


Abstract
Interleukin-1β (IL-1β), a polypeptide immune mediator, is induced within the central nervous system in response to a variety of pathological stimuli, including systemic infection, hypoxia, brain trauma, and seizure. IL-1β action on the γ-aminobutyric acid type A (GABAA) inhibitory neurotransmitter receptor was investigated in whole cell patch-clamped cultured hippocampal neurons. Application of IL-1β at concentrations encountered in pathophysiological conditions (1–10 ng/ml; 59–590 pM) irreversibly decreased the peak magnitude of current elicited by 30 μM GABA. Current inhibition was IL-1β concentration- and time-dependent and was prevented by a specific IL-1β type I receptor antagonist. No significant changes in current kinetics or reversal potential were observed. The IL-1β depression of GABA current was inhibited by high concentrations of nonspecific kinase inhibitors staurosporine (500 nM) and 1-(5-isoquinolinyl-sulfonyl)-2-methylpiperazine (H-7; 50 μM), but not by a protein kinase C selective inhibitor calphostin C (5 μM). We conclude that IL-1β inhibits GABAA receptor function in hippocampal neurons by the involvement of an unidentified kinase. This blockade of the GABAA inhibitory neurotransmitter receptor may underlie the central nervous system hyperexcitability seen in many pathophysiological conditions.
The inflammatory cytokine interleukin-1β (IL-1β) is a 17-kDa polypeptide with a broad spectrum of biological function. Although initially identified as an immunomodulatory molecule that was limited to the peripheral immune cells, it has become clear that IL-1β plays a significant role in the central nervous system (for a review, seeRothwell, 1998). The basal level of brain IL-1β mRNA is low but detectable in the hippocampus and in specific hypothalamic nuclei. Systemic administration of bacterial endotoxin, acute brain injury by focal cerebral ischemia or excitotoxin-induced seizure, and restraint of the animal in itself have been reported to induce brain IL-1β production (Rothwell and Luheshi, 1994). A potential role of IL-1β in neurodegenerative diseases is suggested by an increased amount of this cytokine in the cerebral spinal fluid from patients with acquired immunodeficiency syndrome, Alzheimer's disease, Parkinson's disease, and Down's syndrome (Hopkins and Rothwell, 1995; Rothwell, 1998). However, direct injection of IL-1β into a healthy brain has little effect. The same injection after ischemia enhances neuronal death in the ischemic penumbra, particularly in the striatum (Rothwell, 1998). A recent study demonstrated that long-term potentiation induced IL-1β gene expression in the hippocampus, and IL-1β was found to be critical for the maintenance, but not in the induction, of long-term potentiation (Schneider et al., 1998). At femtomolar concentrations likely to be present under normal physiological conditions, IL-1β inhibits glutamergic synaptic transmission by acting through the release of endogenous adenosine (Luk et al., 1999).
The biological effect of IL-1β is thought to be mediated exclusively through the IL-1 type I receptor (IL-1RI). After IL-1β binds to the IL-1RI, the IL-1β/IL-1RI complex binds to the IL-1 receptor accessory protein. The formation of this trimeric complex results in intracellular signal transduction. G-proteins, cAMP-dependent protein kinase (PKA), protein kinase C (PKC), ceramide, prostaglandins, and diacylglycerol have been reported to be activated directly by this complex (Bankers-Fulbright et al., 1996; Dinarello, 1997). In many systems, IL-1β phosphorylates a serine/threonine residue, although tyrosine phosphorylation also has been reported. In addition, a novel kinase tightly associated with the IL-1RI complex, interleukin-associated kinase, and its associated MyD88 adapter protein recently have been identified as a proximal mediator of IL-1β action (Cao et al., 1996; Muzio et al., 1997). Whether any of these signal transduction pathways play a role in IL-1β action on neurons remains unknown.
Cytokines, including IL-1β, interact with many neurotransmitter systems in the brain (DeSimoni and Imeri, 1998). The GABAA receptor mediates the rapid inhibitory action of GABA in the brain, and previous studies have identified IL-1β effects on the this receptor function. However, IL-1β has been reported to enhance (Miller et al., 1991; Yu and Shinnick-Gallagher, 1994; Luk et al., 1999) or depress (Pringle et al., 1996) GABAA receptor function. GABAA receptors exist as pentameric arrangements of subunit proteins, and it is well recognized that the receptor function is modulated by subunit protein phosphorylation (Moss and Smart, 1996). Both PKA- and PKC-mediated phosphorylation of a specific serine residue on the β1- and γ2-subunits decrease the magnitude and reduce the desensitization of current evoked by GABA application (Sigel et al., 1991; Kellenberger et al., 1992; Moss et al., 1992a,b). In other model systems, PKA activation prevents GABAA current run-down (Stelzer et al., 1988;Kapur and Macdonald, 1996) similar to the c-Src-mediated potentiation of current through tyrosine phosphorylation (Wan et al., 1997). Given the well established modulation of GABAA receptor function by second messengers activated by IL-1β, we investigated the effect of IL-1β on GABAA receptor-mediated current in cultured hippocampal neurons. Specifically, the possible role of intracellular kinases in the IL-1β action on neuronal GABA current was studied.
