


Abstract
Degeneration of dopaminergicrgic neurons in the substantia nigra of the brain is a hallmark of Parkinson's disease and inflammation and oxidative stress are closely associated with the pathogenesis of degenerative neurological disorders. Treatment of rat mesencephalic mixed neuron-glia cultures with lipopolysaccharide (LPS)-activated microglia, resident immune cells of the brain, to release proinflammatory and neurotoxic factors tumor necrosis factor-α, interleukin-1β, nitric oxide, and superoxide and subsequently caused damage to midbrain neurons, including dopaminergic neurons. The LPS-induced degeneration of the midbrain neurons was significantly reduced by cotreatment with naloxone, an opioid receptor antagonist. This study focused on understanding the mechanism of action for the protective effect of naloxone on dopaminergic neurons because of relevance to Parkinson's disease. Both naloxone and its opioid receptor inactive stereoisomer (+)-naloxone protected the dopaminergic neurons with equal potency. Naloxone inhibited LPS-induced activation of microglia and release of proinflammatory factors, and inhibition of microglia generation of superoxide free radical best correlated with the neuroprotective effect of naloxone isomers. To further delineate the site of action, naloxone was found to partially inhibit the binding of [3H]LPS to cell membranes, whereas it failed to prevent damage to dopaminergic neurons by peroxynitrite, a product of nitric oxide and superoxide. These results suggest that naloxone at least in part interferes with the binding of LPS to cell membranes to inhibit microglia activation and protect dopaminergic neurons as well as other neurons in the midbrain cultures from inflammatory damage.
During the development of neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, multiple sclerosis, AIDS dementia complex, and amyotrophic lateral sclerosis, as well as during post-traumatic brain injuries and ischemia, inflammation in the central nervous system is frequently observed (McGeer et al., 1988; Dickson et al., 1993; Raine, 1994; Matyszak, 1998). One of the major characteristics of the neuroimmune responses is the activation of the resident immune cells of the brain, the microglia, through a process termed reactive microgliosis (Dickson et al., 1993; Kreutzberg, 1996). Activated microglia secrete a variety of proinflammatory and cytotoxic factors, including nitric oxide (NO), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), arachidonic acid, eicosanoids, and reactive oxygen species (Merrill et al., 1992; Minghetti and Levi, 1998). Combinations of these glia-released factors were neurotoxic in vitro and are thought to actively participate in the progression of neurodegenerative diseases in vivo (Boje and Arora, 1992; Banati et al., 1993; Raine, 1994; Bronstein et al., 1995; Kreutzberg, 1996; Jeohn et al., 1998). More recently, it has been reported that peroxynitrite, formed from NO and superoxide, may be a more direct and significant cytotoxic intermediate (Beckman et al., 1993). However, the exact mechanisms of action responsible for the cytotoxicity for these numerous factors are still largely unknown.
The loss of the function and subsequently the integrity of a specific subset of neurons, namely, the dopaminergicrgic neurons in the substantia nigra of the brain, is the hallmark of the pathology of Parkinson's disease. Inflammation along with increased oxidative stress in the midbrain appears to precede the eventual loss of dopaminergicrgic neurons (McGeer et al., 1988; Jenner and Olanow, 1996). Therefore, reducing the severity of inflammation and decreasing the intensity of oxidative stress may help to preserve the nigral dopaminergicrgic neurons.
In the course of studying the role of the opioid system in the neuroimmune responses in the brain, we have discovered that (−)-naloxone, a nonselective opioid receptor antagonist, inhibits the lipopolysaccharide (LPS)-induced cytokine release and nitrite production in murine mixed cortical glia cultures (Das et al., 1995;Kong et al., 1997). We set out to investigate effects of naloxone on the inflammation-mediated damage to dopaminergicrgic neurons in the rat mesencephalic neuron-glia culture system. The underlying mechanism of action for the protective effects of naloxone was analyzed in relation to the activity of microglia. In this report, we show that naloxone protects dopaminergicrgic neurons from LPS-induced damage through inhibition of microglia activation and subsequent release of superoxide free radicals.
Materials and Methods
Reagents.
(−)-Naloxone hydrochloride was purchased from NEN-DuPont (Boston, MA) or Research Biochemicals International (Natick, MA). For convenience, (−)-naloxone is called naloxone in this article. The enantiomer (+)-naloxone was a generous gift from Research Triangle Institute (Research Triangle Park, NC). All cell culture ingredients were obtained from Life Technologies (Grand Island, NY). [7,8-3H]Dopamine (50 Ci/mmol) was purchased from Amersham (Arlington Heights, IL). [3H]LPS (1 μCi/μg) was purchased from List Biological Lab., Inc. (Campbell, CA). Monoclonal antibodies against the CR3 complement receptor (OX-42), neuron-specific nuclear protein (Neu-N), and microtubule-associated protein-2 (MAP-2) were obtained from Pharmingen (San Diego, CA), Chemicon (Temecula, CA), and Boehringer Mannheim (Mannheim, Germany), respectively. The polyclonal antibody against glial fibrillary acidic protein (GFAP) was from Dako (Carpinteria, CA). The polyclonal anti-tyrosine hydroxylase (TH) antibody was a gift from Glaxo-Wellcome (Research Triangle Park, NC). Vectastain ABC kit and biotinylated horse anti-mouse and goat anti-rabbit secondary antibodies were purchased from Vector Laboratories (Burlingame, CA). LPS (Escherichia coli 0111:B4) was purchased from Calbiochem (La Jolla, CA). Peroxynitrite was obtained from Upstate Biotechnology (Lake Placid, NY). All other reagents were from Sigma Chemical Co. (St. Louis, MO).
Rat Mesencephalic Neuron-Glia Cultures.
Primary mesencephalic neuron-glia cultures were prepared from the brains of embryonic day 14/15 Fischer 344 rats as previously described (Friedman and Mytilineou, 1987; Casper et al., 1991). The whole brain was removed aseptically and the mesencephalon was dissected. After removing the blood vessels and meninges, the pooled mesencephalic tissues were dissociated by mild mechanical trituration in ice-cold calcium- and magnesium-free W3 buffer (145 mM NaCl, 5.4 mM KCl, 1 mM NaH2PO4, 15 mM HEPES, and 11 mM glucose; pH 7.4). After pelleting by centrifugation, cells were resuspended and plated (7.5 × 105/0.5 ml medium/well) to 24-well cell culture plates precoated with poly(d-lysine) (20 μg/ml). The culture medium consisted of minimum essential medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 10% heat-inactivated horse serum, 1 g/l glucose, 2 mM l-glutamine, 1 mM sodium pyruvate, 100 μM nonessential amino acids, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2, 95% air. Three days later, the cultures were replenished with 0.5 ml of fresh medium. Six-day-old cultures were used for treatment. During the course of treatment, cultures were switched to fresh medium containing 2% each of heat-inactivated FBS and heat-inactivated horse serum. At the time of treatment, the total number of cells per well was 7.7 × 105 (estimated by trypan blue counting of trypsin-EDTA detached cells). The composition of major cell types in the culture was estimated by visual counting of immunostained cells (see below) with antibodies against cell-type specific markers: 40.2% neurons (Neu-N), 11.4% microglia (OX-42), and 48.4% astrocytes (GFAP). Within the population of Neu-N immunoreactive neurons, 1.0 to 1.6% were TH-positive neurons.
