

Abstract
6-Hydroxydopamine, 1-methyl-4-phenyl-pyridinium (MPP+), and rotenone cause the death of dopaminergic neurons in vitro and in vivo and are widely used to model Parkinson's disease. To identify regulated genes in such models, we performed serial analysis of gene expression on neuronal PC12 cells exposed to 6-hydroxydopamine. This revealed a striking increase in transcripts associated with the unfolded protein response. Immunoblotting confirmed phosphorylation of the key endoplasmic reticulum stress kinases IRE1α and PERK (PKR-like ER kinase) and induction of their downstream targets. There was a similar response to MPP+ and rotenone, but not to other apoptotic initiators. As evidence that endoplasmic reticulum stress contributes to neuronal death, sympathetic neurons from PERK null mice in which the capacity to respond to endoplasmic reticulum stress is compromised were more sensitive to 6-hydroxydopamine. Our findings, coupled with evidence from familial forms of Parkinson's disease, raise the possibility of widespread involvement of endoplasmic reticulum stress and the unfolded protein response in the pathophysiology of this disease.
Because the causes of sporadic Parkinson's disease (PD) currently are not understood, much PD research relies on drug models that mimic the selective dopaminergic neuron degeneration that occurs in this disorder. Agents widely used for this purpose include 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenyl-pyridinium (MPP+), and rotenone. These drugs not only selectively destroy dopaminergic neurons but appear to do so by accessing cellular processes relevant to the naturally occurring disease (Ungerstedt et al., 1974; Langston et al., 1983; Betarbet et al., 2000). Thus understanding the mechanisms by which they act is important for uncovering pathophysiological events in PD. All three agents inhibit the mitochondrial electron transport chain, and this, along with the resulting production of reactive oxygen species (ROS), is believed to contribute to neuronal death. However, downstream effectors in this death pathway have not been defined, nor is it clear that mitochondrial events are the sole contributors to cell death by these agents.
One important clue regarding the mechanisms of 6-OHDA and MPP+ is that death induced by these agents appears to require transcription (Itano et al., 1994; Walkinshaw and Waters, 1994; Grunblatt et al., 2000). Therefore, to identify transcriptional responses that underlie the mechanism of PD mimetics, we applied serial analysis of gene expression (SAGE) (Velculescu et al., 1995), an unbiased and quantitative method of gene profiling, to a cellular model of PD. Because various stress-inducing agents activate distinct cell response pathways with “signatory” gene expression profiles, analysis of such genomic responses may provide important clues to a mechanism of action. For instance, oxidative stress, by a mainly undefined pathway, induces the expression of genes including heme oxygenase (Ishii et al., 2000), whereas DNA damage, with nuclear sensors, leads to the activation of effectors such as DNA repair enzymes (Herceg and Wang, 2001). Accumulation of misfolded proteins within the endoplasmic reticulum (ER) induces a highly specific “unfolded protein response” (UPR) (Kaufman, 1999). The key sensors in this case are PERK (PKR-like ER kinase) and IRE1α, ER kinases that respond specifically to a stress signal generated in the lumen of the ER (Bertolotti et al., 2000). When activated, the signal transduction pathways initiated by PERK and IRE1α induce a characteristic set of genes encoding ER chaperones and nuclear transcription factors that ultimately lead either to reduction of ER stress or to death (Mori, 2000).
Here we report on the unexpected finding that the PD mimetics 6-OHDA, MPP+, and rotenone specifically induce ER stress and activate the UPR in cultured neuronal cells. Together with recent genetic evidence implicating ER stress in rare forms of familial PD (Shimura et al., 2000; Imai et al., 2001), our study points to an important role for the ER in the pathophysiology of this common disease.
MATERIALS AND METHODS
Cell culture. PC12 cells were cultured as described previously (Greene et al., 1998) in RPMI 1640 medium (Cellgro, Herndon, VA) supplemented with 1% horse serum, penicillin/streptomycin, and 50 ng/ml recombinant human nerve growth factor (NGF; a kind gift from Genentech, South San Francisco, CA) for 8–10 d. Medium was changed approximately every other day and immediately before treatments. 6-OHDA, rotenone, and MPP+ were prepared as 10 mm stocks immediately before use and diluted in medium to the indicated final concentrations. NGF withdrawal of PC12 cells was performed as described previously (Rukenstein et al., 1991).
Cultures of dissociated superior cervical ganglia were prepared and maintained as described previously (Lee et al., 1980; Troy et al., 2000) from postnatal day 1 (P1) or P2 littermates generated by matingPerk+/− mice (Harding et al., 2001). Ganglia dissected from each mouse pup were kept separate in PBS at 4°C until genotype was determined by PCR as described previously (Harding et al., 2001). Perk+/+,+/−ganglia then were pooled as were ganglia fromPerk−/− animals, and the two pools were used to establish cultures. Dissociated ganglia were plated at approximately one per well in 24-well plates coated with rat tail collagen in RPMI 1640 medium supplemented with 10% horse serum, penicillin/streptomycin, and 50 ng/ml recombinant human NGF. After 24 hr, uridine and 5-fluorodeoxyuridine (10 μmfinal concentration) were added to the cultures to kill non-neuronal cells. On day 3 the medium was changed, and numbers of phase-bright live cells in each well were determined by strip counting as described previously (Rydel and Greene, 1988). 6-OHDA then was added to the cultures, and cell counts were performed after 40 hr of treatment. For NGF withdrawal, on day 4 the cells were washed twice with NGF-free culture medium and then fed with complete medium containing 1:100 monoclonal anti-human NGF antibody (Sigma, St. Louis, MO). Viability of cells was scored before and after 32 hr of antibody treatment. In all cases the cell survival was determined by comparing counts on the same cultures before and after experimental treatment. Triplicate cultures were used for each condition.