Materials and Methods
Primary Hippocampal Culture.
Postnatal 1- to 2-day-old Sprague-Dawley rat pups were decapitated and hippocampi dissected out under a binocular microscope. Meninges were removed and hippocampi minced into ∼1-mm tissue chunks in ice-cold calcium- and magnesium-free Hanks' solution. The tissue suspension was enzymatically treated with papain (1 mg/ml) in a bovine serum albumin-supplemented (200 ng/ml) Hanks' solution for 20 min at 37°C, additionally dissociated by trituration, and plated on Matrigel-coated (Collaborative Research, Bedford, MA) 35-mm tissue culture dishes at an approximate density of 1 hippocampus per dish. The cell-plating medium consisted of minimum essential media withoutl-glutamine (Sigma, St. Louis, MO) supplemented with 10% fetal calf serum (Defined serum; Hyclone, Logan, UT), 10% horse serum, and penicillin/streptomycin antibiotics. After 2 to 3 days of growth in a 5% CO2-containing humidified incubator at 37°C, the plates were treated with 15 μM cytosine arabinoside for 24 h to arrest the growth of background cells. The culture media was replaced with a growth medium (minimum essential media as above, supplemented with 5% horse serum and antibiotics only) and half the media changed every 3 to 4 days thereafter. Neurons at 7 to 9 days postplating were used for electrophysiological experiments.
Electrophysiology and Data Analysis.
Electrophysiological recordings from hippocampal neurons were obtained using the whole cell patch clamp technique. Patch electrodes were fabricated from 1.2-mm outside diameter borosilicate capillary glass (WPI, Sarasota, FL) pulled on a Flaming/Brown micropipette puller (Sutter Instruments, Novato, CA). After fire polishing and filling with an intracellular solution consisting of 4 mM NaCl, 140 mM CsCl, 1 mM CaCl2, 2 mM MgCl2, 10 mM K-EGTA, 10 mM HEPES, and 2 mM Na-ATP, titrated to pH 7.3 with CsOH, the electrode resistance was 4 to 6 MΩ before series resistance compensation. The external solution contained 140 mM NaCl, 2.8 mM KCl, 1 mM MgCl2, 3 mM CaCl2, 10 mM HEPES, and 10 mM glucose, titrated to pH 7.4 with NaOH. Tetrodotoxin (1 μM) was included in the external solution to block voltage-gated sodium channels. GABA-induced currents were recorded with an AxoPatch 200A amplifier, digitized under Clampex V6.0 control, and the magnitude of the current response was analyzed with Clampfit (Axon Instruments, Foster City, CA). The kinetic parameters were obtained by fitting a biexponential function to the activation/desensitization current record during GABA application and by fitting a monoexponential function to the deactivation portion of the record after GABA washout. In some cells, a second, faster (<10 ms) desensitization component was detected; however, this fast component on the same order time scale as the solution exchange time was not included in additional analyses. Statistical significance for the amplitude data was determined by a two-sided t test (P < .01). The statistical significance of the effect of IL-1β on kinetic parameters normalized to the predrug application control values (Table1) was determined by the Wilcoxon one-sample test (P < .01).
Kinetic parameters of GABA-activated currents in hippocampal neurons
Drug Application.
