

Abstract
Aging is associated with learning deficits and a decrease in neuronal excitability, reflected by an enhanced post-burst afterhyperpolarization (AHP), in CA1 hippocampal pyramidal neurons. To identify the current(s) underlying the AHP altered in aging neurons, whole-cell voltage-clamp recording experiments were performed in hippocampal slices from young and aging rabbits. Similar to previous reports, aging neurons were found to rest at more hyperpolarized potentials and have larger AHPs than young neurons. Given that compounds that reduce the slow outward calcium-activated potassium current (sIAHP), a major constituent of the AHP, also facilitate learning in aging animals, the sIAHP was pharmacologically isolated and characterized. Aging neurons were found to have an enhanced sIAHP, the amplitude of which was significantly correlated to the amplitude of the AHP (r = 0.63; p < 0.001). Thus, an enhanced sIAHP contributes to the enhanced AHP in aging. No differences were found in the membrane resistance, capacitance, or kinetic and voltage-dependent properties of the sIAHP. Because enhanced AHP in aging neurons has been hypothesized to be secondary to an enhanced Ca2+ influx via the voltage-gated L-type Ca2+ channels, we further examined the sIAHP in the presence of an L-type Ca2+ channel blocker, nimodipine (10 μm). Nimodipine caused quantitatively greater reductions in the sIAHP in aging neurons than in young neurons; however, the residual sIAHP was still significantly larger in aging neurons than in young neurons. Our data, in conjunction with previous studies showing a correlation between the AHP and learning, suggest that the enhancement of the sIAHP in aging is a mechanism that contributes to age-related learning deficits.
- slow afterhyperpolarization
- aging
- L-type Ca2+ channels
- whole-cell voltage clamp
- current clamp
- neuronal excitability
- plasticity
- nimodipine
Action potentials in hippocampal pyramidal neurons are followed by a post-burst afterhyperpolarization (AHP), which serves to limit further firing in response to sustained excitation in a process known as spike frequency adaptation (accommodation). Reductions in the AHP and accommodation have been observed in neurons from animals trained in various hippocampus-dependent (Moyer et al., 1996, 2000; Thompson et al., 1996b; Oh et al., 2001) and non-hippocampus-dependent tasks (Disterhoft et al., 1986; Coulter et al., 1989; Saar et al., 1998), suggesting that it is a general mechanism to increase neuronal excitability in learning.
The AHP and accommodation are reduced after the acquisition of the hippocampus-dependent trace eyeblink conditioning task in CA1 and CA3 hippocampal pyramidal neurons (Moyer et al., 1996, 2000; Thompson et al., 1996b). These biophysical changes are most likely learning induced, because they are not observed in neurons of pseudoconditioned controls (which receive the same but unpaired stimuli), naive controls, and animals that failed to acquire the task. Furthermore, reductions in the AHP and accommodation return to baseline in 7 d (Moyer et al., 1996; Thompson et al., 1996b), consistent with the hypothesis that the hippocampus functions as an intermediate storage buffer during learning (Cohen and Eichenbaum, 1993; Kim et al., 1995;Disterhoft et al., 1996).
Acquisition of the trace eyeblink conditioned response is impaired in aging animals and aging humans (Thompson et al., 1996a;Weiss et al., 2000; Knuttinen et al., 2001a,b). Interestingly, the AHP and accommodation are enhanced in CA1 neurons from animals at ages that show learning deficits (Landfield and Pitler, 1984; Moyer et al., 1992,2000). Although many aging animals failed to acquire the trace eyeblink conditioned response (Thompson et al., 1996a), those that did learn also showed a reduction in the AHP (Moyer et al., 2000). This inverse correlation between the AHP and learning has led us to hypothesize that AHP enhancement in aging is a mechanism involved in age-related learning deficits. Consistent with our hypothesis, drugs that reduce the AHP in vitro have also been shown to improve learning in aging animals (Deyo et al., 1989; Moyer et al., 1992;Kronforst-Collins et al., 1997; Oh et al., 1999; Weiss et al., 2000,Power et al., 2001).
The AHP is mediated by four outward K+currents (IC,IM,IAHP, and sIAHP), and its time course is modulated by the hyperpolarization-activated current,Ih (Storm, 1990; Maccaferri et al., 1993; Stocker et al., 1999; Oh et al., 2000). Given the kinetics of these currents, previous experiments showing prolonged AHP and enhanced accommodation in aging neurons strongly implicate alterations in the slower currents, particularly the sIAHP, in aging (Landfield and Pitler, 1984; Moyer et al., 1992, 2000; Power et al., 2001). The following experiments were undertaken to determine whether theIAHP–sIAHPcontributes to the enhanced AHP in aging neurons.
