

Abstract
Activity-dependent specification of neuronal architecture during early postnatal life is essential for refining the precision of communication between neurons. In the spinal cord under normal circumstances, the AMPA receptor subunit GluR1 is expressed at high levels by motor neurons and surrounding interneurons during this critical developmental period, although the role it plays in circuit formation and locomotor behavior is unknown. Here, we show that GluR1 promotes dendrite growth in a non-cell-autonomous manner in vitro and in vivo. The mal-development of motor neuron dendrites is associated with changes in the pattern of interneuronal connectivity within the segmental spinal cord and defects in strength and endurance. Transgenic expression of GluR1 in adult motor neurons leads to dendrite remodeling and supernormal locomotor function. GluR1 expression by neurons within the segmental spinal cord plays an essential role in formation of the neural network that underlies normal motor behavior.
Introduction
Although neurons have an innate predisposition to elaborate dendrites, external cues play a decisive role in promoting growth and branching (Whitford et al., 2002; Wong and Ghosh, 2002; Parrish et al., 2007). Components of the local environment such as cell adhesion molecules as well as soluble and substrate bound factors are likely to be the key extracellular determinants of dendrite growth (McAllister et al., 1995; Kim and Chiba, 2004; Xiong et al., 2007). As dendrites elongate, they grow into neuropil containing afferents that will establish specific synaptic contacts and these inputs can further control dendritic elaboration in an activity-dependent manner (Maletic-Savatic et al., 1999; Jontes et al., 2000; Lohmann et al., 2002; Niell et al., 2004). Precisely how synaptic activity is translated into signals for dendrite patterning remains incompletely understood. Insight into this issue may hold promise for promoting nervous system plasticity in adults and thus be relevant to neurorestorative therapeutics.
Because glutamate is the major excitatory neurotransmitter in the vertebrate CNS, glutamate receptors are well positioned to be arbiters of activity-dependent dendrite growth. The family of proteins that assemble into glutamate receptors is widely expressed, molecularly diverse, and developmentally regulated (Hollmann and Heinemann, 1994). The GluR1 subunit (also referred to as GluR-A) is of particular interest because of the central role in plays in use-dependent synaptic plasticity (Carroll et al., 2001; Shi et al., 2001). Insertion of GluR1-containing AMPA receptors into synapses may lead to the stabilization of nascent, otherwise transient synaptic contacts that occur as axons and dendrites first encounter each other (Song and Huganir, 2002; Bredt and Nicoll, 2003; Takahashi et al., 2003). Thus, GluR1-containing AMPA receptors may play a leading role in translating synaptic activity into patterned dendrite growth.
During early postnatal life, GluR1 is expressed at a very high levels by motor neurons and diminishes to low levels (or is lost completely) over the first month of life (Jakowec et al., 1995a,b). During this period, motor neurons elaborate a vast dendritic tree that ordinarily does not, thereafter, undergo large-scale changes in architecture (Westerga and Gramsbergen, 1992; Curfs et al., 1993; Kalb, 1994). The notion that these two events are linked comes from the observation that expressing GluR1 in mature motor neurons (using viral vectors) leads to remodeling of the dendritic tree, with efflorescence of branching without changing the overall size of the tree (Inglis et al., 2002). These observations raise several questions. First, although these studies demonstrate that GluR1 expression is sufficient for dendrite remodeling, whether it is necessary for dendrite growth under normal developmental conditions is not known. Second, only individual neurons could be transduced using the viral vectors; thus, the functional and behavioral consequences of GluR1 expression in motor neurons could not be studied. Third, during the neonatal period of robust motor neuron dendrite growth in vivo, high levels of GluR1 are expressed both by motor neurons and surrounding interneurons (Jakowec et al., 1995a,b), so the degree to which dendrite growth occurs in a cell-autonomous manner is not clear. In the present study, we used in vitro and in vivo model systems to explore these issues.
Materials and Methods
Mixed spinal cord neuron cultures
Mixed spinal cord neuron cultures were prepared as described previously (Jeong et al., 2006; Mojsilovic-Petrovic et al., 2006). Briefly, neocortical astrocyte monolayers were established from postnatal day 2 (P2) rat forebrain of Sprague Dawley rats, and when they were ∼80% confluent, dissociated embryonic day 15 (E15) spinal cord cells were added. AraC was added 1–2 d later (for 24 h) to arrest astrocyte proliferation. The neurons and astrocytes were maintained in the glia-conditioned medium supplemented with six trophic factors (all from Alomone Labs at 1.0 ng/ml): human neurotrophin-3, human neurotrophin-4, human brain-derived neurotrophic factor, human cardiotrophin-1, human glial-derived neurotrophic factor, and rat ciliary neurotrophic factor. One-half of the medium was replaced twice per week.
Constructs were transfected into the mixed spinal cord neuron culture using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Transfections occurred at 5 d in vitro (DIV) unless specified otherwise. The various GluR1 constructs were cotransfected with a green fluorescent protein (GFP) plasmid at a proportion of 3:1 to ensure that all GFP-positive cells also expressed GluR1.
siRNA design
Small interfering RNA (siRNA) was used to knock down the expression of GluR1 in the mixed spinal cord neuron culture. The GluR1 siRNA was designed according to previously published guidelines (Elbashir et al., 2001; Davidson et al., 2004). The target sense sequences were as follows: GluR1, 5′-GAA UGA GUA UAU UGA GCA A-3′; control (scramble), 5′-UUC UCC GAA CGU GUC ACG U-3′.
siRNA coupling with penetratin-1
To deliver siRNA to primary cultures of neurons with high efficiency and low toxicity, we conjugated siRNA to the vector peptide penetratin-1 (Davidson et al., 2004). siRNA with a 5′ thiol group on the sense strand was synthesized by Dharmacon, dissolved in annealing buffer, and treated with Tris (2-carboxyethyl) phosphine for 1 h at room temperature. Penetratin-1 (Q-Biogene) was added, and the mixture was heated at 65°C for 15 min and then incubated at 37°C for 1 h. The effectiveness of conjugation was evaluated by running a sample of this reaction on a 20% Tris borate-EDTA gel and staining with SyBr Gold (Invitrogen; catalog #S-11494). Two bands were detected: a faster migrating species representing the unconjugated RNA, and a slower migrating species representing the protein-RNA conjugate (data not shown). No further purification was performed and the siRNA-penetratin complex was stored at −20°C for further use. Before addition to cultures, siRNA-penetratin complex was heated at 65°C for 15 min to increase its solubility.
Synthesis of the pSUPER GluR1 RNAi construct
The pSUPER RNA interference system is used in this experiment. The pSUPER.neo+GFP vector was purchased from Oligoengine. The 60 nt oligo sequence for GluR1 is 5′-GATCCCCGAATGAGTATATTGAGCAATTCAAGAGATTGCTCAATATACTCATTCTTTTTA-3′. The 60 nt oligo sequence for GluR1 control is 5′-GATCCCCTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTA-3′. The oligos were cloned into the pSUPER vector and the final constructs were sequenced before use.
Site-directed mutagenesis
Three silent nucleotide exchanges were introduced in GluR1(Q)flip/pSGEM to alter the sequence AGT ATA TTG AGC AAC to AAT ATA TCG AGC AGC (nucleotides 2507–2521) by overlap-extension PCR-mediated site-directed mutagenesis using mutagenetic primers as described previously (Hollmann et al., 1994). The inserted point mutations prevented recognition by the siRNA, and the resulting construct was called “resistant GluR1” (resGluR1).
Oocyte electrophysiology
Oocytes were isolated from Xenopus laevis frogs and injected with 10 ng of receptor-encoding [GluR1(Q)flip or resGluR1(Q)flip] cRNA, in-vitro-transcribed as reported previously (Hollmann et al., 1994). Recordings were performed 3–5 d after injection using two-electrode voltage clamp (Turbo Tec 10CX amplifier; NPI Electronic). Electrode resistance was 0.5–2.0 MΩ when filled with 3 mm KCl. The extracellular solution contained (in mm) 115 NaCl, 2.5 KCl, 1.8 CaCl2, and 10 HEPES, pH 7.2. Current–voltage relationships (I–Vs) were obtained by applying 2 s voltage ramps from −150 to +30 mV. I–Vs were corrected for leak current. Agonists (300 μm l-glutamate and 150 μm kainate) were applied for 20 s. Naphthyl acetyl spermine (NASP) (10 μm) was applied as an open channel blocker.
Western blot analysis
Cells were washed twice with cold PBS and harvested in lysis buffer containing 50 mm Tris, pH 7.4, 1.0 mm EDTA, 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, and 1% protease inhibitor cocktail (Sigma P2714-1BTL). Cell lysates were sonicated for 10 s and sat on ice for 20 min before clarification at 12,000 × g for 20 min at 4°C. The supernatants were collected, the concentration was determined, and equal amounts of protein were run in SDS-PAGE in 6× sample buffer (300 mm Tris-HCl, pH 6.8, 12% SDS, 0.6% bromophenol blue, 30% glycerol). After transfer to nitrocellulose membrane and blocking in 5% nonfat dried milk for 1 h at room temperature, membranes were incubated with primary antibodies overnight at 4°C: rabbit anti-GluR1, 1:1000 (Upstate Biotechnology); rabbit anti-GluR2/3, 1:1000 (Upstate Biotechnology); rabbit anti-actin, 1:10,000 (Sigma). After washing, the blots were incubated with secondary antibody: ECL anti-rabbit IgG, horseradish-peroxidase-linked whole antibody (from donkey; GE Healthcare NA934V) and proteins were detected with ECL Plus Western Blotting Detection System (GE Healthcare RPN2132).
Subcellular fractionation of spinal cord tissues for analysis of synaptosomal preparations followed established methods (Gurd et al., 1974; Blackstone et al., 1992). Immunoprecipitations were performed as previously described (Kim et al., 2005; Mojsilovic-Petrovic et al., 2006).
Animal care
GluR1 global knock-out mice: GluR1−/−.