A θ-tube borosilicate glass capillary (Sutter Instruments), pulled to ∼70 μm diameter, was mounted on a Burleigh piezoelectric transducer with a PZ-150 M Amplifier-Driver (Burleigh Instruments, Fishers, NY), and the position was controlled by pCLAMP 6.0 software (Axon Instruments) via square command pulses lowpass filtered at 90 Hz (model 9002; Frequency Devices, Haverhill, MA). One orifice of the barrel continuously perfused the bath solution over the cell, whereas the second orifice contained the drug solution. Characterization of solution exchange time was determined to be ∼20 ms by measuring shifts in holding current upon exchange between a control and a hypo-osmotic salt solution. One-second command steps were issued at 70-s intervals. This transduced the θ-tube ∼40 μm laterally, exposing the cell to GABA-containing solution with or without IL-1β. Thirty micromolar GABA, the approximate EC65 determined for cultured hippocampal cells, was used because we wanted to examine the possible effect of IL-1β on GABA current desensitization. Simultaneous exposure of cells to IL-1β and GABA was chosen versus a more traditional preapplication of IL-1β because the IL-1β depression of GABA-evoked current was irreversible. The gradual decline in the GABA-evoked current with the intermittent coapplication method allowed confirmation that the IL-1β action was graded, thus decreasing the likelihood of a technical artifact. Preliminary experiments confirmed that ∼15 min of intermittent IL-1β exposure attained the same level of GABA current depression as with the 2-min preapplication method of drug delivery. Rat recombinant IL-1β (Sigma) working solutions in the range of 1 to 10 ng/ml (59–590 pM) prepared daily from 10 μg/ml stock were introduced in the drug barrel of the rapid perfusion system together with GABA. All IL-1β-containing solutions were prepared with 2 mg/ml bovine serum albumin to reduce adsorption of the peptide ligand to tubing. In antagonist experiments, IL-1 receptor antagonist (IL-1Ra; R&D Systems, Minneapolis, MN) was introduced in the wash barrel of the rapid perfusion system along with IL-1β. Experiments that attempted to block intracellular kinase activity were accomplished by including 50 μM 1-(5-isoquinolinyl-sulfonyl)-2-methylpiperazine (H-7), 500 nM staurosporine, 20 nM PKA inhibitor (6-22; Calbiochem, San Diego, CA), or 5 μM calphostin (Biomol Lab, Plymouth Meeting, PA) in the internal solution. Calphostin C was exposed to room light before loading the pipette to ensure full activation of the light-dependent anti-PKC pharmacological action (Bruns et al., 1991).
Immunohistochemistry.
Cultured neurons were fixed for 10 min with 4% paraformaldehyde in PBS, blocked in PBST (PBS with 0.2% Tween 20) with 10% normal goat serum (NGS) for 15 min, washed and incubated in PBST with 2% NGS containing anti-rabbit IL-1RI polyclonal IgG antibody (4 μg/ml; Santa Cruz Biotechnology, La Jolla, CA) overnight at 4°C. The next day, cells were washed with three changes of 2% NGS in PBST, then incubated in biotinylated anti-rabbit IgG for 20 min followed by incubation in streptavidin-conjugated horseradish peroxidase for 20 min following the manufacturer's recommended protocol (DAKO LSAB peroxidase kit; DAKO Corp, Carpinteria, CA). Controls lacking incubation in primary antibody were performed concurrently. The stained cells were visualized under bright field microscopy, and the images were captured with a frame grabber.
Western Blot.
The indicated tissue/cells were lysed in NP-40 lysis buffer (10 mM Tris, pH 7.5, 50 mM NaCl, 30 mM sodium pyrophosphate, 50 mM NaF, and 1% NP-40) on ice for 30 min. After centrifugation, the supernatant was removed, and the protein amount was quantified by using Coomassie protein quantitation reagent (Pierce, Rockford, IL). Ten micrograms of total protein were run on a 10% SDS acrylamide gel, transferred overnight to a nitrocellulose membrane, blocked in 5% milk in Tris-buffered saline/0.2% Tween 20 and probed with the primary antibodies [anti-IL-1R antibody (1:500) and anti-GAPDH antibody (1:5000)] for 2 h at room temperature, washed three times, then probed by the secondary antibodies [anti-rabbit horseradish peroxidase (1:5000) and anti-mouse horseradish peroxidase (1:2500)] for 1 h at room temperature. Protein bands were visualized after reacting with SuperSignal chemiluminescent substrate (Pierce) followed by exposure on an X-ray film. For the blocking peptide experiment, anti-IL-1R antibody was preincubated for 1 h with a 5-fold excess of the specific blocking peptide or a nonspecific blocking peptide (specific for PKC-γ) before probing the membrane. Both blocking peptides were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Results