MATERIALS AND METHODS
Subjects. Young (2–3 months old) and aging (>36 months old) female New Zealand albino rabbits (Oryctolagus cuniculus) were chosen as subjects. Animal use procedures were approved by Northwestern University's Animal Care and Use Committee, according to the standards of the United States Department of Agriculture.
Slice preparation. Slices were prepared following procedures similar to those described previously (Moyer et al., 1996). Briefly, two bathing solutions were used during the dissection: (1) normal artificial CSF (aCSF) composed of (in mm): 124 NaCl, 3 KCl, 1.3 MgSO4, 1.24 NaH2PO4, 2.4 CaCl2, 26 NaHCO3, 10d-glucose, and (2) sucrose–aCSF, containing an equiosmolar concentration of sucrose in place of NaCl. Both normal– and sucrose–aCSF solutions were oxygenated (bubbled with 95% O2–5% CO2, pH 7.4).
Rabbits were anesthetized with halothane and killed by decapitation. The brain was rapidly exposed in situ, hemisected, removed within 60 sec, and immediately immersed in oxygenated ice-cold (<1°C) sucrose–aCSF for ∼4 min. Both hippocampi were quickly dissected out, cut into 3–4 mm chunks, and glued onto chilled chambers that were then filled with ice-cold sucrose–aCSF. Transverse slices (300 μm) along the dorsal-ventral gradient of the hippocampus were prepared using Vibratomes (TPI, O'Fallon, MO) and placed in holding chambers filled with normal–aCSF at room temperature (∼23°C) for at least 45 min before any experiment.
Whole-cell electrodes and solutions. Patch electrodes were made from borosilicate glass using a Sutter Flaming-Brown horizontal puller (P-87; Sutter Instrument Company, Novato, CA) and heat-polished with a Narishige microforge (Model MF-930; Narishige International USA, Inc., East Meadow, NY) to a resistance of 1.5–6.0 MΩ. Three pipette solutions were used and consisted of the following (in mm): (1) 2 K-ATP, 10 HEPES, and 150 KMeSO4; (2) 2 K-ATP, 10 HEPES, 140 KMeSO4, and 10 KCl; and (3) 2 K-ATP, 10 HEPES, 130 KMeSO4, and 10 KCl. The pH of these solutions was adjusted to 7.25 with KOH; the osmolarity of these solutions was 290 ± 10 mOsm. The results obtained with these solutions were indistinguishable. MeSO4− was used in place of Cl− to reduce the rundown of the AHP current over time (Zhang et al., 1994). The use of KMeSO4 in the electrode solution resulted in a ∼10 mV junction potential with respect to the aCSF. This junction potential was measured for every batch of electrode solution according to procedures published by Neher (1992) and was corrected before data acquisition.
Data analysis and cell criteria. Hippocampal neurons were visualized using either a Zeiss Axioskop (Carl Zeiss, Inc., Oberkochen, Germany) or a Leica DM LFS microscope (Leica Microsystems AG, Wetzlar, Germany); both were equipped with a long working distance 40× water immersion objective and infrared differential interference contrast optics (Dodt and Zieglansberger, 1990; Stuart et al. 1993).
Two data acquisition systems were developed using either LabVIEW (National Instrument, Austin, TX) or C++ Builder (Borland Software Corporation, Scotts Valley, CA) with National Instrument NIDAQ driver. Data were acquired at 5 kHz and filtered at 2 kHz using a low-pass Bessel filter. Whole-cell recording procedures for young and aging neurons were variants of previously published procedures, using a single pipette to “clean” and patch onto the neuron (Blanton et al., 1989). Recordings were made from the soma of visually identified CA1 pyramidal neurons with seal resistances >1.5 GΩ before break through into the whole-cell mode. A few cells were labeled with Lucifer yellow to monitor the equilibration between the intracellular milieu and the pipette solution. All measurements were made at least 10 min after membrane rupture to allow for adequate solution equilibration. Only neurons with series resistance <15 MΩ, membrane resistance >60 MΩ, resting potential less than −60 mV, and for current-clamp recordings action potential amplitude >90 mV from resting potential were included in the data set.
Current-clamp protocols. The AHP was first measured in current-clamp mode to provide a direct comparison for previous studies. Subsequently, theIAHP–sIAHPwas pharmacologically isolated and characterized in voltage-clamp mode. Slices were individually transferred to small glass-bottom recording chambers and perfused continuously (∼2 ml/min) with oxygenated aCSF heated to 31°C. Our laboratory has evidence for a dorsal-ventral gradient in excitability within the hippocampus (Sametsky et al., 2001). Thus, slices equally distributed across the septotemporal axis of the hippocampus in both age groups were used for recording. During current-clamp recordings, a membrane potential of neurons was maintained at −68 mV with either hyperpolarizing or depolarizing current injection, and the AHP was evoked using a 100 msec depolarizing current step that reliably elicited a burst of four action potentials. The AHP measurements included the following: peak amplitude of the AHP (calculated as the maximum negative voltage deflection from the baseline potential during the first 250 msec after the current offset), duration of the AHP (measured as the time required for the membrane potential to return to the baseline potential for at least 10 msec after the 100 msec depolarizing current step offset), and integrated area of the AHP (calculated from the current offset for the entire duration of the AHP).