Survival assay. Neuronal PC12 cells were cultured in 24-well dishes for 7–9 d and then treated with 10, 50, 100, or 200 μm 6-OHDA. After 24 hr the cells were lysed, and the number of intact nuclei was counted with a hemacytometer as described previously (Rukenstein et al., 1991). Cell survival was expressed as a percentage of the number of living cells in the treated cultures compared with the controls. Triplicate cultures were used for each condition, and the average was reported as the means ± SEM.
SAGE library construction and analysis. The 6-OHDA SAGE library was constructed from NGF-treated PC12 cells exposed to 100 μm 6-OHDA for 8 hr via SAGE protocol version 1.0e and as described previously (Velculescu et al., 1995; Angelastro et al., 2000). SAGE tag data were analyzed with the SAGE 300 software package (Velculescu et al., 1995), and tags were matched to their corresponding gene as described previously (Angelastro et al., 2000). Comparison was made with a previously constructed and analyzed library (Angelastro et al., 2000) from PC12 cells grown under identical conditions, but without 6-OHDA treatment.
Northern blot analysis. Total RNA (20 μg per sample) was analyzed by Northern blotting as described previously (Loeb and Greene, 1993). The CHOP probe was made from a partial mouse CHOP cDNA inserted into pBS (Stratagene, La Jolla, CA) (Wang et al., 1996). The ATF4 (activating transcription factor 4) probe was created by using a PCR product of the open reading frame (ORF) of murine ATF4 (generously provided by Dr. Eric Kandel, Columbia University, New York, NY) ligated into pCR 2.1-TOPO vector (Invitrogen, Carlsbad, CA). The 18S ribosomal RNA DECA probe template was purchased from Ambion (Austin, TX). Other probes were created by PCR amplification, using as the template reverse-transcribed total RNA from NGF-treated PC12 cells treated for 8 hr with 100 μm 6-OHDA and using the following primers: BiP forward primer, 5′-GACCATGGAGAAAGCTGTAGAGGAA-3′; BiP reverse primer, 5′-CCAAGACACGTGAGCAACTGCTA-3′; PDI forward primer, 5′-GAATCTTTCTGAAGCCACAC-3′; PDI reverse primer, 5′-CATACGACCCAGAACCATC-3′; calreticulin forward primer, 5′-CCACCAGTGATTCAAAATCC-3′; calreticulin reverse primer, 5′-TCCTT- CTCATCCTCTTCATC-3′; Hsp70 forward primer, 5′-GGCTGA- GAAAGAGGAGTTC-3′; Hsp70 reverse primer, 5′-TTCGCAGGAA- GGAAACAC-3′, Hsp27 forward primer, 5′-TTCCCGATGAGTGGTCTC-3′; Hsp27 reverse primer, 5′-GTGACCGGAATGGTGATC-3′; α-tubulin forward primer, 5′-ATGAGGCCATCTATGACATC-3′; α-tubulin reverse primer, 5′-TCCACAAACTGGATGGTAC-3′. Probes were ligated into pCR 2.1-TOPO vector (Invitrogen). All probes were excised from vector withEcoRI. The blots were exposed with the use of a Storm PhosphorImager (Amersham Biosciences, Sunnyvale, CA).
Reverse transcription-PCR. Each sample of total RNA was isolated by using TRI reagent (Molecular Research Center, Cincinnati, OH) from three wells of 24-well plates of wild-type murine sympathetic neurons subjected to either 3 or 5 μm 6-OHDA, NGF withdrawal, or no treatment for 8 hr. cDNA was transcribed from total RNA with Superscript RT II (Invitrogen). The primers used for PCR amplification of BiP and α-tubulin are listed above. PCR conditions used were 95°C (for 30 sec), 50°C (for 1 min), and 72°C (for 45 sec) for 35 cycles in PCR buffer containing a final concentration of (in mm): 16.6 ammonium sulfate, 67 Tris, pH 8.8, 6.7 magnesium chloride, 0.6 dNTP, and 10 β-mercaptoethanol plus 6% DMSO, 0.1 μm forward and reverse primer, and 2.5 U platinum Taq DNA polymerase (Invitrogen). The amount of cDNA template used for each PCR sample was adjusted so that levels of α-tubulin were equal among all samples. PCR products were resolved on an agarose gel and photographed.