Primary Microglia-Enriched Cultures.
Rat microglia-enriched cultures were prepared according to a previously described protocol for mouse microglia-enriched cultures (Kong et al., 1997). Briefly, whole brains of 1-day-old pups, with the blood vessels and meninges removed, were triturated in W3 as described above. Cells (2.5 × 107) were seeded in 75-cm2culture flasks in 15 ml of a Dulbecco's modified Eagle's medium/nutrient mixture F12 mixture (1:1) containing 10% heat-inactivated FBS, 2 mM l-glutamine, 1 mM sodium pyruvate, 100 μM nonessential amino acids, 50 U/ml penicillin, and 50 μg/ml streptomycin. The cultures were maintained at 37°C in a humidified atmosphere of 5% CO2, 95% air. Medium (15 ml/flask) was replenished 1 and 4 days after the initial seeding and changed thereafter every third day. On reaching confluence (day 13 or 14), microglia were shaken off (200 rpm for 4 h on an orbital shaker), pelleted at 800g for 10 min, resuspended in fresh medium, and plated (105 cells/well) into 24-well culture plates. Twenty-four hours later, cells were ready for treatment. The enriched microglia were found to be >95% pure as determined by staining with specific markers (see below).
Immunocytochemistry.
Neurons were stained with an antibody against MAP-2, a marker for both the cell body and neurites. For enumeration, neuronal cell bodies but not the neurites were stained with an antibody against Neu-N. Dopaminergic neurons were stained with an antibody against TH, the rate-limiting enzyme in dopaminergic synthesis. Microglia were visualized by staining for the CR3 complement receptor with monoclonal antibody OX-42 and astrocytes were stained with an antibody against GFAP, an intermediate filament protein whose synthesis is restricted to astrocytes.
At the end of the treatment period, the cultures grown in wells of 24-well plates were washed once with PBS and fixed for 20 min at room temperature in 3.7% paraformaldehyde in PBS. After washing (2×) with PBS, the cultures were treated with 1% hydrogen peroxide for 10 min. The cultures were again washed (3×) with PBS and then incubated for 40 min with blocking solution (PBS containing 1% BSA, 0.4% Triton X-100, and 4% appropriate serum: normal horse serum for MAP-2, Neu-N, or OX-42 and normal goat serum for GFAP or TH staining). The cultures were then incubated overnight at 4°C with the primary antibody diluted in blocking solution with appropriate normal serum as stated above (anti-MAP-2, 1:400; anti-Neu-N, 1:2000; anti-TH, 1:20,000; OX-42, 5 μg/ml; or anti-GFAP, 1:1000). Afterward, the cells were washed (3×) for 10 min each in PBS. The cultures were then incubated for 2 h with PBS containing 0.3% Triton X-100 and appropriate biotinylated secondary antibody (MAP-2, Neu-N, or OX-42: horse anti-mouse antibody, 1:227; TH or GFAP: goat anti-rabbit antibody, 1:227). After washing (3×) with PBS, the cultures were incubated for 40 min with the Vectastain ABC reagents diluted according to the manufacturer's suggestion in PBS containing 0.3% Triton X-100. After washing (2×) with PBS, the bound complex was visualized by incubating cultures with 3,3′-diaminobenzidine and urea-hydrogen peroxide tablets (Sigma Chemical Co.) dissolved in water. Color development was terminated by removing the reagents and washing the cultures (2×) with PBS. All washing and incubation procedures were performed at room temperature unless stated otherwise and were done by placing the 24-well plates on an orbital shaker with gentle shaking. The images were analyzed with a Nikon diaphot inverted microscope and recorded onto photographic film with a 35-mm camera mounted to the microscope. For visual counting of Neu-N- or TH-positive neurons, 10 representative areas per well of the 24-well plate were counted under the microscope at 100× magnification.
High-Affinity [3H]Dopamine Uptake Assay.
Cells in each well were washed twice with 1 ml of Krebs-Ringer buffer (16 mM NaH2PO4, 16 mM Na2HPO4, 119 mM NaCl, 4.7 mM KCl, 1.8 mM CaCl2, 1.2 mM MgSO4, 1.3 mM EDTA, and 5.6 mM glucose; pH 7.4). The cells were then incubated with 25 nM [3H]dopaminergic in Krebs-Ringer buffer (0.4 ml/well) for 20 min at 37°C. Nonspecific uptake of dopaminergic was determined in parallel wells receiving both tritiated dopaminergic and 10 μM mazindol, an inhibitor of neuronal high-affinity dopaminergic uptake. Afterward, the cells were washed three times with ice-cold Krebs-Ringer buffer (1 ml/well) and lysed with 1 N NaOH (0.5 ml/well). After mixing the lysate with 15 ml of scintillation fluid (Cytoscint; ICN, Costa Mesa, CA), radioactivity was determined with a liquid scintillation counter (Tri-Carb 4000; Packard, Meriden, CT). The specific dopaminergic uptake was calculated by subtracting the amount of radioactivity observed in the presence of mazindol from that observed in the absence of mazindol.
Nitrite Assay.
The accumulation of nitrite in the culture supernatant, an indicator of the production of NO, was determined with a colorimetric assay with the Griess reagent [0.1%N-(1-naphthyl)ethylenediamine dihydrochloride, 1% sulfanilamide, and 2.5% H3PO4; Green et al., 1982]. Equal volumes of culture supernatant and Griess reagent were mixed, the mixture was incubated for 10 min at room temperature, and absorbance at 540 nm was determined with a UV Max kinetic microplate reader (Molecular Devices, Menlo Park, CA). The concentrations of nitrite in the samples were determined from a sodium nitrite standard curve.
TNF-α and IL-1β Assays.
The levels of TNF-α and IL-1β in the culture medium were determined with commercial enzyme-linked immunosorbent assay (ELISA) kits (Genzyme Diagnostics, Cambridge, MA) according to the manufacturer's instructions.