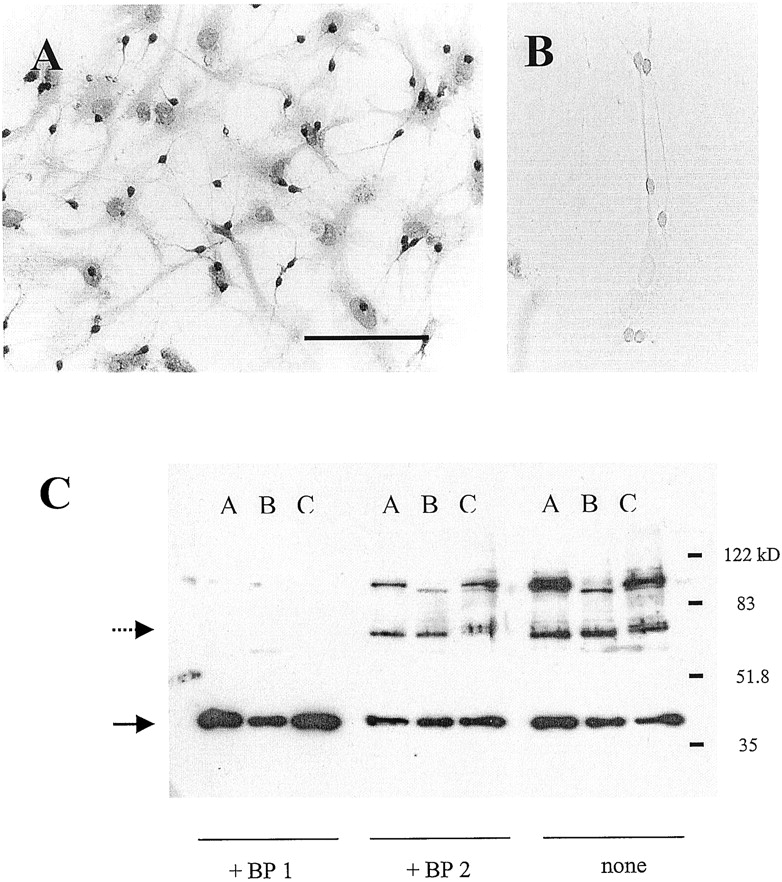
Immunohistochemical studies have documented the presence of IL-1R in the brain, although receptor localization at the cellular level has been controversial. Because the effect of IL-1β in cultured hippocampal neurons was the focus of this study, we first sought to demonstrate the presence of IL-1R in this preparation. Figure1A shows an 8-day in vitro mixed glia-neuron hippocampal culture immunostained with IL-1R-specific antibody. Extensive staining of the punctate neuronal soma and the neurites can be observed. Less dense but still prominent staining of the background glial cells can be observed as well. Control experiments omitting the primary anti-IL-1R antibody resulted in no background staining in a sister culture (Fig. 1B). Western blot of a hippocampal culture reveals a blocking peptide-sensitive signal approximately 80 kDa in mass, confirming the presence of IL-1RI in cultured neurons (Fig. 1C). An identical but weaker signal can be detected from a glial culture consistent with our immunohistochemical data.

Immunohistochemical and Western blot detection of IL-1RIs in postnatal hippocampal culture. Immunohistochemical detection of IL-1RI with (A) or without (B) the primary antibody. Note the presence of staining in background glial cells, although not nearly as pronounced as in the neurons. Calibration bar, 200 μm. C, a Western blot for IL-1RI from membranes prepared from hippocampal culture (lane A), glial culture (lane B), and adult hippocampus (lane C). Replicate membranes were probed in the presence of an IL-1RI-specific blocking peptide (BP1; left), a nonspecific blocking peptide (BP2; middle panel), or with no blocking peptide (right). The identity of the larger molecular mass band is unknown, but it appears to be specific because preincubation with the BP1 blocking peptide eliminates the signal. The dotted and solid arrows denote the expected size of the IL-1RI and the GAPDH protein band, respectively.
Rapid perfusion of GABA (30 μM) resulted in a robust inward current (ECl = 0 mV) in cells held at −60 mV (Fig.2A). The current reached a peak and desensitized during GABA application and rapidly deactivated on washout of GABA. With Mg2+ and ATP present and extensive calcium buffering by EGTA in the pipette solution, the current run-down was approximately 15% over the time course of 15 min for a typical experimental run. With the same patch pipette solution, concurrent application of IL-1β with GABA pulses significantly decreased the peak current magnitude. This decrease in the peak current magnitude was time- and IL-1β concentration-dependent (Fig. 2B). Eighteen minutes after the start of IL-1β application, the inhibition of peak current magnitude was 33 ± 11, 42.7 ± 7, and 79 ± 5% of control for 1, 3, and 10 ng/ml IL-1β, respectively (Fig. 2C). This depression of the GABA-evoked current was not reversible even after extensive 1 h washout with IL-1β-free solution (data not shown). Currents evoked by GABA applications were well described by a biexponential function corresponding to the current activation and desensitization (Fig. 2A, right trace, dashed line). A second, slower desensitization component described by others (Akaike et al., 1986; Frosch et al., 1992) was not seen at 30 μM GABA in our study. Current deactivation was monoexponential (Fig. 2A, right trace, dotted line). Although the experiments were performed on cultures 8 to 11 days in vitro and cells were selected for a typical pyramidal morphology, a wide range of kinetic parameters was obtained, perhaps reflecting a heterogeneous population of neurons. The kinetic time constants from 22 control neurons were (mean ± S.E., range): τact (37.6 ± 2.6 ms, 16.2–66.2 ms), τdesens (1.46 ± 0.16 s, 0.58–2.7s), and τdeact (179 ± 16.8 ms, 77.6–449 ms). However, for a given cell, kinetic parameters of GABA-evoked currents before and after IL-1β exposure were unchanged. The dominant effect of IL-1β was the reduction in current magnitude without changes in current kinetics (Table 1). That the decrease in the evoked current magnitude was not due to a shift in the channel ion selectivity was verified by examining the current-voltage relationship and by estimation of the reversal potential (Erev). Under the symmetrical chloride ion concentration used in this experiment, the Erevwas near 0 mV (−1.8 ± 1.0 mV, n = 8 cells). The current-voltage relationship of IL-1β-treated cells showed a lower slope conductance but unchanged Erev (0.9 ± 1.0 mV, n = 5 cells) (Table 1).