Voltage-clamp protocols. After current-clamp measurements, theIAHP–sIAHPwere isolated by bath perfusion of modified aCSF containing the following: 500 nm tetrodotoxin (TTX) to block Na+ current; 2 mmCsCl to block inward K+ currents andIh; 2 mm4-aminopyridine (4-AP) to block IA andID; 5 mmtetraethylammonium (TEA) to block ICand IM; and 100–500 μm picrotoxin, and 2 mmkynurenic acid or 10 μm6-cyano-7-nitroquinoxaline-2,3-(1H,4H)-dione (CNQX), and 25 μmd-2-amino-5-phosphonovaleric acid (D-AP5) to reduce synaptic current. 4-AP and TEA were substituted for equimolar NaCl. To block the L-type Ca2+ influx, nimodipine (dissolved in dimethyl sulfoxide) was added directly to the modified aCSF to achieve the desired concentration. Because nimodipine is light sensitive, all experiments involving nimodipine were performed in near-darkness. For all pharmacological experiments, slices were exposed to the modified medium for 10–20 min before measurements were taken.
Although both the IAHP and sIAHP coexist in CA1 hippocampus, theIAHP accounts for only 20% of the AHP in rabbit CA1 pyramidal neurons (Oh et al., 2000). The apamin-sensitiveIAHP has a relatively fast onset (1–5 msec) and a slow offset lasting between 50 and several hundred milliseconds (Sah, 1996; Stocker et al., 1999). In contrast, the decay time constant of the sIAHP is ∼1.5 sec (Sah, 1996). Therefore, we differentiated the sIAHP from theIAHP by measuring the tail current 1 sec after pulse offset.
The AHP tail current was evoked by a 100 msec, 50 mV voltage step from a holding potential of −55 mV. This protocol elicited a single, robust, unclamped Ca2+ current followed by the AHP tail current. Although the membrane voltage was not precisely controlled during the depolarizing step because of the gain and space-clamp limitations, voltage control of the AHP tail current was well maintained (Lancaster and Adams, 1986; Constanti and Sim, 1987;Sah and McLachlan, 1991; Zhang et al., 1995). The AHP tail current measurements included the following: peak amplitude, latency to peak amplitude, amplitude of the tail current at 200 msec, 1 sec, and 2 sec after pulse offset, and decay rate. The decay kinetics of the AHP current were determined by measuring the time it took for the current to decay to half of the 200 msec (half decay 200) and 800 msec amplitudes (half decay 800, which revealed the decay of the sIAHP alone). The decay time constant was also determined by fitting a single exponential to the tail current 400–3000 msec after pulse offset.
The voltage dependence and reversal potential of sIAHP were examined by holding the neurons at −55 mV, activating the sIAHP by a 100 msec, 50 mV voltage step, and then varying the holding potential of the tail current from −45 to −95 mV (see Fig. 4A). The voltage-dependence of sIAHP activation was assessed by holding the neurons at −55 mV and varying the amplitude of a 100 msec step potential (see Fig. 3A). The time dependence of sIAHP activation was assessed by holding the neurons at −55 mV and varying the duration of a 15–20 mV subthreshold activation pulse (see Fig. 3B).
Data analysis. All statistical analyses were performed using Statview (SAS Institute Inc., Cary, NC). Correlations were tested using Fisher's r to z. Age-related differences in amplitudes were tested using ANOVA, unpaired t test, or Mann–Whitney U test as appropriate. The F test was used to test for age-related differences in the variance. Data are reported as the mean ± SEM.
Drugs. KMeSO4 was purchased from ICN (Aurora, OH); TTX was from Calbiochem (San Diego, CA); d-AP5 and CNQX were from Tocris (Ellisville, MO). All other drugs were purchased from Sigma (St. Louis, MO).