Immunoprecipitation and Western blot analyses. Whole-cell extracts were prepared as described previously (Loeb and Greene, 1993). For preparation of cytoplasmic and nuclear extracts the cells were washed twice with ice-cold PBS containing 1 mm EDTA and scraped into 1 ml of the buffer. Cells were pelleted at 2000 rpm for 5 min at 4°C, and cytoplasmic lysate was extracted in four cell volumes of Triton X-100 buffer (Harding et al., 2000a). PERK, IRE1α, and eukaryotic initiation factor 2α (eIF2α) protein detection in cytoplasmic lysates was performed by immunoprecipitation/Western blotting (Harding et al., 2000a). The primary antibodies that were used were rabbit anti-PERK (Harding et al., 1999), rabbit anti-IRE1α (Urano et al., 2000), rabbit anti-phospho eIF2α (DeGracia et al., 1997), and monoclonal mouse eIF2α (Scorsone et al., 1987). Nuclei were pelleted at 2000 rpm for 5 min at 4°C and lysed in Laemmli buffer (Loeb and Greene, 1993). For BiP, ATF4, CHOP, and ERK-1 protein detection, either 50 μg of whole-cell extract (BiP) or 40–50 μg of nuclear extract (ATF4, CHOP) was separated on Novex precast 4–20% Tris-glycine gradient gels (Invitrogen). Protein was transferred to nitrocellulose and probed with 1:2000 anti-BiP (StressGen, San Diego, CA), 1:5000 anti-ATF4 (kind gift from Dr. Eric Kandel, Columbia University, New York, NY) (see Figs. 2B,3A), 1:1000 CREB-2 (ATF4) rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) (see Fig. 3B–D), 1:1500 Gadd153 (CHOP) mouse monoclonal antibody (Santa Cruz Biotechnology), or 1:10,000 ERK-1 rabbit polyclonal antibody (Santa Cruz Biotechnology) and then with 1:10,000 goat anti-rabbit horseradish peroxidase (HRP) or 1:5000 goat anti-mouse HRP (Pierce, Rockford, IL) secondary antisera.
RESULTS
Serial analysis of gene expression of 6-OHDA-treated neuronal PC12 cells reveals induction of ER stress genes
To identify genes that mediate neuronal responses in a model of Parkinson's disease, we used SAGE to compare transcripts expressed by neuronally differentiated PC12 cells with or without 8 hr of exposure to 100 μm 6-OHDA. PC12 cells (Greene and Tischler, 1976) were chosen because they are induced by NGF to acquire a neuronal phenotype that resembles sympathetic neurons [that, like CNS substantia nigral neurons, undergo degenerative changes in PD (Wakabayashi and Takahashi, 1997)], are dopaminergic, and undergo apoptotic death in response to 6-OHDA (Walkinshaw and Waters, 1994). The latter agent causes selective damage to substantia nigral neuronsin vivo and produces a Parkinson's-like syndrome (Ungerstedt et al., 1974). In our experiments ∼50% of the 6-OHDA-treated cells died by 24 hr (Fig.1A). Little death or degeneration was apparent at 8 hr, and we reasoned that this time point was sufficient for transcriptional changes to occur before the onset of secondary responses caused by cell deterioration. Approximately 36,000 15 bp SAGE tags from 6-OHDA-treated cells were sequenced (excluding duplicate ditags, tags shorter than 15 bp, repetitive elements, and linker contaminants), representing 14,266 unique transcripts. These data were compared with those from a previously described SAGE library (∼87,000 tags, ∼22,000 unique transcripts) from control NGF-treated cells (Angelastro et al., 2000). With normalization between the two libraries, 296 transcripts were found to be elevated by 12-fold or more in response to 6-OHDA; a total of 1200 transcripts (8%) increased by sixfold or greater.

Dose–response for survival and induction of CHOP in neuronal PC12 cell cultures exposed to various concentrations of 6-OHDA. A, Quantitation of PC12 cell survival at 24 hr after exposure to the indicated concentrations of 6-OHDA. Data are presented as the percentages of surviving cells in treated cultures compared with control cultures and are expressed as the means ± SEM (n = 3 replicate cultures). Similar results were achieved in an independent experiment. B, Western immunoblot of CHOP induction in neuronal PC12 cells after exposure to the indicated concentrations of 6-OHDA for 16 hr. Nuclear extracts were prepared and analyzed by immunoblotting with an anti-CHOP antibody. The blot was stripped and reprobed with anti-ERK1 to indicate equality of loading. Similar results were achieved in an independent experiment.
Identification of transcripts corresponding to the regulated SAGE tags revealed both anticipated and unanticipated changes. For instance, there was marked elevation of transcripts encoding heme oxygenase-1 (84-fold), Zif268/NGFI-A (41-fold), and metallothionein I (36-fold), each of which has been reported to increase either in PD and/or PD models (Rojas et al., 1996; Smith et al., 1997; Schipper et al., 1998). Among unforeseen changes was a striking increase in the expression of transcripts associated with the cellular response to ER stress (Table1). To confirm and extend our SAGE findings, we performed Northern blots for a number of the regulated ER stress-associated genes. This showed a time-dependent regulation of the seven genes that were tested, whereas there was no change in the expression of genes such as α-tubulin that normally are unaffected by ER stress (Fig.2A). Western blots confirmed the changes at the level of protein expression for the ER stress-associated genes BiP (Grp78), ATF4, and CHOP (Gadd153) (Figs.1B, 2B). The degree of induction of CHOP protein at 16 hr closely reflected the degree of death observed at 24 hr (Fig. 1B). This is consistent with the implication of CHOP protein induction in cell death elicited by agents that promote ER stress (Zinszner et al., 1998).