The GluR1 knock-out mice (described by Zamanillo et al., 1999) were crossed with C57BL/6 mice to produce F1 heterozygotes (GluR1 +/−). F1 heterozygotes were intercrossed to produce F2 littermates with mixed genotypes, wild-type (GluR1 +/+), heterozygotes (GluR1 +/−), and homozygous for the knock-out allele (GluR1 −/−), and all experiments were performed on these F2 littermates. This ensured that the variability of genetic backgrounds of mice was distributed randomly between wild-type (GluR1 +/+) and GluR1 knock-out (GluR1 −/−) mice (Inglis et al., 1998).
GluR1 conditional knock-out mice: GluR1deltaHb9 .
Mice bearing a floxed allele of GluR1 (GluR1 LoxP/+) were generated by Dr. Rolf Sprengel (Engblom et al., 2008) and the mice expressing the Cre recombinase under the control of the Hb9 promoter (Cre/+) were provided by Dr. Tom Jessell (Columbia University, New York, NY). GluR1 LoxP/+ mice were crossed with Hb9-Cre/+ mice to produce F1 animals that were heterozygous for both alleles (Cre/+, GluR1 LoxP/+). These F1 heterozygotes were intercrossed to produce F2 littermates with six different genotypes: (1) Cre/+, GluR1LoxP/LoxP ; (2) Cre/+, GluR1 LoxP/+; (3) Cre/+, GluR1 +/+; (4) +/+, GluR1LoxP/LoxP ; (5) +/+, GluR1 LoxP/+; (6) +/+, GluR1 +/+. The anatomical and behavioral experiments were performed on wild-type mice (+/+, GluR1 +/+) and GluR1 conditional knock-out mice in spinal motor neurons (Cre/+, GluR1LoxP/LoxP , referred to hereafter as GluR1deltaHb9 ) of F2 littermates. Limited studies were also performed on mice bearing genetic alteration at a single locus (Cre/+ and GluR1LoxP/LoxP ). All animal experiments were performed in accordance with Children Hospital of Philadelphia Animal Care and Use Committee guidelines.
Genotyping
For each mouse, ∼5 mm of tail was cut and digested with 0.5 ml of embryonic stem cell lysis solution plus 25 μl of proteinase K (10 mg/ml) at 55°C overnight. Genomic DNA was extracted with phenol-chloroform-isoamyl alcohol (25:24:1), precipitated with 100% ethanol plus 10% sodium acetate, pH 6.0, and washed with 70% ethanol. The collected DNA pellet was dissolved in 100 μl of distilled H2O or Tris-EDTA buffer and amplified with PCR. The primer sequences for detection of GluR1 −/− mice were 5′-CTG CCT GGG TAA AGT GAC TTG G-3′ and 5′-AAT GCC TAG TAC TAT AGT GCA CG-3′. The primer sequences for detection of GluR1 +/+ mice were 5′-CTG CCT GGG TAA AGT GAC TTG G-3′ and 5′-CAC TCA CAG CAA TGA AGC AGG AC-3′. The PCR conditions were as follows: 95°C (60 s), 60°C (60 s), and 72°C (40 s) for 40 cycles followed by a 10 min incubation at 72°C. The primer sequences for detection of Hb9-Cre mice were 5′-GTT ATT CGG ATC ATC AGC TAC ACC-3′ and 5′-GTC CAA TTT ACT GAC CGT ACA CC-3′. The PCR conditions were as follows: 95°C (30 s), 60°C (120 s), and 72°C (120 s) for 35 cycles followed by a 10 min incubation at 72°C. The primer sequences for detection of GluR1LoxP/LoxP (250 bp) and GluR1 +/+ mice (200 bp) were 5′-CTG CCT GGG TAA AGT GAC TTG G-3′ and 5′-CAC TCA CAG CAA TGA AGC AGG AC-3′. The PCR conditions were as follows: 95°C (60 s), 60°C (60 s), and 72°C (40 s) for 40 cycles followed by a 10 min incubation at 72°C.
Locomotor activity
Rotarod.
The rotarod treadmill (Ugo Basile) was used to test motor coordination and endurance. The mice were placed on an accelerating rotating rod (3 cm in diameter) and the time to fall off the rod was recorded. The initial speed was 4 rpm and over the subsequent ∼4 min, the spinning accelerates to a maximum of 35 revolutions/min. Each animal was tested three times at a given time point and the mean value was determined.
Grip strength.
The digital grip strength meter (Columbus Instruments) was used to measure the strength of limbs of mice. Each animal was tested nine times at a single age and the three maximum values obtained were averaged.
Treadmill.
The Exer 3/6 treadmill (Columbus Instruments) was used to test the endurance of mice. Mice initially ran on the treadmill at a slow speed (8 m/min) for 20 min and then the belt speed was increased to 12 m/min for 60 min. Finally, belt speed was increased to 17 m/min for up to 60 min. The test was stopped when fatigue was evident (the animals could no longer run) and the time to exhaustion was noted.
Retrograde labeling of spinal motor neurons
Animals were perfused with 4% paraformaldehyde (PFA) and the lumbar spinal cords were dissected out and kept in 4% PFA for post fixation. Crystals of fluorescent tracer DiI (Invitrogen) was applied to the ventral roots of the fixed lumbar spinal cord (L2 to L5). After the application of DiI, the spinal cords were maintained in 4% PFA at 37°C for 14 d. Then, the spinal cords were sectioned with a vibratome at 100 μm thickness. The sections were mounted on slides and examined with fluorescent microscope. The dendritic morphology of the DiI-labeled spinal motor neurons was drawn with the Neurolucida program and analyzed with the Neuroexplorer program (MicroBrightField). The criteria for the neurons included in the study were as follows: within ventral horn, the dendrites are radially distributed (>180°), and no more than one primary dendrite is truncated (less than three times of cell body diameters). Approximately one to three motor neurons were analyzed in each spinal cord. Quantitative descriptors of the dendrites were as follows: “1° dendrites,” the number of primary dendrites emanating from the cell body; “branches, #,” the number of dendritic bifurcations; “Σ length,” the sum of the lengths of all dendritic shafts from a single neuron; “average dendrites,” the sum of the lengths of all dendritic shafts from a single neuron divided by the number of primary dendrites; and “longest dendrite,” the length of the longest dendrite from the cell body to the distal tip.
Numbers of lumbar spinal motor neurons
GluR1 +/+ and GluR1deltaHB9 mice at P10 were perfused with 4% PFA and 100-μm-thick vibratome sections were prepared (n = 5, each group). Every other section was stained with the SMI-32 antibody (Covance), and the number of large immunopositive ventral horn cells (putative motor neurons) was counted. This was used to calculate the total number of motor neurons in the lumbar spinal cord, and the values from the two groups were compared.
GluR1 +/+ and GluR1 −/− mice at P10 were perfused with 4% PFA (n = 5, each group). The lumbar spinal cords were dissected out, postfixed, and 10 μm paraffin sections were prepared (L2 to L5). After Nissl staining, motorneurons were counted in every 10th section, and the total numbers of motor neurons at lumbar spinal cord were calculated and compared.
Muscle analysis
For nicotinamide adenine dinucleotide (NADH)-tetrazolium staining, frozen mouse muscle tissue sections from gastrocnemius were incubated for 45 min at room temperature in a solution of 0.2 m phosphate buffer, NADH (Sigma), nitro blue tetrazolium chloride (Sigma), and Ringer's solution. Sections were then incubated for 10 min with 10% formalin, gel mounted, and analyzed with bright-field microscopy. Morphometry was performed on the muscle fiber type I from four consecutive digital images from the medial gastrocnemius (frontal to dorsal) per mouse (20×) of NADH slides (GluR1 +/+ and GluR1 −/− mice) using the Image-Pro Plus software program (version 6.0; Media Cybernetics).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde and washed with PBS buffer. Goat serum (5%) in 0.1% Triton X-100 in PBS was used to permeate and block cells for 1 h at room temperature. Then, the cells were incubated with primary antibody mouse anti-GFP (1:1000; Sigma) overnight at 4°C. After washing with PBS, the cells were reacted with secondary antibody Alexa Fluor 488 goat anti-mouse (1:500) for 2 h at room temperature. The cells were further incubated with rabbit anti-GluR1 (1:1000; Upstate Biotechnology) overnight at 4°C. After washing, the cells were reacted with secondary antibody Alexa Fluor 594 goat anti-rabbit (1:500) for 2 h at room temperature. Finally, the coverslip with the cells were mounted on the slide and viewed with fluorescent microscopy.
GluR1 cell-surface staining and quantification
The mixed spinal cord neuron culture was cotransfected with resGluR1/GFP and treated with siRNA-penetratin conjugate. Five days later, the cells were fixed with 4% paraformaldehyde and blocked with 5% goat serum in PBS for 1 h. Triton X-100 was not added to retain cell membrane integrity and allow cell-surface GluR1 immunostaining. There was an obvious difference in GluR1 immunostaining between the cells with Triton X-100 treatment and without Triton X-100 treatment (data not shown). After blocking with goat serum, the cells were incubated with primary antibody mouse anti-GluR1 (1:100; gift from Peter Streit, University of Zurich, Switzerland) overnight at 4°C. After washing with PBS, the cells were reacted with secondary antibody Alexa Fluor 594 goat anti-mouse (1:500) for 2 h at room temperature. After GluR1 staining, the cells were treated with 0.3% Triton X-100 for 1 h and incubated with rabbit anti-GFP (1:100; Clontech) overnight at 4°C. After washing, the cells were further reacted with secondary antibody Alexa Fluor 488 goat anti-rabbit (1:500) for 2 h at room temperature. Finally, the coverslip with the cells were mounted on the slide and viewed with fluorescent microscopy. The images were acquired using Olympus Fluoview laser confocal microscope. All image acquisition was performed at the same settings (laser density, scan range, etc.) and analyzed using ImagePro software (Media Cybernetics). Quantified GluR1 and GFP signals for each individual cell were obtained at the cell body and the ratio of these signals was used to compare groups.
