

Abstract
Emerging evidence suggests that cell-based remyelination strategies may be a feasible therapeutic approach for CNS diseases characterized by myelin deficiency as a result of trauma, congenital anomalies, or diseases. Although experimental demyelination models targeted at the transient elimination of oligodendrocytes have suggested that transplantation-based remyelination can partially restore axonal molecular structure and function, it is not clear whether such therapeutic approaches can be used to achieve functional remyelination in models associated with long-term, irreversible myelin deficiency. In this study, we transplanted adult neural precursor cells (aNPCs) from the brain of adult transgenic mice into the spinal cords of adult Shiverer (shi/shi) mice, which lack compact CNS myelin. Six weeks after transplantation, the transplanted aNPCs expressed oligodendrocyte markers, including MBP, migrated extensively along the white matter tracts of the spinal cord, and formed compact myelin. Conventional and three-dimensional confocal and electron microscopy revealed axonal ensheathment, establishment of paranodal junctional complexes leading to de novo formation of nodes of Ranvier, and partial reconstruction of the juxtaparanodal and paranodal molecular regions of axons based on Kv1.2 and Caspr (contactin-associated protein) expression by the transplanted aNPCs. Electrophysiological recordings revealed improved axonal conduction along the transplanted segments of spinal cords. We conclude that myelination of congenitally dysmyelinated adult CNS axons by grafted aNPCs results in the formation of compact myelin, reconstruction of nodes of Ranvier, and enhanced axonal conduction. These data suggest the therapeutic potential of aNPCs to promote functionally significant myelination in CNS disorders characterized by longstanding myelin deficiency.
Introduction
The myelin sheath in the CNS is synthesized by oligodendrocytes in segments separated by nodes of Ranvier, which display a highly organized molecular architecture in which Na+ channels are densely clustered at the node and K+ channels are covered by myelin in the juxtaparanodal zone. These regions are separated by the paranode, which is identified by the presence of contactin-associated protein (Caspr). Myelinating glial cells form septate-like junctional complexes with the paranodal axonal membrane, which appear to be the key to the precise localization of ion channels on axons (Rasband et al., 1999a). We and others (Rasband et al., 1998; Nashmi et al., 2000; Karimi-Abdolrezaee et al., 2004; Eftekharpour et al., 2005; Sinha et al., 2006) have shown that loss or deficiency of myelin attributable to spinal cord injury or genetic mutation alters the distribution of axonal K+ channels, which is associated with axonal functional changes.
The discovery of neural stem cells in the adult mammalian brain (Reynolds and Weiss, 1992) has suggested the possibility of their therapeutic application in CNS diseases (Sugaya, 2005). Stem and progenitor cells from a wide range of sources, including murine, primate, and human fetal or adult tissues, have been used for remyelination studies (Cummings et al., 2005; Keirstead et al., 2005; Nistor et al., 2005; Karimi-Abdolrezaee et al., 2006; Sasaki et al., 2006). Recent work by our group has shown that cellular therapy using adult neural precursor cells (aNPCs) promotes remyelination and functional recovery after spinal cord injury (Karimi-Abdolrezaee et al., 2006)
In this study, we investigated the effect of myelination on axonal structure and function of the Shiverer (shi/shi) mice, which harbor a spontaneous mutation of myelin basic protein (MBP) resulting in lack of central myelin (Bird et al., 1978; Privat et al., 1979), which is involved in myelin compaction in the CNS. The shi/shi mutant displays poorly compacted and sparse myelin sheaths, which are easily distinguishable by light and electron microscopy. Our group has recently reported that spinal cord axons in the shi/shi mice exhibit a dispersed distribution of K+ channel subunits Kv1.1 and Kv1.2 with loss of the characteristic distinction of the juxtaparanodal and paranodal areas. These structural changes are accompanied by functional deficits that can be partially reversed by K+ channel blockers (Sinha et al., 2006). Although previous work has suggested the potential of myelination to improve axonal conductive properties in myelin-deficient (md) rats (Utzschneider et al., 1994) and to reverse the molecular structural alterations in axons because of focal, transient demyelination (Rasband et al., 1998; Sasaki et al., 2006), the ability of cell-based myelination strategies to restore a more normal molecular architecture and function in axons with long-term, chronic deficiency of myelin remains essentially unexplored.
In the present study, we present novel findings that demonstrate that aNPC-mediated myelination of congenitally dysmyelinated spinal cord axons of adult shi/shi mice results in the formation of compact myelin, reconstruction of the nodes of Ranvier, and enhanced axonal conduction.
Materials and Methods
Animal care.
A total number of 56 adult shi/shi mice (C3Fe.SWV-Mbpshi/J; The Jackson Laboratory, Bar Harbor, ME) were used in this study. An inbred colony of shi/shi mice is maintained in our facility. shi/shi littermates were identified by genotyping according to protocol established by Gomez et al. (1990) and their shivering phenotype. All experimental protocols in this study were approved by the animal care committee of the University Health Network in accordance with the policies established in the guide to the care and use of experimental animals prepared by the Canadian Council of Animal Care.
Isolation and culture of adult neural precursor cells.
aNPCs were isolated from enhanced yellow florescent protein (EYFP)-expressing transgenic mice [129-Tg (ACTB-EYFP) 2Nagy/J; The Jackson Laboratory]. The isolation and generation of neurospheres were performed based on methods described previously (Tropepe et al., 1999). Briefly, mice were killed by cervical dislocation, and the brains were excised under sterile conditions and transferred to artificial CSF (aCSF) solution containing 2 m NaCl, 1 m KCl, 1 m MgCl2, 155 mm NaHCO3, 108 mm CaCl2, 1 m glucose, and 1% penicillin/streptomycin (Sigma, St. Louis, MO). After removing the overlying meninges and blood vessels, the subventricular zone of the forebrain was dissected and transferred to a low calcium aCSF solution (10 ml) containing 40 mg of trypsin, 20 mg of hyaluronidase, and 4 mg of kynurenic acid for 1 h at incubator. Then, the tissue was mechanically dissociated into a cell suspension with a fire-polished Pasteur pipette, and, after cell viability was assessed by trypan blue staining, the dissociated cells were plated on uncoated tissue culture flasks (10 cells/μl) in a serum-free medium (200 ml) containing 20 ml of DMEM/F-12, 4 ml of 30%glucose, 3 ml of 7.5% NaHCO3, 1 ml of 1 m HEPES, 200 mg of transferring, 50 mg of insulin, 19.25 mg of putrescine, 20 μl of selenium, 20 μl of progesterone and 1 μg of fibroblast growth medium 2 (FGF2), 2 μg of epidermal growth factor (EGF), and 1% penicillin/streptomycin. The neurospheres were passaged weekly by mechanical dissociation in the same medium (as above).
In vitro immunocytochemistry.