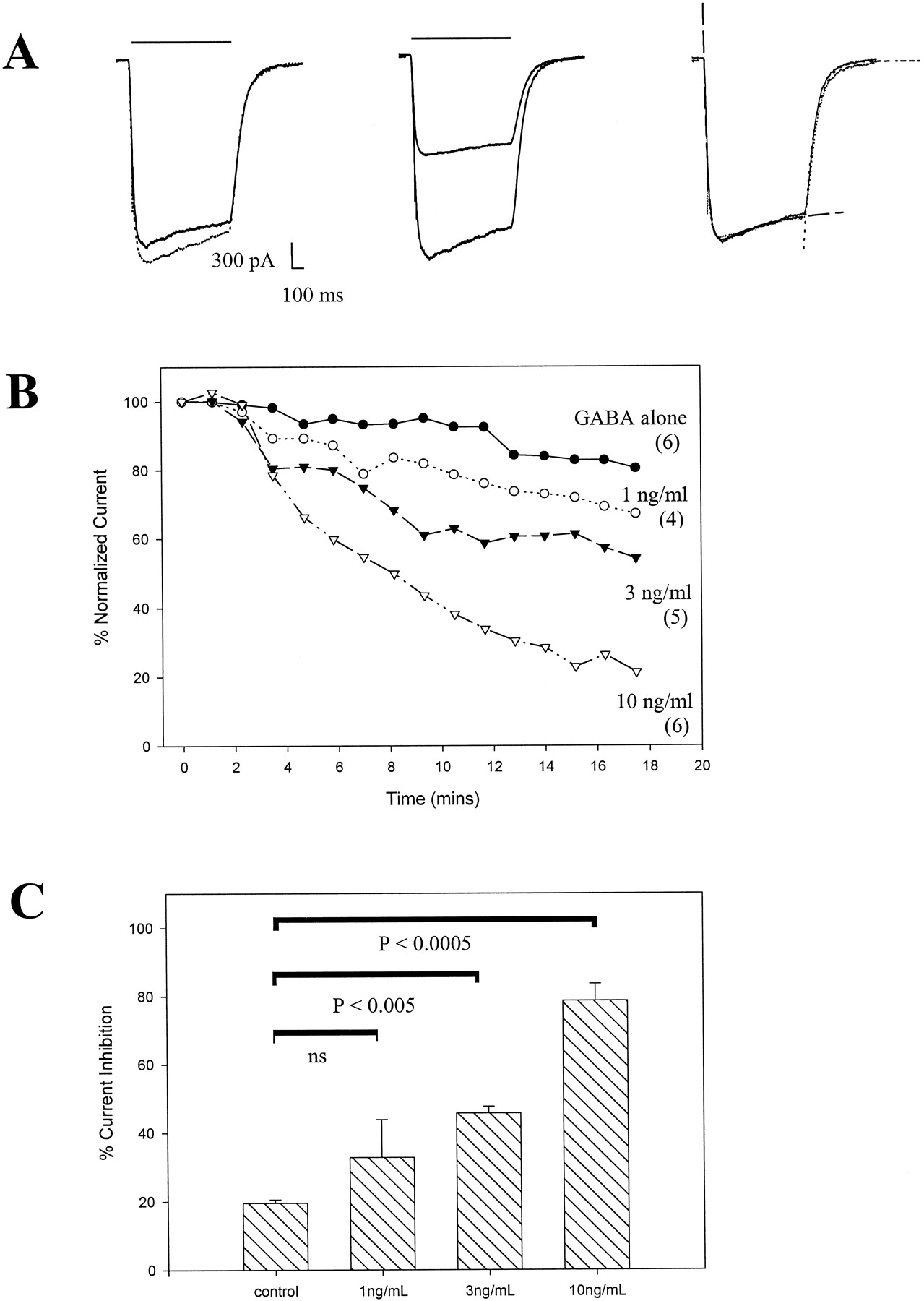
IL-1β concentration- and time-dependent irreversible decrease in GABA-evoked peak current amplitude. A, raw GABA (30 μM)-evoked current traces from whole-cell voltage-clamped neurons. Two superimposed traces corresponding to the 1st and the 16th GABA application for control (left) and a test cell (middle) (14 min after start of IL-1β coapplication) are shown. The right panel is a scaled and superimposed trace from the middle trace to illustrate the identical kinetic time course. The dashed line is a best biexponential fit to the activation/desensitization phase of the current (τact = 32 ms, τdesen = 500 ms) and the dotted line is a best monoexponential fit to the deactivation phase (τdeact = 108 ms). The calibration bar applies to both left and middle traces; the solid bar denotes the drug application duration. B, summary of the time course of peak current magnitude for control and cells exposed to three different IL-1β concentrations with the number of cells depicted within parentheses. All episodes within a given experiment were normalized to their control trace obtained before IL-1β exposure. ●, GABA alone with no IL-1β; ○, 1 ng/ml IL-1β; ▴, 3 ng/ml IL-1β); and ▵, 10 ng/ml IL-1β. C, a bar diagram summarizing percentage of current inhibition (mean ± S.E.) at approximately 16 min postcytokine exposure. The P values for a pair-wise comparison are noted, and a nonsignificant P value is denoted by ns.