Interneuron mapping using PRV-GFP
The generation and characterization of pseudorabies virus (PRV) engineered to express GFP (PRV-GFP) has been described previously (Xiong et al., 2007). To enable quantitative comparisons between groups, all experiments were performed with a single lot of recombinant PRV-GFP (titer 3.4 × 108 pfu/ml). P7 animals were anesthetized by hypothermia and 1 μl of virus was injected into the hamstring muscles. Awake, warm, dry pups were returned to the dam, and 60 h later injected animals were prepared for additional analysis by perfusion fixed with 4% paraformaldehyde. One-hundred-micrometer thick tissue sections were generated with a Vibratome, mounted on slides, and viewed with epifluorescent illumination on a Zeiss Axioskop. In each animal, four segments of spinal cord (L2, L3, L4, and L5) were analyzed separately. For each segment, the number of neurons in each Rexed's lamina were counted in three tissue sections and the average value was determined. Five animal of each genotype were studied, and the values averaged and subjected to Student's t test for statistical comparison.
Patch-clamp recording
Whole-cell patch-clamp recordings were obtained from spinal cord cultures at room temperature using the whole-cell variant of the patch-clamp technique. Cultures were treated with either active anti-GluR1 siRNA-penetratin (320 nm) for 5 d or control siRNA. Electrodes were fabricated from thick-wall borosilicate glass (World Precision Instruments) and pulled to a 5–7 MΩ resistance on a P-30 two-stage vertical puller (Sutter Instrument). Internal pipette solution consisted of (in mm) 135 CsMeSO4, 10 CsCl, 10 HEPES, 5 EGTA, and 2 MgCl2, pH 7.2, 297 mOsm. To isolate AMPA-mediated miniature EPSCs (mEPSCs), cultures were perfused with an external solution consisted of (in mm) 150 NaCl, 3.1 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 0.05 dl-2-amino-5-phosphono-pentanoic acid, 0.005 strychnine, 0.01 bicuculline methiodide, and 0.0004 tetrodotoxin, pH 7.4. Neurons were voltage clamped at −60 mV with an Axopatch ID amplifier and software (Molecular Devices), and mEPSCs were acquired, filtered at 2 kHz, digitized at 5 kHz with pClamp, and saved for subsequent off-line analysis.
To minimize instances of inadequate space clamp, neurons were included in the analysis only if the series resistance was <20 MΩ and at least 80% series-resistance compensation was achieved. In addition, event amplitudes were plotted against rise times to detect inadequate space clamp (r 2 correlation coefficient > 0.5).
Records of mEPSCs were imported into Clampfit software (Molecular Devices) for event analysis. The Mini Analysis (Synaptosoft) procedure was used to select mEPSCs with 10–90% rise times <1.5 ms. All detected events were visually inspected, and any spurious noise that caused the window discriminator to be triggered was rejected.
Single-motor-unit electromyographic recordings
Single motor units from the soleus muscle were made using electromyographic techniques as described previously (Personius and Balice-Gordon, 2001; Personius et al., 2007). Briefly, P14–P16 mice were anesthetized by intraperitoneal injection of 17.4 mg/ml ketamine and 2.6 mg/ml xylaxine along with 2.5 mg/kg diazepam (Abbott Laboratories) to reduce stress. The soleus muscle was exposed via a small incision and mice were positioned in a standing position with hindlimbs and trunk restrained by soft wax. A single 75 μm stainless-steel microelectrode (FHC) was inserted into the soleus near the endplate band, and 125 μm Teflon-coated platinum wire with beveled tip (A-M Systems) was threaded under the muscle. The soleus muscle was bathed in mineral oil to reduce electrical cross talk and potentially increase the signal size. Recordings were initiated when spontaneous motor-unit activity was observed as the animals stood within the wax restraint after recovery from anesthesia. Stable recordings lasted for up to 1 h. EMG signals were filtered (500 Hz high pass), amplified, and digitized through high-impedance preamplifiers and differential amplifiers (TD Technologies). Recordings were performed blind to genotype
Analyses of single-motor-unit activity
Single motor units were discriminated off-line using Spike2 software (Cambridge Electronic Design). The accuracy of discrimination was verified by monitoring interspike intervals and the accuracy of template matching (Nordstrom et al., 1992; Personius and Balice-Gordon, 2001). Mean and median motor-unit firing frequencies were determined from interspike interval histograms (1000 60 ms bins spanning a 60 s period). Overall motor-unit activity was determined by counting the number of spikes occurring during the entire recording period (spikes per second). Cross-correlograms between motor-unit pairs were generated with bin widths of 100 ms over a period of ±25 s to investigate the presence of broad correlations (Meister et al., 1991; Torborg et al., 2005). Normalized custom analyses were used to identify the position and duration of the cross-correlogram peak and the strength of correlation was determined using the index k′ − 1 (Nordstrom et al., 1992). A correlation index between motor-unit pairs was used to determine the presence of narrow corrections over windows of 20 and 100 ms (Wong et al., 1993).
Generation of the Tg mouse
The 6.4 kb promoter region of choline acetyltransferase (ChAT) gene (Naciff et al., 1999) was amplified and cloned into pBluescript upstream in the 5′ end of the multiple cloning site. Bovine growth hormone (BGH) polyadenylation (poly-A) signal from pcDNA 3 was amplified and cloned downstream of ChAT promoter. Myc-tagged GluR1(flip) was then subcloned between ChAT promoter and BGH poly-A signal. The assembled construct containing ChAT promoter, myc-tagged GluR1(flip) gene, BGH poly-A signal (see Fig. 9 A) was excised and purified for pronuclear injection at the University of Pennsylvania core facility to generate transgenic mice. Transgenic animals were identified by PCR and germline transmission was verified during backcrossing into the C57BL/6 background.
Statistics
Student's t test was used in comparisons between two-group data and one-way ANOVA were used (with one exception) in comparisons among three or more groups of data. Significance was set at p < 0.05 in post hoc analysis. Data are expressed as mean ± SD in all figures and Tables 2 and 3. In Table 1, the data are expressed as mean ± SEM. Because the motor-unit data were non-normally distributed, a Mann–Whitney rank sum test was used for comparisons between genotypes.
Electromyographic analysis of wild-type and GluR1 knock-out mice
Results
Knockdown of GluR1 in vitro
Ordinarily mature rodent motor neurons express little or no GluR1 (Jakowec et al., 1995a,b). If a viral vector is used to force expression GluR1 in mature motor neurons in vivo, their dendritic tree becomes more highly branched without alteration in its overall size (Inglis et al., 2002). Overexpression of GluR1 in neurons in vitro similarly promotes dendrite growth and such neurons display increased amplitude of mEPSCs (Jeong et al., 2006). These observations led us to propose that the glutamate-receptor-controlled electrophysiological phenotype of neurons modulates dendrite morphogenesis. According to this view, activation of AMPA receptors of distinct molecular composition control dendrite morphogenesis, i.e., the process is “activity dependent.”
To study the role of GluR1 endogenously expressed by neurons in vitro, we designed a series of siRNAs to GluR1 and linked them to Penetratin (Davidson et al., 2004). Previous work has shown that siRNA-penetratin conjugates are taken up by virtually all neurons in vitro in a nontoxic manner and, if properly designed, can lead to efficient and specific suppression of target gene expression (Fig. 1). When spinal cord cultures were exposed to a siRNA targeting nucleotides 2363–2381 of the open reading frame, we found a dose-dependent reduction in the abundance of the GluR1 protein. The effect of this siRNA was specific for GluR1 (there was no change in the abundance of GluR2/3 or actin) and was not seen with a siRNA-penetratin conjugate with scrambled sequence. These observations parallel the characterization of AMPA receptor subunit expression in the hippocampus of the GluR-A −/− mice (GluR1 −/− mice, alternate nomenclature). Zamanillo et al. (1999) found that the expression of GluR-B, -C and -D (GluR2, GluR3, and GluR4, alternate nomenclature) in the GluR-A −/− mice was no different from wild-type animals.

Reduction in GluR1 expression in neurons with siRNA-penetratin inhibits dendrite growth. Spinal cord cultures were treated with vehicle (no siRNA), a scrambled-sequence siRNA (control), or active siRNA at 80 or 320 nm (linked to penetratin) for 5 d. The top left shows Western blots of lysates from treated cultures. The active siRNA at 320 nm (and to a lesser degree at 80 nm) leads to a specific reduction in the abundance of GluR1, but not GluR2/3 or actin. To the right are immunocytological images of GluR1-stained neurons in cultures treated with the labeled reagents. Below are representative camera lucida drawings of neurons from cultures treated with active siRNA (2 different concentrations), the control siRNA, or vehicle. Scale bar, 20 μm. Quantitative morphometry is displayed in the table below the camera lucida images. In the cell parameter row, n in each column refers to the number of cells drawn. By ANOVA, there are statistically significant reductions of total dendrite length in the 80 and 320 nm active siRNA treatment groups compared with the no siRNA and control siRNA groups. There is a statistically significant reduction in branches and length of longest dendrite in the 320 nm active siRNA group compared with the other groups. There are no statistically significant differences between the no siRNA versus control siRNA groups.
We wondered whether the reduction of GluR1 in siRNA-treated cultures would lead to an alteration in currents evoked by single quanta of glutamate. To examine this issue, we analyzed mEPSCs from neurons in cultures exposed to the active siRNA (n = 6) versus control siRNA (n = 6) from DIV 5 to DIV 10. There were no statistically significant differences between the two culture conditions in mEPSC amplitude (19.9 ± 9.8 vs 13.9 ± 2.2 pA, active siRNA vs control siRNA; p = 0.21), T90 decay time (4.0 ± 2.4 vs 4.7 ± 1.6 ms, active siRNA vs control siRNA; p = 0.25), or 10–90% rise time (0.77 ± 0.32 vs 0.87 ± 0.13 ms, active siRNA vs control siRNA; p = 0.40). We also examined mEPSC frequency (as events per minute) and found no differences between groups (10.8 ± 8.5 vs 11.0 ± 4.8, active siRNA vs control siRNA; p = 0.98). These observations mirror the results obtained previously from hippocampal CA1 pyramidal cells (Zamanillo et al., 1999) and indicate that AMPA-receptor-mediated synaptic currents recorded from spinal cord neurons in vitro are mostly unaffected by a deficiency of GluR1.