Superoxide Assay.
The amount of superoxide (O⨪2) produced was determined by measuring the superoxide dismutase (SOD)-inhibitable reduction of cytochrome c as described in Chao et al. (1994). Briefly, mesencephalic neuron-glia cultures grown in 24-well culture plates were treated with naloxone and/or LPS for the desired time intervals. Afterward, cells were detached by scraping with a rubber policeman or a brief treatment with 5 mM EDTA in saline. Cells were washed twice with Hanks' balanced salt solution (HBSS) without phenol red and 105 cells in 120 μl of HBSS with or without 600 U/ml SOD (Boehringer Mannheim) were seeded into the wells of 96-well culture plates. To each well, 80 μl of ferricytochromec (100 μM) in HBSS containing 200 nM phorbol-12-myristate-13-acetate (PMA; Calbiochem) was added. The cultures were then incubated for 90 min at 37°C and 5% CO2. Afterward, the absorbance at 550 nm was read with a SpectraMax Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA). The amount of SOD-inhibitable superoxide was calculated as described (Chao et al., 1994) with the molar extinction coefficient of cytochrome c at 550 nm (2.11 × 104 M/cm; Massey, 1959) and expressed as nanomoles per 106 cells. The generation of superoxide by microglia-enriched cultures was determined as described above except that 2.5 × 104 cells were plated into the wells of the 96-well plates. Alternatively, the amount of superoxide in the neuron-glia cultures released into the supernatant without further stimulation with PMA was determined by measuring the SOD-inhibitable reduction of cytochrome c according to a protocol described by Crapo et al. (1978). Briefly, 50 μl of culture supernatant was immediately mixed with 1 ml of 50 mM of potassium phosphate buffer (pH 7.8), 0.1 mM EDTA, 50 μM potassium cyanide, and 0.1 mM ferricytochrome c prewarmed to 37°C in a 1.5-ml cuvette. The increase in absorbance at 550 nm was continuously monitored for 3 min with a Beckman DU640 UV-visible wavelength spectrophotometer. The amount of SOD-inhibitable superoxide released was calculated as described above.
[3H]LPS Binding Assay.
Rat mesencephalic glia-neuron cultures in wells of 24-well plates were washed three times with binding medium (minimum essential medium containing 15 mM HEPES, pH 7.4). To each well was added 250 μl of binding medium containing 0.05 μCi [3H]LPS (53 ng) alone or 0.05 μCi [3H]LPS plus 3 to 50 times in excess in mass of unlabeled LPS or naloxone isomers. Cells were then incubated for 2 h at 4°C with gentle shaking. Afterward, the medium was removed and cells were washed four times with ice-cold binding buffer. Cells were lysed with 400 μl of 1 N NaOH and cell lysate was mixed with 10 ml of Ultima Gold scintillation fluid (Packard) and counted for radioactivity. Experiments were performed in triplicate and results are expressed as percentage of total binding observed with [3H]LPS alone.
Statistical Analysis.
The data are expressed as mean ± S.E. Statistical significance was assessed with an ANOVA followed by Bonferroni's t test with the StatView program (Abacus Concepts, Inc., Berkeley, CA). A value of P < .05 was considered statistically significant.
Results
LPS Treatment Impairs High-Affinity Dopamine Uptake by Mesencephalic Cultures in a Dose- and Time-Dependent Manner.
Cells were treated with concentrations of LPS ranging from 0.1 to 100 ng/ml for 9 to 48 h. Afterward, the capacity of the cultures to take up [3H]dopaminergic was examined as a functional indicator of the integrity of dopaminergicrgic neurons. A 9-h treatment with LPS did not significantly affect the ability of the cultures to take up dopaminergic (Table 1). However, a significant reduction in dopaminergic uptake by the cultures was detected at 16 h after the addition of LPS. A 50% reduction was seen with cells treated with 100 ng/ml LPS and a 32.2% reduction with 1 ng/ml LPS. The effect of LPS on dopaminergic uptake was more pronounced with longer treatments with LPS. For example, cultures treated with 0.1 ng/ml LPS for 16 h did not show a significant drop in dopaminergic uptake capacity but exhibited a 35.2% decrease by 24 h. The degree of reduction in dopaminergic uptake by cultures appeared to have reached maximum at 24 h after LPS treatment (Table 1).
Effect of naloxone on high-affinity [3H]dopaminergic uptake by midbrain neurons after LPS treatment
Naloxone Offers Significant Protection against LPS-Induced Reduction in High-Affinity Dopamine Uptake of Mesencephalic Cultures.
Pretreatment of cultures with naloxone (1 μM) significantly prevented the LPS-induced decrease in dopaminergic uptake (Table 1). The extent of the restoration by naloxone of the dopaminergicrgic uptake was both time- and dose-dependent for treatment with LPS. For example, near complete restoration was observed with cultures treated with 1 ng/ml LPS for 16 h, whereas the uptake capacity for cultures treated for 16 h with 100 ng/ml LPS was increased from 50.1 to 85% of the control level (Table 1).
Naloxone Protects Tyrosine Hydroxylase- and MAP-2-Immunoreactive Neurons against LPS-Induced Damage.
Immunocytochemical analysis with an antibody against TH revealed healthy TH-positive neurons with extensive neurites in the control rat midbrain cultures (Fig.1). Treatment of the cultures for 48 h with 100 ng/ml LPS reduced the number of TH-positive neurons to 33.4 ± 2.4% (n = 10, P < .005) of that of the control level. In addition, in the remaining TH-positive cell bodies, a marked shortening and/or a near complete loss of neurites was observed (Fig. 1). Although treatment of the cells with naloxone alone did not have any obvious effects on the number and morphology of the TH-positive neurons (Fig. 1), naloxone offered significant protection against LPS-induced damage to TH-containing neurons (Fig. 1). In cultures that were pretreated with naloxone before treatment with LPS, the number of TH-positive neurons was 70.1 ± 3.5% (n = 10, P < .005) of control level and the TH-positive neurons exhibited a morphology similar to that in the control cultures (Fig. 1).

Immunocytochemical analysis of the effect of naloxone on LPS-induced morphological changes in TH-immunoreactive neurons. Cultures were pretreated with naloxone (1 μM) for 30 min before treatment with 100 ng/ml LPS for 48 h. Cells were fixed and stained for TH as described in Materials and Methods. Scale bar, 100 μm. Images presented are from one experiment and are representative of at least three independent experiments. Healthy TH-positive neurons in the untreated cultures had extensive neurites and were numerous. Following treatment with LPS, a loss of TH-positive neurons was observed and the neurites of remaining TH-positive neurons became much shorter. Naloxone significantly prevented the LPS-induced loss of TH-positive neurons and shortening of their neurites. Naloxone alone had no obvious effect on the morphology of TH-positive neurons.