The IL-1Ra is a member of the interleukin family of gene products with 26% amino acid homology with IL-1β (Dinarello, 1997). Many of the stimuli that trigger the expression of IL-1 also induce the expression of IL-1Ra; however, IL-1Ra inhibits the biological action of IL-1β in a highly specific manner. IL-1Ra, at concentrations 1 million-fold higher than IL-1β, has no agonistic activity (Granowitz et al., 1992). We coadministered IL-1Ra with IL-1β to confirm that the depression of GABA current is an IL-1R-mediated phenomenon. Figure3A shows the time course of peak current magnitude for applications of GABA alone (control), GABA with 3 ng/ml IL-1β, and GABA with the same concentration of IL-1β and 40 ng/ml IL-1Ra. The IL-1β depression of GABA-evoked current was antagonized by IL-1Ra (Fig. 3B). In the presence of IL-1Ra, the peak current magnitude depression was 8.8 ± 4.3% of control and significantly less than 42.7 ± 7% inhibition by the same concentration of IL-1β alone.

IL-1Ra blocks the IL-1β depression of GABA evoked current. A, normalized peak current magnitude for GABA-alone control (●), GABA with 3 ng/ml IL-1β (▴), and the same with 40 ng/ml IL-1Ra (○). B, summary bar diagram of percentage of current inhibition (mean ± S.E.) at 15 min after the start of drug application.
The signal transduction pathway triggered by IL-1β includes activation of PKA and PKC, both with well described inhibitory effects on the GABAA receptors (Moss and Smart, 1996). Therefore, we examined the possibility that the IL-1β inhibition of GABA current may be mediated by an intracellular kinase. First, we examined the effects of including a high concentration of nonspecific kinase inhibitors in the pipette solution. H-7 (50 μM) and staurosporine (500 nM) both prevented the IL-1β-induced time-dependent inhibition of GABA current (Fig.4). In fact, inclusion of H-7 in the internal solution resulted in a potentiation of current magnitude even in the presence of IL-1β. Protein kinase A inhibition through intracellular application of a highly specific PKA inhibitor peptide (Glass et al., 1989) resulted in an accelerated reduction in GABA-evoked current even without IL-1β. This observation is in agreement with earlier reports, suggesting a role of PKA activation in preventing current run-down (Stelzer et al., 1988; Kapur and Macdonald, 1996), but is at odds with other reports (Moss et al., 1992b; Poisbeau et al., 1999). However, in our preparation, it was unlikely that IL-1β reduced current magnitude through activation of PKA. To specifically examine the role of PKC, calphostin C, a kinase inhibitor with greater specificity for PKC (Kobayashi et al., 1989), was used. Inclusion of calphostin (5 μM) in the pipette solution had no effect on the IL-1β inhibition (Fig. 5A). In contrast, phorbol ester [phorbol 12-myristate 13-acetate (β-PMA)] inhibition of GABA current was eliminated completely by the same concentration of calphostin, confirming the expected activity of this PKC antagonist (Fig. 5B). Overall, the pharmacological evidence in primary hippocampal neurons suggests that the IL-1β inhibition of GABA current probably involves a kinase other than PKC.

Nonspecific intracellular kinase inhibitors prevent IL-1β suppression of GABA-evoked current. A, raw current traces showing peak magnitude depression in response to 3 ng/ml IL-1β in cells patch-clamped with a control standard internal solution (top) or with internal solutions supplemented with 500 nM staurosporine (left) and 50 μM H-7 (right). The two superimposed traces are from the 1st and 10th coapplication of GABA and IL-1β (3 ng/ml). The duration of drug application is denoted by the solid bar. B, a summary graph of mean normalized current magnitude for the respective groups of GABA-alone control without IL-1β using standard (○) or PKA-inhibitory peptide containing (▪) internal solutions, or with coapplication of IL-1β using standard (▵), H-7 (●), or staurosporine (▴) internal solutions. C, a bar diagram of percentage current inhibition (mean ± S.E.) at 9 min after the start of IL-1β application.