To determine what happens to dendrite growth in vitro when GluR1 expression is suppressed both in the neuron of interest (the one whose dendrites are labeled and quantitatively analyzed) and its neighbors, we exposed cultures to the active GluR1-siRNA from DIV 5–10, and then on DIV 9 transfected a GFP expression plasmid to label the dendritic tree of an individual neuron.
When the cells were treated with the active GluR1-siRNA-penetratin at 80 nm, versus the control, scrambled-sequence siRNA-penetratin, there was a statistically significant ∼20% reduction in overall dendrite size. When used at 320 nm, the active GluR1-siRNA-penetratin led to statistically significant ∼45% reduction in the number of dendritic branches (F (3,83) = 4.383, p < 0.01), ∼30% reduction in total dendrite length (F (3,83) = 7.745, p < 0.001), and an ∼10% reduction in the length of the longest dendrite (F (3,83) = 2.842, p < 0.05) (Fig. 1). No statistically significant changes were found in the other dendrite morphological parameters (soma area, the numbers of primary dendrites, or average dendrite).
Engineering a short hairpin RNA (shRNA) into a vector suitable for expression in neurons (pSUPER) would permit us to study the effects of reducing GluR1 in single neurons within a background of cells expressing GluR1 at endogenous levels. The target site identified and validated above was incorporated into a shRNA and when cotransfected into human embryonic kidney 293 (HEK293) cells along with a GluR1 expression vector led to a pronounced knockdown of the heterologously expressed GluR1 transgene (Fig. 2). A control, scrambled-sequence shRNA was without effect on GluR1 expression. We next transfected mixed spinal cord cultures with active (or control) shRNA using a pSUPER vector that also expressed GFP (from a second promoter) and, after a 5 d latency, studied the effect on dendrite elaboration. There were no differences between the groups expressing pSUPER empty vector versus pSUPER with a scrambled-sequence siRNA (Fig. 2).

Reduction in GluR1 expression in neurons with pSUPER shRNA inhibits dendrite growth. HEK293 cells were transfected with pSUPER-scrambled shRNA (control) or pSUPER-active shRNA plus the cDNA for GluR1 in a mammalian expression vector (pGW). Western blots of lysates 2 d after transfection (top left) demonstrate the specific reduction in GluR1 abundance cells expressing the active shRNA. To the right and below are representative camera lucida drawings of neurons from spinal cord cultures treated with pSUPER-empty vector, pSUPER-scrambled shRNA (control), or pSUPER-active shRNA for 5 d. Scale bar, 20 μm. Quantitative morphometry is displayed in the table below the camera lucida images. In the cell parameter row, n in each column refers to the number of cells drawn. By ANOVA, there are statistically significant reductions in the number of primary dendrites, branches, overall tree size, average dendrites, and longest dendrites in the pSUPER active shRNA group compared with other two groups.
Compared with neurons transfected with control shRNA, neurons expressing the active shRNA to GluR1 had marked changes in their dendritic tree (Fig. 2). There were statistically significant reductions in all dendrite morphological parameters of spinal motor neurons except the soma area: ∼20% reduction in the numbers of primary dendrites (F (2,65) = 13.27, p < 0.0001), ∼40% reduction in the numbers of branches (F (2,65) = 10.82, p < 0.0001), ∼30% reduction in total dendrite length (F (2,65) = 21.56, p < 0.0001), ∼20% reduction in mean dendrite length (F (2,65) = 7.084, p < 0.005), and ∼20% reduction in the longest dendrite length (F (2,65) = 3.759, p < 0.05). The similarity of effects of the siRNA-penetratin versus shRNA on dendrite parameters emphasizes the cell-autonomous effects of GluR1 expression on dendrite elaboration.
We generated a cDNA that contained silent mutations in the GluR1 open reading frame that should, in theory, resist the ability of the active siRNA to cleave the target (resGluR1). Nucleotides 2362–2381 were changed from G AAT GAG TAT ATT GAG CAA to G AAT GAG TAC ATC GAG CAG (italics denote alterations) but did not affect the amino acid sequence of GluR1 (-N-E-Y-I-E-Q-). When expressed with resGluR1, the siRNA had no effect on the abundance of GluR1 by Western blot, but the siRNA effectively reduced the abundance of the wild-type GluR1 protein (Fig. 3). Under certain circumstances, silent mutations in a gene will not influence the amino acid sequence, but will alter the function of the protein (Kimchi-Sarfaty et al., 2007). To be sure the mutations we introduced did not alter GluR1 function, we studied the electrophysiological properties of AMPA receptors assembled from the cDNA encoding this mutated version of GluR1. Oocytes were injected with cRNA for the wild-type GluR1 and resGluR1, and AMPA receptor currents were recorded. No statistically significant differences were found from the two groups when kainic acid (KA)- or glutamate-evoked currents were studied, when the pore blocker NASP was applied to KA-stimulated receptors, or in the agonist-evoked current–voltage relationships (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results show that resGluR1 assembles into homomeric AMPA receptors that are electrophysiologically indistinguishable from their wild-type counterparts.

Expression of GluR1 in neurons enhances dendrite growth, but only if neighboring neurons also express GluR1. HEK293 cells were transfected with an expression vector containing wild-type (WT) GluR1 or resGluR1 in which the coding sequence has been altered to prevent perfect binding of the active siRNA to the mRNA. Cotransfected with these plasmids was either pSUPER with the inactive shRNA or pSUPER with the active shRNA. In the upper left, Western blots of lysates from transfected cells are shown. The active shRNA knocks down the expression of WT but not resGluR1. There is no effect on the expression of actin. To the right and below are representative camera lucida images of neurons expressing GFP alone or in combination with resGluR1, resGluR1 plus inactive siRNA, or resGluR1 plus active siRNA. Quantitative morphometry is displayed in the table below the camera lucida images. In the cell parameter row, the n in each column refers to the number of cells drawn. By ANOVA, there is a statistically significant increase in branches and overall tree size in the resGluR1 and resGluR1 plus inactive siRNA groups compared with GFP or resGluR1 plus active siRNA groups. There are no statistical differences between the GFP versus the resGluR1 plus active siRNA group.
To investigate whether GluR1 cell-surface expression was changed after the treatment of cultures with resGluR1 and siRNA-penetratin conjugate, immunocytochemical cell-surface GluR1 signal was quantified and normalized to that of cotransfected GFP. Four groups of transfected neurons were compared: (1) GFP alone, (2) GFP plus resGluR1, (3) GFP plus resGluR1 treated with the control siRNA, and (4) GFP plus resGluR1 treated with the active siRNA. Statistical analysis of cell-surface labeling (normalized to the signal from cotransfected GFP), showed significant differences between groups (F (3,78) = 17.19, p > 0.0001). After transfection with resGluR1, the GluR1 signal (endogenous plus transgene) was significantly increased, compared with endogenous GluR1 signal alone (0.54 ± 0.23 vs 0.17 ± 0.07, p < 0.001). Treatment of cultures expressing resGluR1 with the control siRNA had no effect on cell-surface GluR1 expression (0.54 ± 0.23 vs 0.49 ± 0.19, p = 0.44). In contrast, treatment of cultures expressing resGluR1 with the active siRNA led to a decrease in surface GluR1 expression (0.54 ± 0.23 vs 0.34 ± 0.18, p < 0.05). The decreased abundance of cell-surface GluR1 in these later neurons reflects the loss of the endogenously expressed GluR1. These results confirm that the active siRNA reduces the cell-surface expression of the endogenously expressed GluR1 and that cell-surface GluR1 generated from the resGluR1 construct is unaffected by the active siRNA.
We used resGluR1 and the active siRNA-penetratin conjugate together to determine the effects of expressing GluR1 in a neuron of interest when neighboring neurons had reduced GluR1 expression (Fig. 3). In this experimental paradigm, GluR1 expression was suppressed in all neurons in the culture (using siRNA-penetratin) except for the neuron expressing resGluR1 (and a reporter so that the dendritic tree was labeled). Four experimental groups were compared: (1) neurons expressing GFP alone, (2) GFP plus resGluR1, (3) resGluR1 plus control siRNA, and (4) resGluR1 plus active siRNA. Compared with GFP alone, neurons expressing resGluR1 had a statistically significant ∼40% increase in branches (F (3,77) = 4.026, p < 0.05) and ∼20% increase overall size of the dendritic tree (F (3,77) = 5.433, p < 0.005). Treating cultures with the control siRNA did not affect the ability of resGluR1 to cause dendrite growth (e.g., there were no significant differences between groups 2 and 3). In contrast, when cultures were treated with the active siRNA, expression of resGluR1 lost its ability to promote dendrite growth. There were no statistical differences between the dendritic tree of neurons simply expressing GFP versus those resGluR1 plus the active siRNA (e.g., groups 1 and 4).
In summary, the in vitro experiments show that endogenously expressed GluR1 within a network of neurons promotes dendrite growth. Although increasing GluR1 expression in individual neurons enhances dendrite growth, this effect is erased if the neuron overexpressing GluR1 is embedded in a network of neurons that do not express GluR1. Thus, the capacity of a specific neuron in vitro to undergo GluR1-dependent dendrite growth is contextural and not strictly cell autonomous.
Anatomical characterization of the GluR1−/− mice
Previous work on the GluR1 −/− mice has focused on the effects of GluR1 in learning and memory with relatively little inquiry into the effects on motor system development (Zamanillo et al., 1999; Bannerman et al., 2004). The extent to which defects in locomotor behavior contribute to poorer performance of the GluR1 −/− mice on tasks such as the water maze test is unknown. GluR1 is normally expressed in great abundance by neonatal motor neurons and in neighboring neurons throughout the spinal gray matter during early postnatal life (Jakowec et al., 1995a,b). To investigate the potential in vivo relevance of our in vitro findings, we began by studying the morphological and behavioral consequences of germline deletion of GluR1 in mice.