Neurospheres were mechanically dissociated into single cells and plated on matrigel-coated multichamber glass slides (4000 cells per chamber). The cells were grown in a culture medium containing the neurosphere growth medium plus 1% FBS, EGF, and basic FGF (bFGF) (20 ng/ml). To induce differentiation, the medium was replaced with one in which the growth factors were withdrawn. Then, the cultures were grown for another 5 d. On day 8, the cultures were fixed with 4% paraformaldehyde (PFA) and then washed three times with PBS. The cultures were blocked with 1% bovine serum albumin (BSA), 10% normal goat serum, and 0.3% Triton X-100 in PBS for 1 h at room temperature. The following primary antibodies were used to study the differentiation pattern of adult neural precursor cells in vitro: mouse anti-nestin (1:200; Chemicon, Temecula, CA) for neural stem/progenitor cells, mouse anti-GFAP (1:100; Chemicon) for astrocytes, rabbit anti-PDGFα receptor R (PDGFαR) (1:40; Santa Cruz Biotechnology, Santa Cruz, CA) for oligodendrocyte progenitor cells, mouse anti-CNPase (1:100; Chemicon) for mature oligodendrocytes, and mouse anti-microtubule-associated protein 2α,β (MAP-2α,β) (1:200; Sigma) for neurons. The cultures were incubated with the primary antibody in PBS plus 1% BSA, 5% normal goat serum, and 0.3% Triton X-100 for 2 h at room temperature. After washing three times with PBS, the cultures were treated with fluorescent Alexa 594 goat-anti mouse secondary antibody (1:400; Invitrogen, Carlsbad, CA) for 1 h, followed by three washes with PBS. The slides were coverslipped with Mowiol containing 4′-6-diamidino-2-phenylindole (DAPI) to counterstain the nuclei. A Zeiss (Oberkochen, Germany) LSM 510 laser confocal microscope was used for imaging.
Transplantation of adult neural precursor cells.
Young adult mice at 6–8 weeks of age were used for spinal cord transplantation. Under inhalational anesthesia using halothane (1–2%) and a 1:1 mixture of O2/N2O, the vertebral column was exposed at T5–T9, and a T6–T8 laminectomy was performed. For transplantation, the mice were injected with a cell suspension of aNPCs prepared from passage 3–4 neurospheres as described in the previous section. The cells were diluted in the growth medium (50 × 103 cells/μl) and then used for cell transplantation as reported previously by our group (Karimi-Abdolrezaee et al., 2006). Using a Hamilton syringe connected to a microglass pipette (100 μm outer diameter), two injections (a total volume of 6 μl of cell suspension containing 5 × 104 live cells/μl) were made into the dorsal spinal cord, next to the midline at the depth of 1 mm. Injection sites were 2–3 mm apart at the midthoracic (T6) spinal cord. To enhance the survival of transplanted cells, a mixture of growth factors, including bFGF (3 μg/100 μl; Sigma), EGF (3 μg/100 μl; Sigma), and PDGF-AA (1 μg/100 μl; Sigma), was mixed in a solution containing aCSF, BSA (100 μg/ml), and gentamycin (50 μg/ml) and was infused for 7 d using a subarachnoid catheter connected to an osmotic minipump (0.5 μl/h; pump model 1007D; Alzet, Cupertino, CA). The catheter (300 μm outer diameter) was implanted intrathecally at the area of transplantation. Age-matched control shi/shi mice received injection of only growth medium into the spinal cord as well as the subarachnoid infusion of growth factors. Cell transplantation in the brain was performed on neonatal 1-d-old mice anesthetized with brief hypothermia. A total of 6 μl cell suspension was injected bilaterally at 0.5 mm from the midline, 1 mm posterior to bregma, and a depth of 1.5 mm.
Immunohistochemistry.
Animals were killed with an overdose of pentobarbital and then were perfused transcardially with 4% PFA in 0.1 m PBS, pH 7.4. The brain or spinal cords were subsequently postfixed in the fixative plus 10% sucrose overnight at 4°C. After cryoprotection in 20% sucrose in PBS for 24–48 h at 4°C, the whole brain or 1.5 cm of the spinal cord centered at T6 was separated and embedded in tissue embedding media (HistoPrep; Fisher Scientific, Houston, TX) on dry ice. Cryostat sections (15 μm) were cut and mounted onto gelatin-subbed slides and stored at −20°C. For immunostaining, the frozen slides were air dried at room temperature for 10 min and then were washed with PBS for 10 min. Sections were blocked with 1% BSA, 5% nonfat milk, and 0.3% Triton X-100 in PBS for 1 h at room temperature and then were incubated with primary antibody in the same blocking solution for overnight at 4°C. The following primary antibodies were used in this study: mouse anti-nestin (1:200; Chemicon) for neural precursor cells, anti-olig2 for oligodendroglial lineage cells (1:25,000; gift from Dr. C. Stiles, Harvard Medical School, Boston, MA), rabbit anti-PDGFαR (1:40; Santa Cruz Biotechnology) for oligodendrocyte progenitor cells, mouse anti-GFAP (1:100; Chemicon) for astrocytes, mouse anti-adenomatous polyposis coli (APC) (1:40; Calbiochem, La Jolla, CA) and mouse anti-MBP (1:1000; Sternberger Monoclonals, Lutherville, MD) for mature oligodendrocytes, mouse anti-MAP-2α,β (1:200; Sigma) and mouse anti-βIII-tubulin (Tuj1) (1:500; Covance Research Products, Berkeley, CA) for neurons, and mouse anti-neurofilament 200 kDa isoform (NF200) (1:500; Sigma) for axons, rabbit Kv1.2 antibody (1:100; Alomone Labs, Jerusalem, Israel), pan-Na+ monoclonal antibody (1:50; Sigma), rabbit anti-Nav1.2 and Nav1.6 (1:100; Alomone Labs), and mouse anti-Caspr (gift from Dr. E. Peles, The Weizmann Institute of Science, Rehovot, Israel). The slides were washed three times in PBS and then incubated with appropriate fluorescent secondary antibody (Alexa 594 or 647, 1:400; Invitrogen) for 1 h. In triple labeling for YFP/MBP/NF200, YFP/Kv1.2/Caspr, YFP/Kv1.2/pan-Na+, YFP/Nav1.6/Caspr, or YFP/Nav1.2/Caspr, the slides were treated with mouse anti-MBP antibody, rabbit Kv1.2, rabbit Nav1.2, or rabbit Nav1.6 antibody, followed by Alexa 594 secondary antibody incubation. We then incubated the slides with rabbit anti-NF200 antibody, mouse anti-Caspr antibody, or mouse pan-Na+ antibody, followed by incubation with Alexa 647 secondary antibody. The slides were washed three times with PBS and then coverslipped with Mowiol containing DAPI to counterstain the nuclei. The images were taken using a Zeiss 510 laser confocal microscope.
Spinal cords with successful transplantation (presence of YFP-positive cells; n = 20 of 56) were used for immunohistochemistry and electron microcopy. Only samples containing >200 cells per longitudinal section in two sections at least 75 μm apart were used for quantification. To quantify the differentiation pattern of transplanted aNPCs, we used confocal microscopy to count the number of YFP-positive cells that were double-labeled with different cell markers. Quantification of cell differentiation was performed in an unbiased stereological manner according to the stereological principals originally described by Königsmark and Murphy (1970) and used previously by our group (Nashmi et al., 2000; Karimi-Abdolrezaee et al., 2006). Briefly, we chose the spinal cord sections with the highest number of YFP-positive cells from three animals (n = 3). For each cell marker, we immunostained two slides per mouse, which were selected ∼75 μm apart (every five sections). We selected 10 random fields that contained YFP cells and counted the number of YFP double-labeled cells with different cell markers that contained a nucleus verified by DAPI counterstaining. For quantification of nodes of Ranvier, the number of node-like structures double labeled with Nav1.6 and Caspr or Caspr and Kv1.2 subunits in wild-type, control shi/shi, and transplanted shi/shi (five fields per animal; n = 3 per group) were counted using confocal images obtained from the dorsal and lateral white matter tracts. Data were presented as nodal density (number of nodes/100 μm2) (Table 1).
Nodal quantification in wild-type, nontransplanted control shi/shi and transplanted shi/shi mice
Quantitative immunostaining for NF200, Kv1.2, and Caspr distribution.