ER stress- and protein folding-related genes identified from SAGE analysis of 6-OHDA-treated neuronal PC12 cells
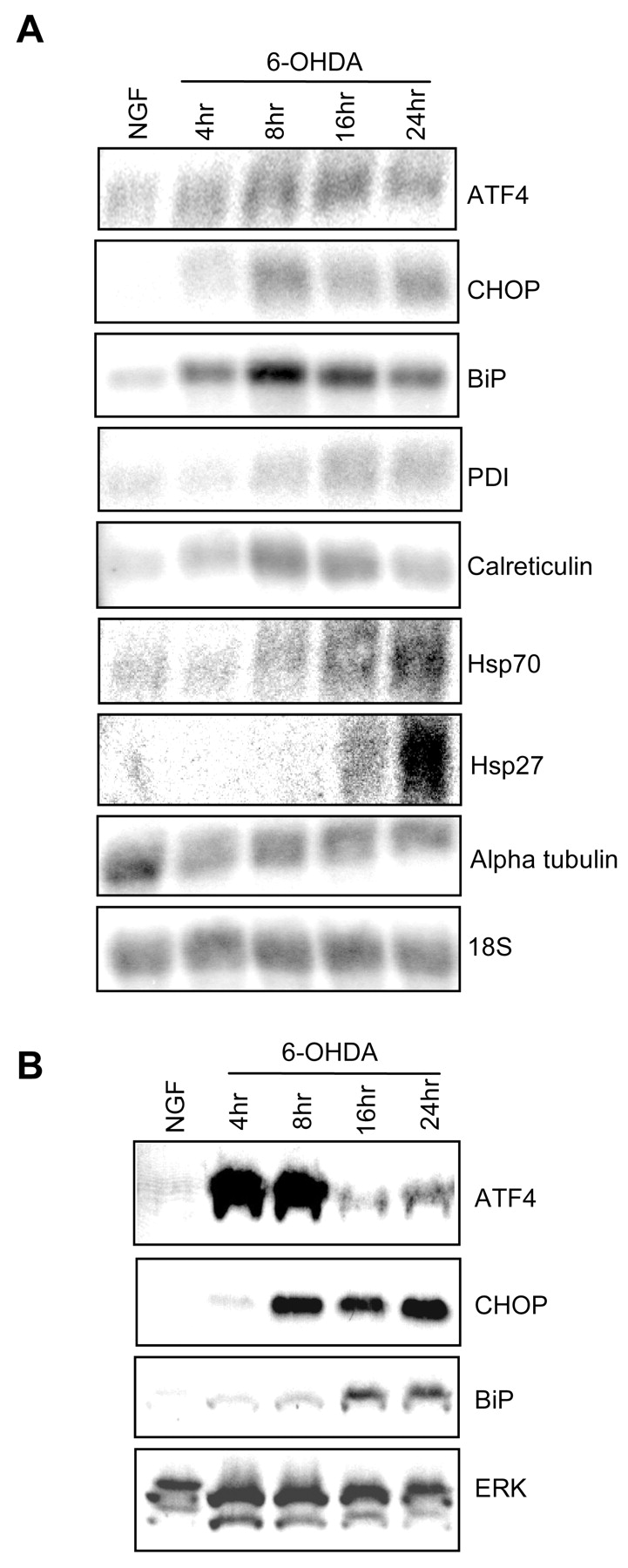
Confirmation of SAGE data by Northern and Western blotting. A, Total RNA from neuronal PC12 cells treated without or with 100 μm 6-OHDA in NGF-containing medium was subjected to Northern blot analysis with the indicated radiolabeled probes. Blots were stripped and reprobed for 18S RNA to assess equality of loading. The 18S blot that is pictured was preprobed for CHOP and calreticulin and is representative of the results obtained with the other blots in the figure. Similar results were achieved in an additional one to three independent experiments. B, Whole-cell (ATF4, BiP) or nuclear (CHOP) extracts from neuronal PC12 cells treated with or without 100 μm 6-OHDA in NGF-containing medium were subjected to Western blotting and probed with antibodies against the indicated proteins. Blots were stripped and reprobed with ERK-1 antiserum to assess equality of loading. The ERK blot that is shown also was probed for BiP and is representative of the results obtained with the other blots in the figure. Similar results were achieved in an additional one to three independent experiments.
ER stress and the UPR are induced by 6-OHDA exposure
A major cause of ER stress is the accumulation of misfolded proteins. This triggers the UPR, leading to induction of ER-associated proteins that aid in protein refolding, the attenuation of protein translation, and activation of protein degradation (Mori, 2000). Such changes can result in rescue from distress or, if the damage is excessive, to cell death (Ferri and Kroemer, 2001). The induction of transcripts like those listed in Table 1 is consistent with the presence of ER stress and the UPR in 6-OHDA-treated cells.
