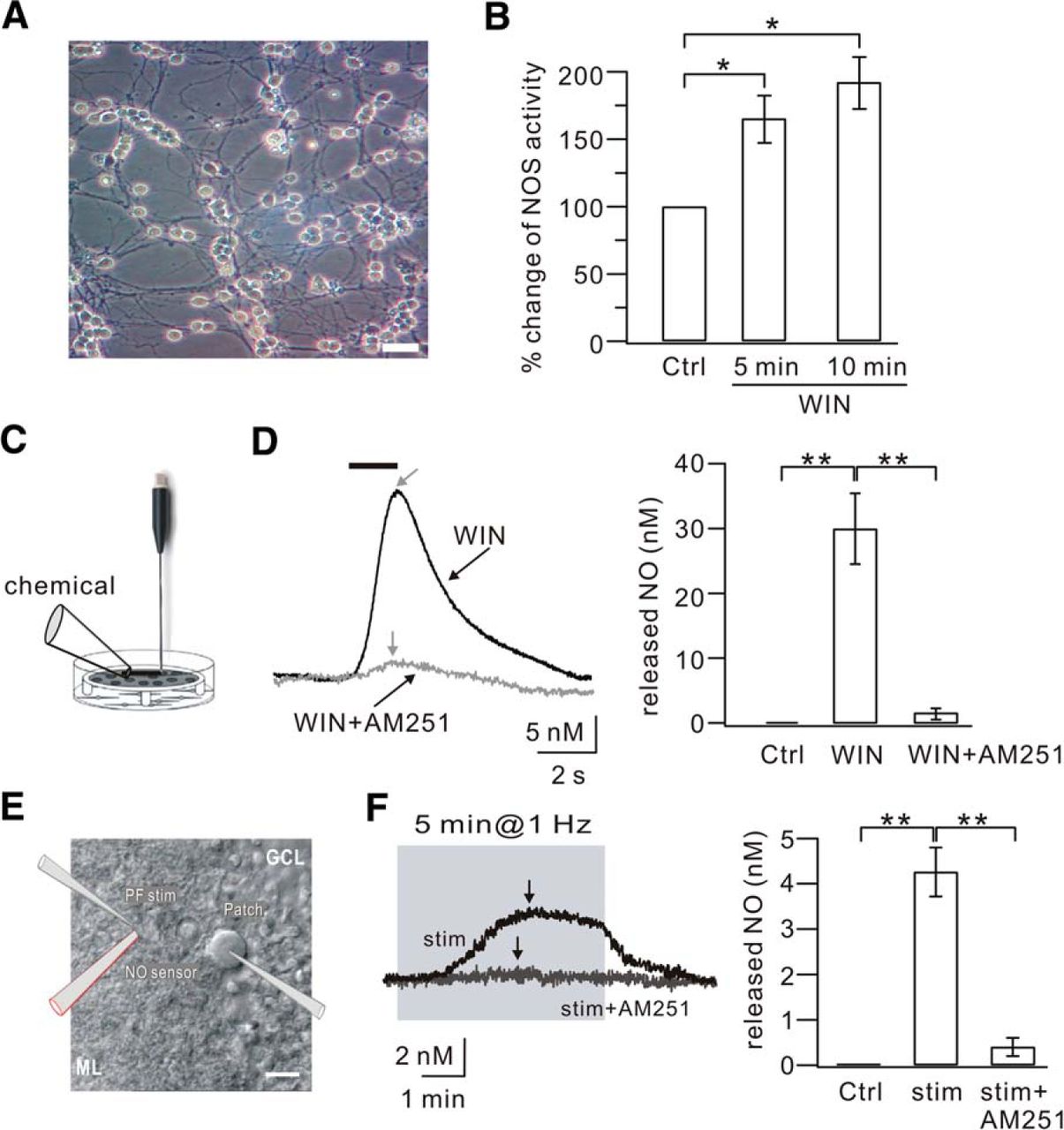
To date, the effector molecules of CB1Rs in brain are not clear. The experiments above gave an interesting clue that CB1R activation may cause the production of NO in granule cells and PF terminals. Several experiments were performed to investigate whether this speculation is correct. First, NOS activity in cultured granule cells (Fig. 5A) was measured in the absence and presence of the CB1R agonist, WIN55 (5 μm) using an ELISA plate reader to assess nitrite concentration. Our results showed that NOS activity increased significantly upon WIN55 application in a time-dependent manner (Fig. 5B), indicating that the activation of CB1Rs increases NOS activity in granule cells. Second, NO efflux in cell cultures was measured using an L-shaped NO electrode placed close to the surface of the cells (Fig. 5C). Continuous monitoring with the NO electrode showed no detectable NO efflux (nanomolar level) at baseline (Fig. 5D). After the cells had stabilized for at least 150 s, 5 μm WIN55 was locally administered. This puff of WIN55 significantly increased NO efflux in a transient and reproducible manner (Fig. 5D; WIN55, 30 ± 5 nm; n = 7, p < 0.01 compared with control). This result was consistent with the NOS assay experiment, but much more efficient in detecting activity of the NOS/NO cascade. Coapplication of 5 μm AM251 with 5 μm WIN55 almost completely blocked the NO efflux (Fig. 5D; WIN55+AM251, 1.1 ± 0.9 nm; n = 6, p < 0.01 compared with the WIN55 group). These experiments demonstrated that CB1R activation of granule cells causes NO production and efflux.