To quantify the distribution of Kv1.2, Caspr, and NF200 along the axons, the total length of continuous immunostaining for each protein was measured using computerized image analysis (ImagePro; Media Cybernetics, Silver Spring, MD), as we have shown previously (Eftekharpour et al., 2005). A total of 20 random fields per animal (n = 4) were measured, and the average length of immunostaining for the protein of interest was compared among the spinal cord segments obtained from the wild-type, nontransplanted shi/shi, and the transplanted segments of shi/shi.
5-Bromo-2-deoxyuridine incorporation studies.
shi/shi mice received 10 injections of 5-bromo-2-deoxyuridine (BrdU) (50 mg/kg, i.p.) before they were killed. Nine injections were made for 3 d (three injections per day) and the last injection was performed 2 h before killing the animals. The animals were perfused transcardially and tissues were collected as described above. The slides were processed for immunostaining against BrdU antibody, and, after washing with PBS, they were incubated in 2N HCl and 1% Triton X-100 for 15 min at room temperature, followed by a 10 min wash in 0.1 m sodium borate in PBS for 10 min. Blocking was performed in 1% BSA, 5% nonfat milk, and 0.3% Triton X-100 in PBS for 1 h at room temperature, and then the sections were incubated with a mouse anti-BrdU antibody (1:100; BD-Immunocytometry Systems, San Jose, CA) in the same blocking solution, overnight at 4°C. The slides were washed in PBS three times and then incubated with fluorescent Alexa 594 goat-anti mouse secondary antibody (1:400; Invitrogen) for 1 h. The slides were washed three times with PBS and then coverslipped with Mowiol containing DAPI to counterstain the nuclei. The images were taken using a Zeiss 510 laser confocal microscope.
Electron microscopy.
The shi/shi mice were perfused transcardially with 4% PFA and 0.15% glutaraldehyde in phosphate buffer (PB). The spinal cords were postfixed in 4% PFA in 0.1 m PB at 4°C. The tissue blocks from the spinal cords were treated with 1% osmium tetroxide in 0.1 m PB for overnight at room temperature. Then they were dehydrated in graded ethanol solutions and embedded in Araldite502/Embed-812 embedding media (Electron Microscopy Sciences, Fort Washington, PA). Thin plastic sections were cut on an ultramicrotome, counterstained with uranyl and lead citrate, and examined with a transmission electron microscope (model 7000; Hitachi, Tokyo, Japan).
Spinal cord evoked potential recordings.
Transplanted shi/shi mice (ages 13–15 weeks) were used (n = 5) for assessing spinal cord evoked potentials (SCEPs). SCEP recording was performed based on methods previously described in our laboratory and used by others (Nashmi and Fehlings, 2001; Eftekharpour et al., 2005). During electrophysiological recordings, mice were maintained in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA) under light halothane anesthesia (1–1%). Concentric electrodes (0.3 mm; Frederick Haer Company, Bowdoinham, ME) insulated except at the tips were used for stimulation of dorsal column axons at T9–T10 and recording of compound action potentials (CAPs) at T5–T6 (see Fig. 8A). The lower segments of the spinal cord between the T10 and L1–L2 from the same animal or thoracic segments from control (nontransplanted) shi/shi were used as controls. Using a stereotaxic apparatus, the electrodes were carefully positioned 5 mm apart at the surface of the midline spinal cord. A Keypoint (Medtronic Keypoint, Skovlunde, Denmark) four-channel unit was used in these studies. The spinal cord was stimulated at 0.13 Hz, 0.1 ms pulse duration, and 1 mA constant current intensity for an average of 20 sweeps. A ground electrode was placed transdermally. The signal from the recording electrode was amplified with filters set at 100–10,000 Hz and collected. Two averaged SCEP traces with a sweep length of 5 ms were obtained to ensure reproducibility of the recordings. The peak–peak amplitude of the CAP was calculated as the value between the positive and negative peaks of the biphasic wave (Fehlings et al., 1989; Nashmi et al., 1997). Response latency was measured as the time between the onset of the stimulus artifact to the first peak, as we described previously (Nashmi et al., 1997; Eftekharpour et al., 2005). The specificity of the response was checked by transection of the cord between stimulating and recording at end of experiment to confirm that a CAP could not be detected. The estimated conduction velocity was calculated by dividing the conduction distance by the conduction time as determined by the latency.
Statistical analysis.
Cell differentiation data are presented using descriptive statistics, based on proportions expressing cell-specific markers. Kruskal–Wallis one-way ANOVA on ranks was used to analyze in the statistical differences in the molecular distribution of Kv1.2 and Caspr between the transplanted and nontransplanted segments of the spinal cord tissue from the same animal. One-way ANOVA was used, followed by a Tukey's post hoc analysis to compare the nodal quantification data. Paired t tests were used to analyze the electrophysiological data (SigmaStat SPSS statistical package; SPSS, Chicago, IL). Data are presented as means ± SD.
Results
Multipotential ability of aNPCs to generate neurons and glia
We confirmed the multipotentiality of the aNPCs by testing their ability to express neuronal and glial markers in vitro as described in recent report from our laboratory (Karimi-Abdolrezaee et al., 2006). Nestin, a marker for neural stem/progenitor cells, was detected in whole neurospheres (Fig. 1A–C) from passages 3/4. Disassociated neurospheres continued to express nestin when cultured on matrigel-coated multichamber glass slides in the presence of EGF and bFGF. After 2 d, all cultured cells showed bipolar morphology with small cell bodies and elongated thin processes that were positive for nestin (Fig. 1D–F). We then withdrew the growth factors and switched to a differentiating medium containing 1% fetal calf serum. After 1 week in culture, the cells displayed a heterogeneous morphology suggesting that they were undergoing differentiation. Immunohistochemistry confirmed that the majority of these cells acquired morphologic and antigenic properties of astroglia as indicated by their immunopositivity for GFAP (∼60%) (Fig. 1G–I). There were some oligodendroglial cells (∼30%) that stained positively for CNPase and PDGFαR (Fig. 1J–L,M–O), which are markers for mature and immature oligodendrocytes, respectively. MAP-2-positive neurons were also identified in our cultures (<10%) (Fig. 1P–R). A very small proportion of the cells remained nestin positive even after 1 week in serum-containing culture. Our results confirm the previous observations that aNPCs have the ability to differentiate into all three neural cell types in vitro (Reynolds and Weiss, 1992; Morshead et al., 1994; Weiss et al., 1996a,b; Martens et al., 2002).

Multipotential ability of aNPCs to generate neural cells in vitro. YFP-derived neurospheres (A–C) from the subventricular zone of adult transgenic mice expressed nestin. After 2–3 passages, the cells were dissociated and then cultured as a monolayer on matrigel-coated multichamber glass slides. For the first 2 d, they were maintained in serum-free growth medium containing EGF and bFGF. Immunocytochemistry on these cultures, 48 h after plating, showed uniform bipolar cells that were positive for nestin (D–F). The nestin-positive stem/progenitor cells were switched to a serum-containing medium. Five days later, immunostaining for neuronal and glial markers showed differentiation toward astrocytes (G–I), oligodendroglia (J–O), and neurons (P–R). Although the majority of YFP-derived aNPCs differentiated into GFAP-positive astrocytes in vitro (G–I), we also observed CNPase-positive mature oligodendrocytes (J–L), PDGF-αR-positive immature oligodendroglia (M–O), and MAP-2-positive mature neurons (P–R).