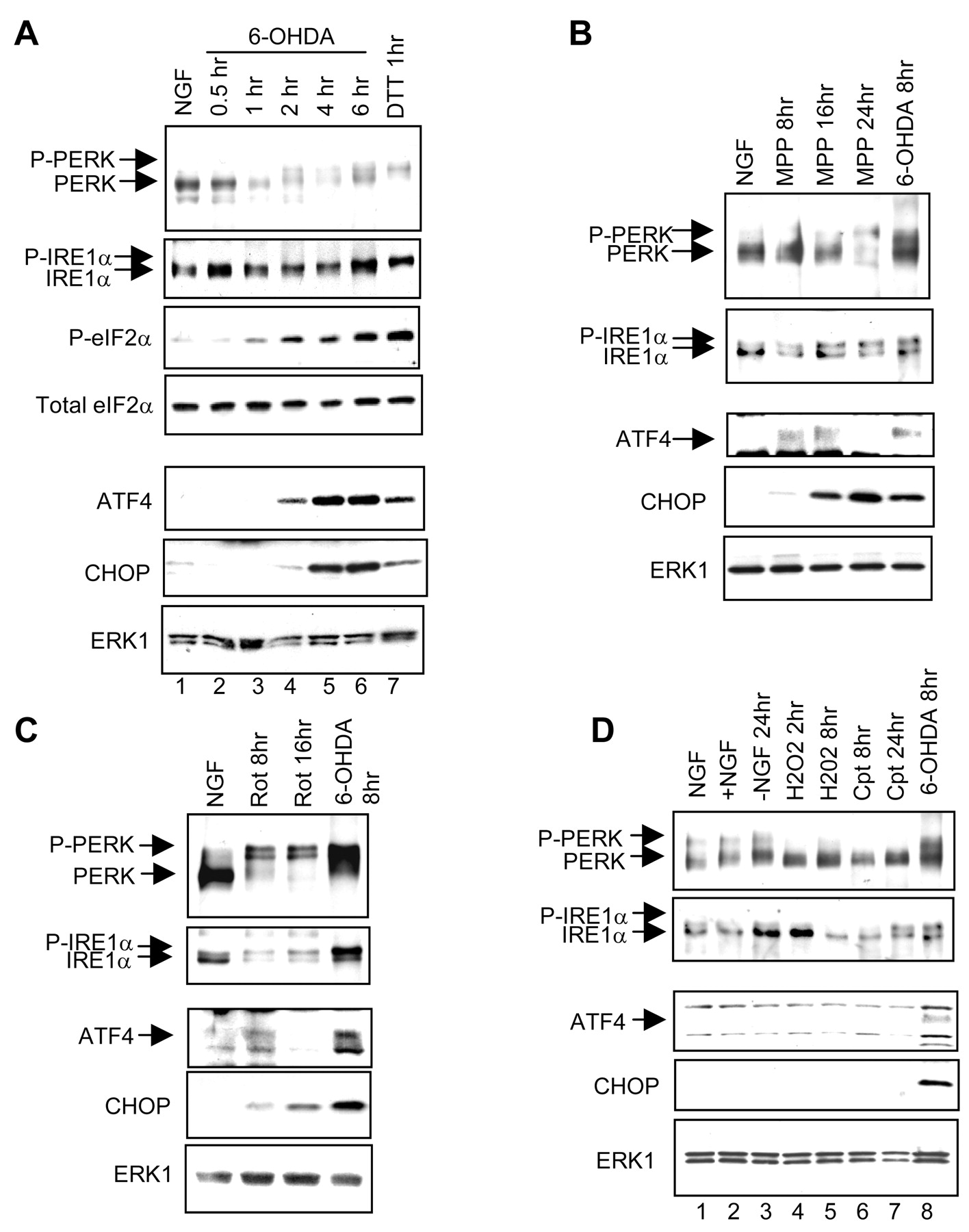
To extend these observations, we examined the activation of ER stress-signaling proteins in 6-OHDA-treated cells. One well characterized UPR pathway involves IRE1 proteins; these ER localized transmembrane protein kinases/endonucleases sense the ER environment through their lumenal domain and are autophosphorylated at multiple sites and activated in response to ER stress (Tirasophon et al., 1998;Wang et al., 1998). The phosphorylation and activation of IRE1 proteins can be detected as a mobility shift on SDS-PAGE. Exposure of cells to 6-OHDA caused a time-dependent shift in the electrophoretic mobility of IRE1α, the IRE1 isoform expressed in PC12 cells (Fig.3A). A similar shift was observed in extracts from cells exposed to dithiothreitol (DTT), a known initiator of the UPR (Bertolotti et al., 2000) (Fig.3A).

Activation of ER stress response proteins by 6-OHDA, MPP+, and rotenone, but not by other inducers of apoptosis. A, Immunoblot analysis of ER stress proteins in neuronal PC12 cells after various times of 6-OHDA treatment. Neuronal PC12 cells were treated without or with 100 μm 6-OHDA in NGF-containing medium or with 2 μm DTT and were used to prepare cytoplasmic (PERK, IRE1α, and eIF2α) or nuclear (ATF4 and CHOP) extracts. PERK and IRE1α were immunoprecipitated and analyzed by Western immunoblotting with antisera that recognize both the phospho and non-phospho forms of these proteins; other proteins were analyzed by Western immunoblotting of extracts. The eIF2α blot was probed with an antiserum specific for phospho-eIF2α and then with an antibody that recognizes total eIF2α. The CHOP blot was reprobed with anti-ERK1 to indicate equality of loading; results were similar with the ATF4 blot (data not shown). Similar results were achieved in an additional one to three independent experiments. MPP+ (B; 200 μm) and rotenone (C; 1 μm) activate ER stress proteins. Experimental details are as inA. D, NGF withdrawal (−NGF), H2O2 (300 μm), and campthothecin (Cpt; 10 μm), under conditions that promote the death of neuronal PC12 cells, show little or no activation of ER stress proteins. Experimental details are as inA.