CB1R activation produces NO in granule cells and at PF terminals. A, DIC image of cultured cerebellar granule cells (DIV7). Scale bar, 10 μm. B, ELISA detection of NOS in cultured granule cells. The NOS activity was calculated as the concentration of nitrite. The percentage changes of NOS activity were 166 ± 18% and 193 ± 20% of the control (Ctrl) when cells were exposed to WIN55 (WIN) for 5 min and 10 min, respectively. C, Schematic illustration showing how NO efflux was detected using the L-shaped NO electrode in cultured granule cells. Chemicals were locally applied onto cells as shown. D, Example traces of NO concentration recorded in the control (Ctrl), 5 μm WIN55 (WIN), and 5 μm WIN55 + 5 μm AM251 (WIN+AM251) groups. Gray arrows indicate the peaks of NO responses in the WIN and WIN+AM251 groups. Right bar graphs represent the averaged peak values in the three groups. E, DIC image illustrating the placement of the PF stimulation pipette, patch pipette, and sharp NO electrode in a cerebellar slice. The clamped cell in the center is a PC. Scale bar, 20 μm. F, Examples of NO trace recorded with 1 Hz stimulation (stim) and 1 Hz stimulation + 5 μm AM251 (stim+AM251). Black arrows indicate the peak of each response. Right bar graphs represent the averaged peak values. *p < 0.05. **p < 0.01.
To assess the role of CB1R in NO production at PF terminals, a sharp NO electrode was placed near the stimulation pipette in cerebellar slices (Fig. 5E), and then 1 Hz LTP and the intercellular efflux of NO were simultaneously recorded at PF-PC synapses. We found that 1 Hz PF stimulation for 5 min increased the concentration of intercellular NO, which reached a peak during the stimulation and slowly decreased as time elapsed (Fig. 5F; peak: 4.2 ± 0.5 nm; n = 10, p < 0.01 compared with control). Coapplication of AM251 (5 μm) during 1 Hz stimulation significantly reduced the concentration of intercellular NO (Fig. 5F; peak: 0.4 ± 0.2 nm; n = 7). Therefore, these results showed that CB1R activation induces NO production and efflux at PF terminals.