Lack of detectable MBP and compact myelin in the spinal cords of shi/shi mice
We confirmed the lack of myelin in the spinal cords of shi/shi mice, using hematoxylin and eosin and Luxol Fast Blue staining of cross sections of spinal cords of wild-type (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) and shi/shi (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material) mice. No detectable myelin was observed at the level of light microscopy. Electron microscopy of the spinal cords of shi/shi mice also showed the lack of multilayered compact myelin in these mutant animals (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). MBP immunostaining of spinal cords sections of wild-type (supplemental Fig. 1D–F, available at www.jneurosci.org as supplemental material) and shi/shi (supplemental Fig. 1G–I, available at www.jneurosci.org as supplemental material) mice confirmed complete absence of MBP in these animals.
YFP-labeled aNPCs survive, integrate, and migrate along the host axons in spinal cord white matter
Adult NPCs were transplanted into the midthoracic spinal cord segments of 6- to 8-week-old shi/shi mice. At the time of transplantation, an osmotic minipump was placed intrathecally at the transplanted area to deliver bFGF, EGF, and PDGF-AA during the first week after transplantation. EGF and bFGF are potent growth factors that expand the subventricular zone cell population in vitro (Reynolds and Weiss, 1992; Vescovi et al., 1993). Previous studies have shown that bFGF induces or enhances the proliferation of neuronal precursor cells (Vescovi et al., 1993; Ray and Gage, 1994) as well as bipotent neuronal/glial precursors (Vescovi et al., 1993), indicating the broad potential of this growth factor in generating a large number of cells. PDGF-AA is a growth factor secreted by type-1 astrocytes that promotes the proliferation of bipotential progenitors and O-2A cells and stimulates the differentiation of oligodendrocytes (Raff et al., 1988). Although we observed cell survival even without the addition of growth factors, our results showed that the growth factor infusion dramatically enhanced the survival of cells in spinal cords. Recent work by our group also showed an increased survival of aNPCs transplanted in the injured spinal cords after transient infusion of this growth factor mixture into the spinal cord at the time of transplantation (Karimi-Abdolrezaee et al., 2006). Growth factor delivery was also performed for the control animals in an identical manner. Six weeks after transplantation, we observed that aNPCs survived, integrated, and migrated in the spinal cord tissue (Fig. 2A). Although cells were transplanted in the dorsal areas of the spinal cords at the gray–white matter interface, we observed a preferential distribution of the cells along white matter tracts (Fig 2B). βIII-Tubulin (Tuj1) antibody was used to immunostain neurons and thereby distinguish between white and gray matter. The majority of YFP-positive cells displayed extended processes along axons and migrated as separate, distinct cells without evidence of clumping (Fig. 2C). The grafted aNPCs were able to migrate 2–3 mm from the injection sites, with no apparent preference along the rostral and caudal axis. Because adult shi/shi mice have a limited lifespan (∼15 weeks), we were not able to monitor the survival of transplanted aNPCs in the spinal cords beyond the 6 weeks after transplantation. Therefore, to examine the ability of long-term survival of the grafted aNPCs, we transplanted the cells into the brain of neonatal shi/shi mice. We observed that aNPCs survived for at least 12 weeks in the brain of shi/shi mice, and, similar to the spinal cord, they mainly resided along the white matter tracts (supplemental Fig. 2A–D, available at www.jneurosci.org as supplemental material).

YFP-labeled aNPCs survive, integrate, and migrate along white matter tracts in shi/shi spinal cord. A, Confocal image of a longitudinal section of transplanted shi/shi spinal cord. Six weeks after transplantation, YFP-labeled aNPCs migrated up to 2–3 mm away from the injection sites (*) along the rostrocaudal axis of the spinal cord. B, Immunostaining for the neuronal marker βIII-tubulin (Tuj1) showed the tendency of YFP-derived cells to reside predominantly in the white matter. C, Higher magnification of YFP-derived cells with extended processes along the white matter tracts.
Preferential differentiation of aNPCs along an oligodendroglial lineage in shi/shi mice after transplantation into the spinal cord
The antigenic properties of YFP–NPCs in transplanted spinal cords were determined using different neural cell markers. Confocal immunofluorescence microscopy with cell-specific markers was used to identify the double-labeled cells at 6 weeks after transplantation. In agreement with previous reports (Mitome et al., 2001; Windrem et al., 2004; Cummings et al., 2005), the aNPCs mainly differentiated along an oligodendroglial lineage with 78 ± 9% of YFP-positive cells staining for the oligodendrocyte transcriptional marker olig2, which is expressed in oligodendrocytes at all developmental stages (Fig. 3A–C). To further characterize the extent of differentiation of the aNPCs into oligodendroglial cells, we found that 15 ± 5% expressed PDGF-αR (Fig. 3D–F), a marker for immature oligodendroglia and 43 ± 7% expressed the mature oligodendrocyte marker APC (Fig. 3G–L). Because APC and PDGFαR are not expressed at the same developmental stage of oligodendrocyte differentiation (Butt et al., 1997), we were able to avoid double counting of the cells. YFP-positive oligodendroglial cells were intermingled with the endogenous oligodendrocytes, suggesting good integration into the host spinal cord tissue. Only 9 ± 3% of aNPCs differentiated into GFAP-positive astrocytes (Fig. 3M–O). In contrast, none of the aNPCs expressed Tuj1 (Fig. 4D–F) and MAP-2 (data not shown), which identify both immature and mature neuronal progenies, respectively. To examine whether YFP–NPCs had the potential to generate Schwann cells, immunohistochemical staining for p75 and P0, markers for nonmyelinating and myelinating Schwann cells, was performed on transplanted shi/shi spinal cord sections. Our immunostaining revealed no immunoreactivity for p75 and P0 among the YFP–NPC-derived cells (Fig. 4G–L). The transplanted cells were also negative for nestin, an intermediate filament protein that labels undifferentiated precursors (Craig et al., 1996) (Fig. 4A–C), suggesting they had acquired a more differentiated phenotype after transplantation into the spinal cord of shi/shi mice.

YFP-positive aNPCs differentiate principally into glial lineage in vivo. Confocal immunohistochemistry of spinal cord sections from transplanted shi/shi mice showed that the majority of YFP-labeled aNPCs expressed olig2, an oligodendroglial transcription factor, 6 weeks after transplantation (A–C). A significant proportion of YFP-derived cells showed colocalization of YFP-positive cells with PDGF-αR, a marker for immature oligodendrocytes (D–F). We also observed immunoreactivity for mature oligodendrocytes as shown by APC labeling (G–L). Our quantitative analysis on three transplanted shi/shi mice at 6 weeks after transplantation showed that ∼78 ± 9% of YFP-positive cells expressed olig2, which is expressed in oligodendrocytes at all developmental stages. To further characterize the extent of differentiation of the aNPCs into oligodendroglial cells, we found that 43 ± 7% expressed APC and 15 ± 5% expressed PDGF-αR. A small proportion of YFP cells (9% ± 4) also showed GFAP immunoreactivity in their cell bodies and were thus considered as astrocytes (M–O).

YFP-positive aNPCs do not express nestin or markers of neurons or Schwann cells. Confocal immunohistochemistry of transplanted shi/shi mice 6 weeks after transplantation showed no nestin expression in aNPCs (A–C). Although some of the YFP-labeled aNPCs were closely associated with Tuj1-positive neuronal processes (D–E), no YFP-positive neuronal somata were observed. We also did not detect any p75 (G–I) or myelinating P0-positive (J–L) Schwann cells among YFP-positive aNPCs. Insets in I and L show control positive staining for p75 and P0 in the peripheral roots of the shi/shi spinal cord.