The correlation between the reduction in the function (dopaminergic uptake) and structural integrity (cell number and neurite network) observed in this study was consistent with that of our previous work (Bronstein et al., 1995). These results and those of others (Friedman and Mytilineou, 1987; Casper et al., 1991) indicated that dopaminergic uptake capacity was a reliable means to gauge the healthiness of dopaminergic neurons.
In addition to dopaminergicrgic neurons, treatment of the mesencephalic cultures also damaged other types of neurons from the midbrain. Staining the cultures with an anti-MAP-2 antibody revealed an extensive and intricate network of neurites in the untreated wells (Fig.2). Treatment of the cultures with 100 ng/ml LPS for 48 h led to a nearly complete loss of the neuritic network and a significant loss of neuronal perikarya (Fig. 2). Although naloxone (1 μM) alone had no effect on the morphology of MAP-2-positive neurons (Fig. 2), naloxone significantly prevented the damage induced by LPS as demonstrated by the apparent restoration of the neuritic network (Fig. 2). Visual counting of Neu-N-immunoreactive neurons indicated that the number of neurons per unit area (1.24 mm2) decreased from 408 ± 30 (mean ± S.E., n = 10) in the control cultures to 275 ± 17 in the cultures treated with 100 ng/ml LPS for 48 h. Treatment with naloxone significantly prevented the LPS-induced neurotoxicity such that the number of neurons per unit area increased to 309 ± 17 (0.1 μM naloxone, P < .05, compared with the LPS-treated cultures) and 322.5 ± 15 (1 μM naloxone,P < .005, compared with the LPS treated cultures), respectively.
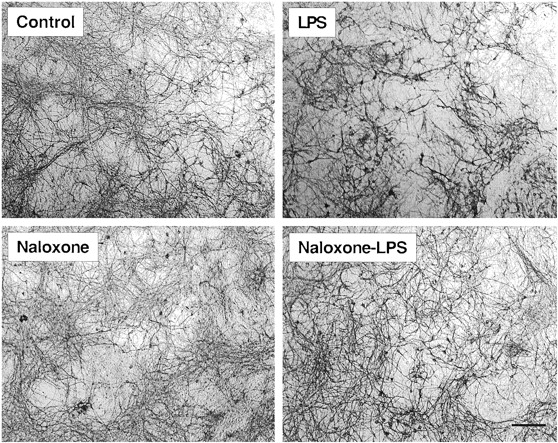
Immunocytochemical analysis of the effect of naloxone on LPS-induced morphological changes in MAP-2-immunoreactive neurons. Cultures were pretreated with naloxone (1 μM) for 30 min before treatment with 100 ng/ml LPS for 48 h. Cells were fixed and stained for MAP-2. Scale bar, 100 μm. In the untreated cultures, an extensive network of MAP-2-positive neurons was revealed. LPS severely damaged the network of the MAP-2-positive neurons and the loss of cell bodies was evident. Treatment with naloxone significantly prevented the LPS-induced damage and markedly restored the network of the MAP-2-positive neurons. Treatment with naloxone alone had no effect on the MAP-2-positive neurons. Images presented are from one experiment and are representative of at least three independent experiments.
Naloxone Stereoisomers Were Equally Effective in Reducing Endotoxin-Induced Damage to Mesencephalic Neurons.
The (−)-naloxone stereoisomer is three to four orders of magnitude more effective than its enantiomer (+)-naloxone in antagonizing the binding of ligands to opioid receptors (Iijima et al., 1978). To compare the effects of (−)-naloxone and (+)-naloxone on the endotoxin-induced damage to dopaminergicrgic neurons, mesencephalic cultures were pretreated (30 min) with either isomer before treatment with 100 ng/ml LPS for 24 h. Cultures were then assayed for high-affinity dopaminergic uptake. As shown in Fig. 3, both (−)-naloxone and (+)-naloxone offered significant protection against LPS-induced damage to dopaminergicrgic neurons. At the same concentrations (0.1 or 1 μM), the enantiomers were equally effective in protecting dopaminergicrgic neurons, whereas neither isomer by itself had any effect (Fig. 3).

Comparison between naloxone stereoisomers for their effect on LPS-induced reduction of [3H]dopaminergic uptake in midbrain cultures. Cells were pretreated with the indicated concentrations of naloxone isomers for 30 min followed by LPS treatment for 24 h. The high-affinity dopaminergic uptake assay was performed as described in Materials and Methods. The results are the mean ± S.E. of three separate experiments performed in triplicate. nal, naloxone; *P < .005, **P < .001 compared with the LPS-treated cultures.
Effect of LPS on Production of NO and Release of TNF-α and IL-1β in Mesencephalic Cultures.
The observation that both the (−)- and (+)-naloxone isomers were equally effective in preventing LPS-induced damage to dopaminergicrgic neurons led us to investigate possible mechanism of action for the neuroprotective effect of naloxone. We first examined the kinetics of LPS-induced production of NO and release of the cytokines TNF-α and IL-1β in the mesencephalic cultures. As shown in Fig.4A, treatment of the mesencephalic cultures with 0.1 to 100 ng/ml LPS for up to 24 h induced a rapid and dose-dependent accumulation of nitrite (a stable metabolite of NO) in the culture medium. The levels of nitrite increased at a faster rate between 6 and 12 h after LPS treatment than between 12 and 24 h (Fig. 4A). The accumulation of nitrite reached the half-maximal level at between 9 and 12 h for all concentrations of LPS tested. Significant nitrite accumulation was detected at 12 h for 0.1 ng/ml LPS, the lowest concentration examined, and equal levels of nitrite were observed for 10 and 100 ng/ml LPS throughout the time frame tested (Fig. 4A).

LPS-stimulated production of NO and release of TNF-α and IL-1β. Mesencephalic cultures were treated with the vehicle control (0) or the indicated concentrations (nanograms per milliliter) of LPS. Culture supernatants were removed at the indicated time intervals, and assayed for the accumulation of nitrite with the Griess reagent (A) and TNF-α or IL-1β with commercial ELISA kits. The results are the mean ± S.E. of three experiments performed in quadruplicate for A and of three experiments done in triplicate for B and C. The levels of TNF-α in the vehicle-treated control cultures and the levels of IL-1β in the vehicle- and 0.1 ng/ml LPS-treated cultures were below the detection limits of the assays. *P < .05 compared with the untreated control cultures.