NMDAR-induced NO release from stellate cells is not involved in 1 Hz LTP
NO is widely distributed in neurons (Bredt and Snyder, 1992; Dawson et al., 1992). In the cerebellum, previous work demonstrated that NMDARs activate nNOS/NO signaling and play important roles in PF-LTD induced by strong PF stimulation (Casado et al., 2002). Convincing evidence from Shin and Linden (2005) showed that this NMDAR/NO cascade is localized to interneurons rather than PFs. Because PF stimulation excites neighboring dendrites over a certain range, a critical question was whether 1 Hz PF stimulation also results in the activation of postsynaptic interneurons, which consequently leads to NO production. To answer this question, high-frequency (100 Hz) and low-frequency (1 Hz) PF stimulation was applied to stellate cells (SCs) during whole-cell recordings (Fig. 6A). To record NMDA currents, SCs were clamped at 40 mV and perfused with Mg2+-free aCSF supplemented with 50 μm GYKI53655, a selective antagonist of AMPARs. Short (5 pulses) and long (60 pulses) trains of PF stimuli at 1 Hz were consecutively delivered to the same cell. As shown in Figure 6B, neither short nor long 1 Hz PF trains elicited NMDAR-mediated currents. Burst stimuli (100 Hz) were then applied to PFs, and NMDAR-mediated currents were induced after 5 pulses. Application of 50 μm d-AP5 was sufficient to eliminate the outward current (Fig. 6C). Thus, NMDAR-mediated currents were only inducible by high-frequency PF stimulation.

NMDAR-mediated NO release from SCs. A, Schematic illustration showing the stimulation protocols at PF-SC synapses and the localization of postsynaptic AMPARs and NMDARs in SCs. PF stimulation (100 Hz or 1 Hz) was applied at PF-SC synapses as high- and low-frequency stimuli, respectively. AMPARs are located in the center, whereas NMDARs are extrasynaptic. B, Representative recordings from one SC in response to 5 (top) and 60 (bottom) PF stimuli given at 1 Hz. Cells were clamped at 40 mV and perfused with GYKI53655 to block AMPAR responses. Time scales: top, 1 s; bottom, 11 s. C, Representative recordings from one SC in response to 5 PF stimuli given at 100 Hz. Cells were clamped at 40 mV and perfused with GYKI53655 to block AMPAR-mediated responses. High-frequency stimulation induced an outward current, which was blocked by the application of 50 μm d-AP5 (bottom). D, DIC image illustrating the recording configuration for NO release from one SC. Glass pipettes for PF stimulation and whole-cell recording in the SC as well as an NO electrode were placed on the slices as shown in the illustration. ML, Molecular layer; GC/PC, granule cells/Purkinje cells. Scale bar, 20 μm. E, Examples of NO recording from one SC evoked by 1 Hz and 100 Hz PF stimulation. Slices were perfused with Mg2+-free aCSF containing GYKI53655. The responses for 1 Hz and 100 Hz PF stimulation are shown in the top and bottom as labeled. Time scales: top, 1 s; bottom, 400 ms. The black arrow indicates the peak of NO release. The gray trace in the bottom represents that 50 μm d-AP5 blocked PF stimulation-induced NO release. F, Examples of NO recording when PFs were given the induction for PF-LTD (PF-LTD stim). Slices were perfused with Mg2+-free aCSF containing GYKI53655. The shadowed background shows the duration of PF-LTD stimulation. The black and gray traces represent the NO responses with PF-LTD stimulation and PF-LTD stimulation plus d-AP5 (stim+AP5), respectively, showing that d-AP5 blocked most of the NO release. The black arrows indicate the peak of NO release in each condition. G, Averaged peak values of NO efflux under different stimulation conditions. Five pulses at 1 Hz (5 stim at 1 Hz): 0, n = 12. Single pulse at 100 Hz (5 stim at 100 Hz): 4.9 ± 1.3 nm, n = 9. Single pulse at 100 Hz with d-AP5 (5 stim at 100 Hz+AP5): 0.3 ± 0.2 nm, n = 6. PF-LTD stimulation (PF-LTD stim): 17.4 ± 1.5 nm, n = 6. PF-LTD stimulation with d-AP5 (PF-LTD stim+d-AP5): 2.3 ± 0.5 nm, n = 4. For comparison, the released NO in response to 5 min PF stimulation at 1 Hz shown in Figure 5F is replotted (5 min at 1 Hz, gray bar). *p < 0.05. **p < 0.01. H, Example traces of PF-EPSCs before (baseline) and after (t = 38 min) 1 Hz stimulation when slices were perfused with d-AP5. I, Time course of percentage change of EPSC amplitude in response to 5 min stimulation at 1 Hz (stim; n = 10) and the overlapping application of 50 μm d-AP5 with 1 Hz stimulation (stim+AP5). Each data point indicates the average of three successive EPSCs evoked at 0.05 Hz. J, PPF ratios from a subset of the cells shown in I.