Transplanted aNPCs do not show any proliferative activity 6 weeks after transplantation into shi/shi spinal cords
To examine the proliferative activity of these cells after transplantation, some animals received injections of BrdU at 1, 3, and 6 weeks after transplantation (n = 2 per group). At 1 week after transplantation, a very low rate (<1%) of YFP-positive cells were BrdU labeled. Moreover, at 3 and 6 weeks after transplantation, none of the transplanted cells were labeled with BrdU, suggesting a low probability of tumor formation by the transplanted aNPCs (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
YFP-labeled oligodendrocytes express MBP in transplanted spinal cord segments
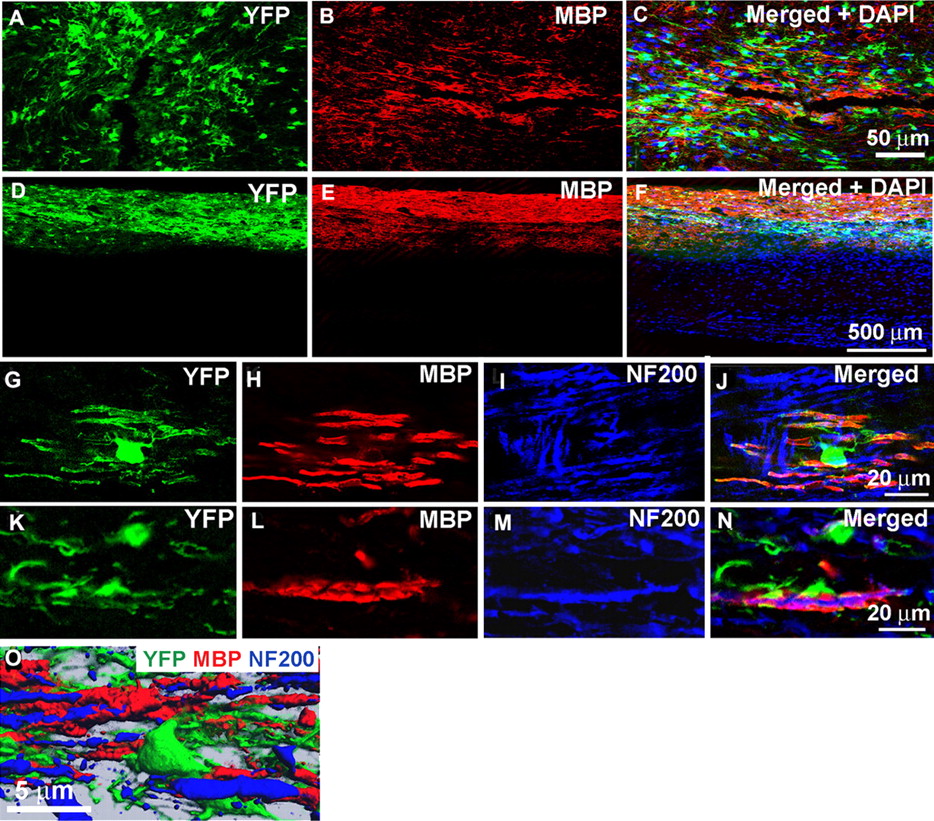
To investigate whether the NPC-derived oligodendrocytes were capable of myelin formation, MBP expression was assessed in transplanted and the age-matched shi/shi control animals, which did not receive aNPC transplantation. In contrast to control shi/shi mice, the aNPC transplanted segments of shi/shi spinal cords contained a large number of YFP/MBP double-positive oligodendrocytes as early as 1 week (Fig. 5A–C) after transplantation. Moreover, the expression of MBP in the spinal cord continued to increase over the 6 week posttransplantation period (Fig. 5D–F). MBP expression by transplanted cells persisted for up to 12 weeks (the duration of the experiment) after transplantation into the brains of newborn shi/shi mice (supplemental Fig. 2C,D, available at www.jneurosci.org as supplemental material). High-power confocal immunohistochemistry showed that the transplanted oligodendrocytes mimicked the normal myelination pattern of oligodendrocytes, because a single YFP–oligodendrocyte could be observed sending MBP-positive processes around several neighboring axons (Fig. 5G–N). A close association of the NPC-derived oligodendrocytes with the NF-200-positive axons was shown with high-power confocal microscopy (Fig. 5G–N) as well as three-dimensional (3D) reconstructed images (Fig. 5O).

YFP-positive oligodendrocytes express MBP. Confocal immunohistochemistry on transplanted shi/shi mice spinal cord sections revealed MBP expression by transplanted aNPCs. Although no MBP was detectable in the naive shi/shi spinal cords, transplanted spinal cord segments of shi/shi mice showed patches of MBP as early as 1 week after transplantation (A–C). After 6 weeks, NPC-transplanted segments displayed extensive MBP expression (D–F). After NPC transplantation, one YFP-positive oligodendrocyte ensheathed several surrounding axons (labeled with NF200) and produced MBP (G–N). Three-dimensional reconstruction of YFP-positive transplanted cells in close association with the host spinal cord axons is shown (O).
NPC transplants promote formation of compact myelin in shi/shi spinal cord axons
The myelinating capacity of the NPC-derived oligodendrocytes was also examined using transmission electron microscopy. Nonmyelinated axons in shi/shi spinal cord were readily recognizable by their thin, 2–3 layered ensheathment by the endogenous oligodendrocytes (Fig. 6A,B). At 6 weeks after transplantation, we observed newly myelinated axons identified by their multilayered myelin figures (Fig. 6C,D). Multilayered compact myelin sheath reached an approximate thickness of 100 nm (Fig. 6E,F). Electron microscopy on longitudinal sections of the spinal cord axons was used to investigate the formation of node of Ranvier and establishment of paranodal junctional complexes by the aNPC-derived cells. The paranodal area in wild-type animals was distinguished by an extensive network of oligodendrocyte processes that bind to the axonal membrane on either sides of the node, known as paranodal junctional complex (Fig. 6G, arrows). Axons in the control regions of shi/shi spinal cord displayed sporadic and irregular contacts with the processes of the endogenous glial cells (Fig. 6H). In aNPC-transplanted areas, we occasionally found node-like structures containing the paranodal junctional complex (Fig. 6I–K). Moreover, the formation of node-like structures was also confirmed by 3D reconstruction of immunoconfocal microscopy of aNPC-derived oligodendrocytes that expressed MBP and ensheathed the spinal cord axons of shi/shi mice (Fig. 7U).

NPC transplants induced formation of compact myelin, appearance of nodes of Ranvier, and paranodal junctional complex. Electron micrographs from a 14-week-old naive shi/shi spinal cord (A) showed a lack of compact myelin. The myelin sheaths in shi/shi mice were characterized by 2–3 layers of noncompacted membrane (B). Six weeks after transplantation, many myelinated axons were observed in transplanted segments of spinal cord axons (C, D) containing multilayered compact myelin (E, F). Electron microscopy on longitudinal sections of spinal cord in wild type (G), naive shi/shi (H), and transplanted (I) to show the paranodal junctional complex. Paranodal junctions (arrows) were detectable on either side of the nodes of Ranvier in wild-type animals (G) marking the paranodal zone. We did not recognize the paranodal zone in control animals, and only sporadic and irregular contacts between the glial cells and axonal membrane were detected (H, arrows). Occasionally, node-like structures were observed in transplanted animals (I). Higher-magnification micrographs confirmed the presence of paranodal zones in which the processes of transplanted aNPC-derived myelinating oligodendrocytes formed paranodal junctional complex with the axonal membrane (J, K, arrows).