A PKC selective inhibitor calphostin C does not prevent the action of IL-1β. A, a plot of peak current magnitude in GABA-alone control (♦), IL-1β alone (●), and IL-1β with calphostin (○). B, a similar experiment except with phorbol ester β-PMA alone (○) or β-PMA with calphostin (●). C, summary bar diagram (mean ± S.E.) for the above experiments at 11 min after initiation of drug application.
Discussion
Our data demonstrated IL-1β-mediated depression of GABA-induced current in cultured hippocampal neurons. The peak current magnitude depression was not associated with changes in current kinetics or Erev. This action of IL-1β was mediated through the IL-1RI because the specific receptor antagonist molecule, IL-1Ra, prevented the attenuation of the current. A prevalent point in all recent in vitro studies has been the requirement of excess amounts of IL-1Ra over IL-1β in various systems to block the cytokine effect. The IL-1R binding affinity for IL-1Ra has been found to be approximately 150 pM in the murine thymoma cell line EL4-6.1, which is equal to the binding affinity for IL-1β (Dripps et al., 1991). However, to block effectively IL-1β action, the ratio of IL-1Ra to IL-1β ranges from ∼10 in rheumatoid synovial cells to >500 for an IL-1β-dependent cell line (Arend et al., 1998). Such a large variation in antagonist sensitivity observed in different systems may be due to the different amounts of nonsignal-transducing IL-1 type II receptors, which could act as a sink for the antagonist. In our cultured hippocampal neurons, a 13-fold excess of IL-1Ra effectively blocked the action of IL-1β.
Results of previous studies of IL-1β on GABA-induced current have been mixed. Miller et al. (1991) demonstrated IL-1β enhancement of GABA-dependent chloride uptake in mice cortical synaptoneurosomes. In the same study, IL-1β was shown to produce a long-lasting potentiation of GABA-induced current in patch-clamped cultured chick cortical neurons. Yu and Shinnick-Gallagher (1994), using a basolateral amygdala brain slice, observed a bicuculline-sensitive hyperpolarization of the cell membrane potential and concluded that IL-1β augments the action of endogenous (spontaneously released) GABA. However, IL-1β had no effect on exogenously applied muscimol in slice neurons, unlike the clear potentiation observed in chick cortical neurons. IL-1β reduced the duration of GABAAreceptor-mediated inhibition of spontaneous action potential firing rate in a cerebellar brain slice (Pringle et al., 1996). This reduction of GABA-mediated inhibition was observed at IL-1β concentrations similar to that used in our study. However, the effect observed by Pringle et al. was reversible after 15 min of washout in contrast to our irreversible inhibition of GABA-gated current. Reasons for the irreversible action of IL-1β observed in our preparation remain unclear, but it appears to be another manifestation of preparation-dependent effects of IL-1β. Irreversibility due to excess IL-1β concentration was unlikely, because even at 1 ng/ml, where GABA current inhibition was minimal, the effect was irreversible. Such diverse effects of IL-1β on GABAAreceptors may be due to a true cell-specific variability in its action. In a recent report, Zeise et al. (1997), using sharp electrode recordings from a hippocampal slice, demonstrated that bath application of IL-1β showed a specific decrease in inhibitory postsynaptic potential amplitude in hippocampal CA3 neurons with no effect on the dentate granule cells.
The reason for the apparent cell-specific action of IL-1β may be, in part, due to the variation in signal transduction initiated by ligand binding to the IL-1RI and, in part, due to the heterogeneity of GABAA receptors expressed in different neurons. IL-1β-triggered activation of PKC in the inhibition of voltage-gated calcium current in hippocampal neurons (Plata-Salaman and ffrench-Mullen, 1994), ceramide-mediated inhibition of L-type calcium current in ventricular myocytes (Schreur and Liu, 1997), and lipid peroxidation-mediated inhibition of long-term potentiation in hippocampal slices (Murray and Lynch, 1998) have been described. In addition, essentially all known signal transduction mechanisms have been reported to be initiated by IL-1β in different tissues (Bankers-Fulbright et al., 1996). Which of these multiple signal transduction pathways is activated by IL-1β in neurons and whether certain pathways are selectively activated in certain populations of neurons are not known.