Rodent motor neuron dendrites grow longer and more highly branched over the first ∼3 weeks of postnatal life and thereafter, under normal circumstances, undergo little change (Kalb, 1994; Inglis et al., 1998). At the approximate midpoint of this postnatal developmental period (e.g., P10), there were specific and significant differences in motor neuron dendrites of GluR1 −/− versus GluR1 +/+ animals (Fig. 4). The GluR1 −/− mice had a statistically significant ∼30% reduction in the mean dendrite length (p < 0.05), an ∼35% reduction in overall tree size (p < 0.05), and an ∼25% reduction in the longest dendrite (p < 0.05) in comparison with GluR1 +/+ animals. There were no other significant changes in the quantitative aspects of the dendritic tree of motor neurons from the two different strains of mice. Over the next ∼2 weeks (∼P23), motor neuron dendrites continue growing and branching until the mature tree is elaborated. At this time, motor neurons from the GluR1 −/− mice had a statistically significant ∼35% reduction in branches (p < 0.0001) and ∼25% reduction in overall tree size (p < 0.01) in comparison with GluR1 +/+ animals, whereas other measures of dendrites did not differ between groups (Fig. 4). Thus, in the absence of GluR1, the early postnatal trajectory of motor neuron development is perturbed and leads to major alterations in dendrite architecture.

Comparison of motor neuron dendrites from GluR1 +/+ versus GluR1 −/− animals at P10 and P23. Representative camera lucida drawings from mice of specified age and genotype are displayed above the tables. Scale bar, 30 μm. At P10, there is a statistically significant (by Student's t test) reduction in overall tree size and longest dendrite and average dendrite length in the GluR1 −/− animals compared with GluR1 +/+ animals. At P23, there is a statistically significant (by Student's t test) reduction in branches and overall tree size in the GluR1 −/− animals compared with GluR1 +/+ animals.
Considering the effects of lacking GluR1 on motor neuron dendrite structure, we wondered whether there was also an effect on the pattern of connections onto motor neurons. To examine this issue, we used the PRV transneuronal labeling system. PRV engineered to express GFP was injected into the hamstring muscles of GluR1 +/+ and GluR1 −/− mice at P7, and 60 h later tissue was prepared for histological analysis. We counted the number of GFP (+) premotor interneurons as a function of (1) laminar location, (2) spinal cord segment level, and (3) ipsilateral versus contralateral to the injection site. We found a significant decrease of the numbers of interneurons in the ipsilateral lumbar segment 4, lamina VIII, in the GluR1 −/− versus GluR1 +/+ mice (3.9 vs 7.6, p < 0.05) (Fig. 5). No other differences ipsilateral to the injection of PRV between the GluR1 −/− versus GluR1 +/+ animals were found. When we looked at the distribution of premotor interneurons contralateral to the injection side, there were statistically significant reductions in the GluR1 −/− compared with the GluR1 +/+ mice in many regions (Fig. 5, table): lamina III (lumbar segment L3), lamina VII (lumbar segment L4), and lamina VIII (lumbar segments L2, L3, L4, and L5). Thus, the smaller and less complex dendritic tree of motor neurons in the GluR1 −/− mice is accompanied by specific alterations in the pattern of innervation onto these motor neurons by premotor interneurons.

A comparison of the number and distribution of premotor interneurons in GluR1 −/− versus GluR1 +/+ mice. A diagram of the Rexed's lamina in the lumbar spinal cord and a low-power image of PRV-GFP-positive cells at P10. Three-dimensional bar graphs summarize the quantitative results (axes: number of cells × Rexed's lamina × lumbar segment). Red columns indicate no differences between the GluR1 −/− and GluR1 +/+ animals. The light blue column indicates a statistically significant increase in the number of neurons in the GluR1 +/+, and the dark blue column indicates a statistically significant decrease in the number of neurons in the GluR1 −/− tissues. The greatest differences between genotypes were found in the number of premotor interneurons contralateral to the injection site, especially in Rexed's lamina VIII. Bottom, The primary data are presented in table format. Ipsi, Ipsilateral; contra, contralateral.
Electrophysiological characterization of the GluR1−/− mice
Considering the alterations in segmental spinal cord circuitry in the GluR1 −/− compared with GluR1 +/+ mice, we wondered whether this led to measurable alterations in neuromuscular unit activity. To examine this issue, we made single-motor-unit recording from the offspring of GluR1 +/− mice at P14–P16. Thirty-four recordings were made of awake animals standing in a restraint, and after unbinding, four and seven mice were subsequently genotyped as GluR1 −/−and GluR1 +/+, respectively. No difference in motor-unit firing frequency was seen between genotypes (Table 1), with values similar to those reported previously in similarly aged mice of different backgrounds (3.6 ± 0.7, CD-1 and 3.1 ± 1.8, C57BL/6J). Because motor units are rarely active for the entire recording period and to determine whether the overall motor-unit activity was different between genotypes, we divided the number of discriminated motor-unit spikes by the time of the recording session (spikes per second). No difference in overall motor-unit activity was seen between genotypes (Table 1). The incidence of temporal correlation between motor-unit pairs was monitored by measuring the index k′ − 1 and correlation index using 20 and 100 ms bins. An index k′ − 1 value >0.3 indicates that a motor-unit pair is temporally correlated (Personius and Balice-Gordon, 2001). Six of 38 motor-unit pairs from GluR1 −/− mice had index k′ − 1 values >0.3 (15.8%), whereas 9 of 79 were >0.3 in GluR1 +/+ mice (11.4%). The low average correlation index values calculated using 20 and 100 ms bin widths indicates that the incidence of temporal correlation between motor neurons is small. An index of 1 indicates that the activity of the two units is independent, whereas an index >1 implies that the units are more likely to fire together within the specific time window than expected by chance (Personius and Balice-Gordon, 2001; Personius et al., 2007). Again, no difference in correlation incidence was seen between genotypes (Table 1). Thus, at this level of analysis, we find no changes in neuromuscular unit activity GluR1 −/− compared with GluR1 +/+ mice.
Anatomical characterization of the motor-neuron-specific GluR1 knock-out mice
Our in vitro experiments indicate that the ability of GluR1 to promote dendrite growth is influenced by the expression of network GluR1 expression. This is relevant to the developing spinal cord because in addition to motor neurons, many interneurons within the segmental spinal cord express GluR1 at high levels during early postnatal life (Jakowec et al., 1995a,b). To examine the in vivo role of GluR1 expression in motor neurons on dendrite growth, we used the LoxP–Cre system. Expression of the Cre recombinase under the control for the Hb9 promoter has been used extensively to manipulate gene expression in embryonic motor neurons (Yang et al., 2001), although the pattern of Cre expression in juvenile and adult animals has not be studied. Thus, we began by mapping Cre recombination using the ROSA26-LacZ reporter mice and examined β-galactosidase-expressing cells at P23 and 3 months of age. Every P23 animal studied had β-galactosidase expression in cervical and lumbar motor neurons, but less than one-half of thoracic motor neurons were β-galactosidase positive (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Interneurons were β-galactosidase positive in approximately one-half of the animals, regardless of the spinal level we studied. When we studied adult animals, all lumbar motor neurons expressed β-galactosidase, although in the cervical and thoracic cord β-galactosidase was found in only approximately one-half of the animals. All adult animals have β-galactosidase-positive interneurons in the lumbar spinal cord, whereas at the cervical and thoracic levels only one-half the animals had β-galactosidase positive interneurons. Regarding β-galactosidase staining above the foramen magnum, none of the neurons devoted to motor function (e.g., motor cortex, basal ganglia, pontine nuclei) expressed β-galactosidase. There were, however, positive neurons seen in the pontine tegmentum and in rare Purkinje cells in the cerebellar hemispheres. Overall, we found that Cre-mediated recombination has occurred in embryonic, juvenile, and adult lumbar motor in neurons. An accretion of Cre-mediated recombination occurs in lumbar interneurons during postnatal life. Although Cre-mediated recombination has occurred in embryonic, juvenile, and adult cervical motor in neurons, there does not appear to be a time-dependent change in the size of the interneuronal population. The loss of β-galactosidase in cervical motor neuron in a subpopulation of animals (three of eight) might reflect epigenetic transgene silencing. Thus, although not exclusively restricted to motor neurons, the Hb9-Cre driver line (used in combination with mice bearing a floxed allele of GluR1) would permit us to examine the role of GluR1 expression in motor neurons and subpopulation of interneurons in motor neuron dendrite growth. Consistent with this, GluR1 immunostaining of spinal cord slices from the GluR1deltaHb9 mice show that P10 motor neurons do not express GluR1 (Fig. 6).