To examine the distinct roles of high- and low-frequency PF stimulation on NO release, a sharp NO electrode was placed near the stimulation pipette in the molecular layer of cerebellar slices, whereas SCs were current-clamped (Fig. 6D). The voltage response of SCs and the intercellular NO concentration at PF-SC synapses were simultaneously recorded. We found that there was no detectable NO release in response to 5 stimuli at 1 Hz (Fig. 6E). On the contrary, 5 pulses at 100 Hz evoked a prominent NO release, which was eliminated by 50 μm d-AP5 (Fig. 6E). These results suggested that high-frequency PF bursts are sufficient to evoke NMDAR-mediated NO release. Previous work demonstrated that the induction of PF-LTD requires 100 Hz PF stimulation repeated 30 times at 2 s intervals (Shin and Linden, 2005; Steinberg et al., 2006). We used this to measure how much NO is released in response to PF-LTD induction. Thirty PF bursts at 100 Hz elicited a gradually increasing NO release (Fig. 6F), which had a peak amplitude much greater than that in response to 1 Hz PF stimulation for 1 min and 5 pulses at 100 Hz. This NO release was almost blocked by 50 μm d-AP5, suggesting that PF-LTD induction is sufficient to elicit a large NMDAR-mediated NO release. The remaining NO after d-AP5 treatment might have been caused by the activation of CB1Rs in PF terminals (Fig. 6F,G). It has been demonstrated that the pharmacological blockade of NMDARs and resulting NO release blocks PF-LTD (Casado et al., 2002; Shin and Linden, 2005). Thus, it was of interest to investigate whether 1 Hz PF-LTP also requires the activation of NMDARs. Cerebellar slices were perfused with aCSF supplemented with 50 μm d-AP5, and the 1 Hz LTP induction protocol was performed (Fig. 6H). Unlike PF-LTD, 1 Hz LTP was normally induced by this stimulation (Fig. 6I). Together, these results showed that NO synthesis and release from SCs and PFs are the result of different mechanisms. Weak stimulation with a long duration evokes a small NO release from PFs, whereas strong PF stimulation evokes a large NO release from SCs.
Discussion
The main finding of this study is that the induction of 1 Hz PF-LTP requires concurrent presynaptic and postsynaptic events. Figure 7 summarizes the essential cascades involved in PF-LTP (red lines) and PF-LTD (black lines). The mechanisms for PF-LTD have been well defined. In brief, it is usually induced with strong PF stimulation or conjunctive CF/PF stimulation (Hansel et al., 2001; Linden, 2003). The released glutamate acts upon postsynaptic mGluRs and AMPARs and results in a high Ca2+ threshold (Coesmans et al., 2004). After these initial signals, there is a requirement for the activation of PKC and αCaMKII (Hansel et al., 2001, 2006; Leitges et al., 2004). The cPLA2α/COX2 cascade is critical for PF-LTD by acting upstream of PKC (Le et al., 2010). The involvement of CB1R and NO/soluble guanylyl cyclase/PKG/phosphatase pathways is also suggested (Hansel et al., 2001; Safo and Regehr, 2005). The required NO is mediated by NMDARs localized to interneurons (Lev-Ram et al., 1995, 1997; Casado et al., 2002; Shin and Linden, 2005) (Fig. 6). PF-LTD is expressed postsynaptically, as a reduction in the number of surface AMPARs (Wang and Linden, 2000; Xia et al., 2000). In contrast to PF-LTD, 1 Hz PF-LTP requires a low Ca2+ threshold (Coesmans et al., 2004), which subsequently leads to cPLA2α activation, AA liberation, and the production of 2-AG that binds to presynaptic CB1R (Figs. 1, 2, and 3). Activated CB1R triggers NOS activation and produces a low level of NO from PF terminals (Figs. 4 and 5). NO anterogradely crosses the intercellular space and is assumed to target phosphatases and AMPAR trafficking. Interestingly, PF-LTP shares several molecules with LTD, including Ca2+, cPLA2α, CB1R, and NO (highlighted in Fig. 7). One notable bifurcation between PF-LTD and PF-LTP is mGluR. It is shown that mGluRs are required for PF-LTD expression by attributing Ca2+ (Takechi et al., 1998; Brasnjo and Otis, 2001; Belmeguenai et al., 2008). However, they do not affect cerebellar 1 Hz LTP (Belmeguenai et al., 2008).