Next, the LPS-stimulated release of proinflammatory cytokines was examined. Treatment of cultures with 0.1 to 100 ng/ml LPS caused a rapid rise in the level of TNF-α in the culture medium. Significant levels of TNF-α were detected in the culture medium as early as 2 h after LPS treatment and the highest levels of TNF-α were seen at the 6-h time point, with slightly reduced levels maintained for up to 24 h (Fig. 4B). Interestingly, the amount of TNF-α released by treatment with 10 ng/ml LPS was significantly higher than that with 100 ng/ml LPS, although at the 2-h time point no difference was obvious (Fig. 4B). Although the exact mechanism remains unknown, preliminary studies implied that reduced ability to release TNF-α might be due to possible cytotoxicity to microglia themselves after treatment with higher concentrations of LPS (100 ng/ml). In addition to TNF-α, LPS treatment of mesencephalic cultures caused a significant release of IL-1β that could be detected as early as 6 h after treatment (P < .05 compared with control; Fig. 4C). The levels of released IL-1β reached a maximal level at 12 h after LPS treatment and slightly lower levels were seen at the 24-h time point. In contrast to TNF-α, the levels of IL-1β induced by 10 and 100 ng/ml LPS were approximately the same (Fig. 4C).
Naloxone Prevents Activation of Microglia and Inhibits Production and Release of Proinflammatory Factors.
To examine the effects of naloxone on the LPS induced-activation of microglia and the generation and release of proinflammatory cytokines, mesencephalic cultures were pretreated with naloxone followed by treatment with LPS. Culture supernatants were taken at optimal time points for determining the levels of proinflammatory cytokines and cells were fixed and immunostained for specific markers of microglial activation. As shown in Fig. 5, immunocytochemical staining of the cultures for the expression of the microglia-specific marker protein revealed predominantly resting microglia in the control cultures. Treatment with LPS (100 ng/ml; 24 h) dramatically altered the morphology of the microglia. Almost all OX-42-positive cells became activated microglia that were characterized by intense OX-42 immunostaining, an increase in size, and changes of shape (Kreutzberg, 1996). Pretreatment with naloxone (1 μM) significantly prevented the LPS-induced activation of microglia, whereas naloxone alone had no effect on their morphology (Fig. 5). In addition to preventing the activation of microglia, naloxone significantly inhibited the production and release of proinflammatory factors. First, naloxone (1 μM) significantly inhibited the accumulation of nitrite induced by 1, 10, and 100 ng/ml LPS at the 12-h time point by 22.2, 15.6, and 15.7%, respectively (Fig. 6A). Second, the amount of TNF-α released into the culture supernatant at 4 and 6 h after treatment with 10 ng/ml LPS was reduced by naloxone (1 μM) from 9.26 ± 0.85 and 11.24 ± 1.01 to 8.05 ± 0.43 and 9.38 ± 0.67 ng/ml, respectively (P < .05; n = 6). Third, naloxone (1 μM) significantly reduced the release of IL-1β from cells treated with 1, 10, and 100 ng/ml LPS (12 h) by 33.0, 19.2, and 18.6%, respectively (Fig. 6B).
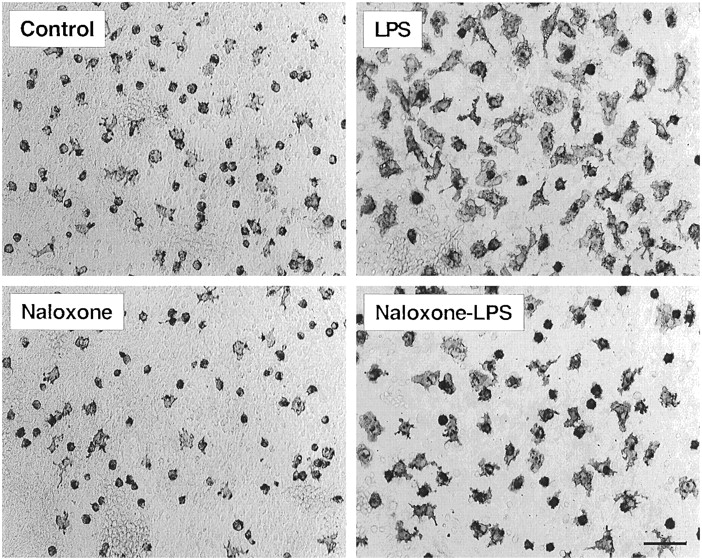
Immunocytochemical analysis for the effect of naloxone on LPS-induced morphological changes in OX-42-immunoreactive microglia. Cultures were pretreated with naloxone (1 μM) for 30 min before treatment with 100 ng/ml LPS for 24 h. Cells were fixed and stained with antibody OX-42 as described in Materials and Methods. Scale bar, 100 μm. Images presented are from one experiment and are representative of three independent experiments. In the control cultures, a significant portion of the OX-42-positive microglia were small in size. Following treatment with LPS, the microglia became activated with a greatly enlarged cell body and the characteristic shapes of activated microglia. Although naloxone alone did not significantly alter the appearance of the microglia, naloxone significantly inhibited the LPS-induced activation of microglia as demonstrated by the return of the OX-42-positive cells to the morphology of untreated cells.

Effect of naloxone on LPS-stimulated production of NO and release of IL-1β. Mesencephalic cultures were pretreated with 1 μM naloxone (Nal) for 30 min followed by treatment with the indicated concentrations of LPS for 12 h. Culture supernatants were collected and assayed for NO and IL-1β. The results are the mean ± S.E. of two to three experiments performed in triplicate. *P < .05 compared with the LPS-treated cultures.
Naloxone Significantly Reduces LPS-Induced Generation of Superoxide in Neuron-Glia Cultures and Microglia-Enriched Cultures.