A second transducer of the UPR is the ER transmembrane kinase PERK that also undergoes autophosphorylation and activation in response to ER stress (Harding et al., 1999). PERK, in turn, phosphorylates eIF2α, thereby attenuating translation of most mRNAs while selectively increasing translation of the mRNA encoding the transcription factor ATF4 (Harding et al., 2000b). This activates the downstream target gene CHOP and also leads to transcriptional activation of ATF4 itself (Fawcett et al., 1999; Harding et al., 2000b). 6-OHDA treatment results in the time-dependent appearance of phospho-PERK (Fig. 3A). The apparent decrease in PERK staining after 1 hr of 6-OHDA treatment may be attributable to the suboptimal recognition of the PERK antibody to all phospho forms of this protein. 6-OHDA treatment also resulted in the appearance of phospho-eIF2α with a time course similar to that for the generation of phospho-PERK (Fig. 3A). In contrast, induction of ATF4 and CHOP proteins was delayed somewhat compared with the onset of PERK, IRE1α, and eIF2α phosphorylation; this is consistent with the known placement of these proteins downstream of PERK and IRE1α in the UPR. Furthermore, ATF4 induction was consistently earlier than CHOP induction, which is in agreement with the known transcriptional regulation of CHOP by ATF4 (Fawcett et al., 1999; Harding et al., 2000b). Additional evidence for activation of the ER stress response is the induction of the ER chaperone protein BiP, which is a transcriptional target of both IRE1α and PERK (Wang et al., 1998; Harding et al., 2000b). 6-OHDA induced BiP at both the RNA and protein levels (Table 1, Fig. 2A,B). Together, our observations indicate that 6-OHDA rapidly triggers ER stress and the UPR in neuronal cells.
MPP+ and rotenone also induce ER stress and the UPR in contrast to other initiators of apoptosis
We next investigated whether the ER stress response observed with 6-OHDA also occurs with additional drug-induced models for PD. MPP+ and rotenone are mitochondrial inhibitors that cause selective degeneration of nigrostriatal neuronsin vivo (Langston et al., 1983; Betarbet et al., 2000) and death of dopaminergic neurons and neuronal PC12 cells in vitro (Seaton et al., 1997). Exposure of neuronal PC12 cells to MPP+ or rotenone (under conditions that cause the death of ∼50% of the cells by 48 hr for MPP+ and 24 hr for rotenone) leads to PERK and IRE1α phosphorylation as well as to subsequent ATF4 and CHOP protein induction (Fig. 3B,C).
6-OHDA as an oxidant has the capacity to produce ROS (Gee and Davison, 1989). 6-OHDA, MPP+, and rotenone also can promote the generation of ROS via the inhibition of mitochondrial complex I. To determine whether the ER stress response we observed with these agents was attributable to ROS alone, we monitored the response of neuronal PC12 cells to the oxidant H2O2. H2O2 has been reported previously to promote the production of ROS and apoptotic death in PC12 cell cultures at concentrations similar to those used in our experiments (Jiang et al., 2001). A dose–response study established that 300 μmH2O2 produced ∼50% death of neuronal PC12 cells after 16 hr under our conditions. We did not detect phosphorylation of PERK or IRE1α or induction of ATF4 or CHOP protein after 2 or 8 hr of H2O2 treatment (Fig.3D). These findings indicate that a variety of agents promoting selective degeneration of dopaminergic neurons triggers an ER stress response, whereas a nonselective oxidant does not.
We next determined whether the ER stress response might be evoked by additional apoptotic stimuli. The DNA-damaging agent camptothecin and the withdrawal of NGF each elicits apoptotic death of neuronal PC12 cells (50% death at ∼72 hr for camptothecin and 24 hr for NGF withdrawal) (Rukenstein et al., 1991; Park et al., 1997). PERK and IRE1α underwent little if any detectably consistent change in phosphorylation with either treatment, except for a weak elevation of IRE1α phosphorylation after 24 hr of camptothecin exposure (Fig.3D). In addition, the two most downstream components of the UPR examined, ATF4 and CHOP, showed no detectable induction in response to camptothecin or NGF withdrawal (Fig. 3D). Thus the UPR is not a general response of neuronal PC12 cells to apoptotic stimuli.