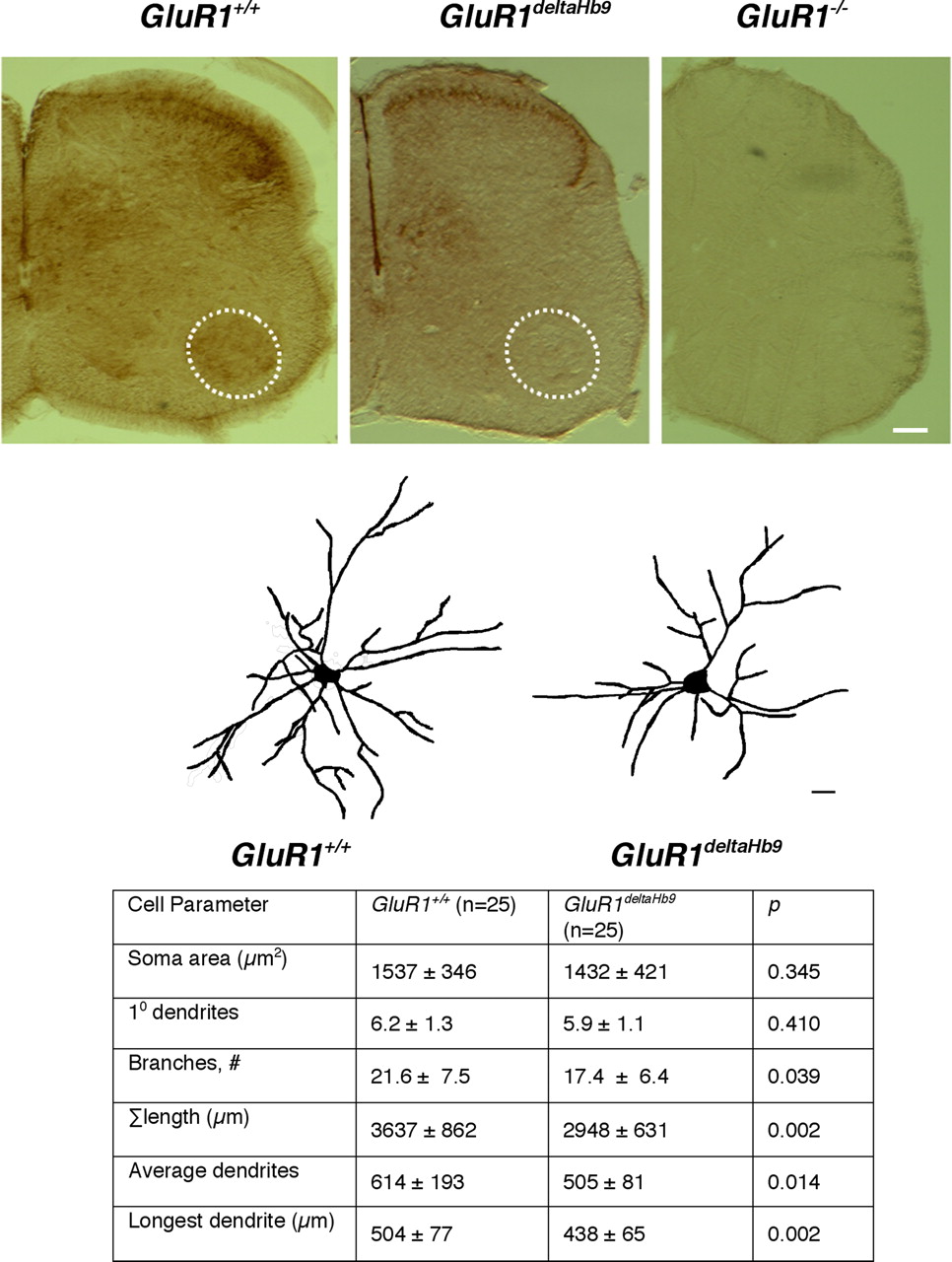
Comparison of motor neuron dendrites from GluR1 +/+ versus GluR1deltaHb9 animals at P23. The top three panels are representative photomicrographs of P10 spinal cords immunostained with anti-GluR1 from GluR1 +/+, GluR1deltaHb9 and GluR1 −/− mice. Motor neurons residing in Rexed's layer IX (dashed circle in the ventral horn) are stained in the GluR1 +/+ animals but not in the GluR1deltaHb9 and GluR1 −/− mice. There are no other clear differences between the GluR1 +/+ and the GluR1deltaHb9 mice. As expected, there is no immunoreactivity in the GluR1 −/− mice. Scale bar, 125 μm. Middle panels, Representative camera lucida drawings from P23 mice of a specified genotype are displayed above the table. Scale bar, 25 μm. Bottom, There is a statistically significant (by Student's t test) reduction in branches, overall tree size, longest dendrite, and average dendrite length in the GluR1deltaHb9 animals in comparison with GluR1 +/+ animals.
Next, we examined the effects of the GluR1LoxP allele or Cre expression, in isolation, on motor neuron dendrite growth. Recent studies indicate that simply expressing Cre in cells can be toxic (Schmidt-Supprian and Rajewsky, 2007). A comparison of P23 GluR1 +/+ animals with each of the singly, genetically modified animals revealed no statistically significant differences in any dendritic measure between the groups (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These results indicate that any observations resulting from the GluR1deltaHb9 mice are likely to be caused by the specific elimination of GluR1 from motor neurons and other Hb9-Cre-expressing neurons.
Compared with GluR1 +/+ animals, the motor neurons from P23 GluR1deltaHb9 mice had a statistically significant ∼20% reduction in branches (p < 0.05), an ∼20% reduction in overall dendritic tree size (p < 0.005), an ∼20% in mean dendritic length (p < 0.05), and an ∼15% shortening of the longest dendrite (p < 0.01) (Fig. 6). The only features of motor neurons not influenced by the loss of GluR1 was the overall soma size and the number of primary dendrites. With the exception of the results on the average dendritic length and longest dendrite, these findings phenocopy the results described above of motor neuron dendrites from the GluR1 −/− mice, although they appear to be quantitatively less. In addition they are consistent with our in vitro findings that GluR1 expression in the cell of interest plays a major role in dendrite elaboration.
Behavioral analysis of the GluR1−/− and GluR1deltaHb9 mice
We next asked whether these alterations in motor neuron dendrite architecture are associated with measurable behavioral consequences. We began by subjecting the GluR1 −/− and GluR1 +/+ animals to a battery of motor system tasks (Fig. 7). At P23, the GluR1 −/− mice (n = 16) performed less well than GluR1 +/+ (n = 17) in three tasks: the GluR1 −/− animals had ∼10% and ∼15%, respectively, less strength the forelimb and hindlimb grip assay (a measure of peak strength), ∼30% less endurance on the rotarod assay, and an ∼50% less endurance on the treadmill assay. If the GluR1 −/− mice were simply delayed in motor system maturation, then a later assessment of motor function might erase these differences. Although animals become stronger over the subsequent 2 months of life, the GluR1 −/− mice continued to have motor defects (Fig. 7). Compared with the GluR1 +/+ animals (n = 20), the GluR1 −/− animals (n = 19) had an ∼15% less strength of both the forelimb and hindlimb grip, ∼30% less endurance on the rotarod, and an ∼40% less endurance on the treadmill. Thus, the lack of GluR1 impairs motor neuron dendrite development and this is accompanied by a variety of persistent defects in motor behavior.
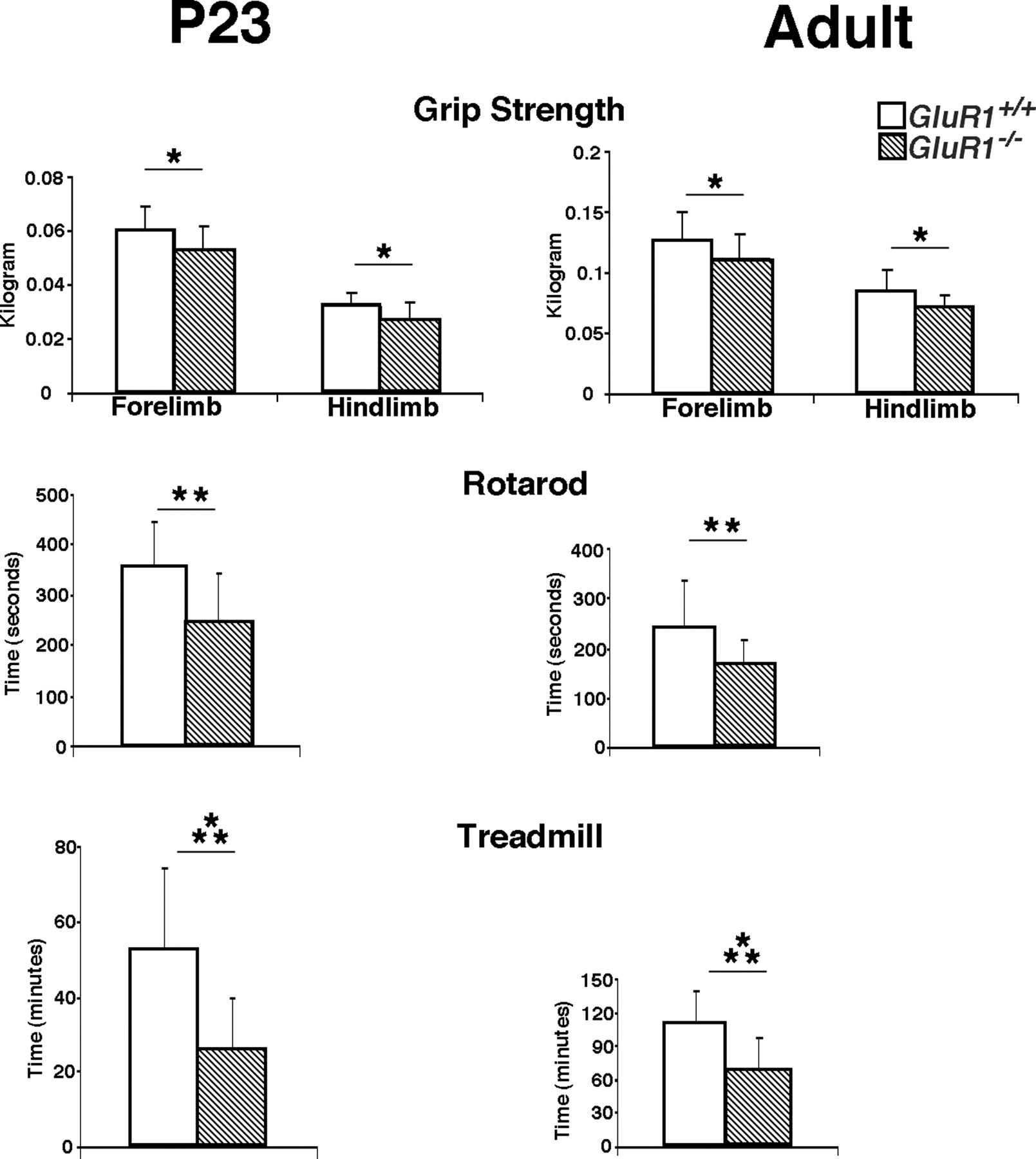
Comparison of motor behavior in GluR1 +/+ versus GluR1 −/− animals. The two genotypes were studied at P23 and adulthood on three motor tasks: grip strength (forelimb and hindlimb), rotarod, and treadmill. Note the change in y-axis scale at different ages. *p < 0.05; **p < 0.01; ***p < 0.001. The GluR1 +/+ animals (open bars) perform better than the GluR1 −/− animals (striped bars) in a statistically significant manner in all tests.