A schematic illustration shows the major signaling molecules involved in the induction of 1 Hz PF-LTP (red lines) and PF-LTD (black lines). Red and black flash signs indicate the stimulations for LTP and LTD, respectively. The stimulation for LTD simultaneously activates postsynaptic AMPARs and mGluRs on PCs and NMDARs on interneurons. Dotted lines indicate the unclear cascades. Bright brown circles represent shared molecules (NO, Ca2+, cPLA2α, and CB1R) in LTP and LTD.
2-AG is a retrograde messenger that mediates the induction of LTD in the hippocampus (Chevaleyre and Castillo, 2003) and cerebellum (Safo and Regehr, 2005). Diacylglycerol lipase (DAGL) is shown to be needed for the formation of 2-AG in cerebellar LTD (Safo and Regehr, 2005). The same paper also showed that blocking DAGL does not affect the 2-AG/CB1R-mediated depolarization-stimulated inhibition of excitation (DSE) (Safo and Regehr, 2005), suggesting that 2-AG may be synthesized through a pathway independent of DAGL. Consistent with this speculation, our previous work showed that cPLA2α/AA signal produces 2-AG during DSE (Wang et al., 2012). This conclusion was further strengthened by the present work that 2-AG is the substrate of cPLA2α/AA signaling in 1 Hz PF-LTP. Together, these data indicate that two independent pathways, phospholipase C (PLC)/DAGL and cPLA2α/AA, may both control the production of 2-AG.
DSE results from a brief depolarization of PCs (Kreitzer and Regehr, 2001). The stimulation strength for both DSE and 1 Hz PF-LTP is much weaker than that for LTD (Hartell, 1996; Ito, 2001; Kreitzer and Regehr, 2001; Shin and Linden, 2005; Steinberg et al., 2006). Thus, the internal Ca2+ evoked by which stimulation may be a decisive factor in PF-PC synaptic plasticity (Fig. 7). Because a prolonged elevation of Ca2+ is needed to activate PLC/DAGL signal (Maejima et al., 2005) and a lower level of Ca2+ activates cPLA2α (Evans and Leslie, 2004), it will be interesting to investigate how Ca2+ differentially modulates the activity of PLC/DAGL and cPLA2α/AA signals, as well as their roles in 2-AG production. In addition, the function of cPLA2α in PF-PC plasticity is complicated. Whereas we found that cPLA2α is required for 1 Hz LTP induction, Le et al. (2010) revealed a crucial role of the cPLA2α-cyclooxygenase-2-PKCα cascade in PF-LTD evoked by a strong surge of intracellular Ca2+. Therefore, cPLA2α may also play a dual role in PF-PC plasticity, depending on how much it is activated (Fig. 7).
Although AEA and 2-AG are both localized in the hippocampus and cerebellum (Bisogno et al., 1999), 2-AG is the only retrograde messenger to mediate DSE, LTD, and LTP (Chevaleyre and Castillo, 2003; Safo and Regehr, 2005; Wang et al., 2012) (Fig. 3). AA metabolism is highly interactive among signaling pathways (Hoggatt and Pelus, 2010). Hence, it is necessary to examine the biosynthesis of AEA and 2-AG after PF stimulation to address why AEA is not the retrograde messenger. Another mystery is the distinct roles of CB1R at PF terminals, as it is required for both LTD and LTP (Fig. 7). It has been suggested that CB1R activation causes the reduced release of neurotransmitter by modulating the activity of ion channels and/or adenylyl cyclases (Levenes et al., 1998; Takahashi and Linden, 2000; Daniel et al., 2004), which may explain the inhibition of presynaptically expressed PF-LTP by CB1Rs (van Beugen et al., 2006) and the requirement of CB1Rs in PF-LTD (Safo and Regehr, 2005; van Beugen et al., 2006). However, the decrease of transmitter release does not account for our finding that CB1Rs stimulated NO production at PF terminals (Fig. 5). Thus, the mechanism underlying CB1R-NOS cascade remains to be explored.