Impairment of the UPR increases sensitivity to 6-OHDA
Cells lacking expression of PERK fail to phosphorylate eIF2α and to attenuate protein translation in response to ER stress (Harding et al., 2000a, 2001). As a consequence, these cells are unable to reduce the load of ER client proteins and are significantly more sensitive to the death-promoting effects of ER stress-inducing agents. Treatment ofPerk null cells with arsenite, an agent that inhibits protein translation via the phosphorylation of eIF2α but does not induce ER stress, had no effect on viability, indicating the specificity of the PERK mutation on the ER stress pathway (Harding et al., 2000a). We therefore reasoned that if ER stress is an important component of the action of 6-OHDA, then suitable neurons fromPerk−/− animals should be more sensitive to this agent. Sympathetic neurons were harvested from the superior cervical ganglia of newborn littermate offspring ofPerk+/− mice and were used to establish dissociated cell cultures containing eitherPerk−/− neurons or a mixture ofPerk+/+,+/− neurons (control). A past study established that Perk+/−cells behave like wild-type cells with respect to sensitivity to agents that cause ER stress (Harding et al., 2000a). Sympathetic neurons were chosen because of their accessibility in suitable numbers, susceptibility to 6-OHDA, and involvement in PD. Moreover, as shown in Figure 4, 8 hr of exposure to 3 and 5 μm 6-OHDA induces BiP mRNA in sympathetic neurons, indicating the presence of ER stress. In contrast, there is no clear increase of BiP mRNA seen at 8 hr of NGF withdrawal.

Induction of BiP in sympathetic neurons exposed to 6-OHDA. Cultured sympathetic neurons from newborn mice were exposed to 3 or 5 μm 6-OHDA or subjected to NGF withdrawal, all for 8 hr. BiP mRNA expression was analyzed by semiquantitative RT-PCR (35 cycles) and agarose gel electrophoresis. The samples were analyzed for the expression of α-tubulin (35 cycles) to ensure the use of an equal amount of template in each case. There was no signal when water was substituted for template. Comparable results were obtained in an independent experiment.
Neurons from Perk−/− animals cultured with NGF for 3 d appeared morphologically indistinguishable from those in control cultures and produced an extensive network of neurites (Fig.5A,C). However, when exposed to 2–4 μm 6-OHDA, thePerk−/− neurons exhibited substantially more neurite degeneration, somal shrinkage, and cell death than control neurons (Fig. 5B,D). Counts of neurons before and after 6-OHDA treatment substantiated the difference in survival. Although there was little or no significant loss of control neurons in response to 2 μm 6-OHDA, a significant proportion of the 6-OHDA-treated PERK null neurons was killed during this time (Fig. 5E). The use of a higher dose of 6-OHDA (4 μm) resulted in ∼50% survival of control neurons compared with 10% survival of PERK null neurons after 24 hr (data not shown) (Fig. 5A–D). In contrast, both control and PERK null neurons showed a similar level of death after 32 hr of NGF deprivation (Fig. 5E). These results indicate that neurons with a defective UPR are more sensitive to the death-promoting actions of 6-OHDA. Thus not only does 6-OHDA provoke ER stress, but neurons lacking the capacity to deal with this by mounting an appropriate UPR are at greater risk of death.

Perk−/−sympathetic neurons exhibit increased sensitivity to 6-OHDA treatment.A–D, Photomicrographs of sympathetic neurons cultured from newborn Perk+/+,+/− (A, B) and Perk−/− (C, D) mice without (A, C) or with (B, D) 8 hr of exposure to 4 μm 6-OHDA. Scale bar, 30 μm. E, Quantitation of effects of 6-OHDA (2 μm for 40 hr; black bars) and NGF deprivation (32 hr; gray bars) on survival of sympathetic neurons cultured from newbornPerk+/+,+/− andPerk−/− mice. Strip counts of phase-bright live cells were performed on each culture just before and after the various treatments. Values are reported as the percentage of neurons present at the end of treatment compared with the numbers present before treatment and are represented as the means ± SEM (n = 3 cultures; p = 0.02 null control vs null 6-OHDA). Comparable results were achieved in an independent experiment.
DISCUSSION
In the present study we set out to identify genes regulated in a cellular model of PD. By using SAGE, an unbiased, quantitative screen for analysis of global gene expression patterns (Velculescu et al., 1995), we were able to implicate the hitherto unsuspected process of ER stress and the UPR in the pathogenesis of 6-OHDA-induced neuronal cell death. Analysis of the SAGE library revealed the induction of many genes involved in various aspects of the UPR including ER chaperone proteins such as BiP, calreticulin, and PDI (Protein disulfide isomerase); components of the PERK-dependent protein translation attenuation pathway (ATF4 and CHOP); and components of the ubiquitin/proteasome degradation machinery (see Table 1). As indicated by the induction of BiP, 6-OHDA evoked ER stress in sympathetic neurons as well as in the PC12 cells used for SAGE evaluation.
These findings are strongly supported by the observed phosphorylation of the key ER stress transmembrane kinases, PERK and IRE1α. Because PERK and IRE1α are highly homologous in their lumenal, but not cytosolic, domains, this suggests that their activation is attributable to perturbation of the ER environment rather than from direct cytosolic stimulation. With accumulation of misfolded proteins in the ER, BiP disassociates from the lumenal domains of both PERK and IRE1α, allowing them to autophosphorylate in trans (Bertolotti et al., 2000). Thus, the early PERK and IRE1α phosphorylation seen by 1 hr of 6-OHDA exposure further indicates that the UPR is triggered by a specific lumenal ER stress response rather than by a late cell degeneration event. In contrast, ER stress was not detected during death elicited by H2O2, NGF withdrawal, or DNA damage. These observations indicate that induction of the UPR in neuronal cells is specific to the PD models and is not attributable to a general cell stress response.