RESULTS
The age-related differences in the AHP and AHP currents are summarized in Table 1. No difference was observed in the membrane resistance or membrane capacitance between the two age groups. Similar to our previous report (Moyer et al., 1992), aging neurons rested at more hyperpolarized potentials (aging = −75.9 ± 1.2 mV; young = −70.2 ± 1.4 mV;p < 0.01) and exhibited greater AHP peak amplitudes (aging = −9.03 ± 0.64 mV; young = - 7.21 ± 0.45 mV; p < 0.05) (Fig. 1) than young neurons. To control for resting membrane potential (RMP) differences, the neurons were maintained at −68 mV with either hyperpolarizing or depolarizing current injection, and the AHP was measured after a 100 msec current step that triggered four action potentials. The current used to hold the neurons at −68 mV was greater for aging neurons than for young neurons (aging = 64 ± 18 pA; young = 6 ± 15 pA; p < 0.05). Similarly, the current required to elicit four action potentials was greater for aging than for young neurons (aging = 966 ± 98 pA; young = 682 ± 52 pA; p < 0.01).
Age-related changes in the AHP and the underlying currents

Current-clamp recordings showing an aging-related enhancement of the post-burst AHP. A, Voltage traces showing representative AHPs from young and aging neurons.B, Mean AHP amplitude was greater in aging neurons than in young neurons (mean ± SEM; unpaired t test; *p < 0.05).
The current-clamp measurements of the AHP may be biased by differences in the depolarizing current pulse that may result in differences in Ca2+ influx or subtle changes in other conductances that control spike broadening. Therefore, we characterized the currents underlying the slow AHP (IAHP–sIAHP) with voltage-clamp protocols in the presence of Na+ and K+channel blockers (TTX, CsCl, 4-AP, and TEA). Recordings of the AHP current also revealed an aging-related increase in the peak current amplitude (mixedIAHP–sIAHPamplitude; aging = 604.7 ± 66.2 pA; young = 374.1 ± 36.3 pA; p < 0.01), current amplitudes at 1 and 2 sec after pulse offset (sIAHPamplitudes; 1 sec: aging = 325.9 ± 39.5 pA; young = 186.8 ± 22.8 pA; p < 0.01; 2 sec: aging = 206.2 ± 30.9 pA; young = 106.3 ± 16.4 pA; p <0.01) (Fig. 2A), and integrated area of the tail current (aging = 830.8 ± 111.2 pC; young = 486.9 ± 57.8 pC; p < 0.01). The amplitude of the AHP showed correlations to all of the amplitude measurements, as well as the integrated area, of the AHP tail current (Table 2), indicating that the enhancements in the IAHP and the sIAHP underlie the enhancement in the AHP in aging.

Voltage-clamp recordings showing an age-related enhancement of the sIAHP. A, Representative sIAHP tail currents from young and aging neurons, underlain with the voltage-clamp protocol for evoking the sIAHP tail current.B, The sIAHP amplitude is greater in aging neurons than in young neurons (mean ± SEM; unpaired t test; **p < 0.01).C, Frequency distribution of the sIAHP (1 sec amplitude) in young and aging neurons.
Correlation between current-clamp and voltage-clamp measurements
Most of the amplitude measurements of the sIAHP showed a greater variance in aging neurons. Although no differences were observed in the peak latency and the decay of the AHP tail current, the time required for the tail current to decay to half its amplitude at 800 msec after pulse offset was significantly longer in aging neurons than in young neurons (p < 0.05). Given that the apamin-insensitive, slow component of the AHP (the sIAHP) accounts for ∼80% of the total AHP in rabbit CA1 pyramidal neurons (Oh et al., 2000) and outlasts theIAHP by seconds (Sah, 1996; Stocker et al., 1999), the correlations between our current-clamp and voltage-clamp measurements suggest that the enhanced sIAHP predominantly underlies the enhanced AHP in aging neurons.
As step potentials were increased, a transient inward current appeared within a few milliseconds of pulse onset, followed by a slow outward current that continued as the AHP tail current. Increasing the amplitude of the step potential increased the amplitude of the sIAHP in both age groups. Although no clear-cut threshold of sIAHP activation could be determined, no significant tail current was observed unless the step potentials elicited an obvious Ca2+ transient (Fig.3A). Increasing the duration of the step potential also increased the amplitude of sIAHP in both age groups (Fig.3B).

Ca2+ influx and sIAHP. A, An example of the sIAHP in response to depolarizing voltage steps from −50 to −10 mV, in increments of 10 mV. For leak subtraction, 2 mV step potentials were used. Note that although increasing the amplitude of the voltage pulse increased the amplitude of the sIAHP, no significant sIAHP was observed until an obvious Ca2+ transient was elicited by the step potentials.B, Increasing the pulse duration also increased the amplitude of the sIAHP. The sIAHP was activated with depolarizing steps from −50 to −30 mV with varying durations. Voltage steps with longer durations allowed for more Ca2+ influx, thereby increasing the sIAHP.
The kinetics and voltage-dependence of the sIAHP were not different in both age groups; these values were consistent with previous recordings from young hippocampal pyramidal neurons (Lancaster and Adams, 1986). The sIAHP showed little voltage dependence from −45 to −85 mV for both age groups (Fig.4C). Likewise, its decay rate was voltage independent (Fig. 4B). In both age groups, the sIAHP reversed at approximately −94 mV, suggesting that the enhancement in the sIAHP in aging was not caused by a change in the driving force for K+.