Coesmans et al. (2004) showed that the induction of PF-LTD versus PF-LTP is governed by a Ca2+ threshold rule that operates inversely to the Bienenstock, Cooper, and Munro model (Bienenstock et al., 1982). PF-LTD has a higher Ca2+ threshold mainly contributed by climbing fibers (Coesmans et al., 2004). Here, we showed that 1 Hz PF stimulation evoked limited NO release from PF terminals (Fig. 5), but PF-LTD stimulation generated a large amount from interneurons (Fig. 6). Combined with previous work (Lev-Ram et al., 2002; Belmeguenai and Hansel, 2005; Shin and Linden, 2005; Schonewille et al., 2010), our results suggested that NO may act as another threshold in the weakening and strengthening of PF-PC synapses (Fig. 7). Interestingly, a strong but brief PF stimulation (8 pulses at 100 Hz) can induce PF-LTP (Piochon et al., 2010). Consistently, we found that 5 PF stimuli at 100 Hz induced a measurable NO rise (Fig. 6E). Together, these results highlight the importance of NO in LTP induction, that is, PF-LTP can be induced by an adequate amount of NO, no matter where it comes from. Nevertheless, NO differs from Ca2+ because it is regulated by CB1Rs at PF terminals during LTP (Fig. 5) and by extrasynaptic NMDARs on interneurons during LTD (Fig. 6). In contrast, the Ca2+ transients required for LTP and LTD induction reflect the excitable state of PCs (Coesmans et al., 2004).
PF-LTP depends on serine/threonine phosphatase activity (Belmeguenai and Hansel, 2005; Schonewille et al., 2010). PF-LTD is PKC- and αCaMKII-dependent (Hansel et al., 2001; Ito, 2001; Hansel et al., 2006). Thus, Ca2+ may bidirectionally control the AMPAR trafficking in LTP and LTD through the kinase/phosphatase switch because both kinase and phosphatase PP2B are sensitive to Ca2+ (Lisman, 1989). On the other hand, it is shown that NO activates soluble guanylyl cyclase and causes phosphatase inhibition of AMPARs in LTD (Shin and Linden, 2005). Nevertheless, how it modulates AMPARs in 1 Hz LTP remains obscure. Interestingly, NO-based S-nitrosylation of cysteine has been shown on G-proteins, transcription factors, transporters, transmitter receptors (Lei et al., 1992), and ion channels (Stamler et al., 1997; Yao et al., 1997). Moreover, NO-dependent S-nitrosylation of N-ethylmaleimide-sensitive factor promotes GluR2 insertion in cultured cortical neurons (Huang et al., 2005). Thus, whether and how the nitrosylation of N-ethylmaleimide-sensitive factor and AMPAR trafficking are affected by NO or PF stimulation in the cerebellum need to be investigated.
Footnotes
Part of the present findings have been reported as a preliminary abstract: Su LD, Wang DJ, Yang D, Shen Y, Hu YH (2013) Retrograde cPLA2α/arachidonic acid/2-AG signaling is essential for cerebellar depolarization-induced suppression of excitation and long-term potentiation. Cerebellum 12:297–299. Abstract for Proceedings of the Fifth International Symposium of Society of Research on Cerebellum in China. This work was supported by the National Basic Research Program of the Ministry of Science and Technology of China Grants 2009CB941400 and 2011CB504400, the National Natural Science Foundation of China Grants 31070945, 31271148, and 31100780, and the Ministry of Education of China New Century Talent Award NCET-07-0751. We thank Dr. Adam Sapirstein (Johns Hopkins University, Baltimore, MD) for kindly providing cPLA2α knock-out mice, Dr. Iain C. Bruce for reading the manuscript, and members of the Y.S. laboratory for helpful advice.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Ying Shen, 866 Yu Hang Tang Road, Hangzhou, Zhejiang 310058, China. yshen{at}zju.edu.cn





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}