Transplanted aNPCs promote the aggregation of K+ channels and the formation of nodes of Ranvier in the spinal cord axons of shi/shi mice. Confocal immunostaining of Kv1.2 subunits (red) and pan-Na+ channels (blue) in the spinal cord of wild-type mice (A–C), control shi/shi mice (G–I), and transplanted shi/shi mice (M–P) is depicted. Kv1.2 subunits were clearly localized to the juxtaparanodal regions of wild-type spinal cord axons (A–C), confirmed with nodal pan-Na+ immunostaining. In shi/shi mice, Kv1.2 immunostaining was abnormally distributed along the axonal internodes (G–I), but Na+ clusters were observed as aberrant nodal aggregates. Six weeks after aNPCs transplantation, spinal cord segments of shi/shi mice showed restoration of Kv1.2 subunit clusters (M–P). YFP-positive processes of transplanted aNPCs were observed in close association with axons containing restored K+ channels aggregates (P). Nodal localization of Na+ channels was further confirmed using Nav1.6 (red) immunostaining in wild-type (D–F), control shi/shi (J–L), and transplanted shi/shi (Q–T). Caspr (blue) immunostaining was used to identify the paranodal area. A 3D reconstruction clearly shows a node of Ranvier that is bordered by an MBP-expressing NPC derived oligodendrocyte. Note that the processes of YFP-labeled oligodendrocytes avoid the nodal region (U).
Myelination of shi/shi spinal cord axons induces the clustering of Caspr and Kv1.2 subunits at the paranodes and juxtaparanodes
Myelinated fibers in the CNS are characterized by clusters of Na+ channels at the nodes of Ranvier and K+ channels in the juxtaparanodal regions. Using pan-Na+ antibody, which recognizes all of the mammalian Na+ channel subtypes, we observed nodal aggregations of Na+ channels in wild-type axons, which was confirmed by juxtaparanodal staining of Kv1.2 (Fig. 7A–C). In control segments of shi/shi spinal cords, aberrant node-like aggregation of Na+ channels were observed, but Kv1.2 K+ channels subunits were irregularly distributed along the internodes of axons with a complete loss of the juxtaparanodal paired localization (Fig. 7G–I). In contrast, in the transplanted segments of the shi/shi spinal cord, paired clusters of Kv1.2 subunits were frequently observed at juxtaparanodal location, which strongly suggests that myelination can reverse the aberrantly dispersed distribution of K+ channel subunits in dysmyelinated axons (Fig. 7M–P). To further characterize the distribution of Na+ channel subtypes, we used specific antibodies against Nav1.2 and Nav1.6 subunits. Previous reports show that Nav1.2 is distributed along the unmyelinated developing axons, and, as myelin is formed, Nav1.6 replaces the Nav1.2 at the nodal area (Boiko et al., 2001). We detected nodal aggregation of Nav1.6 in wild-type (Fig. 7D–F), control shi/shi (Fig. 7J–L), and transplanted shi/shi (Fig. 7Q–T) spinal cords in close association with paranodal marker Caspr. This is in agreement with previous observations by Arroyo et al. (2002) that showed no detectable alteration in the node-like localization of Na+ channels in dysmyelinated md rats. Furthermore, immunolabeling of Nav1.2 in mutant shi/shi mice showed a similar staining pattern to the wild-type counterparts with no detectable nodal aggregations of Nav1.2 (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The localization of Caspr (Fig. 8A–C,D–F) was also altered in the control shi/shi spinal cord axons (Fig. 8G–I,J–L), such that Caspr become abnormally dispersed as patches along the juxtaparanodes and internodes of axons. In contrast to the control shi/shi spinal cords, Kv1.2 and Caspr immunostaining showed a greater reorganization into paired structures surrounding the nodes of Ranvier in the transplanted animals (Fig. 8M–O,P–R). To assess the effect of myelination by transplanted aNPCs on the molecular organization of dysmyelinated CNS axons, we measured the distribution of Kv1.2 subunits and Caspr protein along the spinal cord axons of shi/shi mice. The total length of continuous immunostaining of NF200, Kv1.2 subunit, and Caspr along the spinal cord axons was quantified using computerized image analysis. Nontransplanted adjacent segments of the same animal were used as an internal control. We first investigated whether aNPC transplantation caused any changes in axonal counts, using NF200 as an axonal marker. No apparent differences were observed in the average length of continuous NF200-positive immunoreactive axonal profiles between control and transplanted segments (531.802 ± 101.943 μm in wild type, 472.322 ± 104.571 μm in control shi/shi, and 553.002 ± 123.188 μm in shi/shi transplanted; Student's t test, p = 0.582). We next quantified the total length of Kv1.2 subunit or Caspr distribution along the spinal cord axons of wild-type littermate, control, and transplanted shi/shi mice. We observed a dramatic (70%) reduction in the length of distribution of Kv1.2 subunit [in the spinal cord axons of transplanted shi/shi mice compared with the controls at 6 weeks after transplantation (493.84 ± 296.52 μm in the control segments, 255.30 ± 94.14 μm in NPC-transplanted segments, and 212.34 ± 54.67 μm in wild-type mice; p ≤ 0.001] (Fig. 8S). With aNPC transplants, Caspr also exhibited a tighter and less dispersed axonal distribution (600.89 ± 295.57 μm in control, 440.21 ± 254.06 μm in transplanted segments, and 234.79 ± 55.24 μm in wild type; p ≤ 0.001) (Fig. 8T).
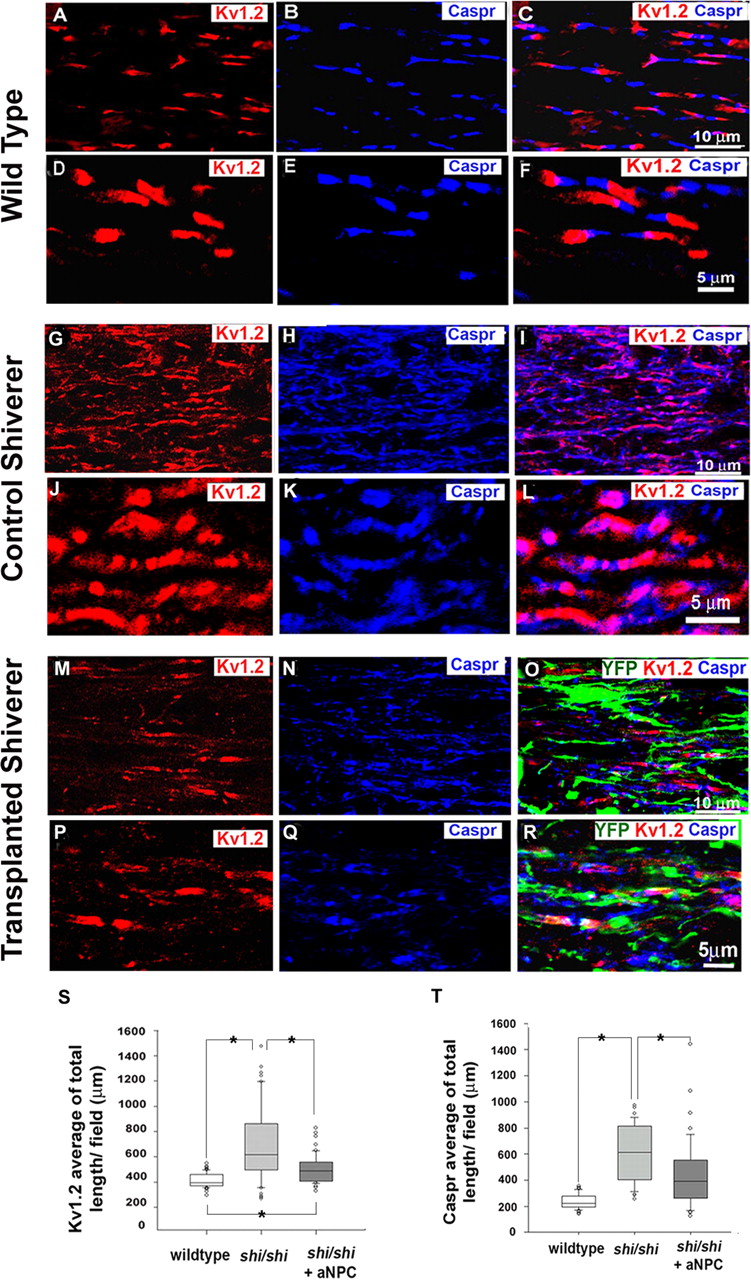
Transplantation of aNPCs leads to shortened distribution of K+ channel and Caspr and reconstruction of paranodal and juxtaparanodal regions in the spinal cord axons of shi/shi mice. The normal juxtaparanodal localization of Kv1.2 channel subunits and the paranodal localization of Caspr (A–F) in wild-type mice were completely disrupted in the spinal cord axons of control shi/shi mice (G–L). In contrast, the transplanted segments of shi/shi mice spinal cords showed evidence of a more normal distribution of Kv1.2 channel subunit and Caspr (M–R). Quantification of the confocal immunostaining data revealed that the distribution of the Kv1.2 K+ channel subunits in the transplanted area was significantly more compact (70%) than in the segments of the spinal cord of the same mice, which was remote to the site of transplantation. Of note, Caspr immunohistochemistry also showed a significant (27%) decrease in the longitudinal distribution along the axons. Box plots S and T show the distribution of total length of Kv1.2 and Caspr in all three groups (Kruskal–Wallis one-way ANOVA on ranks, *p < 0.001; n = 4 per group).