We next asked whether there were motor behavior defects in the GluR1deltaHb9 mice with motor-neuron-specific knock-out of GluR1 expression. Before embarking on these comparisons, we wanted to determine whether the genetic modifications (knock-in of the floxed allele of GluR1 or transgenic expression of Cre under the control of the Hb9 promoter) themselves had any effect on motor behavior. A three-way comparison of GluR1 +/+ animals with animals only bearing the floxed allele of GluR1 or Hb9-Cre showed no differences on any behavorial test (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Next, we examined P23 GluR1deltaHb9 and GluR1 +/+ animals and found the GluR1deltaHb9 mice (n = 18) demonstrated an ∼15% reduction in treadmill time compared with GluR1 +/+ animals (n = 19) (Tables 2, 3). At this age, there were no other differences between the experimental groups. Quantification of motor neurons, identified by SMI-32 immunoreactivity within the ventral horn, revealed no differences between groups (3857 ± 526 vs 3697 ± 798, GluR1deltaHb9 vs GluR1 +/+; n = 5 animals per group; p = 0.72). At the adult time point (2.5 months), GluR1deltaHb9 mice (n = 16) showed an ∼15% reduction in hindlimb grip strength compared with GluR1 +/+ animals (n = 15), and there were no other differences between the experimental groups (Tables 2, 3). Thus, there is a clear disparity between the motor behavior of the GluR1 −/− and GluR1deltaHb9 mice. This suggests that the absence of GluR1 expression in neurons in addition to motor neurons plays an essential role in normal motor system development.
Locomotor activity of P23 mice
Locomotor activity of adult mice
Muscle analysis of the GluR1+/+ and GluR1−/− mice
Considering the prominent defects in motor function in the GluR1 −/− mice, we undertook additional studies to account for this finding. First, we counted the number of motor neurons Nissl stained sections of lumbar spinal cords from GluR1 −/− versus GluR1 +/+ animals. This independent method uses different criteria for identifying motor neurons than SMI-32 staining, and so the two sets of results are comparable but not identical. When we quantified the number of motor neurons in the GluR1 −/− versus GluR1 +/+ animals, no differences were found between the two groups (e.g., 2417 ± 498 vs 2571 ± 368, p = 0.59). Thus, one cannot simply attribute the motor deficits to a numerical reduction in motor neurons driving muscle activity. Next, we examined muscle tissues from GluR1 +/+ versus GluR1 −/− mice and, although we found no evidence for denervation–reinnervation (i.e., fiber type grouping), the muscle from GluR1 −/− mice did appear different from GluR1 +/+ animals. We examined this in greater detail by quantifying the diameter of type I fibers and their number in the gastrocnemius muscle of the two strains of mice (Fig. 8). The gastrocnemius has a mixed population of fiber types, with the ventral portion of the muscle composed mostly of type I fibers and the dorsal portion of the muscle composed mostly of type II fibers. Overall, we found an increase number of type I fibers in the muscle of the GluR1 −/− compared with the GluR1 +/+ animals. There was a leftward shift in the frequency-distribution histogram such that the GluR1 −/− mice have more smaller type I fibers and the GluR1 +/+ animals have more larger type II fibers. When we analyzed the average type I fiber diameter as a function of position along the ventrodorsal axis, we found a statistically significant reduction in type I fiber diameter in the ventral-most regions of the muscle (zones 1 and 2). Thus, there are two differences in the muscles of the GluR1 −/− versus the GluR1 +/+ mice: the GluR1 −/− mice have more type I fibers, and these fibers are smaller in diameter than type I fibers in the GluR1 +/+ animals. These alterations in muscle phenotype are associated with a reduction in motor neuron dendrite tree size as well as the poorer performance of the GluR1 −/− mice on multiple motor behavior tasks.

Comparison of gastrocnemius muscle from GluR1 +/+ and GluR1 −/− mice. Top pictures show stained muscle from animals of each genotype and qualitatively demonstrate the reduction in size and increase in number of darkly stained (type I) fibers in the GluR1 −/− versus GluR1 +/+ animals. Below, a frequency-distribution histogram reveals a leftward shift in the GluR1 −/− versus GluR1 +/+ animals, indicating more smaller type I fibers in the GluR1 −/− animals. The bar graph to the right demonstrates a statistically significant increase in type I fibers in the GluR1 −/− versus the GluR1 +/+ animals. *p < 0.05. Below is a diagram of a cross section of the gastrocnemius muscle and the relative distribution of type I fibers displayed. There are more type I fibers in the ventral (top) versus the dorsal (bottom) portion of muscle, and the location of the four zones we analyzed are shown in boxes. The table below shows the comparison of fiber I fiber diameter of GluR1 +/+ versus GluR1 −/− mice in zones 1–4. Type I fibers were statistically significantly small in the GluR1 −/− animals in the zones enriched with type I fibers (zones 1 and 2).
Effects of overexpression of GluR1 in motor neurons in transgenic mice
Because the lack of GluR1 leads to defects in motor performance, we wondered whether the converse was true: does forced expression of GluR1 in mature motor neurons enhance motor performance? Our aim was to restore GluR1 expression in motor neuron after the point in development when, under normal circumstances (Jakowec et al., 1995a,b), its expression level is very low or absent. To achieve this goal, we generated a transgenic mouse in which a 6-myc-tagged version of rat GluR1(Q)flip was expressed under the control of the ChAT promoter. The 6.4 kB ChAT promoter had been characterized previously as sufficient to drive transgene expression in >50% spinal motor neurons (Naciff et al., 1999).
Eight founder lines were identified, which varied in the level of transgene expression (Fig. 9 B). We were concerned that lines with high levels of transgene expression might have an adverse effect on motor neurons because of the assembly of homomeric GluR1 receptors that were calcium permeable. Thus, we analyzed “line 48” in greater detail because the expression level of the transgene was relatively low. By Western blot, we found that the transgene is first detectable in spinal cord lysates at P45 at the appropriate molecular weight. The level of expression appeared constant over the next 2 months of life. Immunohistology for the myc tag confirmed expression of the transgene in spinal motor neurons at P75 (Fig. 9 C). To determine whether the transgene expression led to large-scale compensatory changes in the expression of genes involved with synaptic transmission, Western blot analyses were performed. When line 48 and wild-type mice were compared at P75, no differences were detected in the spinal cord expression level of GluR1, GluR2/3, GluR4, GluR6/7, gephyrin, synaptophysin, SAP97, PSD97 or actin (Fig. 9 D). Next we looked at the subcellular distribution of the transgene in line 48 (vs wild-type tissues) using a biochemical approach using P75 tissue. Myc immunoreactivity was found in total spinal cord homogenates from line 48, but not wild-type animals. This immunoreactivity was significantly enriched in the synaptosomal membrane (SPM) fraction in comparison with the particulate, P3 fraction (Fig. 9 E). Probing these fractions with anti-GluR1 or anti-GluR2/3 demonstrates the endogenous proteins are present in the total lysates from wild-type and line 48 animals and there is, as expected, enrichment in the SPM fraction. Finally, to confirm that the myc band in these lysates is the transgene, we immunoprecipitated the SPM fraction from line 48 homogenates with anti-myc and probed with anti-myc, anti-GluR1, and anti-GluR2/3. The immunoprecipitated material has strong anti-myc and anti-GluR1 immunoreactivity. A small amount of GluR2/3 immunoreactivity is seen, indicating that the myc-tagged GluR1 assembles into hetero-oligomeric AMPA receptors. Thus, the line 48 mice express GluR1 in adult spinal motor neurons and is likely to assemble into functional AMPA receptors in synaptic membranes.

A transgenic mouse that expresses GluR1 in adult motor neurons. A , The construct used to make the transgenic (TG) mouse uses 6.4 kB of the ChAT promoter, followed by an intron, the 6-myc-tagged GluR1(Q)flip cDNA followed by the BGH polyadenylation sequence. B , Eight mouse lines incorporated the transgene into the germ line, and Western blots of spinal cord tissue for the myc tag showed varying levels of transgene expression. Because of the relatively low level of expression in line 48, these mice were chosen for further study. C , Immunohistology of 3-month-old line 48 mice with anti-myc antibody showed that the transgene is expressed in motor neurons. Scale bars: top, 150 μm; bottom, 50 μm. D , Western blot analysis of spinal cord lysates from wild-type (WT) and line 48 mice showed no large scale changes in the expression of numerous proteins. E , Subcellular fractionation of spinal cord lysate from line 48 and wild-type mice. Myc immunoreactivity is found only in line 48 mice in the total lysate, the P3, and the SPM fraction. Myc immunoreactivity is enriched in synaptic membranes. Both GluR1 and GluR2/3 immunoreactivity are similarly seen in total lysate, the P3, and the SPM fraction with SPM enrichment. Immunoprecipitation of the SPM fraction using anti-myc brought down the myc-tagged protein from line 48 but not wild-type homogenates. The immunoprecipitated material is enriched for GluR1 (consistent with IP of the myc-GluR1 construct) and, to a lesser extent, GluR2/3. WM, White matter; GM, gray matter.
Because the transgene GluR1 does not begin to be expressed until P45, any analysis of motor neuron dendrites from these animals requires a technique that works in older animals. We tried three methods for delineating motor neuron dendrites in these P75 animals: application of DiI to the ventral root, PRV-GFP after intramuscular virus injection, and Golgi staining. The first two methods work well in neonatal and young juvenile mice. Despite extensive effort, none of the three methods yielded the strong and clear labeling required for quantitative analysis of the dendritic arbor.
Finally, we undertook a behavioral characterization of the line 48 mice at P75. Line 48 mice were able to run on the treadmill for ∼20% longer than wild-type mice (p < 0.05). No other statistically significant differences in motor behavior occurred between the two groups (Table 2, 3).