Our findings revealed induction of the transcription factors ATF4 and CHOP. Although induction of ATF4 and CHOP by ER stress is dependent on PERK, these transcription factors also can be activated by other forms of cellular stress that promote eIF2α phosphorylation (Harding et al., 2000b). For instance, ATF4 and CHOP are induced by arsenite as well as other initiators of oxidative stress (Guyton et al., 1996;Fawcett et al., 1999; Harding et al., 2000b). Therefore, although elevation of CHOP and ATF4 in our models is attributable at least in part to PERK and eIF2α activation, it is possible that there is a contribution to their induction independent of the UPR.
We noted a clear correlation between the degree of death evoked by 6-OHDA and the induction of CHOP protein. CHOP induction has been observed in a variety of models of cell stress and apoptotic death, and in some instances its overexpression can elicit apoptosis (Maytin et al., 2001). In addition, overexpression of CHOP sensitizes cells to ER stress, so their long-term survival after exposure to ER-stress-inducing agents is significantly lower (McCullough et al., 2001). Interestingly, CHOP induction has been linked to cellular decreases in glutathione, increased production of ROS, and decreases in the anti-apoptotic protein Bcl-2, all cellular phenomena associated with cell death and PD (Sian et al., 1994; Mogi et al., 1996; Yoritaka et al., 1996; McCullough et al., 2001). Thus regardless of its mechanism of induction, CHOP may promote cell death in these models of PD.
An ER stress response also was evoked by MPP+ and rotenone, indicating a mechanistic commonality of these agents with 6-OHDA beyond their effects on mitochondria. The oxidative stress caused by the effects of these agents on mitochondrial respiration may be responsible for inducing ER stress. One possible mechanism for this is that the accumulation of damaged oxidized proteins interferes with the cellular protein degradation machinery, thereby causing the ER to retain unfolded proteins (Friedlander et al., 2000). In addition, ER stress may contribute further to the inhibition of mitochondrial respiration. A recent report demonstrated that ER stress can produce mitochondrial dysfunction by affecting various components of cytochrome coxidase (Hori et al., 2002).
Our findings indicate not only that ER stress occurs in response to treatment with 6-OHDA, MPP+, and rotenone but also that such stress is likely to play a causative role in neuronal cell death promoted by these PD-mimicking agents. As evidence of this, sympathetic neurons with an impaired ability to respond to ER stress by mounting a full UPR (caused by a loss of PERK expression) were significantly more sensitive to the death-promoting actions of 6-OHDA.
Although we have shown evidence for the involvement of ER stress only in models of PD, there are reasons to suspect ER stress also may play a role in the naturally occurring disease. A juvenile onset autosomal recessive form of PD (AR-JP) is caused by mutation of theParkin gene, which compromises the ubiquitin ligase function of the protein (Shimura et al., 2000). This loss of activity results in overaccumulation of a substrate of Parkin in the ER of neurons, leading to ER stress and, consequently, to cell death (Imai et al., 2001).
A second form of familial PD is attributable to mutation of the gene encoding α-synuclein (Polymeropoulos et al., 1997). Overexpression of mutant forms of α-synuclein in cultured neuronal cells leads to formation of cytoplasmic aggregates, disruption of the ubiquitin-dependent proteasomal degradation system, and cell degeneration (Stefanis et al., 2001; Tanaka et al., 2001). On the basis of these observations and others, it has been suggested that proteasomal dysfunction may play a role in the pathophysiology of PD (McNaught et al., 2001). In this context it is of interest that an important means of removing misfolded proteins from the ER is their translocation to the cytoplasm and degradation by proteasomes. Proteasomal dysfunction therefore may lead to ER stress and play a role in neurodegeneration (Bence et al., 2001; Nishtoh et al., 2002). Environmental toxins, oxidative damage by dopamine itself, and mitochondrial abnormalities are all believed to play a role in sporadic PD (Mouradian, 2002). These affect protein folding in the cytoplasm and may lead to ER stress by impacting the process of ER-associated protein degradation (Bence et al., 2001; Nishtoh et al., 2002). In such instances, neuronal degeneration might involve either excessive ER stress or a failure to mount an effective UPR. Our findings lend credence to such scenarios.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS160636, NS33689, and P50 NS38370; by grants from the Parkinson's Disease Foundation and the Blanchette Rockefeller Foundation to L.A.G; and by NIH Grants NS 436281 and ES08681 to D.R. D.R. is an Ellison Medical Foundation Senior Scholar in Aging. We thank Dr. Michael L. Shelanski for seminal discussions and helpful advice. We also thank Claudine Bitel for her excellent technical assistance.
Correspondence should be addressed to Lloyd A. Greene, Department of Pathology and Center for Neurobiology and Behavior, Columbia University College of Physicians and Surgeons, 630 West 168th Street, New York, NY 10032. E-mail: lag3{at}columbia.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}