Voltage independence and reversal potential of the AHP tail current. A, The protocol for measuring the voltage dependence of the sIAHP tail current. The sIAHP tail current was evoked by a 100 msec, 50 mV pulse from a holding potential of −55 mV. After the voltage pulse, membrane potential was stepped to various test potentials for 2 sec before returning to the −55 mV holding potential. For leak subtraction, the neurons were stepped to the test potentials for 2 sec without the 100 msec, 50 mV prepulse to elicit the sIAHP tail current. B, Representative currents evoked from the voltage-dependence protocol.C, The reversal potential was obtained by linear extrapolation of the voltage versus sIAHPamplitude at 1 sec to the voltage axis. The reversal potential of the sIAHP in this example was −90 mV. Similar to previous reports, the sIAHP conductance did not show any voltage dependence at either age (young:n = 7; aging: n = 11). The test potentials did not alter the decay rate of the current, and amplitude of the sIAHP measured at 1 sec after the various test potentials (labeled as 3s and denoted with an arrow) remained the same.
Our laboratory has reported previously that the AHP enhancement in aging is independent of the resting membrane potential difference (Moyer et al., 1992). Likewise in this study, when we compared the sIAHP from cells with comparable resting membrane potentials (−82 mV < RMP < −77 mV), aging neurons still had significantly larger sIAHP than young neurons (young = 153.63 ± 22.47 pA, n = 14; aging = 269.04 ± 34.64 pA, n = 14; p < 0.01). Thus, the difference in the resting membrane potentials did not contribute to the aging-related enhancement of the sIAHP.
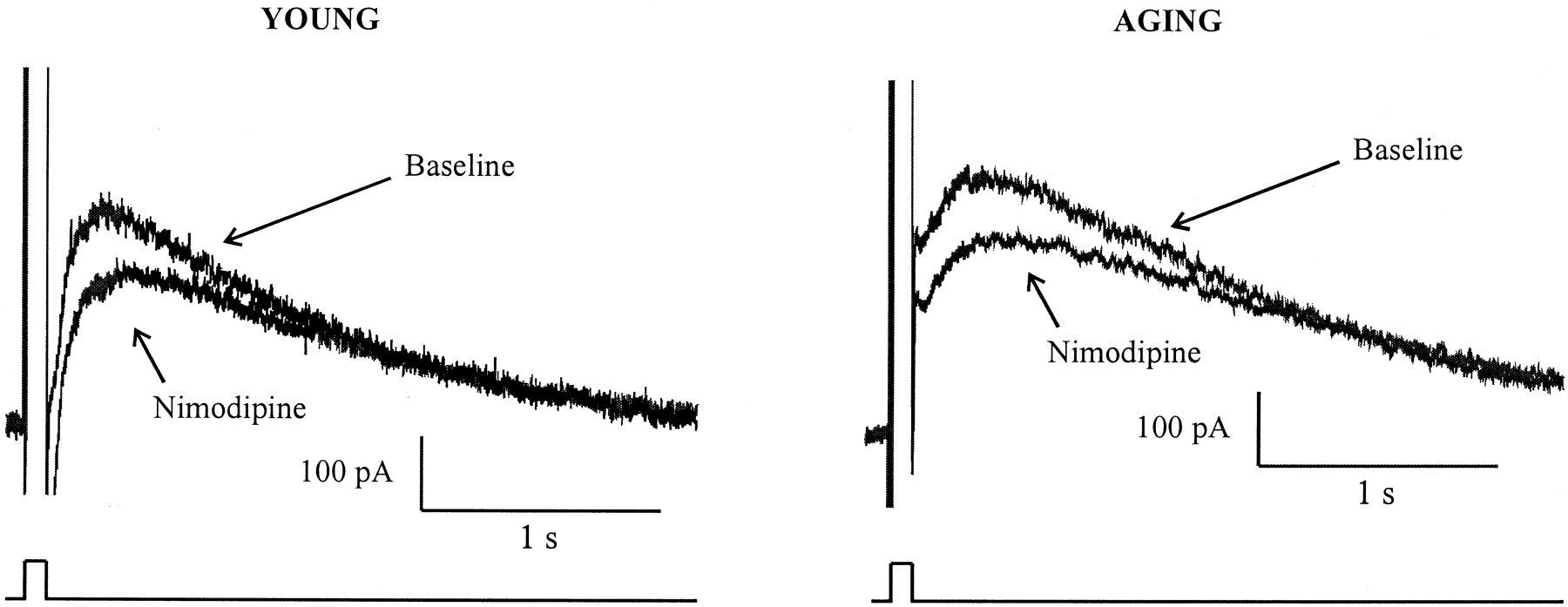
The sIAHP is a Ca2+-dependent K+ current. Thus, its amplitude is affected by the amount of Ca2+ available to cause channel activation. Previous reports have demonstrated an aging-related enhancement in voltage-gated Ca2+ influx (Landfield and Pitler, 1984;Pitler and Landfield, 1990; Moyer and Disterhoft, 1994; Campbell et al., 1996), partially attributable to an increase in the functional L-type Ca2+ channel density (Thibault and Landfield, 1996; Chen et al., 2000), in neurons from aging animals. Furthermore, the enhancements in the AHP and the plateau phase of the Ca2+ action potential in aging neurons are both reduced by nimodipine, an L-type Ca2+channel blocker (Moyer and Disterhoft, 1994), raising a possibility that the enhanced sIAHP in aging neurons is secondary to an enhanced L-type Ca2+ influx. To investigate whether age-related enhancement of the sIAHPis secondary to an enhanced L-type Ca2+influx, we examined the sIAHP before and after bath applications of nimodipine (10 μm). Aging-related enhancement of the sIAHP (p < 0.0005) was replicated and reported in Table3. Bath application of nimodipine significantly reduced the integrated area of theIAHP–sIAHP, as well as the amplitude of the sIAHP, at 1 sec after pulse offset (p <0.0001) (Fig.5). Nimodipine reduced both the area of theIAHP–sIAHPand the amplitude of the sIAHP in young and aging neurons by <25 and <30%, respectively (Fig.6A). Consistent with our previous study (Moyer and Disterhoft, 1994), nimodipine caused quantitatively greater reductions in the integrated area of theIAHP–sIAHPand the amplitude of the sIAHP in aging neurons than in young neurons (p <0.05) (Fig.6B). The residual area of theIAHP–sIAHPand the amplitude of the sIAHP were still significantly larger in aging neurons than in young neurons (amplitude for young = 117.3 ± 17.65 pA; for old, 190.0 ± 21.47 pA; p = 0.01) (Fig. 6C). Hence, our data are consistent with observations of an enhanced L-type Ca2+ influx in aging neurons. However, this enhanced L-type Ca2+ influx cannot fully account for the aging-related enhancement in the sIAHP.
Effect of nimodipine on young and aging neurons

The effect of L-type Ca2+influx on the sIAHP. Shown are representative current traces from young and aging neurons before and after bath applications of nimodipine.

A, Bath application of nimodipine caused a comparable percentage (25–30%) of reduction in the sIAHP of young and aging neurons.B, The amount of reductions in the sIAHP (1 sec amplitude) and the AHP current (integrated area) was significantly larger in aging than in young neurons (unpaired t test; *p < 0.05 and **p < 0.001, respectively;n = 31 for aging; n = 25 for young). C, After eliminating the contribution of the L-type Ca2+ influx on the sIAHP with bath-applied nimodipine, the residual sIAHP and AHP current were still significantly larger in the aging neurons than in the young neurons (unpaired t test; *p<0.05).
DISCUSSION
In hippocampal pyramidal neurons, excitability is controlled by a pronounced AHP. Based on kinetics and pharmacological criteria, the AHP can be separated into fast, medium, and slow components (for review, see Storm, 1990; Sah, 1996). In neurons from trace eyeblink conditioned animals, both the peak amplitude of the AHP and its duration are decreased (Moyer et al., 1996, 2000; Thompson et al., 1996b). In contrast, both parameters are increased in neurons from naive aging animals (Landfield and Pitler, 1984; Moyer et al., 1992). After pharmacologically isolating the slow components of the AHP (IAHP–sIAHP), we found age-related enhancements in the integrated area (mixedIAHP–sIAHP), the peak amplitude (mixedIAHP–sIAHP), and the amplitudes at 1 and 2 sec after pulse offset (sIAHP alone) for the AHP tail current. We did not pharmacologically separate theIAHP and the sIAHP. However, in CA1 pyramidal neurons from rabbits and rats, the apamin-sensitiveIAHP accounts for only a small percentage (∼20%) of the total AHP (Stocker et al., 1999; Oh et al., 2000). Furthermore, blocking IAHP with saturating concentrations of apamin had only small effects on accommodation (Oh et al., 2000). Thus by inference, the reduced neuronal excitability observed in aging neurons can mostly be attributed to an enhanced sIAHP. Consistent with this hypothesis, the amplitude measurements of the sIAHP correlated with the integrated area, as well as amplitude measurements, of the AHP, further supporting the possibility that an enhanced sIAHPunderlies an enhanced AHP in aging.