Besides the release of proinflammatory cytokines and NO, LPS is known to induce the generation of oxidative free radicals that are associated with LPS-induced cytotoxicity. To study the LPS-induced generation of superoxide free radicals in the rat mesencephalic cultures, LPS-treated mesencephalic cultures (a mixture of neurons and glial cells) were reseeded into 96-well plates and challenged with the phorbol ester PMA to measure the LPS-primed, PMA-stimulated, and SOD-inhibitable reduction of ferricytochrome c. Alternatively, supernatants from LPS-treated cultures were directly used for measurement of the SOD-inhibitable reduction of ferricytochrome c by superoxide. When mesencephalic cultures were treated with 10 ng/ml LPS for 4 to 24 h, stimulated with PMA, and then measured for their ability to reduce ferricytochrome c, significant amounts of superoxide were generated as early as 9 h after LPS treatment (Fig. 7A). The amount of superoxide generated as a result of LPS (10 ng/ml) treatment peaked at 12 h and by 24 h superoxide generation appeared to have slightly subsided (Fig. 7B). The effect of naloxone on the LPS-induced generation of superoxide was, therefore, examined by measuring the superoxide generation at 12 h after LPS treatment. Pretreatment of cultures with 1 μM naloxone for 30 min before addition of LPS significantly inhibited the generation of superoxide. At the 12-h time point, naloxone (1 μM) reduced superoxide formation by 72 and 68%, respectively, in cells exposed to 10 and 100 ng/ml LPS when the amounts generated by untreated control cells were subtracted (Fig. 7B). When a series of concentrations of naloxone (0.1–1.0 μM) was evaluated, a naloxone concentration-dependent inhibition of superoxide formation was observed with an apparent EC50 of 0.7 μM (Fig.7C).

Effect of naloxone on LPS-induced generation of superoxide. Mesencephalic cultures (A and B) were treated with naloxone and/or LPS and the treated cells were further stimulated with PMA for 90 min. Superoxide generation, measured as the SOD-inhibitable reduction of ferricytochrome c, was performed as described in Materials and Methods. A, time course for superoxide generation induced by 10 ng/ml LPS. The results are the mean ± S.E. of triplicate determinations and are representative of two separate experiments. **P < .005 compared with time-matched controls. B, effect of naloxone (Nal) on LPS-induced generation of superoxide. Mesencephalic cultures were pretreated with 1 μM naloxone for 30 min followed by treatment with the indicated doses of LPS for 12 h. The results are the mean ± S.E. of two separate experiments performed in triplicate. **P< .005 compared with the LPS-treated cultures. Similar results were obtained when supernatants from LPS- and/or naloxone-treated cultures were directly assayed for reduction of ferricytochrome c. C, naloxone concentration-dependent inhibition of LPS-induced superoxide formation. Mesencephalic cultures were pretreated with indicated concentrations of naloxone for 30 min followed by treatment with the indicated doses of LPS for 12 h. The results are the mean ± S.E. of two separate experiments performed in triplicate. D, effect of naloxone on LPS-induced superoxide generation in rat microglia-enriched cultures. Microglia were prepared from the brains of 1-day-old rat pups as described in Materials and Methods. Enriched microglia (105 cells) were cultured in 24-well plates for 24 h and then treated with the indicated naloxone isomers (1 μM) followed by LPS (10 ng/ml) for 12 h. Cells were then transferred to 96-well plates and superoxide generation was determined. The data are the mean ± S.E. of two experiments performed in triplicate. **P < .005 compared with LPS-treated cultures.
The effect of naloxone on the LPS-induced generation of superoxide was further analyzed in microglia-enriched cultures. As shown in Fig. 7D, microglia-enriched cultures were particularly responsive to LPS and significantly larger quantities of superoxide were generated at the 12-h time point compared with neuron-glia cultures (Fig. 7A). Again, naloxone (1 μM) significantly inhibited the LPS-induced generation of superoxide in microglia-enriched cultures (Fig. 7D). More importantly, both naloxone stereoisomers inhibited the LPS-induced superoxide production with equal potency (Fig. 7D).
Naloxone Does Not Prevent Peroxynitrite from Damaging Dopaminergic Neurons.
To further determine the site of action for naloxone, we first tested the effect of naloxone on the cytotoxicity of peroxynitrite on dopaminergic neurons. Peroxynitrite is a downstream product through the reaction of NO and superoxide and is thought to be much more cytotoxic than NO or O⨪2 (Beckman et al., 1993). Cultures were treated with various concentrations of peroxynitrite and high-affinity dopaminergic uptake was determined 24 h later. As shown in Fig. 8, 50 μM and higher concentrations of peroxynitrite showed a concentration-dependent impairment of the function of the dopaminergicrgic neurons and a complete loss of function was observed with peroxynitrite at 500 μM. Pretreatment of cells with 1 μM naloxone followed by treatment with peroxynitrite for 24 h did not prevent peroxynitrite from damaging dopaminergic neurons (Fig. 8).

Effect of naloxone on peroxynitrite neurotoxicity in mesencephalic cultures. Mesencephalic cultures were pretreated with 1 μM naloxone for 30 min. The peroxynitrite stock solution was added directly to the cultures to achieve the indicated final concentrations. The high-affinity dopaminergic uptake of the cultures was determined 24 h after the addition of peroxynitrite. The results are the mean ± S.E. of triplicate determinations from two separate experiments. *P < .05 compared with the untreated control cultures.
Naloxone Reduces Association of [3H]LPS to Cells in Mesencephalic Neuron-Glia Cultures.
The failure of naloxone to reverse peroxynitrite cytotoxicity further suggested that the site of action for naloxone was upstream of that for peroxynitrite and was probably at the step of microglia activation. To this end, we determined the effect of naloxone on the binding of radiolabeled LPS to the cell surface of glia in the mesencephalic cultures. Cultures were incubated for 2 h at 4°C with medium containing [3H]LPS. The effect of naloxone on the binding of LPS was determined by comparing the ability to inhibit the binding of radiolabeled LPS to cells of equal quantities (3, 10, or 50× in excess in mass of radiolabeled LPS) of naloxone with unlabeled LPS. As shown in Fig. 9, unlabeled LPS reduced the binding of [3H]LPS in a concentration-dependent manner such that a 65% reduction was observed in the presence of the highest amount of unlabeled LPS (50-fold; 26.6 μM). Amounts of (−)-naloxone or (+)-naloxone equivalent to those of unlabeled LPS competed with unlabeled LPS to significantly reverse its reduction of the binding of [3H]LPS to cell membranes (Fig. 9). Significant inhibition of [3H]LPS binding also was seen with the lowest amount of naloxone used (3-fold; 1.6 μM) and no significant difference was observed in the ability to inhibit LPS binding between (−)-naloxone or (+)-naloxone (Fig. 9).

Effect of unlabeled LPS, (−)-naloxone, or (+)-naloxone on binding of [3H]LPS to cells of the mesencephalic cultures. Cells in each well of 24-well plates were incubated for 2 h at 4°C with 250 μl of medium containing 0.05 μCi [3H]LPS (53 ng) with and without amounts of unlabeled LPS, (−)-naloxone, or (+)-naloxone at 3, 10, or 50× (1.6, 5.3, and 26.6 μM, respectively) the mass of [3H]LPS. After washing off unbound [3H]LPS, cells were solubilized with 1 N NaOH and radioactivity was determined by liquid scintillation counting. The results are the mean ± S.E. of triplicate determinations from two separate experiments. *P < .05 compared with the cultures incubated with [3H]LPS only. †P < .05 compared with the cultures incubated with [3H]LPS plus corresponding amount of unlabeled LPS.