Myelination of shi/shi spinal cord axons induces formation of node-like clusters along the axons
To investigate the effect of myelination on the formation of nodes of Ranvier, we first quantified the node-like structures identified by Caspr stained regions flanked by focal Kv1.2-immunopositive zones in the spinal cord of the shi/shi control, transplanted shi/shi, and wild-type mice (Fig. 7, Table 1). Nodal structures were easily and frequently observed in wild-type animals, whereas in the control shi/shi mice such structures were infrequently identifiable on spinal cord axons. A significant increase in the node frequency was observed between the transplanted and control shi/shi mice (p < 0.001, one-way ANOVA). We also used double labeling for Nav1.6 and Caspr for nodal quantification. Similar to md rats (Arroyo et al., 2002), in shi/shi mice, Nav1.6 subunits were found as isolated patches with no association with Caspr (naked) or were flanked by Caspr on one side (hemi-node) or both sides (node-like) (Fig. 7J–L). We only quantified the node-like structures in wild-type, transplanted, and control shi/shi mice (Table 1). We detected a significant decrease in frequency of these node-like structures in shi/shi mice compared with the wild-type animals (p < 0.001). No detectable changes in nodal density of Na+ channels were observed after NPC myelination (p = 0.335).
Myelination of spinal cord segments in shi/shi mice results in improved axonal conduction
To assess the effect of myelination on axonal conduction, we used in vivo SCEP recordings. Briefly, the dura was opened using a pair of fine scissors, and the stimulating and recording electrodes were placed precisely 5 mm apart on the dorsal surface of the cord in the transplanted area. Several controls were used in these experiments. We used the upper lumbar (nontransplanted) segments of the same animals as an internal control for the recordings. As an additional control, recordings were performed in control shi/shi mice. These data were similar (p = 0.237, student's t test) to the recordings derived from the nontransplanted spinal cord segments (internal controls) (Fig. 9C,D). Our results revealed improved axonal conduction in transplanted spinal cord segments as shown by an increase in the amplitude and decreased latency of SCEP responses (Table 2). To estimate the conduction velocity, the recording distance was divided by the conduction time (response latency). Our results showed an increase in the estimated conduction velocity for the transplanted cord segments (nontransplanted, 3.35 ± 0.95 m/s; the transplanted segments, 6.21 ± 2.15 m/s). However, the estimated conduction velocity of transplanted shi/shi spinal cord axons was still significantly different than the age-matched wild-type spinal cords (18.59 ± 1.27 m/s) (Fig. 9G).

Transplantation of aNPCs improves axonal conduction in shi/shi mouse as shown by increased amplitude and decreased latency. A, Electrodes were positioned 5 mm apart on the dorsal surface of the cord at the transplanted area. Control recordings were done from either nontransplanted segments (T10–L1) of the transplanted animal or from thoracic segments of control (nontransplanted) shi/shi mice: B, wild-type mouse; C, control shi/shi spinal cord; D, nontransplanted segments of cord; and E, transplanted shi/shi mouse cord. F, At the end of the experiment, the cord was cut to confirm the specificity of the CAP. Electrophysiological measurements showed reduced latency, increased amplitude, and enhanced estimated conduction velocity of transplanted spinal cord segments when compared with nontransplanted cord segments (paired t test, *p = 0.04; n = 5) in transplanted spinal cords.
Improved axonal conduction properties after NPC transplantation in shi/shi mice
Discussion
In this study, we show that adult NPCs differentiate preferentially along an oligodendroglial lineage, induce myelination, initiate the formation of the nodes of Ranvier, and promote enhanced axonal conduction of dysmyelinated spinal cord axons in adult shi/shi mice. Although previous work has revealed that cell-based myelination strategies can promote structural and functional restoration after experimental demyelination (Sasaki et al., 2006) or in dysmyelinated neonatal md rats (Utzschneider et al., 1994), our work indicates that long-term molecular abnormalities can be successfully reversed in the adult dysmyelinated CNS axons with aNPC transplantation.
Effect of dysmyelination on structural and molecular organization of node of Ranvier in CNS
Alteration of axonal membrane proteins occurs with myelin disruption because of trauma, disease, or in a variety of experimental or genetic models (Dugandzija-Novakovic et al., 1995; Nashmi and Fehlings, 2001; Arroyo et al., 2002; Karimi-Abdolrezaee et al., 2004; Woodruff et al., 2004; Eftekharpour et al., 2005; Sasaki et al., 2006; Sinha et al., 2006). Genetic models of dysmyelination such as shi/shi and jimpy mice and md rats have been used to study the axoglial interactions and the molecular consequences of myelin deficiency. jimpy mice and md rats die within 3–4 weeks after birth and are characterized by lack of central myelin and progressive oligodendrocyte death. These changes result in a lack of paranodal junctional complex, dispersed axonal expression of Caspr and K+ channels, whereas the nodal localization of Na+ channels remains unaltered (Mathis et al., 2001; Arroyo et al., 2002). shi/shi mice represent a less severe CNS dysmyelination model and live longer than comparable mutants. In contrast to md and jimpy mutants, shi/shi mice contain abundant oligodendrocytes (Brundin et al., 2003) with aberrant paranodal junctions (Rosenbluth, 1981), which may explain the patchy staining of Caspr along spinal cord axons observed in this study and reported previously in optic nerve (Tait et al., 2000). To our knowledge, our study is the first to demonstrate a dispersed distribution of Caspr along the spinal cord axons of shi/shi mice. We also observed a diffuse axonal distribution of Kv1.1 and Kv1.2 subunits, in agreement with previous reports (Rasband et al., 1999a; Sinha et al., 2006).