An additional reason for divergence in IL-1β action on neuronal GABAA receptors is the fact that neuronal GABAA receptors are heterogeneous. Functional GABAA receptors are pentaoligomeric assemblies of different receptor subunits, and the distribution of different receptor subunits vary depending on the areas of the brain. Even within a well defined anatomical region such as the hippocampus, in situ hybridization and immunoprecipitation experiments have demonstrated the presence of multiple receptor subunits (reviewed in Rabow et al., 1995). Consensus sequences for PKA, PKC, and tyrosine kinase can be found on the intracellular loop region of most receptor subunits and protein phosphorylation of the β1- and β2-subunits by PKA, β1 phosphorylation by PKC, and γ2L phosphorylation by tyrosine kinase have been demonstrated directly by phosphopeptide mapping (Moss and Smart, 1996). The presence of various kinase consensus phosphorylation sequences on different subunits vary and because different GABAA receptor isoforms consist of different subunit combinations, action of even a specific kinase on hippocampal neurons could be variable. A recent report suggests that the basal phosphorylation status of GABAA receptors and consequently the action of kinases differ between the pyramidal and dentate gyrus neurons of the hippocampus (Poisbeau et al., 1999). It is likely that any phosphorylation-mediated modulation of GABAA receptor function in any neuronal population depends on the precise receptor subunit composition and the basal level of receptor phosphorylation. In this respect, the consistent depression of GABA current observed in our dissociated hippocampal culture preparation is puzzling. Because our neuronal population is clearly heterogeneous, if the cell population-specific action of IL-1β observed in a brain slice preparation persisted in culture, GABA current potentiation in some neurons and inhibition in others might have been observed. The unidirectional effect of IL-1β observed in our study may be due to a selection bias because larger pyramidal neurons are preferentially selected at the time of electrophysiological recording. Alternatively, neurons in culture exist in a physiologically different state from neurons in a brain slice.
In our experiments, inclusion of protein kinase inhibitors H-7 or staurosporine prevented the IL-1β attenuation of GABA-induced current. The same experimental protocol was used by Plata-Salaman and ffrench-Mullen (1994) in their study of IL-1β inhibition of voltage-gated calcium current in hippocampal neurons. They concluded that PKC was likely the kinase mediating the IL-1β action. Although the half-inhibitory concentration (Ki) value of H-7 for PKC is 6.0 μM, and that of staurosporine is 1 to 3 nM (Hidaka and Kobayashi, 1992), Kivalues for other kinases such as PKA and cGMP-dependent kinase are not significantly different (Hidaka and Kobayashi, 1992). Furthermore, H-7 and staurosporine inhibition of newer IL-1R-associated kinase is not known. Therefore, H-7 and staurosporine, at least at high concentrations, are better regarded as nonspecific kinase inhibitors. The role of PKA activation in mediating the IL-1β action was excluded because PKA inhibition, rather than activation, decreased the GABA current magnitude in our preparation. Our results, which show that intracellular application of calphostin C, a selective PKC inhibitor, did not prevent the IL-1β action, support the idea that a kinase other than PKC is likely the signal transduction molecule. Our kinetic analysis gives additional support for non-PKA- or non-PKC-mediated attenuation of GABA current magnitude. IL-1β failed to alter current kinetics. Both PKA- and PKC-mediated decreases in GABA current magnitude are accompanied by slowing of desensitization (Sigel et al., 1991; Kellenberger et al., 1992; Moss et al., 1992b), although the effect on both current magnitude and desensitization may depend on the method used to modulate the kinase activity (Krishek et al., 1994; Kapur and Macdonald, 1996; Lin et al., 1996). IL-1β may reduce the total number of activatable surface receptors, resulting in a reduced current magnitude without a change in kinetics by enhancing receptor internalization similar to that described for PKC (Chapell et al., 1998).
Additional definition of the specific GABAAreceptor subunits involved in the IL-1β in native preparations, such as the primary hippocampal neurons, may be difficult due to receptor heterogeneity present even in a single cultured neuron (Brooks-Kayal et al., 1998). A systematic study of subunit sensitivity to IL-1β action on heterologously expressed GABAA receptors of defined subunit composition will help define the signal transduction molecules mediating cytokine modulation of GABAAreceptor function. Further studies should shed light on the complex interaction of immune mediators and the neurotransmitter receptor function, which is undoubtedly important in immune-mediated neurological diseases.
Acknowledgments
We thank Nancy Ward for technical assistance and Christine Seccombe for help with manuscript preparation.
Footnotes
-
Send reprint requests to: Dr. Jay Yang, Department of Anesthesiology, Box 604, University of Rochester Medical Center, 601 Elmwood Ave., Rochester, NY 14642. E-mail: jyang{at}anes.rochester.edu
-
1 This work was supported by National Institutes of Health Grants GM52325 (to J.Y.) and T32-DA07232 (to S.M.).
- Abbreviations:
- IL-1β
- interleukin-1β
- IL-1RI
- interleukin-1 type I receptor
- IL-1Ra
- interleukin-1 receptor antagonist
- Erev
- reversal potential
- GABA
- γ-aminobutyric acid
- GABAA
- γ-aminobutyric acid type A
- PKA
- cAMP-dependent protein kinase
- PKC
- protein kinase C
- β-PMA
- β-phorbol 12-myristate 13-acetate
- H-7
- 1-(5-isoquinolinyl-sulfonyl)-2-methylpiperazine
- PBST
- PBS with 0.2% Tween 20
- NGS
- normal goat serum
- Received August 9, 1999.
- Accepted October 12, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}