Discussion
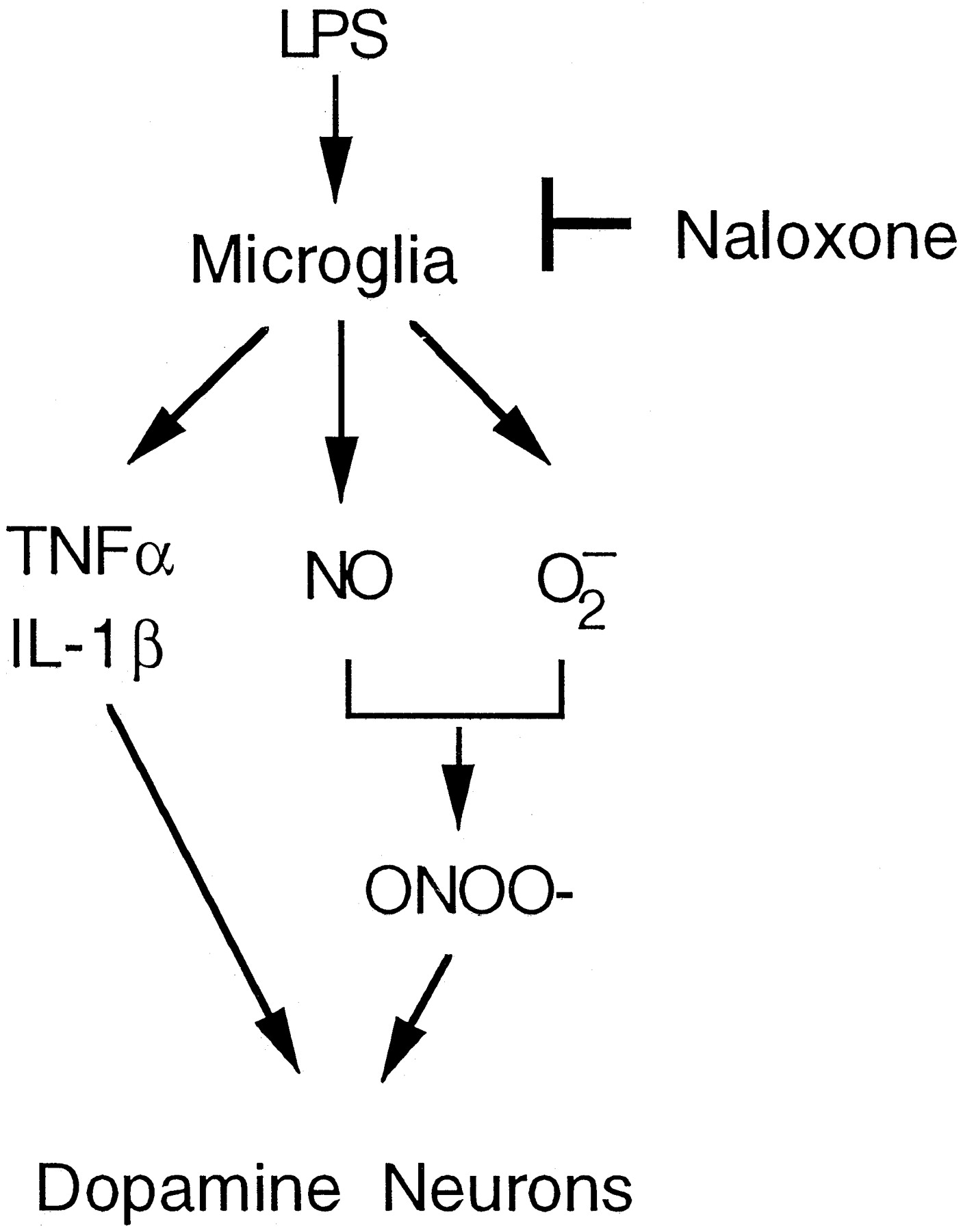
In this study with rat mesencephalic neuron-glia cultures as a model and LPS as a tool, we have demonstrated that naloxone effectively protects dopaminergic neurons from inflammatory damage. Second, both (−)-naloxone and its stereoisomer, (+)-naloxone, an ineffective opioid receptor antagonist, were equally potent. Third, naloxone significantly inhibits the activation of microglia and their release of proinflammatory and cytotoxic factors, including NO, TNF-α, and IL-1β, and most importantly superoxide free radical. Fourth, both naloxone isomers compete with LPS to bind to its cell surface receptors. These results demonstrate that the mechanism(s) of action underlying the neuroprotective effect of naloxone may be closely related to its ability to interfere with activation of microglia and their production of proinflammatory and neurotoxic factors (Fig.10).

Proposed mechanism of action for the protective effect of naloxone on LPS-induced damage to dopaminergicrgic neurons in the rat mesencephalic neuron-glia cultures.
Activation of microglia has been reported when brain glia or glia-neuron cultures are treated with LPS, the HIV-1 coat protein gp120, or β-amyloid peptides (Boje and Arora 1992; Chao et al., 1992;Dawson et al., 1993; Ii et al., 1996; Kong et al., 1997). In animal models and humans, microglia activation, a process often referred as reactive gliosis, is frequently observed during the development of neurodegenerative diseases, including Parkinson's, Alzheimer's disease, amyotrophic lateral sclerosis, and multiple sclerosis (McGeer et al., 1988; Compston, 1992; Rogers et al., 1992). Activated microglia release a wide spectrum of proinflammatory and potentially cytotoxic factors, including NO, TNF-α, IL-1β, and free radicals (Merrill et al., 1992; Minghetti and Levi, 1998). It is well accepted that proinflammatory and cytotoxic factors produced by activated microglia contribute to neurodegeneration and that prevention of microglia activation serves to reduce neuronal injury (McGeer and McGeer, 1995;Epstein, 1998).