Discussion
In the present work, we investigated how the GluR1 protein can control the developmental processes leading to the acquisition of motor behaviors. We focus on the elements of the spinal motor unit because, under normal circumstances, calcium-permeable AMPA receptors assembled with GluR1 are enriched in neonatal motor neurons and subpopulations of spinal cord interneurons (Bar-Peled et al., 1999; Vandenberghe et al., 2000). This is temporally coincident with the period of specification of connectivity within the segmental spinal cord and the transformation of mostly uncoordinated limb movements into functionally useful patterned output (Altman and Sudarshan, 1975; Pellis et al., 1991). We provide evidence that during postnatal development, the repertoire of AMPA receptors expressed by distinct populations of spinal cord neurons participate in the process by which synaptic activity sculpts the neuronal circuitry that underlies motor system behavior.
The first major goal of the present work was to examine the requirement of GluR1 for dendrite growth. Reducing the level of endogenously expressed GluR1 in all neurons within an ensemble in vitro inhibits dendrite growth, whether measured by overall tree size or the degree of branchiness. Similarly motor neurons of GluR1 −/− mice have a reduction in overall dendritic tree size and branching. Even in the absence of GluR1, neurons are synaptically connected in vitro and interneuronal communication (as monitored by mEPSC frequency and kinetics) is unaltered. Likewise, in GluR1 −/− mice, many aspects of segmental spinal cord network activity must be normal because these animals are viable and mobile, although they display behavioral defects. Our in vivo electromyographic recordings of standing animals also support this conclusion. Although no differences were noted in the EMG recordings from GluR1 +/+ versus GluR1 −/− mice, it is important to recognize that the recordings were obtained while animals engaged in low levels of activity (standing), and this is unlikely to be representative of the entire repertoire of motor behaviors. One interpretation of these observations is that the lack of GluR1 in cell-surface AMPA receptors leads to a subtle alteration in the electrophysiological properties of the neuron, a change that we have failed thus far to detect, and this inhibits dendrite growth. We favor this interpretation for several reasons. First, overexpression of GluR1 containing an arginine in the Q/R editing site assembles into AMPA receptors that, after activation, pass very little current, and we find that this version of GluR1 has no effect on dendrite growth in vivo (Inglis et al., 2002). Second, GluR1-dependent dendrite growth is blocked when AMPA receptor activation is pharmacologically antagonized (using 6-cyano-7-nitroquinoxaline-2,3-dione) (Jeong et al., 2006). Third, overexpression of an electrophysiologically inactive version of GluR1 (M599R), which acts in a dominant negative manner (Robert et al., 2002), has no dendrite-growth-promoting activity (Prithviraj et al., 2008). Fourth, there is a rich literature, particularly in the visual system field, showing that patterns of activity within a neuronal network control a developmental trajectory (Wiesel and Hubel, 1963; Hubel et al., 1977; Constantine-Paton and Law, 1978; Crair et al., 1998; Rajan and Cline, 1998; Debski and Cline, 2002; Haas et al., 2006). We find that the lack of GluR1 alters the pattern of segmental spinal circuit interneuronal connectivity (compare Fig. 5), and it is plausible this could lead to new patterns of network activity. The extent to which GluR1, in this scenario, acts as a permissive factor or plays an instructive role is unknown. Nevertheless, our findings are consistent with the special role played by GluR1 in synaptic stabilization and process growth (Inglis et al., 2002; Haas et al., 2006).
Although we propose that changes in network activity perturb the normal elaboration of motor neuron dendrites, it is possible that cell-surface GluR1 promotes dendrite growth via a nonelectrophysiological effect. In this regard, it is interesting that the extracellular, N-terminal domain (NTD) of GluR2, and not the channel properties of AMPA receptors assembled with GluR2, promote dendritic spine formation in vitro (Passafaro et al., 2003). It is possible the GluR2 NTD at the cell surface is a component of a receptor–ligand interaction that promotes spine growth or stability. Although the GluR1 NTD did not have the same effects in this assay system, it remains a formal possibility that the extracellular portion of GluR1 has dendrite growth promoting actions.
Does the loss of GluR1 lead to a smaller dendritic tree in a cell-autonomous manner or does the loss of GluR1 in an afferent cell population cause a “trans-synaptic” reduction in the dendrites of a neuron? The results of the in vitro (using pSUPER to express siRNA) provide clear support for the cell-autonomous mechanism. The in vivo studies (using mice with a conditional knock-out of GluR1) are more complex because, while Cre-mediated recombination occurs in virtually every lumbar motor neuron, it also occurs in lamina VIII excitatory interneurons that innervate motor neurons (Hinckley et al., 2005; Wilson et al., 2005). So either the loss of GluR1 in a motor neuron leads to the development of a smaller dendritic tree, or the loss of GluR1 in Hb9-expressing interneurons controls motor neuron dendrite growth. Of course, these alternatives are not mutually exclusive and both may be true. In any event, these results do permit us to eliminate the contribution of other motor system neurons (in the cerebral cortex and brainstem, for example) that impact motor neuron activity but do not control (in a GluR1-dependent manner) motor neuron dendrite growth.
Subsequent experiments (using penetratin-linked siRNA plus resGluR1) more sharply define the role of GluR1 expression in a network of interconnected neurons and reveal that GluR1-dependent dendrite growth is not strictly cell autonomous. Although overexpression of GluR1 in a neuron promotes dendrite growth, this only occurs if the neurons comprising the endogenous network within which it is embedded also express GluR1.
The second major aim of these studies was to investigate role of GluR1 in motor behavior and attempt to relate it to the anatomical findings. At the gross level, GluR1 +/+ and GluR1 −/− mice are indistinguishable except that the wild-type mice are generally larger than the nulls. On all of our quantitative tests of motor behavior, however, the GluR1 −/− mice are subnormal. The correlation between changes in motor neuron dendrite architecture and locomotor performance may indicate a functional link. We hypothesize that the activity-dependent development of motor system spinal cord circuitry is perturbed in the absence of GluR1 (this is reported by changes in dendrite architecture and afferent input onto motor neurons). As a result of this, we posit an alteration in the pattern of motor neuron firing and muscle usage. This is consistent with our observation of a transformation of muscle phenotype in the GluR1 −/− mice because muscle fibers types are dictated by the innervating motor neurons and a variety of experiments indicate that the motor neuron action potential firing rate is the main determinant of muscle fiber type (Lomo et al., 1974; Eken and Gundersen, 1988; Ausoni et al., 1990). The weakness in GluR1 −/− mice is likely to result from the transformation of muscle fiber types.
Elimination of GluR1 from motor neurons and a subpopulation of spinal cord interneurons (in the GluR1deltaHb9 mice) also leads to the production of a smaller motor neuron dendritic tree. The behavioral deficits displayed by these animals, however, are less marked than seen in the GluR1 −/− mice. How can one account for the differences in motor behavior between the GluR1 −/− and GluR1deltaHb9 mice? Considering the widespread of expression of GluR1 during development (Monyer et al., 1991), it is unlikely we delete GluR1 in all of the neurons devoted to motor system function (Brooks, 1986) in the GluR1deltaHb9 mice. Our results suggest that the expression of GluR1 in these non-Hb9-Cre-expressing neurons play a behaviorally significant role in the development of motor system function. Although reduction in the size and complexity of the motor neuron dendritic tree may be a prerequisite for alterations in motor function, it (in isolation) is not sufficient to impair motor function.
Whereas the lack of GluR1 leads to stunted growth of the motor neuron dendrite tree and weakness, forced expression of GluR1 in mature motor neurons appears to have the opposite effect. We showed previously that overexpression of GluR1 using herpes simplex virus causes a remodeling of the P33 motor neuron dendritic tree (Inglis et al., 2002). Here, we show that abrogating the normal downregulation of GluR1 expression in motor neurons (in our line 48 transgenic mouse) leads to supernormal locomotor performance. This is particularly remarkable because the transgene is only expressed in 50% of the motor neurons (Naciff et al., 1999). Although quantitative study of the motor neuron dendritic tree of 2.5-month-old line 48 transgenic mice was desirable, for technical reasons we could not do so. At the biochemical level, the transgene traffics normally into synaptosomal membranes and oligomerizes with appropriate AMPA subunit subunits. It is very likely this alters the electrophysiological properties of mature motor neurons and activity within the segmental spinal cord circuitry in a manner that alters muscle usage and locomotor performance.
After spinal cord injury (SCI), the portion of the spinal cord below the lesion site receives insufficient instructions for behaviorally useful output. Yet, if the circuitry within the deafferented spinal cord remains mostly intact, it is capable of remarkable plasticity (Kalb, 2003). Training programs after SCI in rats, cats, and humans can restore, albeit incompletely, weight bearing and walking (Lovely et al., 1986; Wernig et al., 1995; Edgerton et al., 1997; De Leon et al., 1998; de Leon et al., 1999; Wirz et al., 2001). This activity-dependent restoration of motor function is attributable to electrophysiological and anatomical reorganization of interneuronal communication within the segmental spinal cord (Raineteau and Schwab, 2001; Edgerton et al., 2004; Gazula et al., 2004). The present observations could be directly relevant to recovery of function after SCI. We propose that propitious manipulation of GluR1 expression by specific populations of spinal neurons will enhance spinal learning in the setting of SCI and enhance the functional benefits of training programs. We believe that a greater understanding of the cellular and molecular events occurring during the activity-dependent development of spinal cord circuitry will inform our approach to treatment of patients with SCI.
Footnotes
-
This work was supported by the United States Public Health Service, National Institutes of Health Grant NS-29837, and a grant from the Pennsylvania Department of Health. The Department specifically disclaims responsibility for any analyses, interpretation, and conclusions. We thank Carol Troy for help with siRNA linkage to penetratin, Tom Jessell for the gift of the Hb9-Cre mice, Cristian Perez for the gift of PRV-GFP, Peter Streit for the gift of anti-GluR1 antibody, and Erin Tennyson, Jacob Kalb, Angie Chung, and Sophia Khan for technical assistance.
- Correspondence should be addressed to Dr. Robert Kalb, Joseph Stokes Jr. Research Institute, Children's Hospital of Philadelphia, 3615 Civic Center Boulevard, Philadelphia, PA 19104. kalb{at}email.chop.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}