Many factors can lead to an enhanced sIAHP in aging. Increased leak conductance is not likely to be one of them, because neurons of both age groups shared similar membrane resistances. Although aging neurons rested at more hyperpolarizing potentials, the reversal potentials for the sIAHP were similar in both young and aging neurons. Furthermore, when the cells were grouped by resting membrane potentials, aging neurons still had significantly larger sIAHP than young neurons, suggesting that changes in the driving force for K+ions do not contribute to an enhanced sIAHP in aging. The slow AHP is also affected by conductances that control spike broadening, and hence the amount of Ca2+ influx, such as the A-type voltage-dependent K+ current (Giese et al., 1998). The enhanced sIAHP that we observed in aging neurons is not caused by alterations in these conductances, because all voltage-clamp recordings were performed in the presence of 4-AP and TEA.
The sIAHP is a Ca2+-dependent K+ current that is modulated by many neurotransmitters. Therefore, its amplitude also depends on (1) the amount of Ca2+ available to cause channel activation, (2) the degree of modulation, and (3) channel conductance and functional density of the sIAHPchannels. Many studies have shown altered Ca2+ homeostasis in aging neurons. Changes in voltage-gated Ca2+ influx (Landfield and Pitler, 1984; Moyer et al., 1992; Thibault and Landfield, 1996) and intracellular Ca2+ release (Martini et al., 1994), as well as an elevation in the free cytosolic Ca2+ concentration, most likely resulting from a combination of disrupted Ca2+handling, buffering, sequestration, and efflux mechanisms (Khachaturian, 1989; Disterhoft et al., 1993, 1994; Thibault et al., 1998; Verkhratsky and Toescu, 1998), have all been implicated in aging. Exactly how these changes affect the sIAHP is unclear. For example, it has been hypothesized that the age-related enhancement of the AHP is secondary to an enhanced Ca2+ influx (Pitler and Landfield, 1990), particularly that through the L-type Ca2+ channels (Campbell et al., 1996;Thibault and Landfield, 1996; Chen et al., 2000). In this study, we addressed this possibility by examining the effect of nimodipine on the sIAHP in the context of aging. Saturating concentrations of nimodipine caused quantitatively greater reductions in the sIAHP in aging neurons than in young neurons, suggesting that the contribution of the L-type Ca2+ influx to activate the sIAHP is enhanced in aging. However, after blocking the L-type Ca2+ influx, the residual sIAHP from aging neurons remained significantly larger than that of the young neurons, indicating that the enhanced L-type Ca2+influx alone cannot account for the aging-related enhancement of the sIAHP.
Although nimodipine is a highly lipophilic compound, and its diffusion through slices can be problematic, it is not likely that the enhanced sIAHP in aging neurons observed in the presence of nimodipine is caused by differences in the amount of unblocked L-type Ca2+ currents. In this study, saturating concentrations of nimodipine were applied for at least 10 min before recording. In addition, recordings were typically made from CA1 pyramidal neurons at no more than 100 μm below the surface of the slice. Previously our laboratory has shown that the L-type Ca2+ current in aging neurons can be blocked with lower concentrations of nimodipine than that of the young neuron (Moyer et al., 1992). In the unlikely event of a diffusion problem, these data suggest a greater likelihood for residual L-type Ca2+ current to exist in young neurons than in aging neurons. In such a case, the aging-related enhancement of the sIAHP that is independent of the L-type Ca2+ influx reported here would be an underestimation.
Ca2+ that activates the sIAHP also comes from intracellular Ca2+ store release through a process known as Ca2+-induced Ca2+ release (CICR) (Sah and McLachlan, 1991; Torres et al., 1995, 1996; Tanabe et al., 1998; Shah and Haylett, 2000). Whether altered CICR contributes to an enhanced sIAHP in aging remains speculative.
The amplitude of the sIAHP also depends on the degree of neuromodulation it receives. The sIAHP is reduced by metabotropic glutamate agonists (Liu et al., 1993), acetylcholine (Madison et al., 1987), serotonin (Colino and Halliwell, 1987), histamine (Haas and Greene, 1986), dopamine (Malenka and Nicoll, 1986), noradrenaline (Haas and Konnerth, 1983; Madison and Nicoll, 1986), corticotropin releasing factor (Fox and Gruol, 1993), vasoactive intestinal peptide (Haas and Gahwiler, 1992), and calcitonin gene-related peptide (Nohmi et al., 1986). Many of these molecules were shown to suppress the sIAHP through protein kinase activities (Gerber et al., 1992; Müller et al., 1992; Pedarzani and Storm 1993, 1995, 1996; Torres et al., 1995; Abdul-Ghani et al., 1996; Haug and Storm, 2000). Changes in many of these neurotransmitter systems (Luine et al., 1990; Fischer et al., 1992; Smith and Booze, 1995; Richter-Levin and Segal, 1996; Miguez et al., 1999; Stemmelin et al., 2000; Segovia et al., 2001), as well as their effector kinases (Colombo et al., 1997; Pascale et al., 1998), have been implicated in aging. Conceivably, altered neurotransmission in aging, coupled with altered kinase functions, can shift the balance between kinase and phosphatase activities that normally maintain the sIAHP (Pedarzani et al., 1998) and alter this current. The enhanced sIAHPin aging neurons that we report in this study is a postsynaptic phenomenon and does not reflect changes in basal neurotransmission, because our slices were perfused in aCSF containing TTX as well as glutamatergic and GABAergic antagonists.