Oxidative stress has long been thought to be closely associated with the pathology of neurodegenerative diseases (Jenner and Olanow, 1996). Overproduction of free radicals is thought to cause an imbalance in the oxidation/reduction capacity of a cell that is primarily maintained by high intracellular concentrations of glutathione. Depletion of glutathione can either render cells more susceptible to insults or result in activation of key pathways in the cells, leading to eventual cell death (Liu et al., 1998). In this study, enriched-microglia as well as neuron-glia cultures generate significant quantities of superoxide free radical in response to LPS treatment, in addition to release of TNF-α, IL-1β, and NO. Naloxone, regardless of its isomeric form, effectively inhibits the LPS-induced generation of superoxide free radical, as well as TNF-α, IL-1β, and NO. The concentration of naloxone required for half-maximal inhibition of LPS-induced superoxide generation in rat neuron-glia and microglia-enriched cultures is ∼1 μM, which is the same concentration needed for >50% protection of dopaminergicrgic neurons against LPS-induced damage (Table 1 and Fig. 3). The effective concentration range (0.1–1 μM) of naloxone in this system is significantly lower than that (1–1000 μM) required to inhibit superoxide generation in human neutrophils stimulated withN-formyl-methionyl-leucyl-phenylalanine with an ED50 of ∼12.5 μM (Simpkins et al., 1985).
In addition to being cytotoxic by themselves, superoxide and NO can form a more potent cytotoxic intermediate, peroxynitrite (Beckman et al., 1993). Formation of peroxynitrite has been detected in various injured cells or tissue as evidence for its participation in pathological processes. Peroxynitrite is clearly very toxic to rat mesencephalic neurons and naloxone does not protect against peroxynitrite-induced cytotoxicity, further supporting our hypothesis that the site of action for naloxone is upstream of peroxynitrite formation. More importantly, regardless of the role of peroxynitrite, it is clear that NO and possibly oxygen free radicals are needed for nitration of various cellular proteins (Goodwin et al., 1998). Because naloxone inhibits both the production of NO and especially the generation of superoxide in the rat mesencephalic cultures, it will be of great interest to determine the potential effect of naloxone on nitration of key cellular proteins.
The fact that naloxone inhibits microglia activation and production of proinflammatory factors but fails to prevent damage evoked by a downstream product of two of the factors released (i.e., NO and superoxide) led us to examine the possibility for naloxone to interfere with upstream events of LPS signal transduction pathways. LPS signaling across the cell membrane involves the binding of LPS to a receptor complex consisting of LPS, LPS binding protein, CD14, and one of the Toll-like receptors, leading to nuclear localization of the transcription factor nuclear factor-κB and subsequent activation of genes for proinflammatory factors (Hoffmann et al., 1999). Interference of LPS binding to cell surface receptors is a potential site of action of naloxone. Indeed, we have demonstrated that naloxone partially inhibits the binding of [3H]LPS to cell surface receptors. This result provides a possible mechanism of action for the neuroprotective effect of naloxone for dopaminergic neurons. Furthermore, this finding also offers a possible explanation for previous reports describing the beneficial effect of naloxone in the treatment of septic shocks (Holaday and Macolm, 1986; Lysle and How, 1999). Naloxone may possibly be interfering with the association of bacterial endotoxins to macrophages or other immune cells to reduce their ability to induce inflammatory and toxic responses.
Naloxone is a potent antagonist of classic opioid receptors. For instance, it has a similar affinity for μ-type opioid receptor as morphine (Knapp et al., 1995). The opioid receptor antagonistic property of naloxone is stereospecific: only (−)-naloxone is effective and the (+)-enantiomer is considered inert (Iijima et al., 1978;Marcoli et al., 1989). In animal models of stroke, myocardial and brain ischemia, and traumatic injuries of the brain and spinal cord, (−)-naloxone has been found to have significant protective effects (Hosobuchi et al., 1982; Fallis et al., 1983; Faden and Salzman, 1992;Kan et al., 1992). However, several groups have subsequently reported on the efficacy of the (+) isomer of naloxone in central and peripheral nervous systems. For example, Dunwiddie et al. (1982) found no difference in the ability of the (+) and (−) isomers of naloxone to provoke depressions of spontaneous hippocampal activity in rats.Chatterjie and associates (1996, 1998) also reported that dextro-naloxone [i.e., (+)-naloxone] effectively reduced cocaine- and amphetamine-induced hyperactivity in mice. High concentrations (0.1–3 mM) of (+)-naloxone were, in fact, slightly more effective in protecting murine cortical neurons fromN-methyl-d-aspartate-mediated neurotoxicity (Kim et al., 1987). These previous observations and this study call for further investigation into the mechanism(s) of action for these effects of naloxone.
It should be pointed out that in this study naloxone protected both dopaminergic neurons and other types of neurons against LPS-induced degeneration (Figs. 1 and 2). The seeming lack of selectivity in its neuroprotective effect may actually suggest a broader spectrum of efficacy in combating various neurodegenerative disorders. In view of its relatively low toxicity and the fact that naloxone and its analog naltrexone have long been used clinically for treating patients of drug abuse, it is highly conceivable that further research along this line may pave a new path for therapeutic intervention of neurodegenerative diseases. Furthermore, because both the (−) and (+) forms of naloxone are equipotent in neuroprotection, our findings suggest certain advantages of using (+)-naloxone over the (−) form for potential therapeutic purposes that should minimize, if not avoid, any side effects in relation to the opioid systems.
Acknowledgments
We thank Drs. J. L Maderdrut and R. Mohney for critical reading of the manuscript and R. P. Mason, B. Sturgeont, and H.-C. Kim for suggestions about the superoxide assay. We also thank B. Wilson and P. Hudson for suggestions on immunocytochemical and ELISA assays and J-W Jiang for assistance in preparing the cell cultures.
Footnotes
-
Send reprint requests to: Bin Liu, M.D., Ph.D., National Institute of Environmental Health Sciences, Laboratory of Pharmacology and Chemistry, MD: F1-01, P.O. Box 12233, Research Triangle Park, NC 27709. E-mail: liu3{at}niehs.nih.gov
-
↵1 This study was supported in part of by National Institute of Environmental Health Sciences/National Institutes of Health intramural research fund.
-
↵2 B.L. is a recipient of the National Institutes of Health Research Excellence Award.
-
↵3 L.D. is on leave from Shanghai Medical University, State Key Laboratory of Medical Neurobiology, Shanghai, China.
- Abbreviations:
- NO
- nitric oxide
- TNF-α
- tumor necrosis factor-α
- IL-1β
- interleukin-1β
- LPS
- lipopolysaccharide
- FBS
- fetal bovine serum
- OX-42
- anti-CR3 complement receptor antibody
- Neu-N
- neuron-specific nuclear protein
- MAP-2
- microtubule-associated protein-2
- GFAP
- glial fibrillary acidic protein
- TH
- tyrosine hydroxylase
- SOD
- superoxide dismutase
- ELISA
- enzyme-linked immunosorbent assay
- HBSS
- Hanks' balanced salt solution
- PMA
- phorbol-12-myristate-13-acetate
- Received October 5, 1999.
- Accepted January 14, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}