In contrast to Caspr and K+ channels, Na+ channels are less affected by congenital dysmyelination. Using both a pan anti-Na+ channel antibody and an Nav1.6-specific antibody, we showed that the focal nodal expression of Nav1.6 is not disrupted in dysmyelinated spinal cord axons of adult shi/shi mice. This is in agreement with findings in md rats (Arroyo et al., 2002). We also detected a decreased nodal frequency in shi/shi mice spinal axons compared with their wild-type counterparts, which confirms previous reports (Rasband et al., 1999b). Although axoglial interactions during early development appear to induce Na+ channel clustering at the nodes (Baba et al., 1999; Ishii et al., 1999; Poliak et al., 2001), the regulatory mechanisms underlying their axonal localization (Boiko et al., 2003) remain unclear.
Myelination approaches to restore axonal molecular structure and function
Recent studies have shown that transplanted cells such as olfactory ensheathing glia (Sasaki et al., 2006) or Schwann cells (Black et al., 2006) can remyelinate axons, restore axonal conduction and promote reconstruction of nodal regions of the CNS after experimental demyelination, in which the endogenous glial cells are selectively killed but axons are spared. These studies show that the existing molecular machinery in healthy axons after experimental demyelination can be successfully redirected to form such complex molecular architecture as early as 3 weeks (Sasaki et al., 2006) after transplantation, which can be maintained up to 1 year after remyelination (Black et al., 2006). To investigate whether it is possible to reverse such altered molecular structures in developmentally dysmyelinated models, we used shi/shi mice, which lack the myelinating ability of the endogenous oligodendrocytes.
Various cellular myelination strategies have been studied in shi/shi mice (Gumpel et al., 1989; Baron-Van Evercooren et al., 1992; Vignais et al., 1993; Yandava et al., 1999; Liu et al., 2000; Mitome et al., 2001; Windrem et al., 2004; Cummings et al., 2005; Nistor et al., 2005; McKenzie et al., 2006). shi/shi mice represent a more clinically relevant model compared with models in which demyelination is induced. In experimental models of transient demyelination, the residing oligodendrocytes and astrocytes are lost and the transplanted cells face no competition with the endogenous cells to ensheath normal axons and establish the junctional complex necessary for node formation. In contrast, in shi/shi mice, endogenous oligodendrocytes are present in large numbers (Brundin et al., 2003), which may interfere with the function of transplanted cells. In the present study, we transplanted aNPCs into the neonatal brain (postnatal day 1) or the spinal cords of young adult mice (6–8 weeks old). Animals did not receive any immunosuppression and were allowed to survive to the age of 13–14 weeks. Transplantation of aNPCs in neonatal shi/shi brain was chosen to study the maximum survival of the transplanted cells. We noticed that, although >80% of brain transplantations were successful, only 30% of the transplanted spinal cords contained sufficient cell numbers for histological analysis. The technical challenges of cell transplantation, the use of adult mice (Roser, 1989), and lack of immunosuppression might be responsible for the lower success rate in the spinal cord. With technical refinements in microsurgical technique and the use of less toxic immunosuppressants, we anticipate improving the success of spinal cord transplantation in the shi/shi mice.
Myelination of shi/shi spinal cord tracts, in our study, was associated with a 70% normalization of K+ channel distribution along the axons. The decreased distribution of Caspr staining was more modest (∼27%). The differential impact of myelination on Caspr and Kv1.2 subunit restoration may reflect different signaling mechanisms for their localization along axons. Although K+ channel juxtaparanodal localization is dictated by compact myelin, as was suggested previously (Baba et al., 1999), the formation of axoglial junctions is sufficient for Caspr paranodal localization. Reorganization of Caspr immunostaining in our studies suggests that transplantation of aNPCs results in formation of normal axoglial junctions but decreases the aberrant axoglial contacts. Despite increased node formation as confirmed by frequent K+ channel clusters around the nodes of Ranvier, some overlap was still observed in Kv1.2 and Caspr expression along axons, which is an indication of formation of new nodes. Of note, similar transient interactions between Caspr and K+ channels have been reported during the early stages of myelination, which is corrected by further maturation of axoglial binding. Considering the relatively short lifespan of shi/shi mice, an examination of the long-term axoglial interactions that could lead to a more focal distribution of Caspr in the paranodal zone would be impractical.
In this study, we observed that aNPC-induced myelination promoted enhanced spinal cord axonal conduction in the shi/shi mice as evidenced by an increased amplitude, reduced latency, and enhanced estimated conduction velocity. This work confirms previous reports in md rats with in vitro recording techniques (Utzschneider et al., 1994). In our in vivo work, similar to Lo et al. (2003), we used a one-point recording technique to elicit SCEPs in shi/shi mice because of the technical challenges posed by their small size. Although the observed shift in latency in transplanted shi/shi mouse spinal cord axons could also reflect an alteration in axonal excitability, the use of supramaximal stimulation would suggest that the latency shift most likely reflects an enhanced conduction velocity. Nonetheless, to be conservative, we referred to our calculations of conduction velocity as “estimates.” A two-point recording method would more accurately measure the conduction velocity; however, the application of such a technique in the shi/shi mouse in vivo proved to be technically formidable. Of note, our conduction velocity estimates in shi/shi mice are comparable with those reported by Lo et al. (2003) in a murine model of encephalomyelitis. Moreover, our data are congruent with in vitro recordings in isolated dorsal column segments of md rats (Utzschneider et al., 1994). Confirmation of our in vivo recording estimates will require more detailed analysis in vitro by methods such as the sucrose gap technique (Sinha et al., 2006).
In conclusion, we have provided evidence that transplanted aNPCs can successfully integrate among the endogenous shi/shi oligodendrocytes, migrate along the axons, establish anatomically normal axoglial junctions, and myelinate the CNS axons. To our knowledge, our study is the first to report the potential of aNPCs for myelination and restoration of the molecular structure of the node of Ranvier in spinal cord axons of adult shi/shi mice. These data along with those reported previously (Rasband et al., 1998; Black et al., 2006; Sasaki et al., 2006) indicate that formation of myelin through endogenous or transplanted glial cells, in a variety of experimental models, results in restoration of axonal molecular structure and organization of functional nodes of Ranvier with appropriate nodal distribution of Nav1.6, paranodal aggregation of Caspr, and juxtaparanodal localization of Kv1.2. These results are of potentially major translational significance, because restoration of a normal axonal molecular architecture is of critical importance in ameliorating neurological deficits secondary to loss of myelin in the CNS.
Footnotes
-
This work was supported by operating grants from the Canadian Institutes of Health Research (CIHR) to M.G.F (CIHR-MT14459) and Stem Cell Network to C.M.M. M.G.F. and C.M.M. are also partially supported by a CIHR New Emerging Teams Grant in Regenerative Medicine and Nanotechnology. M.G.F. holds the Krembil Chair in Neural Repair and Regeneration. S.K.-A. was supported by fellowships from CIHR and Ontario Neurotrauma Foundation. E.E. was supported by a fellowship grant from the Christopher Reeve Paralysis and Sam Schmidt Paralysis Foundations. We thank Dr. E. Peles for providing us with the Caspr antibody and Dr. C. Stiles for the gift of Olig2 antibody.
- Correspondence should be addressed to Dr. Michael G. Fehlings, Division of Neurosurgery, University of Toronto, Toronto Western Hospital, University Health Network, Room 4W-449, 399 Bathurst Street, Toronto, Ontario, Canada M5T 2S8. michael.fehlings{at}uhn.on.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}