Age-related changes in the functional sIAHP channel density as well as the channel properties can also affect the sIAHP. Whether these mechanisms contribute to the enhanced sIAHP in aging remains to be addressed.
Molecules that affect the sIAHP have also been implicated in other forms of plasticity. For example, kinases known to modulate the sIAHP (PKC, PKA, and calcium-calmodulin kinase II) are also important for the induction of long-term potentiation (LTP), a model for cellular mechanisms of learning and memory (for review, see Soderling and Derkach, 2000). Currently, the sIAHP channels are thought to be located on apical or basal dendrites close to the soma (Sah and Bekkers, 1996; Bekkers, 2000), although a recent study byLancaster et al. (2001) questions the basal dendritic location, suggesting a role for the sIAHP in controlling dendritic excitability and synaptic integration (LoTurco et al., 1988; Andreasen and Lambert, 1995). Consistent with this hypothesis, activation of the sIAHPdampens temporal summation of the EPSPs as well as speeds up their decay rate (Lancaster et al., 2001). Pharmacological manipulations that facilitated LTP have also been shown to reduce the AHP (Cohen et al., 1999), suggesting that the AHP and its underlying currents can serve as an adjustable gain control, variably hyperpolarizing and shunting synaptic potentials arising in the distal dendrites and controlling the induction of further plasticity (Sah and Bekkers, 1996). Accordingly, the enhanced sIAHP in aging can hamper the formation of further plastic alterations important for learning and memory (Dunwiddie et al., 1992; Sah and Bekkers, 1996; Giese et al., 2001).
The correlation between the AHP (and thus, accommodation) and learning have led us to hypothesize that the enhancement in the AHP is involved in aging-related memory deficits. Here we report that this AHP enhancement in aging is attributable to an enhanced sIAHP. In addition, the variance of the sIAHP amplitude increases with age, strikingly similar to the age-related heterogeneity of learning observed in rabbits of the same age (Thompson et al., 1996a). The data presented in this study have allowed us to refine our initial hypothesis: enhancement of the sIAHPis involved in age-related learning deficits. In support of our hypothesis, we have shown that nimodipine facilitates acquisition of the trace eyeblink conditioned response and reduces the AHP in animals (Deyo et al., 1989; Moyer et al., 1992; Kowalska and Disterhoft, 1994). In this study, we have further shown that nimodipine reduces the sIAHP in young and aging animals. Similarly, chronic treatment with metrifonate, a cholinesterase inhibitor, or CI1017, an M1 muscarinic agonist, both reduce the sIAHP and the slow AHP and facilitate trace eyeblink conditioning in aging rabbits (Kronforst-Collins et al., 1997; Oh et al., 1999; Weiss et al., 2000; Power et al., 2001). The cholinergic reduction of the sIAHP is independent of Ca2+ entry (Muller and Connor, 1991), suggesting that the cognitive benefits of procholinergic treatments are caused by a reduction of the sIAHP itself. We are unaware of experiments designed to disrupt learning by enhancing the sIAHP. However, treatment with the central cholinergic blocker scopolamine, which should eliminate cholinergic reduction of the sIAHP, disrupts trace eyeblink conditioning in young rabbits (Kaneko and Thompson, 1997). The results of the present study provide additional support for an involvement of the enhanced sIAHP in age-related learning deficits, and further suggest that key modulators of this current are good candidates for future therapeutic interventions in age-related cognitive impairments.
Footnotes
↵* J.M.P. and W.W.W. contributed equally to this study.
This work was supported by National Institutes of Health Grants AG08796 (J.F.D.), AG17139 (J.F.D.), MH11737 (M.M.O.), and MH11858 (W.W.W.). We thank Dr. David Mogul for his guidance.
Correspondence should be addressed to John F. Disterhoft, Department of Physiology, Northwestern University Medical School, 303 East Chicago Avenue, Chicago, IL 60611-3008. E-mail: jdisterhoft{at}northwestern.edu.
J. M. Power's present address: Division of Neuroscience, John Curtin School of Medical Research, Australian National University, ACT 2601, Australia.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}